

Supplemental Digital Content is available in the text
Keywords: acute liver failure, bioinformatical analysis, coexpression network analysis, hepatitis B virus, protein–protein interaction network
Abstract
Hepatitis B virus-associated acute liver failure (HBV-ALF) is a rare but life-threatening syndrome that carried a high morbidity and mortality. Our study aimed to explore the possible molecular mechanisms of HBV-ALF by means of bioinformatics analysis. In this study, genes expression microarray datasets of HBV-ALF from Gene Expression Omnibus were collected, and then we identified differentially expressed genes (DEGs) by the limma package in R. After functional enrichment analysis, we constructed the protein–protein interaction (PPI) network by the Search Tool for the Retrieval of Interacting Genes online database and weighted genes coexpression network by the WGCNA package in R. Subsequently, we picked out the hub genes among the DEGs. A total of 423 DEGs with 198 upregulated genes and 225 downregulated genes were identified between HBV-ALF and normal samples. The upregulated genes were mainly enriched in immune response, and the downregulated genes were mainly enriched in complement and coagulation cascades. Orosomucoid 1 (ORM1), orosomucoid 2 (ORM2), plasminogen (PLG), and aldehyde oxidase 1 (AOX1) were picked out as the hub genes that with a high degree in both PPI network and weighted genes coexpression network. The weighted genes coexpression network analysis found out 3 of the 5 modules that upregulated genes enriched in were closely related to immune system. The downregulated genes enriched in only one module, and the genes in this module majorly enriched in the complement and coagulation cascades pathway. In conclusion, 4 genes (ORM1, ORM2, PLG, and AOX1) with immune response and the complement and coagulation cascades pathway may take part in the pathogenesis of HBV-ALF, and these candidate genes and pathways could be therapeutic targets for HBV-ALF.
1. Introduction
Hepatitis B virus infection is a global health problem. There are approximately 2 billion people infected with hepatitis B virus (HBV) worldwide, of which 350 million were chronic infected.[1–3] The Chinese national HBV epidemiological survey in 2006 showed that 7.18% of the population were HBV carriers. It is estimated that the HBV-infected patients in China have reached a staggering number of 93 million. And 15% to 40% of HBV chronic-infected patients would develop cirrhosis, or even liver failure and cancer.[4,5] Therefore, the HBV infection-associated liver failure, which is a lethal disease with a high morbidity, should be studied intensively.[6,7]
Acute liver failure is a rare but life-threatening syndrome with a rapid deterioration of liver function and failure of multiorgan.[8–10] A variety of diseases, including ischemia, metabolic disorders, Wilson's disease, autoimmune hepatitis, and intoxications, could lead to the associated acute liver failure (ALF).[11,12] In the developing world including China, HBV infection is the main cause of ALF.[12,13] Patients with ALF would develop a number of complications, including pulmonary or cardiac failures, renal failures, hepatic encephalopathy, and so on.[14] There is still an absence of effective therapies for ALF, in addition to liver transplantation. However, shortage of donor organs, difficulty of the transplant operation, and substantial postoperative complications limit the application of liver transplantation. Therefore, the present knowledge of hepatitis B virus-associated acute liver failure (HBV-ALF) is still insufficient.
The gene array technologies have been a powerful and widespread tool to study the pathogenesis of complex human diseases for years. Nevertheless, there are rare studies of HBV-ALF with gene expression profiling. The present study was performed to explore the molecular mechanism of HBV-ALF by bioinformatical analysis. The genes expression microarray datasets of ALF from Gene Expression Omnibus (GEO) were collected, and then we identified differentially expressed genes between ALF samples and normal samples. Subsequently, we performed the functional enrichment analysis and constructed the protein-protein interaction (PPI) and coexpression networks to explore the underlying genes and pathways associated with HBV-ALF.
2. Materials and methods
2.1. Data source
The microarray datasets (GSE38941, GSE62029, GSE62030, and GSE14668), which were related to HBV-ALF, were extracted from GEO for analysis. Search terms included “hepatitis B virus” and “acute liver failure.” Platforms of all 4 sets of gene expression profiles were the same (GPL570 Affymetrix Human Genome U133 plus 2.0 Array) that could make the analysis process convenient and credible. Three microarray datasets (GSE38941, GSE62029, and GSE14668) were utilized for the analysis of differentially expressed genes (DEGs) between HBV-associated acute liver failure tissue and normal tissue. Another set of gene expression profile (GSE62030) was added for the construction of the microRNA-gene network. All the microarray data were summarized into gene-level information, and the probe names were transformed into gene symbols. If one gene was detected by >1 probe, we would take the mean value as its expression level. Then all expression data were log2-transformed in the normalization process.
The limma package in R was utilized to screen out the DEGs in the 3 microarray datasets (GSE38941, GSE62029, and GSE14668), respectively. It is the most popular method for the DEGs analysis. Only the genes met the criteria of |fold change| ≥2 and false discovery rate (FDR) <0.01 could be defined as the DEGs. Then we picked out the overlapped DEGs from 3 different datasets for the subsequent analysis by the means of Venn analysis.
2.2. Gene Ontology annotation and pathway analysis
Gene Ontology (GO) analysis, which comprises 3 independent ontologies (cellular component, molecular function, and biological process), is increasingly applied for functional studies of transcriptomic data. The Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.8 was utilized for the GO analysis. DAVID was an easy-to-use software with multifunction tools that integrated specific functions of the genes. The Kyoto Encyclopedia of Genes and Genomes (KEGG) knowledge database is an online databases of biochemistry pathways. The clusterProfiler package in R was utilized to annotate and visualize the KEGG pathways of DEGs. We selected GO terms and pathways that the DEGs mainly enriched in with a cutoff criteria of FDR <0.05.
2.3. Construction of PPI network
The Search Tool for the Retrieval of Interacting Genes (STRING) database is a global resource of PPI information (http://string-db.org). The PPI network was constructed with the DEGs mapped into STRING database. The species for PPI analysis was set as human with the interaction score ≥0.4. The nodes in the PPI network represented the genes, and the edges between the nodes represented the interactions between the genes. The number of edges linked to a given gene was defined as the degree of that gene, and only those experimentally validated interactions (edges) were utilized in this analysis. The gene with a high degree was deemed as the hub gene with an essential biological function.
