

Abstract
Phthalate esters are plasticizers that impart flexibility to polvinylchloride plastics. As they are not covalently bound, they can leach from a wide range of products, including food containers, medical devices, clothing, and toys, leading to widespread environmental exposure. Phthalate toxicity has been linked to male infertility by disrupting testosterone production and testis development. Phthalates also impair proliferation and viability of spermatogonial stem cells (SSC), the role of which is to support lifelong spermatogenesis. To elucidate cellular mechanisms in spermatogonia affected by long-term phthalate exposure, we grafted primate testis tissue into mice. Grafts treated with di-n-butyl phthalate showed an increase in autophagy compared to controls. Short term in vitro exposure of porcine germ cells to mono(2-ethylhexyl) phthalate, also resulted in an increase in autophagy. Viability was lower in cells exposed to phthalates, but treatment with rapamycin to induce autophagy significantly increased viability. The data suggests autophagy is triggered in spermatogonia as a response to a toxic insult, which may constitute a survival mechanism in spermatogonia.
Keywords: testis, germ cells, phthalates, autophagy, mammalian
Introduction
Phthalate esters are commonly used as plasticizers and additives in a wide range of products, such as medical devices, household items, and personal care products, because they confer flexibility and durability. Testosterone production and testis development are susceptible to disruption by phthalate exposure and it has been proposed [1] that the cellular targets of phthalates in the male reproductive organs include Sertoli and Leydig cells [2–4]. Phthalates are not covalently bound to the plastic matrix in which they are used, and therefore can easily leach into the surrounding environment [2, 5], causing human and animal exposure [6]. Di-n-butyl phthalate (DBP) and di-(2-ethylhexyl) phthalate (DEHP) are two of the most commonly used phthalates [2, 6, 7], and widely used in medical devices [8].
Several studies have linked phthalate toxicity with male infertility. Very few of these however, have looked into the effects on spermatogonial stem cells (SSC). Spermatogenesis is sustained by a constant supply of undifferentiated spermatogonia maintained the pool of SSCs.
Using a xenografting assay, we previously reported an indirect effect of phthalates on spermatogonia where exposure of immature Rhesus monkey testis to DEHP and DBP resulted in disturbance of the steroidogenic pathway and a reduction in Sertoli cell numbers [9]. A reduction in Sertoli cell number may have caused a reduction in numbers of spermatogonia in the phthalate exposed tissue.
Lucas et al. [4], found that mono(2-ethylhexyl) phthalate (MEHP), DEHP’s active metabolite, impairs the proliferation and viability of a mouse spermatogonia-like cell line. It was found that MEHP interferes with the GDNF signaling pathway, essential for self-renewal of SSCs, by downregulating ERK1/2 phosphorylation.
Processes maintaining SSC homeostasis and regulating response to cellular stressors, such as toxins, are not well understood. Macroautophagy (hereafter referred to as autophagy) is an evolutionarily conserved process of cellular self-degradation. Autophagy has a number of vital roles such as maintenance of the amino acid pool during starvation, antiaging, tumor suppression, regulation of innate, and adaptive immunity [10], and clearance of toxic accumulation of damaged or dysfunctional components [11, 12]. It is negatively regulated by serine/threonine kinase mTORC 1(a target of rapamycin complex 1 in mammalian cells) which is a key regulator of cellular growth and metabolism [13]. Upon induction of autophagy, a small vesicular sac called the isolation membrane or phagophore elongates and then encloses a portion of cytoplasm, to form a double membrane vesicle, the autophagosome [14, 15]. The outer membrane of the autophagosome then fuses with a lysosome, leading to the degradation of the enclosed materials together with the inner autophagosomal membrane [10]. Important for autophagosome biogenesis is LC3 (light chain 3 of microtubule-associated protein), which will generate LC3II. LC3II integrates into autophagosomes selecting cargo for degradation and the LC3B-II isoform correlates with increased levels of autophagic vesicles. LC3BII is widely used to monitor autophagy [16].
Mancilla et al. [17] investigated the importance of autophagy as an adaptative stress response in spermatogonia. They depleted glutathione in a spermatogonia-like cell line, GC-1, which stimulates autophagy and appeared to promote survival. When autophagy was blocked by bafilomycin A1, caspase 3–7 activity was increased in the glutathione depleted germ cells. Their conclusion was that autophagy promoted cell survival during stressful conditions by antagonizing apoptotic cell death and by removal of damaged organelles.
Liu et al. [18] found that tri-ortho-cresyl phosphate (TOCP), increased the level of autophagy. Thus, while TOCP reduced spermatogonial viability, and inhibited proliferation there was no effect on cell cycle progression or levels of apoptosis. Similarly, formaldehyde exposure in rats induced autophagy in the testis, and the levels of autophagy correlated with the severity of testicular injury [19]. These results suggest the autophagic cell response acts as a survival mechanism in the presence of a toxicant.
