

Abstract
The mineral component of bone and other biological calcifications is primarily a carbonate substituted calcium apatite. Integration of carbonate into two sites, substitution for phosphate (B-type carbonate) and substitution for hydroxide (A-type carbonate), influences the crystal properties which relate to the functional properties of bone. In the present work, a series of AB-type carbonated apatites (AB-CAp) having varying A-type and B-type carbonate weight fractions were prepared and analyzed by Fourier transform infrared spectroscopy (FTIR), powder X-ray diffraction (XRD), and carbonate analysis. A detailed characterization of A-site and B-site carbonate assignment in the FTIR ν3 region is proposed. The mass fractions of carbonate in A-site and B- site of AB-CAp correlate differently with crystal axis length and crystallite domain size. In this series of samples reduction in crystal domain size correlates only with A-type carbonate which indicates that carbonate in the A-site is more disruptive to the apatite structure than carbonate in the B-site. High temperature methods were required to produce significant A-type carbonation of apatite, indicating a higher energy barrier for the formation of A-type carbonate than for B-type carbonate. This is consistent with the dominance of B-type carbonate substitution in low temperature synthetic and biological apatites.
Keywords: carbonated apatite, A-type carbonate, B-type carbonate, microstructural analysis, XRD, Infrared
Graphical legend
A series of differently substituted high temperature AB-type carbonated apatites show interesting structural changes, an additional B-carbonate site with the presence of A-carbonate in the apatite, and crystal domain sizes that correlate only with the amount of A-site carbonate. Our results provide an explanation for the dominance of B-type carbonate in biominerals.
1. Introduction
Bone, tooth, and other biological calcifications play an important role in the proper functioning of the human body and vertebrate animals. Bone is mostly comprised of a calcium phosphate mineral, apatite, in a nanocomposite cross-linked with type I collagen and serves as a support for the body [1]. Age-related or pathological bone fragility is a significant health problem and a relevant contributor to healthcare costs [2]. While different components of bone have a prominent role in determining its properties, the mineral phase provides stiffness to the overall composite, as well as storage of essential elements, and it is the focus of our work.
Hydroxyapatite (HAp) is the main inorganic constituent that is found in different substituted forms in human and animal bone, enamel, and dentin [3] (Figure 1). The structure of HAp, Ca10(PO4)6(OH)2, can readily accommodate substitution(s) by a large variety of ions [4, 5]. Chemical analysis shows that most biomineral forms of apatite contain carbonate as a predominant substituent, which is typically 2–8% by weight, depending on the source (bone, tooth, pathological calcification), species, and age [6, 7]. The biocompatibility of carbonated apatite can be attributed to the tendency of the carbonate to reduce crystallinity within the apatite structure, thereby increasing its solubility which enhances bone reformation or turnover [8]. Carbonated apatite is also a dominant mineral in phosphorites, natural apatitic rocks found in the earth [9].
Figure 1.
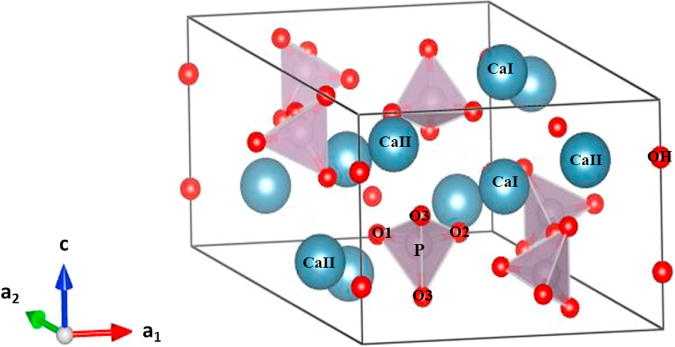
Hexagonal unit cell of a hydroxyapatite crystal Ca10(PO4)6(OH)2 (Drawn using VESTA [10] from the CIF obtained from Sudarsanan and Young [11].
Carbonate can substitute into two anionic sites of the hydroxyapatite structure: at PO43− sites (B-type carbonated apatite, B-CAp) and OH- sites located within a channel, along the crystallographic c-axis (A-type carbonated apatite, A-CAp). Carbonate in bone mineral is primarily B-type; while a concomitant presence of both A-type and B-type carbonate has been reported [6, 12, 13]. The fraction of A-type carbonate in biological apatites in all instances reported is very low while in apatites produced by synthetic routes, 4 – 6 wt.% A-type carbonate is reported [14, 15]
Many studies on carbonated apatites have focused on determining the structural position(s) and mechanism(s) of the carbonate substitution, either in natural [16,17,18,19] or synthetic [20,21,22,23] samples. This aspect of apatite mineralization has received attention in biological studies [24]. What remains to be clarified in AB-type carbonated apatite, however, is the interaction of carbonate ions substituted in each of the crystallographic sites available and the resultant changes in the apatite crystal structure with respect to non-carbonated hydroxyapatite.
The conventional technique to assess the degree of crystallinity of a solid phase is based on X-ray diffraction measurements. Such analyses confirm that the introduction of carbonate into the crystal structure of apatite causes a change in the lattice parameters of HAp. The A-type carbonate substitution causes a dilation of the apatitic channel and therefore an increase in the a lattice parameter. An expansion of the c and a contraction of the a lattice constants were found for the B-type carbonated apatites [17, 21, 22, 25]. The orientation of the carbonate group in the A-type apatites is generally accepted as approximately parallel to the a/c plane [22, 25], although other angles with the a-axis were also proposed [26]. The orientation of the trigonal planar CO32− group in B-type substitution is, however, more controversial. Most of the attention is dedicated to hydroxyapatite, where B-type carbonate has been found to occupy one of the sloping faces of the PO43− tetrahedron, making it approximately parallel to the c-axis [21, 22].
Synthetic carbonated apatites are common model compounds used for investigating chemical and structural properties of materials for biomedical applications. The biological response to synthetic apatites, both in vitro and in vivo, partly depends on the type and degree of carbonation which affects the crystallinity and solubility of the apatite. To this end, we have carried out studies on AB carbonated apatites and compared them to other types of substituted apatites. A series of apatite samples with a broad range of A-type and B-type carbonate substitution, synthetically produced at high temperature, were well-characterized, and for each sample the individual levels of A-type and B-type carbonate were determined. The samples were explored to investigate the resulting effects of substitution on the apatitic structure and how the carbonate ion in one site influences the carbonate in the other site. In our study, we provide a detailed characterization of a series of high-temperature AB carbonated apatites using XRD and FTIR spectra.
2. Methods
2.1 Synthesis of apatite powders
Apatite samples, with carbonate substituted in both the phosphate site (B-type) and hydroxide site (A-type) of hydroxyapatite, were prepared in a two-step process. First, B-CAp samples were prepared at moderate temperature (80 – 85 °C) via an aqueous precipitation method at high pH (11–13) conditions with ammonium carbonate as the source of carbonate, primarily substituting for phosphate (B-site substitution). These samples were dried and their carbonate content determined. A portion of each sample was further carbonated by high temperature treatment (950 °C, under CO2) producing carbonate substitution for hydroxide (A-site substitution). This method produced apatite samples with variable amounts of both A-type and B-type carbonate substitution (AB-CAp). For comparison, a hydroxyapatite (HAp) sample was prepared (room temperature, under N2) to produce an apatite sample with negligible carbonate. A portion of the HAp was subsequently carbonated in the A-site by the same high temperature treatment to produce A-CAp.