2.4. Meta-analysis of expression level of the hub DEGs
To confirm the reliability of the identified hub genes, the relative expression level of the hub genes between the disease and normal group was compared by the means of meta-analysis. This meta-analysis was conducted by RevMan software version 5.3 (the Nordic Cochrane Centre, Cochrane Collaboration, Copenhagen, Denmark). Microarray data were converted into a log2-transformed scale, subsequently the mean and standard deviation of the data were pooled for analysis. Heterogeneity among the included data was qualitatively evaluated using x2-based Q test. P <.05 showed that there was statistically significant heterogeneity across the studies. Random effects model was applied for all analysis process.
2.5. Weighted genes coexpression network analysis
To further investigate the biological functions of the DEGs, we performed the genes coexpression network analysis through the WGCNA package in R. WGCNA, as a typical systemic biological arithmetic in the construction of genes coexpression network, has been widely used in bioinformatical analysis. The theory of how the coexpression network constructed was similar to that of previous studies.[15] First, we calculated the Pearson's correlation coefficient value of the gene–gene pairs of all samples. Second, we got an unsigned similarity matrix of gene coexpression with the threshold of the correlation coefficient set as 0.8. Third, the similarity matrix was transformed into a weighted adjacency matrix before that was transformed into a topology matrix. Then, the gene modules of highly correlated genes were detected based on the topology matrix. The coexpression modules were named as the color assigned by WGCNA. The degree of the genes was defined by the way that similar to the PPI analysis and the genes with a high degree of each module were defined as the hub genes. To better understand the biological function of each module, we performed GO or pathway analysis in the genes that belong to the same module.
2.6. The construction of microRNAs-genes network
The construction of microRNAs-genes network was completed by 4 steps. First, we used the gene expression profile (GSE62030) to screen out the differentially expressed microRNAs (DEmiRNAs) between the disease and normal group. The inclusion criteria were set as that |fold change| ≥1 and FDR <0.01. Second, we explored the target genes of DEmiRNAs from 3 different database (starBase, Targetscan, and miRDB), and only the overlapped target genes of 3 database were picked out. Third, we found out the overlapped genes between the target genes of DEmiRNAs and the DEGs that selected above as the target DEGs. Finally, the DEmiRNAs and the target DEGs were utilized to construct the microRNAs-genes network through the Cytoscape software.
2.7. Connectivity Map analysis
To date, studies for the treatment of acute liver failure were still insufficient. DEGs could be utilized to identify new therapeutic effect of drugs by the software of Connectivity Map. The Connectivity Map could pick out the drugs which induced the same or opposite gene expression patterns with DEGs by therapeutic scores. Drugs with negative scores had opposite gene expression patterns comparing with our microarray data, therefore might have a therapeutic effect. The inclusion criteria for the drugs selection were set as P < .05.
2.7.1. Ethics
All analyses were based on the data from public database, so ethics approval and patient consent were not required.
3. Results
3.1. Datasets characteristics and screening of differentially expressed genes
Four sets of microarray data (GSE38941, GSE62029, GSE62030, and GSE14668) were obtained from the GEO database with the detail in Table 1. The GSE62029, GSE62030, and GSE14668 contained liver samples from the patients with HBV-ALF, liver donors, and subjects that underwent resection for liver angioma. But the GSE38941 only contained samples from the patients with HBV-ALF and liver donors. Therefore, the data of samples from subjects that underwent resection for liver angioma were excluded. After the data preprocessing and normalization, data from 3 datasets (GSE38941, GSE62029, and GSE14668) were used for the screening of DEGs. Eventually, 624 DEGs, 585 DEGs, and 1395 DEGs were selected from GSE38941, GSE62029, and GSE14668, respectively (Fig. 1). A total of 423 DEGs with 198 upregulated genes and 225 downregulated genes were commonly changed in the 3 microarray datasets (Fig. 2). The DEGs were listed in Table 2. In addition, we presented a list of validated genes from previous studies in the Supplementary Table 1.
Table 1.
Four datasets included in the present study.
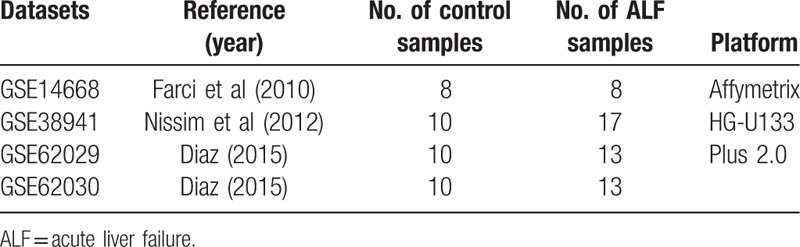
Figure 1.

Volcano plots representation of differential expression analyses. The criteria for selection of DEGs was set as |fold change| ≥2 and adjusted P <.01.
Figure 2.
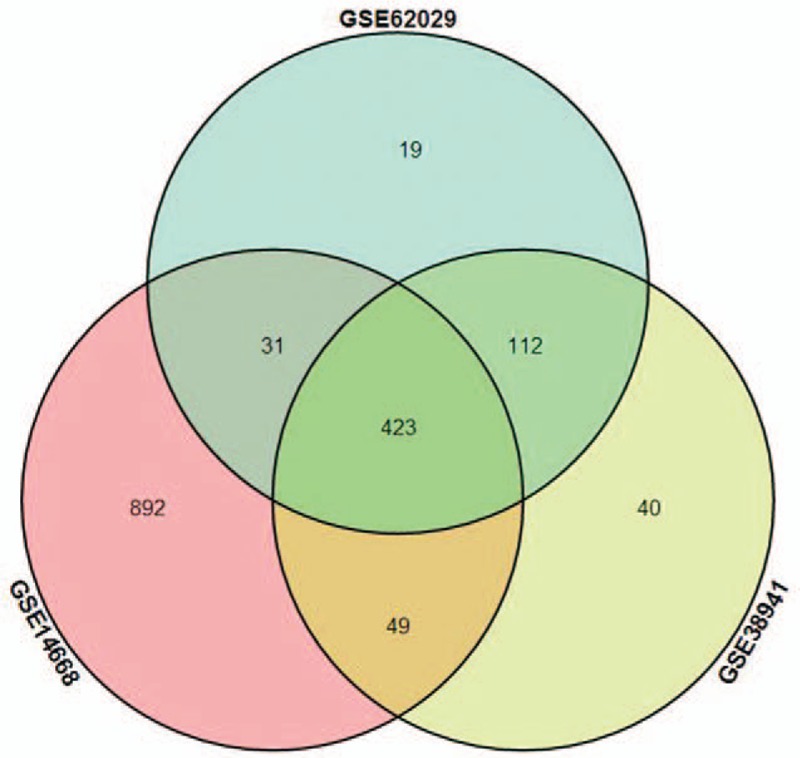
The Venn plot of DEGs. There were a total of 423 overlapped genes from 3 different datasets.