Therefore, we further investigated the hypothesis that spermatogonia undergo autophagy as a stress response mechanism by phthalate exposure of in vivo and in vitro cells.
Results
Exposure to DBP in Vivo Induced Autophagy in Primate Testis Tissue
To evaluate the effect of exposure to DBP on autophagy in germ cells in vivo, LC3BII puncta were counted in UCH-L1 positive spermatogonia in rhesus monkey tissue xenografted to mice. Treated mice received 500 mg/kg/day for 28 weeks DBP (obtained from [9]). As LC3BII associates both with the inner and outer membranes of the autophagosome, puncta are formed and then quantified to measure levels of autophagy. We observed a robust formation of puncta in all the cells of the treated tissue, compared to controls (Fig. 1A). In each of the treated (n = 4) and untreated tissue samples (n = 4), 20 UCH-L1 positive cells were identified and the LC3BII puncta counted. In vivo exposure of immature primate testis tissue to DBP in a xenograft assay resulted in higher levels of autophagy in germ cells (Fig. 1B).
Figure 1:

induction of autophagy by DBP exposure in primate testis tissue. (A) Fluorescence microscopy analysis showing LC3BII expression in DBP treated tissue in comparison to the vehicle treated control. Arrows show cells in the inserts which are UCH-L1 positive cells showing LC3BII puncta. Nuclei are stained with DAPI (blue). Scale bars = 10 μm. (B) Number of LC3BII puncta per cell. Data shown is mean ± SEM from 20 different cells from 4 different samples from each group. Comparison made by unpaired t-test. (C) Immunofluorescence showing an accumulation of P62 protein in the cytoplasm of cells in DBP exposed tissue compared to the control. Asterisks mark UCH-L1 positive germ cells with P62 positive protein aggregates in the cytoplasm. Nuclei are stained with DAPI. Scale bars= 10 μm. (D) The area of fluorescence of the p62 cytoplasmic aggregates was quantified in the UCH-L1 positive germ cells. The data shown is mean ± SEM from 20 cells from 4 samples per treatment. Comparison made by unpaired t-test.
As another measure of autophagy, p62 levels in spermatogonia were assessed using immunofluorescence. The DBP treated tissue displayed an increase in aggregated proteins, as evidenced by the accumulation of p62 in the cytoplasm (Fig. 1C) and there was a significantly greater area of p62 fluorescence in UCH-L1 positive germ cells after DBP exposure (Fig. 1D). These results indicate that DBP exposure caused protein aggregation, and that these aggregates are tagged for degradation through autophagy, which is indicative of cytotoxicity.
Increasing Concentrations of MEHP Induced Autophagy in Germ Cells In Vitro
Isolated porcine spermatogonia were exposed to MEHP concentrations ranging from 0 to 1 μM for 24 h and accumulation of LC3BII and the formation of LC3BII puncta were assayed. The experiment was repeated 3 times, and 20 cells were analyzed in each of the treatments. Fluorescence microscopy and autophagosome analysis identified more LC3BII puncta in cells treated with 0.5 and 1 μM MEHP compared to controls (Fig. 2A and B). The number of LC3BII puncta peaked at 0.5 μM MEHP, with fewer puncta observed in the 1 μM MEHP treatment group (Fig. 2B). Western blot analysis confirmed this observation, as the amount of LC3BII protein also decreased in the 1 μM MEHP treatment group compared to the 0.5 μM treatment (Fig. 2C and D). The data indicated that exposure of germ cells to MEHP at a concentration of 0.5 μM induced a marked autophagy response in germ cells.
Figure 2:

autophagy in porcine germ cells exposed to increasing concentrations of MEHP. (A) Double immunolabeling of LC3BII and VASA in porcine germ cells. LC3BII puncta were increased in MEHP exposed cells (inferior panels) compared to the control cells (control panels). Nuclei are stained with DAPI. Scale= 5 μm. (B) Number of LC3BII puncta per cell is shown, for four different MEHP concentrations. 60 cells per treatment were analyzed, and 3 independent experiments were performed. The data is mean ± SD, comparison made by one-way ANOVA. (C) Immunoblot of lysates from the porcine germ cells exposed to increasing concentrations of MEHP. (D) The relative intensity of LC3BII bands were calculated from the scanned intensities of bands of 3 independent experiments exemplified by the Western blot shown in (C). Data is mean ± SD, comparisons made by one-way ANOVA.