2.1.1 B-type carbonated apatite synthesis (B-CAp)
Four samples of B-CAp were synthesized, with a carbonate content ranging from 1 to 6 wt.%. The synthesis procedure was based on aqueous precipitation, under basic conditions and moderate temperature, as described by G. Penel et al [27]. A small modification to the procedure involves the use of ammonium salts instead of sodium salts (since sodium ion can complicate the IR ν3 region [28]) to minimize cationic substitution for calcium in apatite. In brief, a phosphate solution with variable amount of ammonium carbonate ((NH4)2HPO4: 10.0 g, (NH4)CO3: 1.80 – 2.50 g, NH4OH: 25 mL, H2O: 150 mL) was slowly added to a calcium solution (Ca(NO3)2.4H2O: 23.60 g, NH4OH: 50 mL, H2O: 150 mL) maintained at 80–85 °C, while continuously stirring. A white precipitate formed as the two solutions were mixed. The precipitate was left to “mature” for one hour while maintaining the temperature at 80–85 °C under continuous stirring. The precipitate was subsequently filtered and washed with 2 L of water to remove nitrates and ammonia (until nitrate ion was not detected by FTIR (1384 cm−1)), and then dried overnight in a conventional oven at 90 °C.
2.1.2 Hydroxyapatite synthesis (HAp)
Stoichiometric hydroxyapatite (HAp) with negligible amounts of carbonate was prepared based as described by Hayek [29], under N2 atmosphere, by bubbling N2 gas through the solutions participating in the reaction, then slowly adding a phosphate solution ((NH4)2HPO4: 6.605 g, NH4OH: 50 mL, H2O: 20 mL) to a calcium solution (Ca(NO3)2·4H2O: 19.72 g, NH4OH: 35 mL, H2O:40 mL) at room temperature. The solution was then heated to boiling for 10 min, and subsequently left to cool to room temperature, while stirring under N2 atmosphere. The precipitate was filtered and washed with water and then dried in the same manner as the B-CAp samples.
2.1.3 High temperature carbonation of apatite A-site (A-CAp and AB-CAp)
High temperature treatment of HAp and B-CAp samples produced the A-CAp and AB-CAp samples, respectively. The synthesis follows a method developed by Bonel et al and Gibson et al [30, 15]. In brief, stoichiometric HAp (Section 2.1.1), and the four samples of well-characterized B-CAp (Section 2.1.2) were introduced into a sealed tube furnace under a constant flow of 500 SCCM (standard cubic centimeters per minute) of dry CO2 (Airgas Instrumental grade 4.0 – Coleman grade) while the temperature was slowly increased from room temperature to 300°C. After three hours at 300 °C the temperature was increased to 950 °C and maintained for 48 hrs. After cooling to room temperature, the final carbonated products were retrieved from the furnace and analyzed.
2.2 Carbonate concentration determination
The total carbonate content of thus synthesized B-CAp, AB-CAp, HAp, and A-CAp samples was determined via coulometric titration after heating and collecting CO2 gas in an absorption cell containing lithium hydroxide (Galbraith Laboratories, Knoxville, TN). From the total weight percent of carbonate reported and the locally measured FTIR carbonate ν2 A/B peak area ratio (B-type 872 cm−1: A-type 879 cm−1), respective weight fractions of the A–type and B-type carbonate in the AB-CAp samples were determined (equation 1).
| (1) |
2.3. FTIR analysis
Fourier transform infrared (FTIR) spectra were acquired using a Nicolet iS50 FTIR spectrometer (Thermo Electron Corp., Madison, WI) with single-bounce diamond-attenuated total reflectance (ATR) sampling and a deuterated triglycine sulfate (DTGS) detector. A thin layer of apatite sample powder was directly spread on the ATR diamond and pressed to distribute the sample uniformly. 128 scans at a resolution of 4 cm−1 were ratioed against a nitrogen-purged background. Omnic software (ver.9, Thermo Scientific Corp.) was used to process the spectra where a linear baseline was manually set in the region of interest. ATR sampling was used for all the FTIR analyses due to its advantage of faster analysis because no sample preparation was necessary. Studies showing an evaluation of FTIR spectra, suggest ATR sampling to be more reliable and have comparable results to that of transmission FTIR. [31]
Spectra were analyzed using GRAMS/AI software (ver. 7.00 – Thermo Galactic, Waltham, MA). Gaussian and Gaussian-Lorentzian peaks and a linear baseline were summed to match small regions of the spectrum using a nonlinear peak-fitting routine that employs the Levenberg – Marquardt algorithm to minimize the chi-squared value [32].
2.4. XRD
Powder X-ray diffraction (XRD) data were acquired using a Rigaku Miniflex II diffractometer (Rigaku Corp., The Woodlands, TX), equipped with a scintillation counter detector, and a Cu Kα X-ray source (λ = 1:5418 Å, 30 kV, 15 mA, 0.3 mm slit). The samples were finely ground before analysis, and gently pressed in the sample holder to obtain a smooth and leveled surface. Diffraction patterns were recorded from 4° to 60° 2θ at a step size of 0.04° with a scan rate of 0.25° per minute in a Bragg-Brentano geometry. The XRD analysis of lattice constants and crystallite size was carried out by using the FullProf.2k suite (https://www.ill.eu/sites/fullprof/) [33]. Specifically, the Thompson, Cox, and Hastings (TCH) function and the Le Bail method were used to do a whole pattern profile matching from which it was possible to extract lattice constants and crystallite sizes [34, 35]. For determination of the diffraction peak positions (2θ-values), refined profile matching was performed using pre-ground silicon powder (NIST SRM 640b) as an internal standard. The instrumental resolution was derived from an average of two separate scans of silicon diffraction lines obtained at scan speeds of 0.01 2θ/min and 0.02 2θ/min. The average of the measurements was supplied as a Voigt function in the input file. This helped us perform the microstructural analysis for the profiles studied.
3. Results and Discussion
3.1. Characterization of AB-CAps with variable carbonate concentration
FTIR spectroscopy was used to study the AB-CAps of a range of compositions synthesized at high temperature. The influence of the location of the respective carbonate ions on the crystal structure with regard to (002) and (100) reflection planes was determined from the powder X-ray diffraction patterns and the refinement data obtained. This contribution to the existing discussions on carbonate substitution in hydroxyapatites starts with the characterization of A- and AB-CAp crystals synthesized at high temperatures.
3.1.1 FTIR analysis of carbonated apatite
The substitution of carbonate in apatite is most readily detected by FTIR and to a lesser extent by Raman spectroscopic analysis. The application of the IR techniques has been demonstrated in the pioneering studies by Elliott, LeGeros, Bonel, Fleet, and Tacker through the years [12, 36, 37, 38, 39, 40]. FTIR spectra of the synthesized HAp, A-CAp, B-CAp, AB-CAp are shown in Figure 2, with an enlargement of the ν3 carbonate region. (Figure 2)
Figure 2.
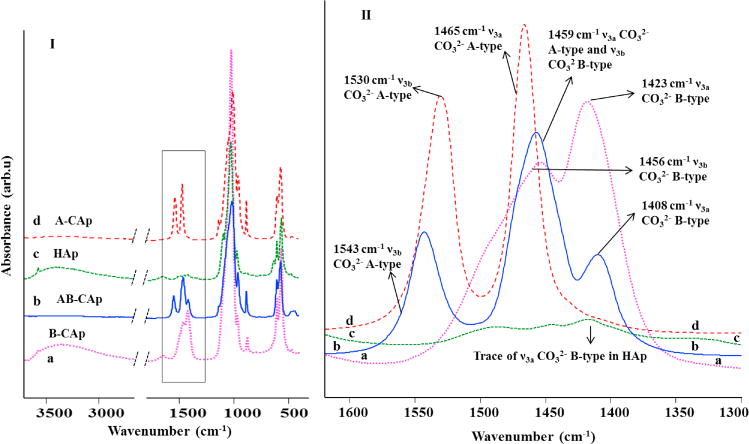
FTIR spectra of the apatite samples (I. full spectrum and II. expanded ν3 region) of a) B-CAp b) AB-CAp c) HAp, d) A-CAp.