Table 2.
The list of 423 DEGs (198 upregulated genes and 225 downregulated genes).

3.2. GO annotation and pathway analysis
As shown in Table 3 and Figure 3, the top 10 GO terms which upregulated genes majorly enriched in were immune response, defense response, extracellular region, antigen processing, and presentation of peptide or polysaccharide antigen via MHC class II, cellular defense response, immune effector process, extracellular region part, inflammatory response, positive regulation of immune system process, and antigen processing and presentation. GO analysis illustrated that immune response which contained humoral and cellular immunity played an important role in mediating or inducing the damage to the liver due to hepatitis B in ALF. The same way with DAVID, the top 10 GO terms which downregulated genes majorly enriched in were response to wounding, acute inflammatory response, extracellular region, extracellular space, inflammatory response, oxidation reduction, electron carrier activity, complement activation, extracellular region part, and activation of plasma proteins involved in acute inflammatory response.
Table 3.
The top 10 most enriched GO terms in this study.
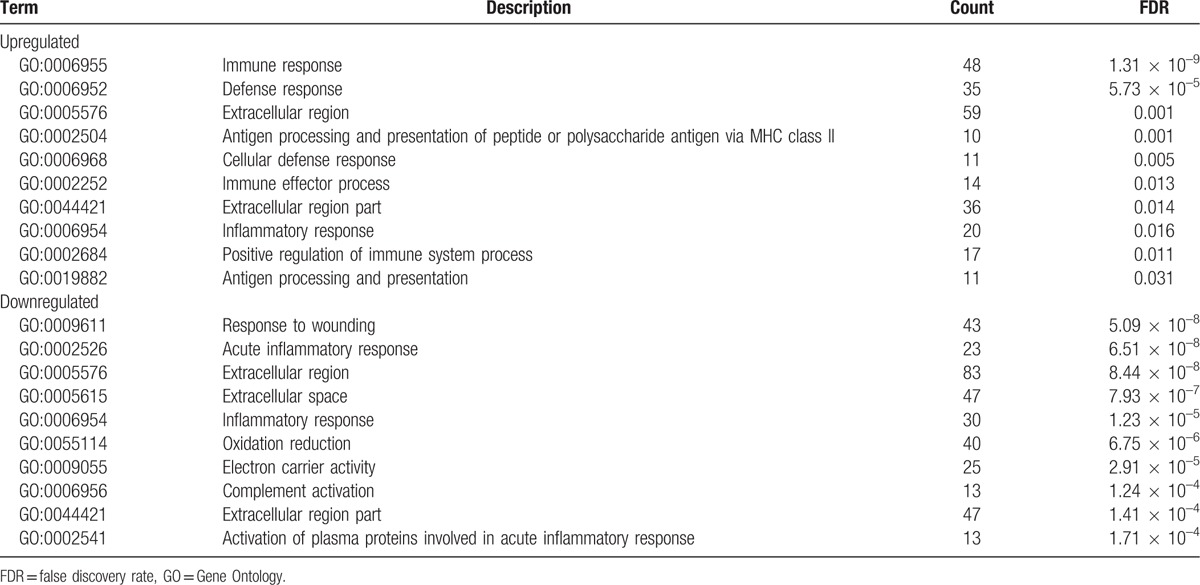
Figure 3.
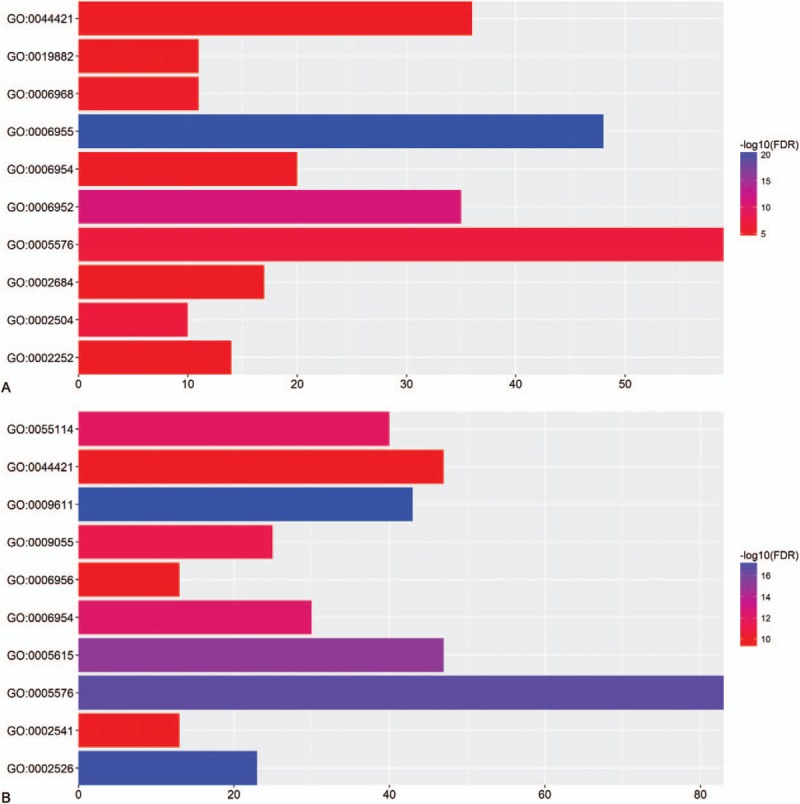
The top 10 most enriched GO terms of (A) upregulated genes and (B) downregulated genes.