To quantify the autophagic flux in the cells exposed to MEHP, cells were treated with bafilomycin A1, which prevents lysosome degradation, and thus increased LC3BII puncta in cells undergoing active autophagy. As shown in Fig. 3, the cells treated with bafilomycin A1 and 0.5 μM MEHP accumulated LC3BII puncta and thus displayed a marked increase in the autophagic flux. This data confirms that autophagy is increased in germ cells exposed to MEHP.
Figure 3:
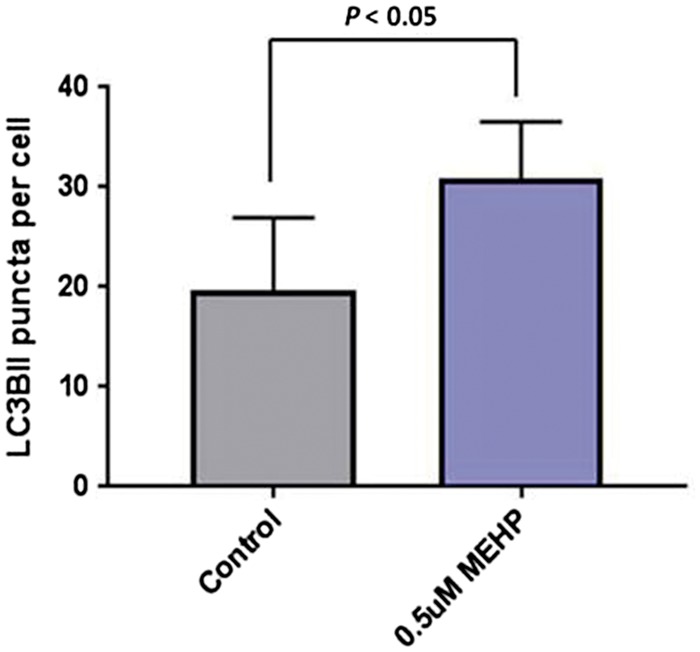
autophagic flux in porcine germ cells exposed to MEHP. Cells exposed to 0.5 μM MEHP were treated with bafilomycin A1 for 4 h previous to collection. LC3BII puncta per cell are shown. Sixty cells per treatment were analyzed from 3 independent experiments. Data shown is mean ± SEM, analyzed by unpaired t-test.
Autophagy and Cell Viability In Vitro
To quantify the effects of MEHP on cell viability and autophagy, cells were analyzed in the presence of rapamycin and wortmannin. More LC3BII puncta and higher cell viability were observed in the rapamycin treated cells compared to the controls (Table 1). Autophagy was further increased in the MEHP treated cells, while viability decreased. Viability and autophagy levels were unchanged in the cells treated with wortmannin. Therefore, as germ cells exposed to rapamycin had higher levels of autophagy and higher viability than the control, it is possible that autophagy is acting as a survival mechanism for these cells.
Table 1:
autophagy and viability in porcine germ cells in vitro
| Treatment | LC3BII puncta per germ cell (n = 60) | Cell viability (n = 3) (%) |
|---|---|---|
| Control | 6.1 ± 2.1a | 83 ± 2a |
| MEHP | 19.2 ± 3.4b | 69 ± 8.3b |
| MEHP/RAPA | 40.9 ± 7.7c | 78 ± 3.5a |
| MEHP/WORT | 11.5 ± 5.7b | 76 ± 4.1a |
| RAPA | 27.6 ± 6.3d | 90 ± 2.5c |
| WORT | 4.5 ± 2.2a | 80 ± 2.3a |
Average number of LC3BII puncta per germ cell and cell viability after 48 h MEHP exposure. Data are mean ± SD. Different letters within columns indicate significant difference (P < 0.05, one way ANOVA).
Effect of MEHP on Apoptosis in Porcine Germ Cells
To further elucidate the effects of the MEHP exposure on germ cells, cells were stained with an antibody against cleaved caspase 3, which is an early indicator of apoptosis, and VASA, a germ cell marker (Fig. 4A). As a positive control, germ cells were treated with 5 μM staurosporine, a microbial alkaloid, for 24 h. The staurosporine treatment promoted the apoptotic process in VASA positive and negative cells; there was an 81 ± 4% co-localization of cleaved caspase3/VASA in these cells (Fig. 4B). In contrast, there was no significant difference in the percentage of co-localization in the control and MEHP treated cells, 8 ± 3 and 10 ± 5%, respectively (Fig. 4B). The data indicates that exposure to MEHP did not trigger the apoptotic pathway.
Figure 4:

apoptosis in porcine germ cells treated with MEHP and staurosporine. (A) Double immunolabeling of cleaved caspase 3 (green) and VASA (red) in porcine germ cells. Cells were treated with staurosporine 5 μM for 24 h as positive control. Arrowheads show double positive cells, arrows show VASA positive only cells. Nuclei were stained with DAPI (blue). Scale bars = 20 μm. (B) Percentage of double positive cells from (A). 100 cells from 3 independent experiments were counted and the double positive cells for cleaved caspase 3 and VASA were quantified. Comparisons made by a one way ANOVA. Data are mean ± SD.