Assignments of all the peaks are based on literature studies of a variety of synthetic and biological apatites [12, 37, 41, 42, 43, 44]. Independent studies of Vignoles and Baxter et al [43, 44] on synthetic B-type carbonated apatites and bone, suggest that in B- CAp the ν3 antisymmetric stretching vibration splits into ν3a and ν3b peaks at 1423 cm−1 and 1456 cm−1, respectively (Figure 2a). An additional broad shoulder at ~1480 cm−1 can be attributed to the presence of surface labile carbonate [45]. HAp (Figure 2c) shows traces of ν3a B-type carbonate incorporated as evidenced from a very weak peak at 1410 cm−1. Studies by Elliott and Bonel on synthetic A-type carbonated apatites [12, 37] found that A-CAp can be characterized by peaks at 1530 and 1465 cm−1 in the IR spectrum (Figure 2d) attributed to the carbonate ν3b and ν3a vibrations, respectively. Dental enamel and synthetic AB-type carbonated apatites studied by Elliott and Fleet et al [12, 39], respectively, suggest an assignment of the AB-CAp (Figure 2b) IR peak at 1543 cm−1 to the ν3b vibration of A-type carbonate (a shift of +13 cm−1 compared to the same peak in A-CAp), while the smaller AB-CAp peak at 1408 cm−1 is assigned to the ν3a vibration of B-type carbonate (a shift of −15 cm−1 compared to the same peak in B-CAp). Due to the presence of both A and B carbonate in this structure, the strong peak centered at 1459 cm−1 can be a combination of the A-type ν3a vibration and the B-type ν3b vibration. The concomitant presence of A and B carbonate affected the positions of the peaks in the ν3 region of carbonate as compared to apatite with only A-type carbonate or only B-type carbonate.
Peak fitting studies provided additional information on the ν3 region of the AB-CAp samples. The number of deconvolution bands chosen was based on the studies performed by Fleet, Pleshko et al and Comodi et al [16, 28, 39] on a varied class of carbonated apatites. Several sets of initial parameters that differed in peak centers and heights were tried until consistent results with minimum chi-squared values were obtained. Deconvolution results for the ν3 region of A-type and AB-type carbonated apatites (A-CAp and AB-CAp2) are shown in Figure 3-I and 3-II, respectively.
Figure 3.
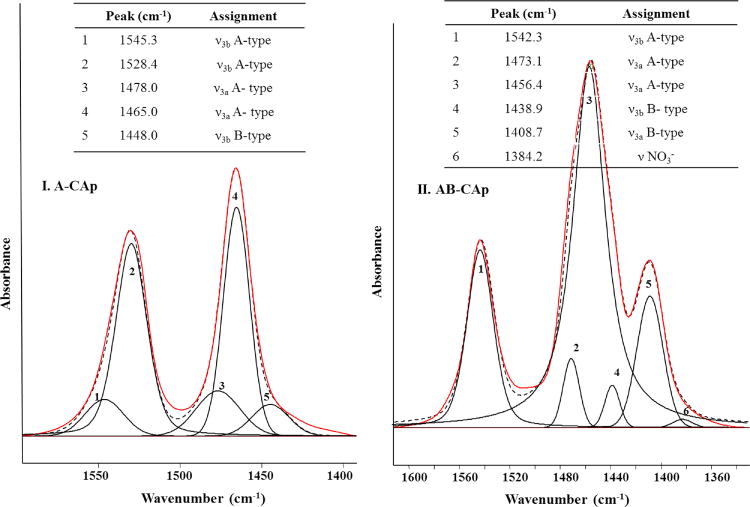
Experimental (solid/red) FTIR spectrum and calculated (dashed/black) FTIR spectrum from the sum of the fitted curves (numbered). I. A-CAp in the 1400 – 1600 cm−1 carbonate ν3 region. II. AB-CAp2 in the 1320–1600 cm−1 region. The position and assignment of the fitted curves are reported (inset tables).
Peak fitting of the ν3 FTIR spectral region of A-CAp (Fig. 3-I) produced a best fit (Standard Error (SE) = 0.002) by using 5 deconvolution bands. All the bands used in the fitting were Gaussians, except for band 2 which was a mixed Gaussian/Lorentzian. Bands 1 and 2 can be assigned to ν3b, while bands 3 and 4 correspond to ν3a of A-type carbonate [26, 39]. The less intense bands, 1 and 3 account for the asymmetry towards higher wavenumbers in the observed peaks, and bands 1 and 3 can be due to the presence of a second, less common orientation of the A-type carbonate, within the apatitic channel [46]. The peak centered at 1465 cm−1 in Fig. 3-I also showed a slight asymmetry towards smaller wavenumber which is accounted for by the Gaussian curve 5, centered at 1448 cm−1. This peak had a very low intensity and it can be assigned to the ν3b vibration from a very small amount of B-type carbonate (Figure 3).
The FTIR spectrum of the AB-CAp (Fig. 3-II) shows a ν3 region that is more complex than in A-CAp (Figure 3-I). The best fit was obtained by using 6 curves (standard error (SE) = 0.0004). Most were Gaussians except curves 1 and 3 which were mixed Gaussian/Lorentzian curves. In Figure 3-II, curves 1, 2 and 3 correspond to the A-type carbonate, while curves 4 and 5 can be assigned to the B-type carbonate vibrations; curve 6 is a residual impurity of nitrate from the apatite synthesis. The peak corresponding to the A-type ν3b vibration was fitted with only one curve, labeled 1, centered at 1542 cm−1. This is a very close value to the fitted curve 1 in the A-CAp FTIR spectrum (Figure 3-I), indicating that in the AB-CAp sample the orientation of the carbonate relative to the c axis giving rise to this signal is preferred. The fitted curves corresponding to the A-type ν3a vibration (curves 2 and 3, Figure 3-II) were shifted by 8 cm−1 towards smaller wavenumbers with respect to pure A-CAp. This finding suggests that the presence of a substantial amount of B-type carbonate in the lattice strongly affects the A-type ν3a vibrational frequencies. Finally, the curves at 1438 and 1408 cm−1 can be assigned to the ν3b and ν3a vibrations of B-type carbonate, respectively. These peaks are found at lower wavenumbers in this sample, with respect to the pure B-CAp, where they are usually found at 1450–1456 cm−1 and 1422–1427 cm−1 [29, 30, 38, 42] (in transmission FTIR spectra), respectively.
This downshift in the frequencies for B-type carbonate when A-type is also present, suggests that the carbonate may be positioned differently in the unit cell with respect to pure B-type. An example is natural carbonated fluorapatite [22, 26] where two types of A and two types of B carbonates, slightly different orientations of carbonate in the sites, were detected within the same sample. The authors suggested that the ν3a peak found at 1408 cm−1 can be associated to the most “stable” form of B-type carbonate, lying perpendicular to the c axis, while the ν3a at higher wavenumbers corresponds to the carbonate lying almost parallel to the c axis. The new peak positions observed for A-type and B-type carbonate in our AB-CAp samples indicate that the presence of a significant amount of B-type carbonate in the AB-CAp samples affects the vibrational frequencies of both A and B-type carbonate. The peak shifts can be attributed to slightly different orientations of the carbonate groups within the crystal structure.