As shown in Table 4 and Figure 4, the pathways that the upregulated genes majorly enriched in were antigen processing and presentation, cell adhesion molecules, cytokine–cytokine receptor interaction, hematopoietic cell lineage, and chemokine signaling pathway. And the pathways that the downregulated genes majorly enriched in were complement and coagulation cascades, metabolism of xenobiotic by cytochrome P450, drug metabolism, arachidonic acid metabolism, and tryptophan metabolism.
Table 4.
The top 5 most enriched pathway in this study.
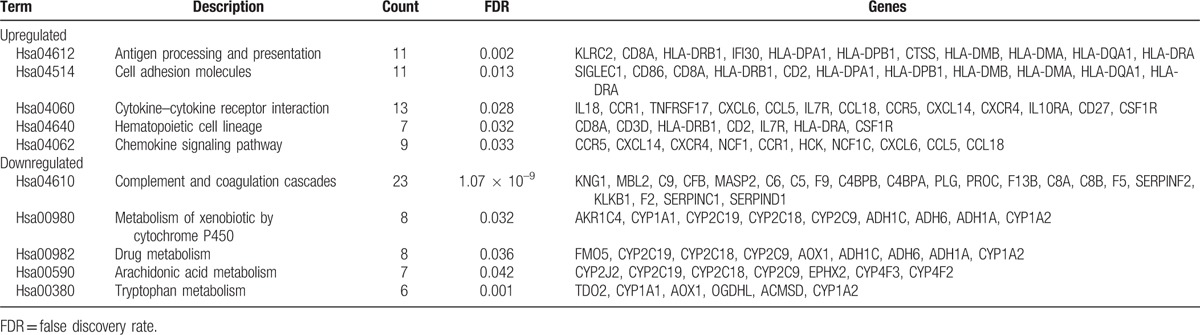
Figure 4.
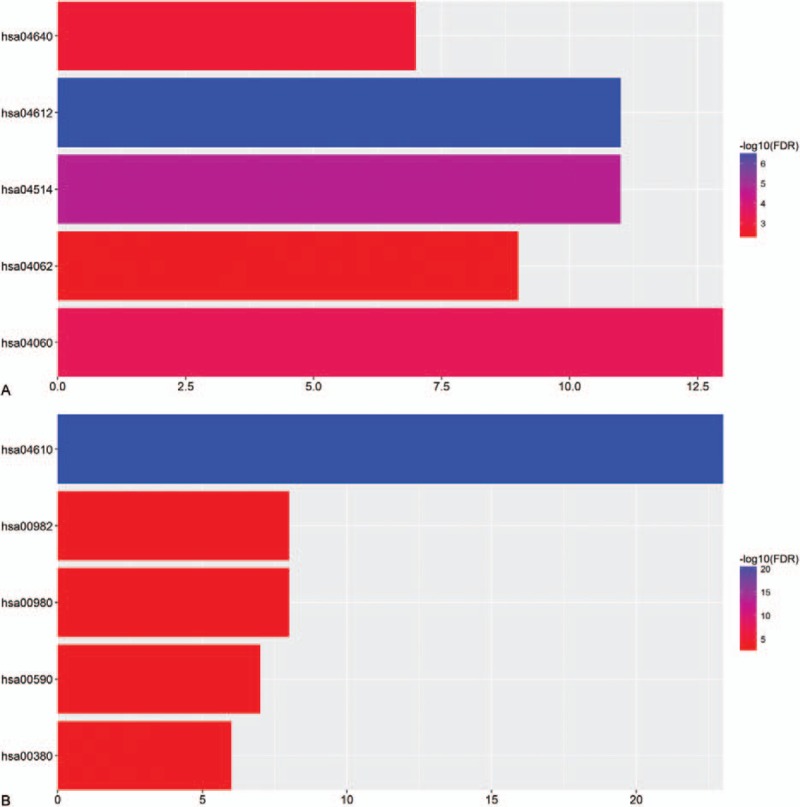
The top 5 most enriched KEGG pathways of (A) upregulated genes and (B) downregulated genes.
3.3. The PPI network analysis and meta-analysis of expression level of the hub DEGs
All the 423 DEGs were utilized to construct the PPI network. After excluded the disconnected genes in the network, a total of 195 genes with 454 pairs of interactions were mapped in the PPI network by the used of the STRING database (Fig. 5). The number of upregulated genes were similar to that of downregulated genes. The most significant 10 node-degree genes were kininogen 1 (KNG1), coagulation factor V (F5), insulin-like growth factor 1 (IGF1), plasminogen (PLG), major histocompatibility complex, class II, DR alpha (HLA-DRA), major histocompatibility complex, class II, DR beta 1 (HLA-DRB1), serpin family F member 2 (SERPINF2), aldehyde oxidase 1 (AOX1), complement C5 (C5), and complement factor D (CFD). As shown in Figure 6, we used the means of meta-analysis to test the expression level of the 10 genes between the disease and normal group. The expression levels of all the 10 genes in HBV-associated ALF group were significantly higher than that in the control group.
Figure 5.
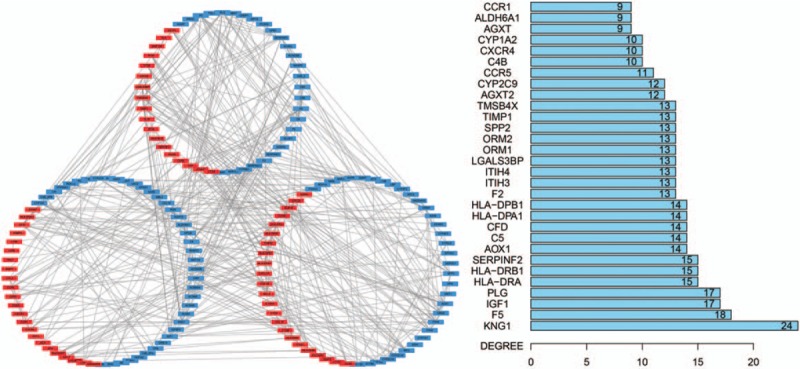
The PPI network. The left panel showed the interactions between the upregulated (red nodes) and downregulated (blue nodes) genes. The right panel showed the degree of the genes, only top 30 genes were listed out.