Discussion
Autophagy is an important homeostatic process which allows cells to cope with stressful conditions, including toxic insults. Little is known about the presence and role of autophagy in germ cells. Here, we demonstrated that exposure to phthalate esters in vivo and in vitro resulted in increased levels of autophagy in primate and porcine germ cells. While induction of autophagy is considered to occur in response to cellular stress, it was unknown if increased levels of autophagy are an indication of cellular damage or constitute a survival response. Interestingly, we found that the viability of germ cells increased when the level of autophagy was increased by treatment with rapamycin. This suggests that autophagy is promoting germ cell survival.
Germ cells exposed to MEHP did not undergo apoptosis based on analysis for cleaved caspase 3, an early indicator of apoptosis. This is in agreement with the findings by Lucas et al. [4], where C-18 cells treated even with high doses of MEHP did not undergo apoptosis. Similarly, Liu et al. [18], reported that treatment of rat SSCs with TOCP, another plasticizer also implicated to cause reproductive toxicity, lowered cell viability and induced autophagy without apoptosis, as analyzed by increased LC3 vesicles visualized by electron microscopy and LC3 protein analyzed by Western blotting, and Annexin V/PI staining, respectively. Taken together, these studies imply that autophagy rather than apoptosis may be involved in germ cell death. Shen and Codogno [20] have proposed three criteria to classify cell death as autophagic cell death: (1) Cell death occurs without the involvement of the apoptosis machinery; (2) there is an increase of autophagic flux; (3) suppression of autophagy is able to rescue or prevent cell death. The first two criteria are met in the current experiments: first, there was no increase in apoptosis, shown by the lack of caspase 3; second, there was a marked increase in autophagic flux in the treated cells compared to the control, as seen in the increase in the number of LC3II puncta in the bafilomycin A1 treated cells, in the 0.5 μM MEHP treated cells compared to the control cells (Fig. 3). The third criterion was not completely met, since there was still some cell death in the spermatogonia analyzed. However, the autophagic puncta decreased in the cells exposed to 1 μM compared to the cells treated with 0.5 μM MEHP; this could mean that the cells with higher levels of autophagy are dying, and therefore the number of autophagic puncta counted decreased.
There is still much discussion as to which conditions are required for the autophagic death, but there is enough evidence to conclude that while autophagy initially supports germ cell survival in response to cytotoxic stress, excessive stress can cause autophagy to become cytotoxic.
Supporting the relationship between autophagy and apoptosis we observed in germ cells exposed to MEHP in vitro, we previously observed that testis tissue that was chronically exposed to 500 mg/kg DBP showed the lowest number of seminiferous tubules with apoptotic structures [9]. Here, we demonstrated that germ cells in this tissue also had increased autophagy, compared to the controls. It is known that there is an interaction between autophagy and apoptosis, where autophagy is usually stimulated during non-lethal stress, while apoptosis is activated when stress exceeds a critical duration or intensity threshold [21]. It is therefore possible that in germ cells exposed to phthalates, autophagy served as an inhibitor of apoptosis. Autophagy is known to inhibit apoptosis by mitophagy, the specific autophagy of damaged mitochondria which are likely to cause the initiation of apoptosis [22]. Autophagy is also known to attenuate apoptosis by selectively reducing pro-apoptotic proteins in the cytoplasm, such as caspase 8 [21].
Previous studies [23, 24] have identified that the mechanism through which phthalates damage cells is oxidative stress. Oxidative stress induces protein aggregation when damaged proteins adopt conformations that expose regions of hydrophobic amino acids [25]. Aggregated proteins become less susceptible to proteolytic cleavage [26], and thus autophagy is required to remove the protein aggregates to prevent further damage to the cell. Accordingly, the DBP treated monkey testis tissue contained protein aggregates identified with an antibody to p62, which could support this assumption. However, there was no evidence of protein aggregates in the germ cells exposed to MEHP in vitro, which could mean that germ cells in vitro were able to break down the protein aggregates and eliminate them via autophagy or the proteasome pathway, given the shorter exposure time. It was reported in previous studies that there are differences in potency between different phthalates [23, 27], and species differences in sensibility to the different phthalates have been reported [28, 29]. These two factors, together with the different length of exposure, could account for differences found between the primate and the porcine germ cells analyzed.