The FTIR spectra are useful to establish the relative concentrations of both the types of carbonate ions present in AB-CAp. Peak intensities and peak areas have been considered to carry out this quantification in samples synthesized under different conditions [29, 47, 48]. Peak heightratios of the ν3a or the ν3b vibrations for the B-type and the A-type carbonates, have been used to estimate relative amounts present in the samples. But, changes in peak width and peak center observed in this series of AB-CAps, for the ν3 region make it less accurate for determining the fractions of the two carbonate substitutions present. In addition, with attenuated total reflectance (ATR) FTIR the penetration depth of the infrared radiation into the sample is wavelength dependent. This results in peak absorbance at long wavelengths that is higher than from peaks at shorter wavelengths [31]. Therefore, the ν2 carbonate out-of-plane bending region (880–870 cm−1) was used to determine the relative amounts of A-type to B-type carbonate because the peaks are narrow, with very little change in peak position among samples, and the peak area ratio is most accurately measured from these very close FTIR peaks at 879 cm−1 (A-type carbonate) and 872 cm−1 (B-type carbonate). The best peak fit (standard error SE = (0.0004) was obtained by deconvoluting into three totally Gaussian bands (Figure 4 – I). The peak at 866 cm−1 (labeled 3 in Fig, 4I) has been attributed to either A- or B – type carbonate with slightly different orientation, denoted as A2 and B2 by Fleet [45, 49]. This assignment varies depending on the presence of other substituents in the apatitic structure as well as the heating and annealing conditions applied. Other studies have attributed an 866 cm−1 peak to a ‘labile non-apatitic surface carbonate’ in low temperature synthetic apatites [48, 50]. However, we don’t expect labile carbonate to remain given the temperature our samples were exposed to (950°C) (Figure 4).
Figure 4.
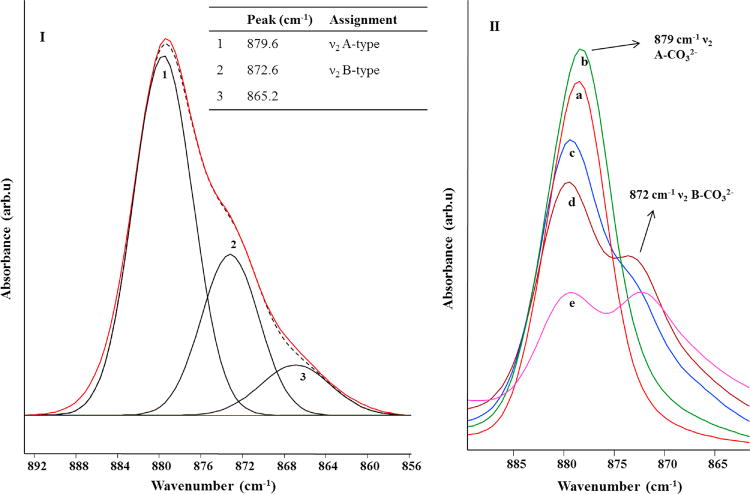
I. Experimental (solid/red) and deconvolved (dashed/black) FTIR spectrum of AB-CAp2 in the 890–860 cm−1 carbonate ν2 region. The 3 fitted curves are also reported, together with position and assignment (inset table). II. FTIR spectra of the carbonate ν2 region showing the trend in the relative A-type and B-type carbonate in a) A-CAp, b) AB-CAp1, c) AB-CAp2, d) AB-CAp3, e) AB-CAp4.
FTIR absorbance spectra of the AB-type apatites with different ratios of A-type and B-type carbonate substitution are shown in Figure 4-II. Peak fitting on each spectrum yielded peak heights for the ν2 A-type and ν2 B-type carbonate peaks. From the total carbonate content of the AB-CAp samples, determined by coulometric analysis (Table 1), and the FTIR ν2 A/B peak height ratios, equation 1 was solved for the weight fraction of B-type carbonate in the AB-CAp samples. Subtracting the amount of B-type carbonate from the total carbonate, the weight fraction of A-type carbonate in the AB-CAp samples was determined (Table 1).
Table 1.
Analysis of A-type and B-type carbonate substitution (wt.%) in AB-CAp.
| Sample Name | Before heating | After heating |
|||
|---|---|---|---|---|---|
| Total B-typea CO32− wt.% |
Totala
CO32− (wt.%) |
ν2 A/B CO32− peak area ratiob | B-typec
CO32− (wt.%) |
A-typec
CO32− (wt.%) |
|
| A-CAp | 0.43 | 4.90 | 24.3 | 0.19 | 4.71 |
| AB-CAp1 | 1.14 | 5.45 | 14.1 | 0.36 | 5.09 |
| AB-CAp2 | 2.08 | 5.85 | 2.11 | 1.88 | 3.97 |
| AB-CAp3 | 3.50 | 6.05 | 1.42 | 2.50 | 3.55 |
| AB-CAp4 | 5.73 | 6.90 | 1.00 | 3.45 | 3.45 |
Total carbonate content determined by coulometric analysis.
FTIR peak area 879 cm−1/peak area 872 cm−1 of the heated A-CAp and AB-CAp samples.
A-type and B-type carbonate in the heated samples calculated with equation 1.
As reported by Gibson et al [15], some loss of B-type carbonate is observed after heating apatite samples at 950°C in CO2, (Table 1). Previous studies on thermal analysis of the carbonated apatites shows that B-CAp suffers a complete loss of carbonate when heated beyond 700°C [51, 52]. In the present work, only partial loss of the B-type carbonate was observed when the apatites were heated at 950°C in the presence of CO2 gas flowing through the reaction cell. Formation of A-type carbonate by heating hydroxyapatite is expected to follow a mechanism whereby the incoming CO2 gas reacts with 2 OH− in the apatitic channel forming A-site carbonate and water [12, 15, 53]. The hydroxide available for reaction with CO2 in the apatite channel is less in B-CAps prepared in low (80–85°C) temperature hydrothermal conditions due to the charge compensation necessary for substitution of carbonate (CO32−) for phosphate (PO43−). One mechanism is the loss of a Ca2+ ion and an OH− ion for each carbonate gained [54, 55]. In fact, studies have shown that the amount of B-type CO32− in apatite is inversely correlated to the amount of OH− along the c-axis [13]. We observed that apatite samples with the lowest initial B-type carbonate resulted in the highest levels of A-type carbonate after heating. (Table 1); i.e., if there was more OH− in the apatite channel (less B-type carbonate within the apatite before heating in CO2), then more A-type carbonate was incorporated.
In an experiment to test whether carbonate would remain in the B-sites if heated without flowing CO2, our B-CAp samples were heated at high temperature (950°C) in an inert gas medium, argon, instead of carbon dioxide. This resulted in the loss of all B-type carbonate and interestingly, a new hydroxyl peak at 3645 cm−1 (FTIR spectra) and at 3615 cm−1 (Raman spectra) appeared, which can be attributed to the formation of calcium hydroxide – Ca(OH)2 in the heated samples [56, 57]. More details are provided in the supporting materials.
3.1.2. Microstructural analysis – powder XRD
The structural role of carbonate in apatite, its systematic behavior in the crystal structure, and the measurable physical parameters in the high-temperature synthetic apatite crystals are further revealed by X-ray powder diffraction studies. Analyses were performed on our A-CAp and AB-CAp samples (Figure 5). They indicate that the apatitic structure was retained, although deformations of the unit cell occurred due to carbonate incorporation (Figure 5).
Figure 5.
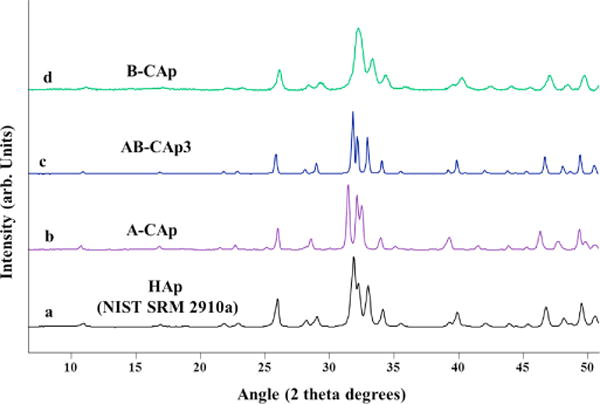
Powder XRD diffractograms of a) HAp (NIST standard 2910a), b) A-CAp (4.90 wt.% CO32−), c) AB-CAp3 (A-type 3.55 and B-type 2.50 wt.% CO32−) and d) B-CAp (5.73 wt.% CO32−).