Figure 6.
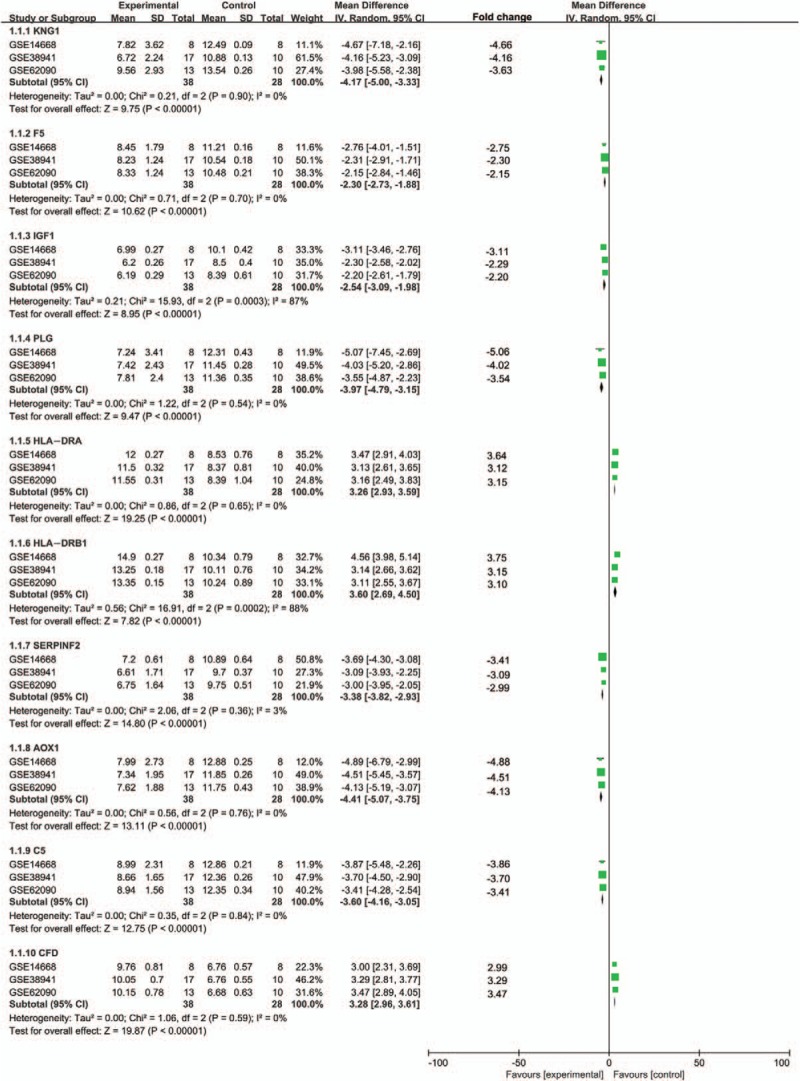
Meta-analysis of expression level of top ten hub genes in PPI network (KNG1, F5, IGF1, PLG, HLA-DRA, HLA-DRB1, SERPINF2, AOX1, C5, and CFD).
3.4. Construction of the weighted genes coexpression network
The WGCNA analysis for the DEGs was performed in all HBV-ALF samples. As shown in Figure 7, the upregulated genes were majorly enriched in 5 modules. The hub genes were put in the center of each module. However, the hub genes in the coexpression network were not similar to those in the PPI network analysis. The reason might be that the hub genes in the PPI analysis were majorly downregulated ones; therefore, the hub upregulated genes were not better represented in the PPI analysis. To understand the biological meaning of the modules, we performed functional enrichment analyses in each module. The upregulated genes were majorly enriched in the antigen processing and presentation pathway or GO terms that were immune response related. The downregulated genes were enriched in only 1 module, the blue module. As shown in Figure 8, the hub genes of this module were serpin family D member 1 (SERPIND1), hydroxysteroid 17-beta dehydrogenase 6 (HSD17B6), orosomucoid 1 (ORM1), PLG, orosomucoid 2 (ORM2), solute carrier organic anion transporter family member 1B1 (SLCO1B1), AOX1, and hydroxyacid oxidase 1 (HAO1). And ORM1, ORM2, PLG, and AOX1 were the hub genes of the PPI network at the same time. Therefore, we defined this 4 genes as the hub genes of this study. The functional enrichment analyses revealed that the genes of the blue module were majorly enriched in the complement and coagulation cascades pathway.
Figure 7.
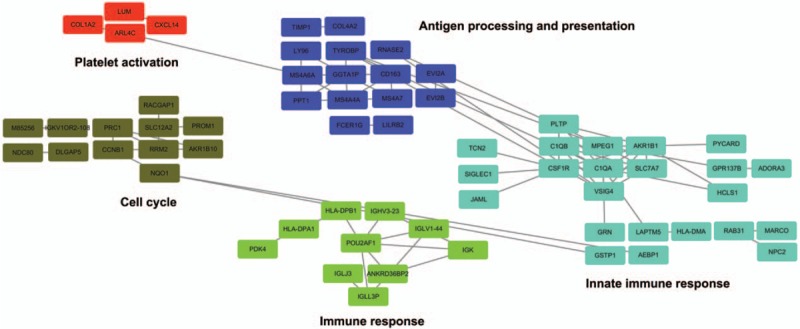
The weighted genes coexpression network of upregulated DEGs. There were 5 modules that the upregulated DEGs enriched in. Each module was named as the color assigned by the WGCNA analysis.
Figure 8.