During the prenatal period, the primordial germ cells undergo a series of epigenetic modifications required for germ cell development, such as the erasure of parental imprinting and demethylation. These reprogramming phases turn the prenatal period into a sensitive time for environmental induced abnormalities [30]. Evidence from animal studies suggests that both phthalates and bisphenols may exert important epigenetic effects [31]. In utero and neonatal exposure of bisphenol and/or phthalates may cause alteration in DNA methylation, histone modifications, and expression of non-coding RNAs. These epigenetic marks can induce gene alterations that may persist throughout a lifetime. In the human, bisphenol and phthalates have been related to methylation changes in over 20 genes [32]. Therefore, the exposure to these chemicals could be causing adverse health effects and infertility in adults.
In conclusion, we have shown in this study that exposure of germ cells to phthalates in vitro and in vivo promotes autophagy, which in turn, allows the germ cells to survive the toxic insult. The interplay between autophagy and apoptosis as well as the autophagic cell death in germ cells require further study.
Materials and Methods
Reagents
MEHP, rapamycin, and bafilomycin A1 were purchased from Sigma-Aldrich (St. Louis, MO). Wortmannin and staurosporine were purchased from Cell Signaling Technology Inc. (Danvers, MA). Chemicals were dissolved in DMSO (Sigma Aldrich, St Louis MO), to a final concentration of 0.1% DMSO. Dulbecco’s modified Eagle’s medium (DMEM), DMEM/F12, StemPro medium, and fetal bovine serum (FBS) were purchased from Thermo-Fisher Scientific Inc. (Waltham, MA). Collagenase Type IV with trypsin activity, hyaluronidase, trypsin, and PBS were obtained from Sigma-Aldrich. The enhanced chemiluminescence (ECL) substrate was purchased from Thermo-Fisher Scientific Inc. The polyvinylidene difluoride (PVDF) membranes were obtained from Bio-Rad.
Testis Tissue and Cells
Primate testes tissue from 6-month-old rhesus macaques was obtained from a previous study where it had been xenografted to mouse hosts that were exposed to phthalates as reported in Rodríguez-Sosa et al. [9]. Porcine spermatogonia were isolated by a two-step enzymatic digestion modified from Honaramooz et al. [33] from testes harvested by castration of 8-week-old pigs. Briefly, the tunica albuginea and visible connective tissue were removed. The exposed seminiferous tubules were then dissociated with collagenase (2 mg/ml; Type IV) in DMEM medium at 37 °C for 30 min. For the second digestion, tissue was exposed to collagenase (2 mg/ml; Type IV) and hyaluronidase (1 mg/ml; Type IV) for 30 min at 37 °C. The tissue was further digested with 0.25% trypsin/1 mM EDTA in the presence of DNAse I (1 mg/ml) at 37 °C for 20–30 min. FBS was added to stop the enzymatic digestion. The resulting cell suspension was filtered through both 70 and 40 μm pore size nylon mesh cell strainers, the cells collected by centrifugation at 500 g for 5 min, and resuspended in DMEM. Isolated cell populations were then enriched for spermatogonia by differential adhesion culture [34]. Briefly, 15–25 million cells/dish in 10 ml DMEM/F12 with 5% FBS and 100 mg/ml penicillin and 100 μ/ml streptomycin, were plated in 100 mm tissue culture dishes and incubated overnight at 37 °C. After 16–18 h, cells remaining in suspension were recovered, and then to recover the slightly adhered cells, dishes were agitated with 2 ml of a 1:20 dilution of 0.25% trypsin/1 mM EDTA in PBS at room temperature for 5 min. These two cell fractions were then pooled together and used for the subsequent experiments. Cell populations used in all experiments contained 72 ± 6% UCH-L1 positive germ cells.
Exposure of Primate Testis Tissue to DBP In Vivo
Primate testis tissue xenografts obtained from mice treated with DBP (500 mg/kg/day) for 28 weeks and from the vehicle treated control group were analyzed by immunofluorescence as described below. Spermatogonia were identified based on expression of ubiquitin carboxyterminal hydrolase L1 (UCH-L1) [4, 35], and autophagy was measured based on the presence and number of LC3BII puncta/cell [10, 36]. Immunofluorescent images were visualized with a Leica TCS SP5 confocal microscope. The images were then processed to identify UCH-L1 positive cells and LC3BII puncta were counted [37]. Four treated and four control samples were used for immunohistochemical evaluation, and 20 randomly selected UCH-L1 positive spermatogonia from each were analyzed. Immunohistochemistry on tissue was also performed for p62, which is a scaffold protein responsible for the selective sequestration of polyubiquitinated and damaged cellular proteins and organelles into aggregates, targeting them for degradation via autophagy [38].