The lattice constants for the a and c axes of apatite were obtained from the refinement of the diffractograms. In Table 2, these values are compared among three differently substituted carbonated apatite samples (A-CAp, B-CAp and AB-CAp) discussed in the present work as a percentage change from the lattice parameters obtained for the HAp (NIST SRM 2910a) sample with negligible carbonate. The reflection angles obtained from the profile fitting of the NIST sample were within +/− 0.01 degrees 2θ of those published by NIST [58]. The diffraction peak shifts of A-CAp are consistent with an increase in the a lattice parameter and a slight contraction of c relative to HAp. AB-CAp3 showed only a small increase in the c value, with nearly no change in the a lattice parameter. Finally, a B- CAp displayed a trend opposite to that of A- CAp for the a lattice parameter, with a modest decrease, whereas c remained nearly unchanged relative to HAp. These trends agree with the results typically obtained for the A-CAp and B-CAp apatites [21, 26] (Table 2).
Table 2.
Apatite lattice spacing along the a-axis and the c-axis for apatite samples with different types of carbonate substitution. A-type, B-type, and combined AB-type are presented, as well as the percentage change with respect to the non-carbonated standard HAp (NIST).
| Sample | Lattice parameters (Å) | |
|---|---|---|
|
| ||
| a | c | |
| HAp (NIST) | 9.423 | 6.889 |
| A-CAp | 9.552 (+ 1.4%) | 6.865 (− 0.3%) |
| AB-CAp3 | 9.449 (+ 0.3%) | 6.893 (+ 0.06%) |
| B-CAp | 9.383 (− 0.4%) | 6.893 (+ 0.06%) |
Carbonate wt%: A-CAp (4.90%); B-CAp (5.73%); AB-CAp (A-type 3.60%, B-type 2.45%)
The A-CAp and series of AB-CAp samples were characterized by powder XRD; all the data were consistent with the hydroxyapatite crystal structure, and no other phases were detected. The XRD patterns of the synthesized AB-CAp samples were analyzed for trends correlated to the level of B-type CO32− and A-type CO32− substitution in hydroxyapatite. Changes in the diffraction peak widths and lattice parameters along the a and c axes relative to the composition of carbonate in A and B sites are tabulated (Table 3).
Table 3.
XRD peakwidths (FWHM) along the (100) and (002) planes and lattice constants of the AB-type carbonated apatites and their carbonate distributions.
| Total
CO32− (wt.%) |
A-type
CO32− (wt.%) |
B-type
CO32− (wt.%) |
Peak width (2θ degrees) |
Lattice
Constant (Å) |
|||
|---|---|---|---|---|---|---|---|
|
|
|||||||
| (002) | (100) | c | a | ||||
| A-CAp | 4.90 | 4.71 | 0.19 | 0.22 | 0.26 | 6.865 | 9.552 |
| AB-CAp1 | 5.45 | 5.09 | 0.36 | 0.29 | 0.32 | 6.863 | 9.529 |
| AB-CAp2 | 5.85 | 3.97 | 1.88 | 0.21 | 0.24 | 6.891 | 9.474 |
| AB-CAp3 | 6.05 | 3.55 | 2.50 | 0.19 | 0.20 | 6.893 | 9.449 |
| AB-CAp4 | 6.90 | 3.45 | 3.45 | 0.17 | 0.19 | 6.904 | 9.423 |
Changes in the lattice constant parameters were observed along the a and c axes of the series of AB-CAps (Table 3). The samples with the highest amount of A-type carbonate have the smallest c lattice parameter indicating a decreasing c-axis length. Other studies on A-CAp have noted the same trend [22, 59], which is consistent with reduced occupancy of the apatitic channel as CO2 reacts with two hydroxides to form carbonate. Suetsugu et al [26] proposed that the carbonate plane is aligned parallel with the apatite c-axis. This type of orientation of CO32− in the channel is consistent with a shrinking c-axis as the amount of A-type carbonate increases.
With increasing in B-site CO32− substitution in the series of AB-CAp samples, the a lattice parameter decreases by 1.1 %, indicating a slight contraction along the a-axis. This trend is consistent with a smaller trigonal planar carbonate substituting for a larger tetrahedral phosphate [45, 60]. The contraction in the a-axis length also correlates with decreasing A-site substitution (Table 3). Since carbonate in the A-site will have at least one oxygen that is not as near the c-axis as the other one or two oxygen atoms in the c- axis channel will be dilated, resulting in a small increase along the a- axis [21]. Thus, contraction along the a-axis is consistent with lower A-site carbonate and with higher B-site carbonate concentration.
The effect of carbonate orientation in apatite on crystal axial lengths can also be seen in by comparing among samples with carbonate primarily in one site vs significant amounts in both, (A-CAp, B-CAp and AB-CAp) in Table 2. Relative to HAp (no carbonate) the a lattice parameter has contracted in B-CAp and expanded in A-CAp, but in AB-CAp, the competing effects are balanced such that for the AB-CAp3 sample the a lattice parameter is intermediate between A-CAp and B-CAp. The c lattice parameter, relative to HAp, has contracted in A-CAp and is slightly expanded in B-CAp (Table 2). The AB-CAp3 sample has nearly the same c lattice parameter as B-CAp although other samples in the AB-CAp series have c-axis lengths that are intermediate between those of the primarily A-CAp and B-CAp samples (Figure 6).
Figure 6.
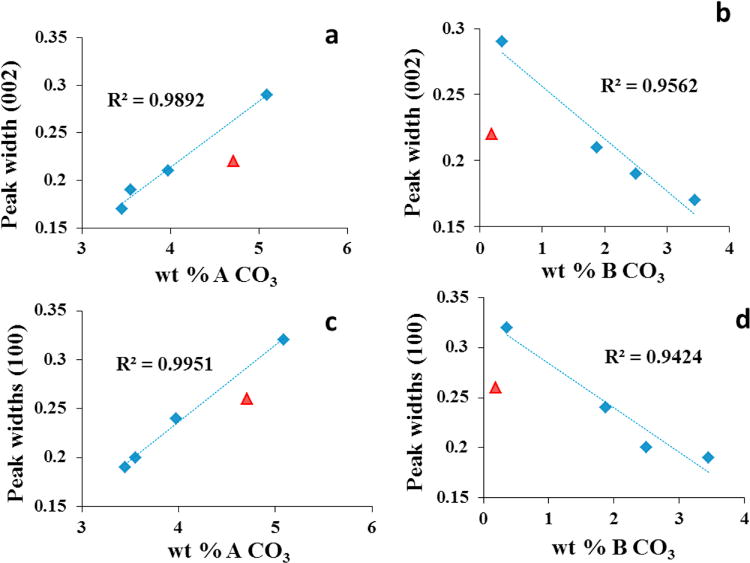
Trends in the XRD peakwidths (FWHM) of the (002) lattice planes perpendicular to the c-axis relative to a) wt.% A-type carbonate and b) wt.% B-type carbonate in the AB-CAp samples. Trends in the XRD peakwidths of the (100) lattice planes perpendicular the a-axis relative to c) wt.% A-type carbonate and d) wt.% B-type carbonate in the AB-CAp samples. The triangle-shaped (▲) data point corresponds to the A-CAp sample while the diamond-shaped (◆) data points correspond to the AB-CAp samples. Trend lines with the R2 values were calculated for the series of AB-type apatites, but not including A-CAp.