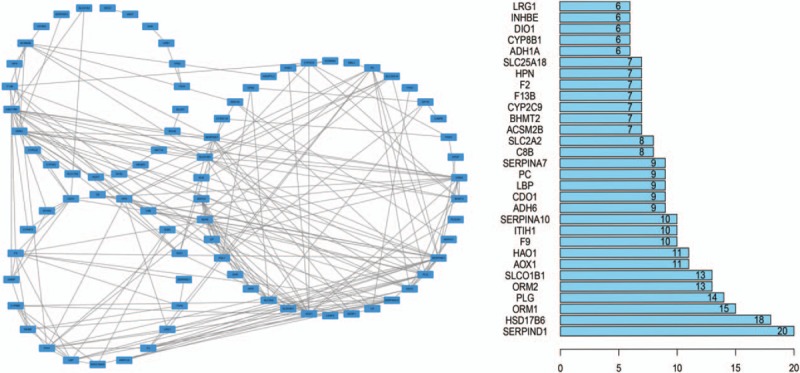
The weighted genes coexpression network of downregulated DEGs (the blue module of WGCNA analysis). The left panel showed the coexpression interactions between the genes. The right panel showed the degree of the genes, only top 30 genes were listed out.
3.5. The microRNAs-genes network and Connectivity Map analysis
With the use of the GSE62030, we selected out 114 DEmiRNAs. There were 70 upregulated miRNAs and 44 downregulated miRNAs among the DEmiRNAs (Table 5). The target genes of DEmiRNAs were identified by the simultaneous use of 3 databases. However, only 20 genes among the target genes were belong to the DEGs. Therefore, those 20 DEGs and 23 DEmiRNAs were utilized to construct this microRNAs-genes network (the DEmiRNAs that did not have regulatory relationships with the DEGs were excluded). Figure 9 revealed how the DEmiRNAs regulate their target genes.
Table 5.
The list of 114 DEmiRNAs (70 upregulated miRNAs and 44 downregulated miRNAs).

Figure 9.

The microRNAs-genes network. The green nodes represented the DEmiRNAs and the blue nodes were the target genes of the DEmiRNAs.
For the Connectivity Map analysis, a total of 29 drugs met the inclusion criteria, Figure 10 showed the top 20 drugs that the DEGs enriched in. The drugs with positive scores had a better rank than drugs with negative scores. The drugs such as ciprofibrate and thiamine could induce the same gene expression patterns as HBV-ALF; therefore, those drugs should be avoid in the treatment of HBV-ALF. And the drugs such as chlorphenesin that with the opposite gene expression patterns might have a therapeutic effect to the HBV-ALF.
Figure 10.
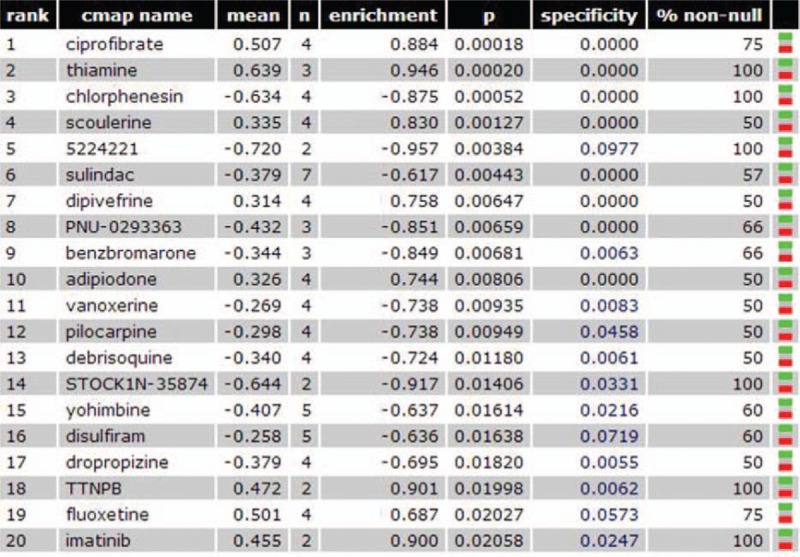
Connectivity Map analysis. The mean was the computed score. A positive score indicates that the drug exhibits an expression pattern that is synergistic with the disease. A negative score indicates that the drug exhibits an expression pattern that is opposite to the disease.
4. Discussion
In this study, the genetic profiles of the HBV-ALF samples and normal samples were compared with select the DEGs. Eventually, 423 DEGs with 198 upregulated genes and 225 downregulated genes were selected out. The enrichment analysis revealed that the upregulated DEGs majorly enriched in immune response. Meanwhile, 2 of the hub genes of the PPI network (HLA-DRA, HLA-DRB1) have been evaluated to play central roles in the immune system. And the weighted genes coexpression network analysis found out 3 of the 5 modules that upregulated genes enriched in were closely related to immune system. Several previous studies have evaluated the primary role of immune response in the pathogenesis of HBV-ALF.[10,16] Farcia et al demonstrated ALF resulted from HBV was mediated by intrahepatic B-cell response against the core antigen of HBV. And in their study, massive IgG and IgM secreted by plasma cells accumulated in liver showed unsubstitutive role of humoral immunity in HBV-ALF.[16]
The enrichment analysis of downregulated DEGs revealed that the downregulated genes majorly enriched in the complement and coagulation cascades pathway. And most of the hub genes in the PPI network were downregulated and coagulation related.
The downregulated genes enriched in only one module in the weighted genes coexpression network analysis, and the genes in this module also majorly enriched in the complement and coagulation cascades pathway. Meanwhile, PLG, which were hub genes in both PPI network and weighted genes coexpression network, was closely related to the coagulation. Therefore, the complement and coagulation cascades pathway might be necessary in the pathogenesis of HBV-ALF. The focus of the early studies regarding HBV-ALF was the viral factors and virus–host interactions. The relationship between the genomic variant of HBV DNA (Pre-C and BCP mutants) and pathogenesis of the HBV-ALF has been widely studied.[17–21] Subsequently, the vital role of hosts’ immune response (host factors that containing HBV-specific cytokine, chemokine profiles, and plasma cells) in the occurrence of HBV-ALF was discovered and studied.[10,22,23] The present study revealed the complement and coagulation cascades might have a future filed in the study of HBV-ALF.
