

Abstract
A recent study investigated sperm-mediated inheritance of diet induced metabolic phenotypes, reported underlying regulation of the target genes of the endogenous retroelement MERVL and the ribosomal protein genes in embryos, and suggested that the altered regulation observed may cause placentation defects which can secondarily result in abnormal metabolism. A reanalysis of available transcriptomic data however shows that MERVL targets and the developmentally altered genes are themselves enriched for metabolic pathways, thus connecting embryonic gene expression with offspring phenotypes, and providing an alternative interpretation of the reported findings. This is consistent with a similar study suggesting a contribution of embryonic transcriptional change-induced gene expression reprogramming in altered offspring metabolism.
Keywords: epigenetic inheritance, transcriptome, embryonic development, offspring phenotype, bioinformatics
Introduction
Accumulating evidence suggests that parental exposure of certain environmental factors can cause altered phenotypes in subsequent generation(s). Mechanisms underlying this unconventional mode of transmission however remain elusive. Importantly, Sharma et al. [1] demonstrated sperm-mediated transfer of phenotypic information in a mouse model of paternal Low Protein diet induced metabolic perturbations in the offspring. The transgenerational effects that characterize Sharma et al.’s mouse model include altered levels of cholesterol or cholesterol esters, saturated cardiolipins, saturated free fatty acids, and saturated and monounsaturated triacylglycerides, and altered expression of genes associated with lipid or cholesterol biosynthesis and metabolism, steroid biosynthesis, oxidation-reduction process, and insulin-like growth factor 1 (Igf1) levels [1, 2]. Upon finding altered small RNA (sRNA) populations including increased levels of 5′ tRNA fragment (tRF)-Gly-GCC in sperm obtained from males reared on Low Protein diet, termed henceforth as Low Protein sperm, the authors examined the transcriptomic effect of antisense oligos targeting the tRF on embryonic stem cells and embryos, and obtained evidence suggesting tRF-Gly-GCC regulation of the target genes of the endogenous retroelement MERVL. This evidence was further supported by RNA sequencing (RNA-seq) analysis of embryos cultured to various stages of development following in vitro fertilization (IVF) with Control or Low Protein sperm, intracytoplasmic sperm injection (ICSI), and embryos generated through zygotic injection of sperm small RNA populations or synthetic tRF-Gly-GCC oligos. Besides, Sharma et al. also found that altered transcripts in embryos obtained from Low Protein sperm IVF are enriched for ribosomal protein genes. The authors, instead of examining if a correlation exists between embryonic gene expression changes and offspring phenotypes, explained these findings by speculating that regulation of MERVL targets and ribosomal protein genes may lead to altered placentation which in turn can cause downstream effects on metabolism. The present reanalysis of Sharma et al.’s gene expression data however directly links the altered gene expression in the embryos with the offspring phenotypes, thus providing an alternative to the authors’ explanation.
Results
First, a gene ontology analysis of the previously identified MERVL targets [3], that formed the basis of Sharma et al.’s evidence, shows that these targets do overrepresent various processes relevant to the offspring phenotypes [1, 2] described for the mouse model (Fig. 1A). For example, the phospholipid and the glycerophospholipid metabolic process that are found enriched are children of lipid metabolic process in gene ontology. Likewise, the primary and the regulation of primary metabolic process are ancestors of lipid metabolic process. Similarly, cellular response to insulin-like growth factor stimulus is a biological process term associated with Igf1.
Figure 1:
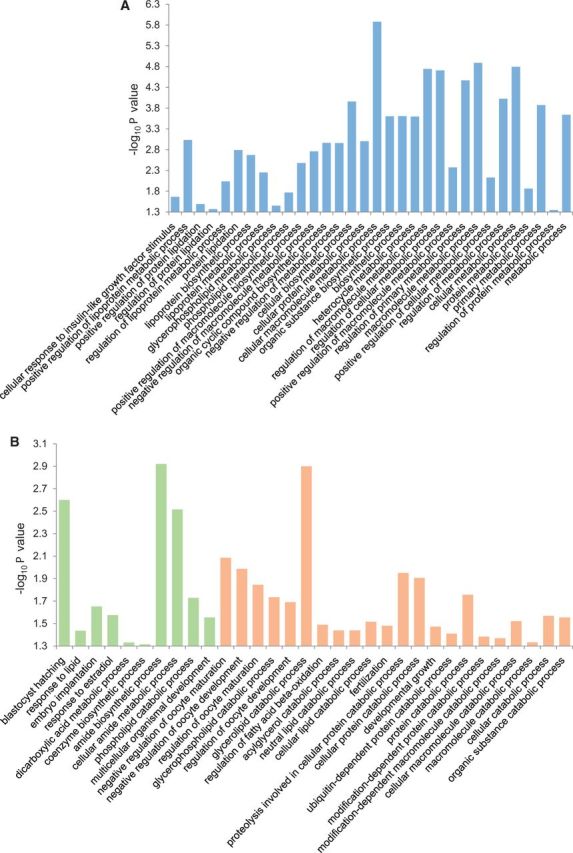
Enriched processes in (A) MERVL targets, and (B) tRF-Gly-GCC inhibition induced differentially expressed genes in embryos. Green and orange bars represent down- and up-regulated genes, in that order. Nominal significance P values (y axis, −log10) for enrichment are shown. Not all enriched processes are shown.