The diffraction peak widths of the AB-type carbonated apatites for the (100) and (002) planes were compared relative to the experimentally measured weight fractions of the A-type and B-type carbonate in the samples (Table 3). The changes in the peakwidths (Figure 6) show a linear correlation (R2 > 0.98) with respect to the carbonate content in the AB-CAps. Figures 6a and 6c show that an increase in the A-type CO32− from 3.5 to 5.1 wt.% increases the width of the diffraction peaks corresponding to planes perpendicular to the c-axis (002) and the a-axis (100) by a factor of 0.12 and 0.13, respectively When the peakwidths are compared against the amount of B-type carbonate in AB-CAps, a decrease is observed (Figure 6b and 6d) with increasing B-site carbonate content. The A-CAp sample slightly deviates from the observed trendline for the AB-CAp samples. This could be because of the slightly different synthesis procedures followed for preparing the HAp and B-CAp samples before heating to form A-CAp as the HAp was prepared in a completely carbonate free atmosphere (Table 4).
Table 4.
Average crystal domain size, obtained from the microstructural analysis of XRD profile matching.
| Sample | Total CO32− (wt.%) |
A-type CO32− (wt.%) |
Avg. crystallite domain size (nm) |
|---|---|---|---|
| AB-CAp1 | 5.45 | 5.09 | 34.3 |
| AB-CAp2 | 5.85 | 3.97 | 74.6 |
| AB-CAp3 | 6.05 | 3.55 | 106.9 |
| AB-CAp4 | 6.90 | 3.45 | 141.0 |
Decreasing peakwidths observed for the two reflection planes also correspond to increasing average crystallite domain sizes, the distance between grain boundaries or significant defects within the crystallites. Profile matching of diffractograms provides calculation of an average apparent crystallite domain size, of the AB-CAp samples (Table 4). Upper limits of the lattice strain effects corresponding to one-fourth of the apparent strain defined by Stokes and Wilson [61] are taken into consideration in the microstructural analysis. In other words, both Gaussian and Lorentzian broadening of the TCH function [34] are refined, allowing the independent calculation of apparent size and strain. The crystallite domain dimensions are in the nanometer range and decrease with increasing A-type carbonate content in the apatite samples. A similar trend in crystallite size is reported for B-CAp samples with carbonate levels in the same range as our samples [45]. In our AB-CAp samples the trend is consistent with the reduction of the long-range order in the material with increasing amounts of carbonate in the A-site. In fact, as seen in Table 4, the apparent crystal domain size increases in the samples exhibiting the trend from the highest to the lowest A-type carbonate concentration. This is consistent with the view of A-site carbonate as a localized defect in the lattice, which disrupts the long-range order as the number of defects increases. Surprisingly, the total amount of carbonate (from the A and B sites) does not correlate inversely with crystal domain size. Sample AB-CAp4 has the highest total carbonate (6.90 wt.%) and yet the largest crystal domain size. However, among the samples studied here, the total carbonate concentrations differ by only 2 wt.%, and by less than 1.5 wt.% among the four AB-CAp samples. Thus, this interpretation of a relation between the apatite crystallite size and respective carbonate incorporated could be strengthened by studying more samples of greater differences in carbonate levels.
Within the four AB-CAp samples observed, the connection of the crystal domain size exclusively to the amount of A-type carbonate indicates that the two sites of carbonate substition affect the apatite crystallinity very differently. Carbonate substitution in the A-sites, along the c-axis channel, must have a greater disruptive effect on the long-range order of apatite than carbonate in the B-sites of our AB-carbonated apatites. Model calculations of carbonated apatite have shown that A-CAp and AB-CAp are more energetically favorable than B-CAp [57]. Yet only by high temperature methods have significant A-site carbonation of apatite been shown [15,38], indicating a higher energy barrier to A-site carbonation in the apatitic c axis channel than to B-site carbonation of phosphate sites. This finding contributes to an understanding of the dominance of B-site carbonate in biological apatites (bone, teeth) and carbonated apatite prepared at low to moderate temperature (20 – 100℃) by aqueous methods [37], where the majority of apatitic carbonate is B-type (typically 10% or less of the carbonate is in the A-site). In these conditions, B-type carbonation of apatite is kinetically favored, perhaps because the substitution of carbonate in a phosphate site causes less pertubation to the hydroxyapatite unit cell. The trigonal planar carbonate ion in B-type sites is reported to be oriented with its oxygens very close to the phosphate oxygen positions along one of the faces that the tetrahedral phosphates occupy [21, 22]. In contrast, A-type carbonate resides in the c-axis channel and has its plane oriented in the apatite a/c plane [26], where it is bulkier than the hydroxides that it replaces. The high temperature needed for significant levels of A-type carbonation provides the energy required for the reaction of CO2 with hydroxide and the accomodation of carbonate in the apatite channel.
While A-type carbonate controls crystal domain size at high temperature, apatite with primarily B-type carbonate, formed in aqueous conditions and close to physiological temperatures, has nanometer range crystallite sizes that are clearly correlated with B-type carbonate content. Deymier et al, [45] showed that the formation of apatite crystallites of bone-like nanometer size and plate-like morphology can be controlled simply by carbonate concentration (primarily B-type) in the absence of protein. The conditions that affect the nanometer size of bone mineral crystal support the structural optimization and thereby mechanical strength as well as resistance to fractures of biological apatite [62].
4. Conclusions
A series of mixed AB-type carbonate apatite powders with variable carbonate content were synthesized by heating well characterized HAp and B-type carbonated apatites at 950°C under flowing CO2 gas. Characterization of the synthesized AB-type apatites was carried out by FTIR and XRD. The assignment of the carbonate peaks in the ν3 region of the FTIR spectra was strongly affected by the concomitant presence of both A and B carbonate in AB-CAp. Deconvolution of the ν3 region of the AB apatites indicate more than one orientation for B-type carbonate in the phosphate site of the apatite crystal structure. A-type and B-type carbonate weight fractions in the AB-type carbonated apatites were determined based on the total weight percent carbonate and the FTIR peak area ratios of the ν2 carbonate peaks. A-site carbonate levels correlate with the availability of hydroxide, which inversely correlates with B-site carbonate in the apatite prior to heating. Apatite with more A-type carbonate can therefore be prepared from samples with less initial B-type carbonate.
Separate and combined effects of carbonate in the A and B sites of AB carbonated apatites were examined. We found that substitutional doping of carbonate in apatite causes changes in the interatomic distances, which are reflected by changes in the lattice constants. For apatite samples prepared as described here, the crystal domain size is strongly affected by A-site carbonate substitution rather than by the total amount of A- and B-site carbonate in the apatite samples. Only increasing A-type carbonate resulted in smaller crystallite domains. We propose that with both A-type and B-type carbonate in the apatitic structure, the carbonate in the A-sites, within the c-axis channel, has a greater disruptive effect than B-type carbonate on the long range order of apatite. In the typical formation environment of biological apatites, below 100 °C, the dominance of B-type carbonate can be explained by a higher energy barrier associated with the formation of A-type carbonate.
We conclude that the two sites for carbonate substituition in apatite each control the crystal’s perfection and crystallite domain size, affecting material properites, in different environmental conditions. B-type carbonate dominates the apatitic formation of biominerals, whereas A-type carbonate controls the properties of apatitic minerals in non-physiological conditions.
Supplementary Material
Highlights.