The important role of inflammatory cytokines in liver injury has been revealed in several studies,[24–26] and quite a few of cytokines-related genes were contained in DEGs such as CCL5, CCR1, CCR5, and IL18. Previous studies demonstrated the significant influence of cytokines on hepatocyte regeneration, extrahepatic complications, and hepatocellular death.[27,28] FasL has been evaluated to be associated with liver injury by induction and triggering of destruction of liver in chronic and acute-on-chronic liver diseases,[24,29,30] whereas the expression level of that in our HBV-ALF samples was not upregulated significantly. The same situation was also observed in the expression of some other genes which were related to the encoding of interleukin (IL-1, IL-6, and IL-10). Several studies reported the CXC subfamily of chemokine involved in the inflammatory processes by triggering of the accumulation of neutrophils and caused acute injury.[31,32] However, whether that plays an important role in acute liver injury remains elusive. What give us some implications in our study is the upregulation of the CXC subfamily encoding-associated genes (CXCL14, CXCL16, and CXCR4).
In recent years, new perspective on T cell-mediated liver injury has arisen along with the emergence of T-helper 17 cells (Th17), a new subset of CD4+ T cells. Several studies elucidated the Th17 cells increased significantly in HBV-associated acute-on-chronic liver failure patients that could recruite immune cells to mediate inflammation and immune injury via IL 17.[33] The differentiation and maturation of Th17 are majorly rely on IL6, IL21, TGF-β, and mTOR/STAT3.[34,35] Nevertheless, we did not find out the appropriate environment for the differentiation of Th17, owing to neither IL6 nor IL21 was upregulated in HBV-ALF samples. Therefore, it is not weird that IL17 did not emerge in the DEGs and the Th17 seem to matter less in HBV-ALF.
The miRNAs can bind to specific target sites of messenger RNAs and silence or destabilize them.[36] The present study selected out the aberrant regulated miRNAs between the HBV-ALF and normal samples and constructed the microRNAs-genes network. However, our microRNAs-genes network analysis evaluated that the hub genes were not directly regulated by the DEmiRNAs. Therefore, the role of miRNAs in HBV-ALF should be further studied. The Connectivity Map analysis identified several drugs which could induce the opposite or similar gene expression patterns as HBV-ALF. But the effect of reversing the gene expression patterns by drugs cannot be determined, due to the genes that might be protective in liver failure. And if the genes were protective in liver failure need further experiments to find out. This is a limitation of the Connectivity Map analysis for this study. Therefore, whether those drugs will be able to assist the treatment of liver failure in the future remains to be verified. In addition, due to an absence of non-HBV-ALF as a control in this study, it is difficult to evaluate which genes are HBV related and which are ALF related. This is another limitation of the present study. What is more, we could not figure out if the interactions in the PPI network were direct or indirect. This was also one of the limitations of the present study.
In summary, the present study found out 4 genes (ORM1, ORM2, PLG, and AOX1) with immune response, and the complement and coagulation cascades pathway may take part in the pathogenesis of HBV-ALF, and these candidate genes and pathways could be therapeutic targets for HBV-ALF. However, it is not sufficient to explore the possible molecular mechanisms of HBV-associated ALF by means of bioinformatics merely. Functional experiments should be added to verify our results, including western blot confirmation, luciferase report assay, and gain or loss of genes function.[37] Thus, the follow-up experiment of us will aim to pick up the hub genes and carry experiments to unveil the mechanisms involved in the HBV-ALF.
Supplementary Material
Footnotes
Abbreviations: AOX1 = aldehyde oxidase 1, C5 = complement C5, CFD = complement factor D, DAVID = Database for Annotation, Visualization and Integrated Discovery, DEGs = differentially expressed genes, DEmiRNAs = differentially expressed microRNAs, F5 = coagulation factor V, FDR = false discovery rate, GEO = Gene Expression Omnibus, GO = Gene Ontology, HAO1 = hydroxyacid oxidase 1, HBV-ALF = hepatitis B virus-associated acute liver failure, HLA-DRA = major histocompatibility complex, class II, DR alpha, HLA-DRB1 = major histocompatibility complex, class II, DR beta 1, HSD17B6 = hydroxysteroid 17-beta dehydrogenase 6, IGF1 = insulin-like growth factor 1, KEGG = Kyoto Encyclopedia of Genes and Genomes, KNG1 = kininogen 1, ORM1 = orosomucoid 1, ORM2 = orosomucoid 2, PLG = plasminogen, PPI = protein–protein interaction, SERPIND1 = serpin family D member 1, SERPINF2 = serpin family F member 2, SLCO1B1 = solute carrier organic anion transporter family member 1B1, STRING = Search Tool for the Retrieval of Interacting Genes, Th17 = T-helper 17 cells.
This research was supported by the Foundation for Talents of the Affiliated Hospital of Southwest Medical University (12263).
The authors have no conflicts of interest to disclose.
Supplemental Digital Content is available for this article.