Second, the genes identified by Sharma et al. as differentially expressed in embryos generated following zygotic microinjection of antisense tRF-Gly-GCC oligos show enrichment of offspring phenotype related processes (Fig. 1B). For example, response to fatty acid, steroid hormone, or triglyceride are all children of the enriched process response to lipid, in gene ontology. Likewise, lipid metabolic or catabolic process is an ancestor of phospholipid, glycerophospholipid, glycerolipid, acylglycerol, neutral lipid, or cellular lipid catabolic process, and regulation of fatty acid beta-oxidation, terms that show enrichment. Similarly, response to lipid or steroid hormone is an ancestor of the enriched process response to estradiol.
Third, the genes showing expression change in embryos obtained from Low Protein sperm IVF at 2-fold cut-off, a criterion used by the authors for analyzing differential expression in embryos (Fig. 4E of Sharma et al.), enrich several gene ontology categories relevant in epigenetic inheritance of metabolic phenotypes (Fig. 2A). These categories include, for example glycerolipid or phospholipid biosynthetic process, primary metabolic process, regulation of insulin-like growth factor receptor signaling pathway, oxidation-reduction process, cholesterol metabolic process, lipid catabolic or metabolic process, positive regulation of fatty acid biosynthetic, or metabolic process, regulation of cholesterol esterification, and steroid metabolic process.
Figure 2:

Heatmap clustering of enriched processes in differentially expressed genes in (A) various stages of embryos generated through Low Protein sperm IVF, and (B) 2-cell stage embryos obtained using zygotic sperm small RNA or synthetic tRF-Gly-GCC injection, and T. sperm or C. sperm ICSI. Red, green, and black represent enriched processes in up- and down-regulated genes, and both, in that order. Grey represents no enrichment. C. sperm, cauda sperm; T. sperm, testicular sperm.
Fourth, enrichment of processes related to offpsring phenotypes is observed in the genes that show 2-fold or greater expression change in embryos generated through Low Protein sperm ICSI, zygotic injection of sperm small RNA population, or zygotic injection of synthetic tRF-Gly-GCC oligos (Fig. 2B). These processes include, for example, cardiolipin biosynthetic or metabolic process, cardiolipin acyl-chain remodeling, glycerolipid biosynthetic or catabolic process, lipid or glycerophospholipid biosynthetic process, oxidation-reduction process, negative regulation of fatty acid metabolic process, regulation of cholesterol homeostasis, response to cholesterol, and steroid biosynthetic process. Cumulatively, the above reanalysis establishes embryonic gene expression-offspring phenotype correlation.
Discussion
Like Sharma et al.’s article, a recently published paper separately reported sperm tRF mediated inheritance of diet induced metabolic disorder [4]. In this study, RNA-seq analysis of 8-cell and blastocyst stage embryos, generated through zygotic injection of tRFs obtained from sperm of males reared on High Fat diet, identified differentially expressed genes that were enriched for gene ontology processes related to metabolic regulation, besides others. It was suggested that these transcriptional changes in the embryos may lead to reprogrammed gene expression and result in altered offspring phenotypes. The present reanalysis is consistent with this hypothesis. Besides tRFs, these studies, as also others reporting recently epigenetic inheritance through the male line [5–7], also found altered levels of various small noncoding RNAs including let-7 miRNAs in sperm. Interestingly, a role of miRNAs, specifically let-7 species, in epigenetic inheritance was predicted previously on the basis of a bioinformatic analysis [8] that tested the concept [9–11] that small RNAs transfer heritable information from soma to germline and mediate transmission of environmental effects across generations through gene networks. Emerging evidence [1, 4–7] seems consistent with this model. First reported in Drosophila as late as only seven years ago [12], transgenerational spermatogenic inheritance of acquired characteristics has now been widely demonstrated in mammals. Given the current pace of discovery in epigenetics, it seems possible that a fairly reasonable understanding of the mechanisms involved in this newly discovered mode of inheritance will emerge sooner rather than later.