Series of AB-type carbonated apatites with varying carbonate levels synthesized
FTIR indicates additional orientations for B-site CO32− with A-type substitution
Crystal domain lengths correlate only to A-site CO32− in high-temp AB-apatites
Acknowledgments
We thank Tommaso Costanzo for the XRD profile refinements using FullProf.2k suite, an unnamed reviewer for the helpful comments, and the National Institutes of Health for financial support under the grant AR056657.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.GLIMCHER MJ. Bone: nature of the calcium phosphate crystals and cellular, structural, and physical chemical mechanisms in their formation. Reviews in Mineralogy and Geochemistry. 2006;14(1):223–282. [Google Scholar]
- 2.BUDHIA S, MIKYAS Y, TANG M, BADAMGARAV E. Osteoporotic fractures: a systematic review of U.S. healthcare costs and resource utilization. Pharmacoeconomics. 2012;30:147. doi: 10.2165/11596880-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 3.WOPENKA B, PASTERIS JD. A mineralogical perspective on the apatite in bone. Mater Sci Eng C. 2005;25(2):131–143. [Google Scholar]
- 4.LEGEROS RZ, TRAUTZ OR, LEGEROS JP, KLEIN E, SHIRRA WP. Apatite crystallites: effects of carbonate on morphology. Science. 1967;155(3768):1409–1411. doi: 10.1126/science.155.3768.1409. http://doi.org/10.1126/science.155.3768.1409. [DOI] [PubMed] [Google Scholar]
- 5.PAN Y, FLEET ME. Compositions of the apatite-group minerals: substitution mechanisms and controlling factors. Rev Mineral Geochem. 2000;48(1):13–49. [Google Scholar]
- 6.LANDI E, CELOTTI G, LOGROSCINO G, TAMPIERI A. Carbonated hydroxyapatite as bone substitute. J European Ceramic Society. 2003;23(15):2931–2937. [Google Scholar]
- 7.BOSKEY AL, COLEMAN R. Aging and bone. J Dent Res. 2010;89(12):1333–1348. doi: 10.1177/0022034510377791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.ISON IC, FULMER MT, BARR BM, CONSTANTZ BR. Synthesis of dahlite: the mineral phase of bone. In: Brown PW, Constantz B, editors. Hydroxyapatite and related materials. CRC Press; 1994. p. 215. [Google Scholar]
- 9.MCCLELLAN GH, LEHR JR. Chemical investigation of apatites. Am Mineral. 1969;54(9–10):1374–1391. [Google Scholar]
- 10.MOMMA K, IZUMI F. Vesta: a three-dimensional visualization system for electronic and structural analysis. Journal of Applied crystallography. 2008;41:653–658. [Google Scholar]
- 11.SUDARSANA K, YOUNG RA. Significant precision in crystal structural details, Holly Springs hydroxyapatite. Acta Crystallographic Section B: Structural Science. 1969;B25:1534–1543. [Google Scholar]
- 12.ELLIOTT JC. vol. 18 of Studies in Inorganic Chemistry. Amsterdam: Elsevier; 1994. Structure and Chemistry of the Apatites and Other Calcium Orthophosphates. [Google Scholar]
- 13.KRAJEWSKI A, MAZZOCCHI M, BULDINI PL, RAVAGLIOLI A, ANNA T, TINTI P, FAGNANO C. Synthesis of carbonated hydroxyapatites: Efficiency of the substitution and critical evaluation of analytical methods. J Mol Structure. 2005;744–747:221–228. [Google Scholar]
- 14.LEGEROS RZ, TRAUTZ OR, KLIEN E, LEGEROS JP. Two types of carbonate substitution in the apatite structure. Experientia. 1969;25(1):5–7. doi: 10.1007/BF01903856. [DOI] [PubMed] [Google Scholar]
- 15.GIBSON I, BONFIELD W. Novel synthesis and characterization of an AB-type carbonate-substituted hydroxyapatite. Journal of Biomedical Materials Research. 2002;59(4):697–708. doi: 10.1002/jbm.10044. [DOI] [PubMed] [Google Scholar]
- 16.COMODI P, LIU Y. CO3 substitution in apatite: further insight from new crystal-chemical data of Kasekere (Uganda) apatite. Eur J Mineral. 2000;12(5):965–974. [Google Scholar]
- 17.LEVENTOURI T, CHAKOUMAKOS B, MOGHADDAM H, PERDIKATSIS V. Powder neutron diffraction studies of a carbonate hydroxyapatite. Thermal decomposition. J Mater Res. 2000;15(2):511–517. [Google Scholar]
- 18.ANTONAKOS A, LIAROKAPIS E, LEVENTOURI T. Micro-Raman and FTIR studies of synthetic and natural apatites. Biomaterials. 2007;28(19):3043–3054. doi: 10.1016/j.biomaterials.2007.02.028. [DOI] [PubMed] [Google Scholar]
- 19.TACKER RC. Carbonate in igneous and metamorphic fluorapatite: Two type A and two type B substitutions. Am Mineral. 2008;93(1):168–176. [Google Scholar]
- 20.REGNIER PL, BERNER RA, HAN O, ZIM K. Mechanism of (CO3) O2 Substitution in Carbonate-Fluorapatite- Evidence from FTIR spectroscopy, C-13 NMR, and Quantum Mechanical Calculations. Am Mineral. 1994;79(9–10):809–818. [Google Scholar]
- 21.IVANOVA T, FRANK-KAMENETSKAYA O, KOL’TSOV A, UGOLKOV V. Crystal structure of calcium deficient carbonated hydroxyapatites. Thermal decomposition. J Solid State Chem. 2001;160(2):340–349. [Google Scholar]
- 22.FLEET M, LIU X, KING P. Accommodation of the carbonate ion in apatite: An FTIR and X-ray structure study of crystals synthesized at 2–4 GPa. Am Mineral. 2004;89(10):1422–1432. [Google Scholar]
- 23.FLEET ME, LIU X. Accommodation of carbonate ion in fluorapatite synthesized at high pressure. Am Mineral. 2008;93(8–9):1460–1469. [Google Scholar]
- 24.CUISINIER FJG. Bone mineralization, Current Opinion in Solid State & Materials Science. 1996;1:436–439. [Google Scholar]
- 25.TONEGAWA T, IKOMA T, YOSHIOKA T, HANAGATA N, TANAKA J. Crystal structure refinement of A-type carbonate apatite by X-ray powder diffraction. Journal of Materials Science. 2010;45(9):2419–2426. [Google Scholar]
- 26.SUETSUGU Y, TAKAHASHI Y, OKAMURA F, TANAKA J. Structure analysis of A-type carbonate apatite by a single-crystal X-ray diffraction method. Journal of Solid State Chemistry. 2000;155(2):292–297. [Google Scholar]
- 27.PENEL G, LEROY G, REY C, BRES E. MicroRaman spectral study of the PO4 and CO3 vibrational modes in synthetic and biological apatites. Calcified Tissue International. 1998 Dec;63(6):475–481. doi: 10.1007/s002239900561. [DOI] [PubMed] [Google Scholar]
- 28.FLEET ME. Infrared spectra of carbonated apatites: Evidenced for a connection between bone mineral and body fluids. Am Mineral. 2017;102:149–157. [Google Scholar]
- 29.HAYEK E, NEWSELY H. Pentacalcium monohydroxyorthophosphate. Inorg Syn. 1963;7:63–65. [Google Scholar]
- 30.TROMBE J, BONEL G, MONTEL G. Structural analysis of carbonated apatites prepared at high temperatures. Bull Soc Chim Fr. 1968:1708–1711. [Google Scholar]
- 31.BEASLEY MM, BARTELINK EJ, TAYLOR L, MILLER RM. Comparison of transmission FTIR, ATR, and DRIFT spectra: implications for assessment of bone bioapatite diagenesis. J Archaeol Sci. 2014;46:16–22. [Google Scholar]