Exposure of Porcine Germ Cells to MEHP In Vitro
To identify the effects of acute exposure to phthalates on germ cells in vitro, isolated porcine germ cells were exposed to MEHP for 24 h. Initially, a dose–response curve was performed with doses ranging from 0 to 10 μM MEHP for 24 h. However, concentrations higher than 1 uM resulted in lowered cell viability (<40%). Therefore, cells were treated with concentrations ranging from 0 to 1 μM MEHP. In all the experiments reported, the concentration of MEHP used was 0.5 μM. After exposure, 5 × 105 cells per treatment were fixed in methanol for staining and 1 × 106 cells were used for Western blotting.
Germ cells were exposed to MEHP for 24 and 48 h before analysis. As a positive control for autophagy, cells were treated with rapamycin (1 μM), an activator of autophagy [10]. Rapamycin is an inhibitor of mTOR which is a potent suppressor of autophagy [39, 40]. To decrease autophagy germ cells were treated with wortmannin (10 μM), a phosphoinositide 3-kinase (PI3K) inhibitor [41]. Also, to analyze the autophagic flux with these treatments, cells were treated with the lysosomal inhibitor Bafilomycin A1 (10 nM) for 4 h. This chemical blocks the autophagosomal degradation in lysosomes to show the total amount of converted LC3BII during the blockade [36], thus increasing the number of LC3BII puncta exclusively when autophagy is active [42].
Immunohistochemistry
Tissue sections were deparaffinized and rehydrated, and then subjected to antigen retrieval by boiling in 10 mM citrate buffer (pH 6) for 30 min and cooled down to room temperature. Then the sections were washed in PBS and permeabilized in 0.5% Triton X in PBS for 10 min at room temperature. Afterwards, sections were washed in PBS for 5 min and permeabilized in ice cold methanol at −20 °C for 10 min. Sections were washed for 5 min in PBS and blocked with CAS Block (Thermo-Fisher Scientific Inc.) for 15 min at room temperature. For double immunofluorescence, two primary antibodies raised in different species were mixed in 10% CAS Block in PBS and incubated at 4 °C overnight. Sections were washed in PBS for 5 min and incubated in the dark with the secondary antibodies conjugated with Alex Fluor for 1 h at room temperature. Slides were washed in PBS and mounted in Vectashield mounting medium with DAPI (Vector Laboratories, Burlingame, CA).
Single cells were fixed in ice cold methanol for 15 min at −20 °C, resuspended in PBS and kept at 4 °C until staining.
The primary antibodies used were: mouse monoclonal anti-PGP 9.5/UCH-L1 (1:200; cat# 8189 Abcam, Cambridge, MA), mouse monoclonal anti-DDX4/MVH (1:500; cat#27591, Abcam, Cambridge, MA), rabbit polyclonal anti-LC3BII (1:100; cat# 2775 Cell Signaling Technology Inc., Danvers, MA), rabbit polyclonal anti-p62/SQSTM1 (1:300; P0067 Sigma-Aldrich, St Louis, MO), rabbit polyclonal anti-cleaved caspase 3 (1:200; cat#9661 Cell Signaling Technology Inc., Danvers, MA).
The secondary antibodies used were: Alexa Fluor488 donkey anti-rabbit IgG and Alexa Fluor546 goat anti-mouse IgG (1:500; Invitrogen Corporation, Carlsbad, CA).
Protein Extraction
After recovery, cells were washed twice in PBS and then lysed with radioimmunoprecipitation assay buffer [RIPA buffer: 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulphate (SDS), 50 mM Tris-HCl pH 8.0, and 1% protease inhibitor cocktail (Sigma-Aldrich)], and kept at −20 °C until used. Protein concentration was determined by a DC protein assay (Bio-Rad).
Western Blotting
Lysates were thawed and 4× SDS loading buffer (4% SDS, 10% 2-mercaptoethanol, 20% glycerol, 0.004% bromophenol blue, 0.125 M Tris-HCl) was added, and boiled for 10 min, and then loaded (at least 10 μg of lysate per lane) onto an SDS-PAGE gel for electrophoresis, for 1 h at 0.05 A. Then, proteins were transferred to a PVDF membrane by electrophoretic transfer, during 1 h at 0.15 A. The membranes were blocked for 1 h in a 5% solution of skim milk and TBST. After blocking, the primary antibodies were diluted as per the manufacturer’s instructions, and incubated overnight at 4 °C on a shaker. The next day, the membranes were washed 3 times for 10 min in TBST, the secondary antibodies were added, and incubated for 1 h at room temperature on the shaker. Afterwards, the membrane was washed again 3 times, 10 min each on the shaker with TBST. The signal was visualized by incubating the membrane with ECL, and developed in the ChemiDoc XRS+ imaging system (Bio-Rad).