References
- [1].Franco E, Bagnato B, Marino MG, et al. Hepatitis B: epidemiology and prevention in developing countries. World J Hepatol 2012;4:74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Liu J, Fan D. Hepatitis B in China. Lancet 2007;369:1582–3. [DOI] [PubMed] [Google Scholar]
- [3].Zheng MH, Wu SJ, Shi KQ, et al. Conditional survival estimate of acute-on-chronic hepatitis B liver failure: a dynamic prediction based on a multicenter cohort. Oncotarget 2015;6:23261–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Escorsell A, Mas A, de la Mata M. Acute liver failure in Spain: analysis of 267 cases. Liver Transpl 2007;13:1389–95. [DOI] [PubMed] [Google Scholar]
- [5].Koskinas J, Deutsch M, Kountouras D, et al. Aetiology and outcome of acute hepatic failure in Greece: experience of two academic hospital centres. Liver Int 2008;28:821–7. [DOI] [PubMed] [Google Scholar]
- [6].Wu Z, Han M, Chen T, et al. Acute liver failure: mechanisms of immune-mediated liver injury. Liver Int 2010;30:782–94. [DOI] [PubMed] [Google Scholar]
- [7].Polson J, Lee WM. AASLD position paper: the management of acute liver failure. Hepatology 2005;41:1179–97. [DOI] [PubMed] [Google Scholar]
- [8].Bernuau J, Rueff B, Benhamou JP. Fulminant and subfulminant liver failure: definitions and causes. Semin Liver Dis 1986;6:97–106. [DOI] [PubMed] [Google Scholar]
- [9].O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet 1993;342:273–5. [DOI] [PubMed] [Google Scholar]
- [10].Nissim O, Melis M, Diaz G, et al. Liver regeneration signature in hepatitis B virus (HBV)-associated acute liver failure identified by gene expression profiling. PLoS One 2012;7:e49611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ostapowicz G, Fontana RJ, Schiodt FV, et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002;137:947–54. [DOI] [PubMed] [Google Scholar]
- [12].Volkmann X, Anstaett M, Hadem J, et al. Caspase activation is associated with spontaneous recovery from acute liver failure. Hepatology 2008;47:1624–33. [DOI] [PubMed] [Google Scholar]
- [13].Lee WM. Etiologies of acute liver failure. Semin Liver Dis 2008;28:142–52. [DOI] [PubMed] [Google Scholar]
- [14].Stravitz RT, Kramer DJ. Management of acute liver failure. Nat Rev Gastroenterol Hepatol 2009;6:542–53. [DOI] [PubMed] [Google Scholar]
- [15].Guo X, Xiao H, Guo S, et al. Identification of breast cancer mechanism based on weighted gene coexpression network analysis. Cancer Gene Ther 2017;24:333–41. [DOI] [PubMed] [Google Scholar]
- [16].Farci P, Diaz G, Chen Z, et al. B cell gene signature with massive intrahepatic production of antibodies to hepatitis B core antigen in hepatitis B virus-associated acute liver failure. Proc Natl Acad Sci USA 2010;107:8766–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Carman WF, Fagan EA, Hadziyannis S, et al. Association of a precore genomic variant of hepatitis B virus with fulminant hepatitis. Hepatology 1991;14:219–22. [PubMed] [Google Scholar]
- [18].Kosaka Y, Takase K, Kojima M, et al. Fulminant hepatitis B: induction by hepatitis B virus mutants defective in the precore region and incapable of encoding e antigen. Gastroenterology 1991;100:1087–94. [DOI] [PubMed] [Google Scholar]
- [19].Liang TJ, Hasegawa K, Rimon N, et al. A hepatitis B virus mutant associated with an epidemic of fulminant hepatitis. N Engl J Med 1991;324:1705–9. [DOI] [PubMed] [Google Scholar]
- [20].Ehata T, Omata M, Chuang WL, et al. Mutations in core nucleotide sequence of hepatitis B virus correlate with fulminant and severe hepatitis. J Clin Invest 1993;91:1206–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Laskus T, Rakela J, Nowicki MJ, et al. Hepatitis B virus core promoter sequence analysis in fulminant and chronic hepatitis B. Gastroenterology 1995;109:1618–23. [DOI] [PubMed] [Google Scholar]
- [22].Tripathy AS, Das R, Chadha MS, et al. Epidemic of hepatitis B with high mortality in India: association of fulminant disease with lack of CCL4 and natural killer T cells. J Viral Hepat 2011;18:e415–22. [DOI] [PubMed] [Google Scholar]
- [23].Zhang Z, Zhang JY, Wherry EJ, et al. Dynamic programmed death 1 expression by virus-specific CD8 T cells correlates with the outcome of acute hepatitis B. Gastroenterology 2008;134: 1938–1949, 1949.e1931–1933. [DOI] [PubMed] [Google Scholar]
- [24].Doughty L, Clark RS, Kaplan SS, et al. sFas and sFas ligand and pediatric sepsis-induced multiple organ failure syndrome. Pediatr Res 2002;52:922–7. [DOI] [PubMed] [Google Scholar]
- [25].Ambrosino G, Naso A, Feltracco P, et al. Cytokines and liver failure: modification of TNF- and IL-6 in patients with acute on chronic liver decompensation treated with Molecular Adsorbent Recycling System (MARS). Acta Biomed 2003;74(Suppl. 2):7–9. [PubMed] [Google Scholar]
- [26].Riordan SM, Williams R. Mechanisms of hepatocyte injury, multiorgan failure, and prognostic criteria in acute liver failure. Semin Liver Dis 2003;23:203–15. [DOI] [PubMed] [Google Scholar]
- [27].Li J, Zhu X, Liu F, et al. Cytokine and autoantibody patterns in acute liver failure. J Immunotoxicol 2010;7:157–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Liu Q. Role of cytokines in the pathophysiology of acute-on-chronic liver failure. Blood Purif 2009;28:331–41. [DOI] [PubMed] [Google Scholar]
- [29].Sen S, Davies NA, Mookerjee RP, et al. Pathophysiological effects of albumin dialysis in acute-on-chronic liver failure: a randomized controlled study. Liver Transpl 2004;10:1109–19. [DOI] [PubMed] [Google Scholar]
- [30].Guo LM, Liu JY, Xu DZ, et al. Application of molecular adsorbents recirculating system to remove NO and cytokines in severe liver failure patients with multiple organ dysfunction syndrome. Liver Int 2003;23(Suppl. 3):16–20. [DOI] [PubMed] [Google Scholar]
- [31].Luedde T, Liedtke C, Manns MP, et al. Losing balance: cytokine signaling and cell death in the context of hepatocyte injury and hepatic failure. Eur Cytokine Netw 2002;13:377–83. [PubMed] [Google Scholar]
- [32].Guicciardi ME, Gores GJ. Apoptosis: a mechanism of acute and chronic liver injury. Gut 2005;54:1024–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Harrington LE, Mangan PR, Weaver CT. Expanding the effector CD4 T-cell repertoire: the Th17 lineage. Curr Opin Immunol 2006;18:349–56. [DOI] [PubMed] [Google Scholar]
- [34].Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem 2007;282:20059–63. [DOI] [PubMed] [Google Scholar]
- [35].Zhang JY, Zhang Z, Lin F, et al. Interleukin-17-producing CD4(+) T cells increase with severity of liver damage in patients with chronic hepatitis B. Hepatology 2010;51:81–91. [DOI] [PubMed] [Google Scholar]
- [36].Diaz G, Zamboni F, Tice A, et al. Integrated ordination of miRNA and mRNA expression profiles. BMC Genomics 2015;16:767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Luo Y, Wen X, Wang L, et al. Identification of MicroRNAs Involved in Growth Arrest and Apoptosis in Hydrogen Peroxide-Treated Human Hepatocellular Carcinoma Cell Line HepG2. Oxid Med Cell Longev 2016;2016:7530853. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.