- 32.DODD JG, DENOYER LK. Curve-fitting: Modelling Spectra. In: CHALMERS J, GRIFFITHS P, editors. Handbook of vibrational spectroscopy. Vol. 3. J Wiley; New York: 2000. pp. 2215–2223. [Google Scholar]
- 33.RODRÍGUEZ-CARVAJAL J. Recent advances in magnetic structure determination by neutron powder diffraction. Phys B Condens Matter. 1993;192(1–2):55–69. [Google Scholar]
- 34.THOMPSON P, COX DE, HASTINGS JB. Rietveld refinement of Debye Scherrer synchrotron X-ray data from Al2O3. J Appl Crystallogr. 1987;20:79–83. [Google Scholar]
- 35.LE BAIL A. Whole powder pattern decomposition methods and applications. A retrospection, Powder Diffr. 2005;20(4):316–326. [Google Scholar]
- 36.LEGEROS RZ. Two types of carbonate substitution in the apatite structure. Experientia. 1969;25(1):5. doi: 10.1007/BF01903856. [DOI] [PubMed] [Google Scholar]
- 37.BONEL G. Contribution to study of apatites carbonation. 1. Synthesis and physiochemical properties of type A carbonated apatites. Annales de Chimie. 1972;7:65. [Google Scholar]
- 38.ELLIOTT JC. The interpretation of the infrared absorption spectra of some carbonate containing apatites. In: STACK MV, FEARNHEAD RW, editors. Tooth enamel. Its composition, properties and fundamental structure. Bristol: John Wright & Sons, Ltd; 1965. pp. 20-2–50-58. [Google Scholar]
- 39.FLEET ME. Infrared spectra of carbonate apatites. Biomaterials. 2009;30(8):1473–1481. doi: 10.1016/j.biomaterials.2008.12.007. http://doi.org/10.1016/j.biomaterials.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 40.TACKER RC. Hydroxyl ordering in igneous apatite. Am Mineral. 2004;89:1411–1421. [Google Scholar]
- 41.REN FZ, LENG Y. Carbonated Apatite, Type-A or Type-B? Key engineering materials. 2011;(493–494):293–297. [Google Scholar]
- 42.REY C, COLLINS B, GOEHL T, DICKSON IR, GLIMCHER MJ. The carbonate environment in bone mineral: a resolution-enhanced Fourier transform infrared spectroscopy study. Calcif Tissue Int. 1989;45:157–164. doi: 10.1007/BF02556059. [DOI] [PubMed] [Google Scholar]
- 43.Vignoles C. thése. L’ université de Paul Sabatier; Toulouse, France: 1973. Contribution à l’étude de l’influence des ions alcalins sur la carbonatation dans les sites de type B des apatites phosphor-calciques. [Google Scholar]
- 44.BAXTER JD, BILTZ RM, PELLEGRINO ED. The physical state of bone carbonate: A comparative infra-red study in several mineralized tissues. The Yale Journal of Biology and Medicine. 1966;38:456–470. [PMC free article] [PubMed] [Google Scholar]
- 45.DEYMIER AC, NAIR AK, DEPALLE B, QIN Z, ARCOT K, DROUET C, YODER CH, BUEHLER MJ, THROMOPOULOS S, GENIN GY, PASTERIS J. Protein-free formation of bone-like apatite: New insights into the key role of carbonation. Biomaterials. 2017;127:75–88. doi: 10.1016/j.biomaterials.2017.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.FLEET ME, LIU X, LIU XIAOYANG. Orientation of channel carbonate ions in apatite: Effect of pressure and composition. Am Mineral. 2011;96:1148–1157. [Google Scholar]
- 47.FLEET ME, LIU X. Coupled substitution of type A and B carbonate in sodium bearing apatite. Biomaterials. 2007;28(6):916–926. doi: 10.1016/j.biomaterials.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 48.FLEET ME, LIU X. Type A-B carbonate chlorapatite synthesized at high pressure. J Solid State Chem. 2008;181(9):2494–2500. [Google Scholar]
- 49.BARROUG A, REY C, TROMBE JC. Precipitation and formation mechanism of Type AB Carbonate apatites analogous to dental enamel. Adv Mater Res. 1994;1–2:147–154. http://doi.org/10.4028/www.scientific.net/AMR.1-2.147. [Google Scholar]
- 50.REY C, COMBES C, DROUET C, GROSSIN D. Bioactive ceramics: physical chemistry. In: DUCHEYNE P, HEALY KE, HUTMACHER DW, GRAINGER DW, KIRKPATRICK CJ, editors. Comprehensive Biomaterials. Elsevier; 2011. pp. 187–191. [Google Scholar]
- 51.LAFON JP, CHAMPION E, BERNACHE-ASSOLLANT D, GIBERT R, DANNA AM. Thermal decomposition of carbonated calcium phosphate apatites. J Therm Anal and Calorim. 2003;72(3):1127–1134. [Google Scholar]
- 52.TÕNSUAADU K, GROSS KA, PLUDUMA L, VEIDERMA M. A review on the thermal stability of calcium apatites. J Therm Anal and Calorim. 2012;110(2):647–659. [Google Scholar]
- 53.TACKER RC. Hydroxyl ordering in igneous apatite. Am Mineral. 2004;89:1411–1421. [Google Scholar]
- 54.MCELDERRY JDP, ZHU P, MROUE KH, XU J, PAVAN B, FANG M, ZHAO G, MCNERNY E, KOHN DH, FRANCESCHI RT, BANASZAK HOLL MM, TECKLENBURG MMJ, RAMAMOORTHY A, MORRIS MD. Crystallinity and compositional changes in carbonated apatites: Evidence from 31P solid-state NMR, Raman, and AFM analysis. J Solid State Chem. 2013;206:192–198. doi: 10.1016/j.jssc.2013.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.CAZALBOU S, EICHERT D, RANZ X, DROUET C, COMBES C, HARMAND MF, REY C. Ion Exchange in apatites for biomedical applications. J Mater Sci Mater Med. 2005;16:405–409. doi: 10.1007/s10856-005-6979-2. [DOI] [PubMed] [Google Scholar]
- 56.TÕNSUAADU K, PELD M, BENDER V. Thermal analysis of apatite structure. J Therm Anal and Calorim. 2003;72:363–371. [Google Scholar]
- 57.BARINOV SM, RAU JV, CESARO SN, ĎURIŠIN J, FADEEVA IV, FERRO D, TRIONFETTI G. Carbonate release from carbonated hydroxyapatite in the wide temperature range. J Mater Sci Mater Med. 2006;17(7):597–604. doi: 10.1007/s10856-006-9221-y. [DOI] [PubMed] [Google Scholar]
- 58.MILENKO M, BRUCE OF, MING ST. Preparation and Comprehensive characterization of a calcium hydroxyapatite reference material. J Res Natl Inst Stand Technol. 2004;109:553–568. doi: 10.6028/jres.109.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.PEROOS S, DU Z, DELEEUW NH. A computer modelling study of the uptake, structure and distribution of carbonate defects in hydroxy-apatite. Biomaterials. 2006;27(9):2150–2161. doi: 10.1016/j.biomaterials.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 60.SCHEVILL WE, WATKINS WA. Effect of carbonate on the lattice parameters of apatite. Nature. 1965;206:403. doi: 10.1038/206403a0. [DOI] [PubMed] [Google Scholar]
- 61.STOKES AR, WILSON AJC. The diffraction of x-rays by distorted crystal aggregates – I. Proc Cambridge Philos Soc. 1944;56(3):174–181. [Google Scholar]
- 62.GAO HJ, JI BH, JAGER IL, ARTZ E, FRATZL P. Materials become insensitive to flaws at nanoscale: lessons from nature. PNAS. 2003;100(10):5597–5600. doi: 10.1073/pnas.0631609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.