Cell Viability/Apoptosis
Cell viability was evaluated by trypan blue (Sigma-Aldrich) exclusion. Apoptosis was evaluated by Western blotting and immunocytochemistry using a cleaved caspase 3 antibody. Staurosporine (5 μM) treated cells were used as positive control.
Statistical Analysis
Values are from three independent experiments or as examples of representative experiments performed on at least three separate occasions. Data were analyzed by unpaired t-tests to compare two groups, and one way ANOVA for more than two groups with Tukey’s multiple comparisons test. A value of P < 0.05 was set as limit of statistical significance. GraphPad Prism 7.02 software (La Jolla, CA, USA) was used for all statistical analyses.
Acknowledgements
This work was supported by NIH/ORIP (9 R01 OD016575-12) and NIH/NICHD (1 R01 HD091068-01) to I.D. and a CONACyT doctoral scholarship to P.V.L. We would like to thank Dr Jonathan Hill, Dr Whitney Alpaugh, and Alla Bondareva for their help in reviewing the paper.
Author Contributions
P.V.L. designed and performed experiments, analyzed data and wrote the manuscript. I.D. oversaw experimental design and analysis and reviewed and edited the manuscript.
Conflict of interest statement. None declared.
References
- 1. Howdeshell KL, Rider CV, Wilson VS, Gray LE.. Mechanisms of action of phthalate esters, individually and in combination, to induce abnormal reproductive development in male laboratory rats. Environ Res 2008;108:168–76. [DOI] [PubMed] [Google Scholar]
- 2. Martino-Andrade AJ, Chahoud I.. Reproductive toxicity of phthalate esters. Mol Nutr Food Res 2010;54:148–57. [DOI] [PubMed] [Google Scholar]
- 3. Foster PMD. Disruption of reproductive development in male rat offspring following in utero exposure to phthalate esters. Int J Androl 2006;29:140–7. [DOI] [PubMed] [Google Scholar]
- 4. Lucas BEG, Fields C, Joshi N, Hofmann MC.. Mono-(2-ethylhexyl)-phthalate (MEHP) affects ERK-dependent GDNF signalling in mouse stem-progenitor spermatogonia. Toxicology 2012;299:10–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Benson R. Hazard to the developing male reproductive system from cumulative exposure to phthalate esters-dibutyl phthalate, diisobutyl phthalate, butylbenzyl phthalate, diethylhexyl phthalate, dipentyl phthalate, and diisononyl phthalate. Regul Toxicol Pharmacol 2009;53:90–101. [DOI] [PubMed] [Google Scholar]
- 6. Lyche JL, Gutleb AC, Bergman A, et al. Reproductive and developmental toxicity of phthalates. J Toxicol Environ Health B Crit Rev 2009;12:225–49. [DOI] [PubMed] [Google Scholar]
- 7. Latini G, Del Vecchio A, Massaro M, Verrotti A, De Felice C.. Phthalate exposure and male infertility. Toxicology 2006;226:90–8. [DOI] [PubMed] [Google Scholar]
- 8. Halden RU. Plastics and health risks. Annu Rev Public Health 2010;31:(1):179–194. [DOI] [PubMed] [Google Scholar]
- 9. Rodriguez-Sosa JR, Bondareva A, Tang L, et al. Phthalate esters affect maturation and function of primate testis tissue ectopically grafted in mice. Mol Cell Endocrinol 2014;398:89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mizushima N, Yoshimorim T, Levine B.. Methods in mammalian autophagy research. Cell 2010;140:313–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Levine B, Kroemer G.. Autophagy in the pathogenesis of disease. Cell 2008;132:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang S, Niu Q, Gao H, et al. Excessive apoptosis and defective autophagy contribute to developmental testicular toxicity induced by fluoride. Environ Pollut 2016;212:97–104. [DOI] [PubMed] [Google Scholar]
- 13. Phadwal K, Watson AS, Simon AK.. Tightrope act: autophagy in stem cell renewal, differentiation, proliferation, and aging. Cell Mol Life Sci 2013;70:89–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Feng Y, Yao Z, Klionsky DJ.. How to control self-digestion: transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol 2015;25:354–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu Y, Shoji-Kawata S, Sumpter RM, et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci USA 2013;110:20364–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barth S, Glick D, Macleod KF.. Autophagy: assays and artifacts. J Pathol 2010;221:117–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mancilla H, Maldonado R, Cereceda K, et al. Glutathione depletion induces spermatogonial cell autophagy. J Cell Biochem 2015;116:2283–92. [DOI] [PubMed] [Google Scholar]