Methods
MERVL target genes were identified from a previous report [3], the same source that was used in Sharma et al.’s analyses. These genes, around 320 in number, were used for enrichment analysis (Fig. 1A). All other genes used in the present reanalysis relate to the single-embryo RNA-seq data presented by Sharma et al. The 72 genes identified by the authors as differentially expressed in embryos generated following zygotic microinjection of antisense tRF-Gly-GCC oligos were used for examining overrepresentation of gene ontology categories (Fig. 1B). Expression data provided by the authors for 2-cell, 4-cell, morula and blastocyst stages of Low Protein sperm IVF embryos, and for 2-cell stage of Low Protein sperm ICSI embryos, and sperm small RNA or synthetic tRF-Gly-GCC oligo injected embryos were processed further to identify differentially expressed genes at a cut-off of 2-fold change, a criterion used by the authors for identifying differential expression in single-embryo RNA-seq data (Sharma et al.’s, Fig. 4E). The average gene counts, normalized to sum of expected counts, were used to identify <0.5- or >2-fold expression in Low Protein diet associated embryos, as downregulation or upregulation, respectively. The total number of differentially expressed genes identified were around 345, 525, 610, and 585, for 2-cell, 4-cell, morula and blastocyst stages associated with IVF, and 170, 80, 45, and 370 for 2-cell stage associated with synthetic tRF-Gly-GCC oligo injection, and sperm small RNA injection, C. sperm ICSI, and T. sperm ICSI, in that order. Gene ontology enrichment analysis [13] was carried out for all differentially expressed genes (Fig. 1) or for down- and up-regulated genes (Fig. 2), with mouse genes as reference list, and all biological processes as annotation dataset, and without Bonferroni correction for multiple testing (http://geneontology.org/). A subset of enriched processes were synthetically clustered using 2.5, -2.5, 0, and blank cell values for enriched gene ontology terms in upregulated genes, downregulated genes, and both upregulated and downregulated genes, and for no enrichment, in that order (Fig. 2). City-block distance was used as similarity metric and average linkage as hierarchical clustering method, in Cluster 3.0 [14]. The results were graphically represented using Java TreeView 1.1.6r2 [15].
Supplementary data
Supplementary data is available at EnvEpig online.
Funding
The laboratory of A.S. is supported by the research grant BSC0122 of the Council of Scientific and Industrial Research, India.
Conflict of interest statement. None declared.
Supplementary Material
Supplementary Data
References
- 1. Sharma U, Conine CC, Shea JM, et al. Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science 2016;351:391–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Carone BR, Fauquier L, Habib N, et al. Paternally induced transgenerational environmental reprogramming of metabolic gene expression inmammals. Cell 2010;143:1084–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Macfarlan TS, Gifford WD, Driscoll S. et al. Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature 2012;487:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen Q, Yan M, Cao Z. et al. Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science 2016;351:397–400. [DOI] [PubMed] [Google Scholar]
- 5. Cropley JE, Eaton SA, Aiken A. et al. Grand paternal inheritance of an acquired metabolic trait induced by ancestral obesity is associated with sperm RNA. bioRxiv 2016; doi: 10.1101/042101 [DOI] [Google Scholar]
- 6. de Castro Barbosa T, Ingerslev LR, Alm PS. et al. High-fat diet reprograms the epigenome of rat spermatozoa and transgenerationally affects metabolism of the offspring. Mol Metab 2015;5:184–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schuster A, Skinner MK, Yan W. Ancestral vinclozolin exposure alters the epigenetic transgenerational inheritance of sperm small noncoding RNAs. Environ. Epigenet 2016;2:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sharma A. Systems genomics analysis centered on epigenetic inheritance supports development of a unified theory of biology. J. Exp. Biol 2015;218:3368–73. [DOI] [PubMed] [Google Scholar]
- 9. Sharma A. Transgenerational epigenetic inheritance requires a much deeper analysis. Trends Mol. Med 2015;21:269–70. [DOI] [PubMed] [Google Scholar]
- 10. Sharma A. Transgenerational epigenetic inheritance: resolving uncertainty and evolving biology. Biomol. Concepts 2015;6:87–103. [DOI] [PubMed] [Google Scholar]
- 11. Sharma A. Transgenerational epigenetic inheritance: emerging concepts and future prospects. J. Reprod. Health Med 2015;1:60–3. [Google Scholar]
- 12. Sharma A, Singh P. Detection of transgenerational spermatogenic inheritance of adult male acquired CNS gene expression characteristics using a Drosophila systems model. PLoS One 2009;4:e5763.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ashburner M, Ball CA, Blake JA. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet 2000;25:25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. de Hoon MJ, Imoto S, Nolan J. et al. Open source clustering software. Bioinformatics 2004;20:1453–54. [DOI] [PubMed] [Google Scholar]
- 15. Saldanha AJ. Java Treeview–extensible visualization of microarray data. Bioinformatics 2004;20:3246–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data