- 18. Liu ML, Wang JL, Wei J, et al. Tri-ortho-cresyl phosphate induces autophagy of rat spermatogonial stem cells. Reproduction 2015;149:163–70. [DOI] [PubMed] [Google Scholar]
- 19. Han S-P, Zhou D-X, Lin P, et al. Formaldehyde exposure induces autophagy in testicular tissues of adult male rats. Environ Toxicol 2015;30:323–31. [DOI] [PubMed] [Google Scholar]
- 20. Shen HM, Codogno P.. Autophagic cell death: Loch Ness monster or endangered species? Autophagy 2011;7:457–65. [DOI] [PubMed] [Google Scholar]
- 21. Marino G, Niso-Santano M, Baehrecke EH, Kroemer G.. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol 2014;15:81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Youle RJ, Narendra DP.. Mechanisms of mitophagy. Nat Rev Mol Cell Biol 2011;12:9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kasahara E, Sato EF, Miyoshi M, et al. Role of oxidative stress in germ cell apoptosis induced by di(2-ethylhexyl)phthalate. Biochem J 2002;365(Pt 3):849–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee E, Ahn MY, Kim HJ, et al. Effect of di(n-butyl) phthalate on testicular oxidative damage and antioxidant enzymes in hyperthyroid rats. Environ Toxicol 2007;22:245–55. [DOI] [PubMed] [Google Scholar]
- 25. Bolt AM, Klimecki WT.. Autophagy in toxicology: self-consumption in times of stress and plenty. J Appl Toxicol 2012;32:465–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Davies KJ. Degradation of oxidized proteins by the 20S proteasome. Biochimie 2001;83:301–10. [DOI] [PubMed] [Google Scholar]
- 27. Foster PMD. Mode of action: impaired fetal Leydig cell function—effects on male reproductive development produced by certain phthalate esters. Crit Rev Toxicol 2005;35:713–9. [DOI] [PubMed] [Google Scholar]
- 28. Gray TJ, Rowland IR, Foster PM, Gangolli SD.. Species differences in the testicular toxicity of phthalate esters. Toxicol Lett 1982;11:141–7. [DOI] [PubMed] [Google Scholar]
- 29. Tomonari Y, Kurata Y, David RM, Gans G, Kawasuso T, Katoh M.. Effect of di (2-ethylhexyl) phthalate (DEHP) on genital organs from juvenile common marmosets: I. Morphological and biochemical investigation in 65-week toxicity study. J Toxicol Environ Heal Part A 2006;69:1651–72. [DOI] [PubMed] [Google Scholar]
- 30. Dores C, Alpaugh W, Dobrinski I.. From in vitro culture to in vivo models to study testis development and spermatogenesis. Cell Tissue Res 2012;349:691–702. [DOI] [PubMed] [Google Scholar]
- 31. Ponsonby AL, Symeonides C, Vuillermin P, Mueller J, Sly PD, Saffery R.. Epigenetic regulation of neurodevelopmental genes in response to in utero exposure to phthalate plastic chemicals: how can we delineate causal effects? Neurotoxicology 2016;55:92–101. [DOI] [PubMed] [Google Scholar]
- 32. Singh S, Li SSL.. Epigenetic effects of environmental chemicals bisphenol A and phthalates. Int J Mol Sci 2012;13:10143–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Honaramooz A, Megee SO, Dobrinski I.. Germ Cell Transplantation in Pigs 1. Biol Reprod 2002;66:21–8. [DOI] [PubMed] [Google Scholar]
- 34. Luo J, Megee S, Dobrinski I.. Asymmetric distribution of UCH‐L1 in spermatogonia is associated with maintenance and differentiation of spermatogonial stem cells. J Cell Physiol 2009;220:460–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang Y, Liu W, Sun Y, et al. Overexpression of ubiquitin carboxyl‐terminal hydrolase L1 arrests spermatogenesis in transgenic mice. Mol Reprod Dev 2006;73:40–9. [DOI] [PubMed] [Google Scholar]
- 36. Xie R, Nguyen S, McKeehan WL, Liu L.. Acetylated microtubules are required for fusion of autophagosomes with lysosomes. BMC Cell Biol 2010;11:89.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kuma A, Matsui M, Mizushima N.. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy 2007;3:323–8. [DOI] [PubMed] [Google Scholar]
- 38. Seibenhener ML, Geetha T, Wooten MW.. Sequestosome 1/p62–more than just a scaffold. FEBS Lett 2007;581:175–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 2004;36:585–95. [DOI] [PubMed] [Google Scholar]
- 40. Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC.. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ 2009;16:46–56. [DOI] [PubMed] [Google Scholar]
- 41. Wu YT, Tan HL, Shui G, et al. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem 2010;285:10850–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. García-Prat L, Martínez-Vicente M, Perdiguero E, et al. Autophagy maintains stemness by preventing senescence. Nature 2016;529:37–42. [DOI] [PubMed] [Google Scholar]