

Abstract
Background:
Diastolic dysfunction develops in response to hypertension and estrogen (E2) loss and is a forerunner to heart failure (HF) in women. The cardiac renin–angiotensin system (RAS) contributes to diastolic dysfunction, but its role with respect to E2 and blood pressure remain unclear.
Methods:
We compared the effects of ovariectomy (OVX) or sham surgery on the cardiac RAS, left ventricular (LV) structure/function, and systemic/intracardiac pressures of spontaneously hypertensive rats (SHRs: n = 6 intact and 6 OVX) and age-matched Wistar-Kyoto (WKY: n = 5 intact and 4 OVX) controls.
Results:
WKY rats were more sensitive to OVX than SHRs with respect to worsening of diastolic function, as reflected by increases in Doppler-derived filling pressures (E/e′) and reductions in myocardial relaxation (e′). This pathobiologic response in WKY rats was directly linked to increases in cardiac gene expression and enzymatic activity of chymase and modest reductions in ACE2 activity. No overt changes in cardiac RAS genes or activities were observed in SHRs, but diastolic function was inversely related to ACE2 activity.
Conclusion:
Endogenous estrogens exert a more significant regulatory role upon biochemical components of the cardiac RAS of WKY versus SHRs, modulating the lusitropic and structural components of its normotensive phenotype.
Keywords: Angiotensin converting enzyme-2, chymase, diastolic dysfunction, estrogen, ovariectomy, renin angiotensin system, SHR, Wistar-Kyoto rats
Introduction
Approximately 2.5 million women in the United States have heart failure (HF),1 which accounts for a third of all disease-related mortality in this group.2 Impaired diastolic dysfunction and HF with preserved left ventricular (LV) ejection fraction (HFpEF) are seen more often in women, and women develop HFpEF twice as often as men.3 The clinical implications for sex differences in HF are significant, affecting risk factor screening and targeting of sex-specific interventions. Recent studies show that HFpEF is almost as lethal as systolic HF (HF with reduced ejection fraction; HFrEF), yet there is a significant gap in our knowledge regarding HFpEF and how to treat it.4 The renin–angiotensin system (RAS) is a central regulator of cardiovascular function and plays a key role in the pathophysiology of HF. While angiotensin II (Ang II) receptor blockers (ARBs) and angiotensin converting enzyme (ACE) inhibitors have been extensively studied in patients with HFpEF,4–7 strong evidence of a superior effect of these agents on reducing morbidity and improving survival in these patients has not been confirmed. Large trials suggest that the effects of ACE inhibitors may be less pronounced in women than in men receiving treatment for hypertension and HF.8–11 Whether this is due to an ACE-escape phenomena,12 activation of an alternate pathway for Ang II formation via degradation of the dodecapeptide angiotensin-(1-12) [Ang-(1-12)] by chymase13,14 and/or the inability of these drugs to access the intracellular sites where Ang II is generated,15,16 is unclear.
Uncertainty also remains regarding the action of estrogen on the heart17,18 and the influence of estrogen on the cardiac RAS. Indeed, the dramatic increase in HF incidence in women after 55 years of age supports a protective effect of estrogen that is lost after menopause.19,20 We previously reported that estrogen (E2) deprivation and replacement in a congenic rodent model of hypertension due to increased tissue renin gene expression (the mRen2.Lewis female rat) could modulate cardiomyocyte and interstitial remodeling, thereby altering lusitropic function and ventricular compliance.21 In this model, surgical loss of ovarian estrogens correlated with an upsurge in cardiac Ang II expression and an increase in cardiac chymase and mast cell number that were attenuated following chronic E2 replacement.22 Because the cardiovascular effects of estrogen loss in the mRen2.Lewis model manifested only when the gonads were removed very early in life (4-weeks of age),23–27 a time before commencement of ovarian cycling, it remains unclear as to how estrogens affect the progression of diastolic dysfunction. Moreover, given that the mRen2.Lewis rat is a tissue renin-driven model of hypertension,28 a more clinically relevant model is needed to address how deprivation of ovarian estrogens influence the cardiac RAS and modify diastolic function.
With these considerations in mind, we characterized the effect of estrogen loss on the expression and activity of the canonical and non-canonical16 cardiac RAS and subsequent development of diastolic dysfunction in sexually mature sham-operated (intact) and ovariectomized (OVX) adult spontaneously hypertensive rats (SHRs) and their age-matched normotensive genetic counterparts, the Wistar-Kyoto (WKY) rat. The ability to investigate the estrogen/cardiac RAS interaction in the context of hypertensive versus normotensive heart disease is clinically relevant since diastolic dysfunction often occurs in postmenopausal women without pre-existing hypertension29,30 as well as in normotensive women with Turner’s syndrome.31
Methods
Animals
Female SHR (n = 12) and WKY (n = 9) rats were obtained from breeding colonies within the Division of Biological Sciences at Federal University of Rio de Janeiro, Brazil. In vivo studies were performed at the Federal University of Rio de Janeiro, Brazil and all biochemical and molecular studies were performed at Wake Forest School of Medicine, Winston Salem, North Carolina, USA. All procedures were carried out in accordance with the Guide for the Care and Use of Laboratory Animals, published by National Institute of Health, and the study was approved by the Ethics and Research Committee of the Federal University of Rio de Janeiro, Rio de Janeiro, Brazil (protocol # 01200.001568/2013-87) and Wake Forest School of Medicine’s Animal Care and Use Committee (protocol # A14-064). All possible steps were taken to avoid animal suffering at each stage of the experiment. All animals were bred and exposed to the same paired housing conditions including a 12h:12h light:dark cycles, room temperature of 22 ± 2°C, and 60–70% humidity, and provided food and water ad libitum.
Experimental protocol
At 12–14 weeks of age, SHR and WKY rats were randomly assigned to undergo either bilateral ovariectomy (SHR-OVX: n = 6; WKY-OVX n = 4) or sham surgery, referred as gonadal-intact (SHR-intact: n = 6 ; WKY-intact: n = 5). Bilateral ovariectomy was performed under 2% isoflurane anesthesia, as previously described.32 The absence of ovaries and decrease in uterine weight upon postmortem examination were used to confirm the efficacy of ovariectomy.33 We have previously documented reduced circulating estradiol levels in sham-operated and OVX hypertensive rats.22,24
At 8 weeks after OVX or sham surgery, four rats per group were submitted to a maximal exercise capacity test using a customized rodent treadmill (EP-131, Insight, São Paulo, Brazil). At 9 weeks after surgery, all rats underwent transthoracic echocardiograms to collect cardiac morphometrics and function data. After 1 week, or experimental week 10, rats were prepared for terminal invasive hemodynamic studies prior to euthanasia via exsanguination by cardiac puncture. Whole hearts were isolated and weighed prior to dissection of the left ventricle, right ventricle, and both atria. Left ventricles were cut horizontally into thirds and stored in −80°C for real-time polymerase chain reaction (PCR) and immunoblot studies. The left tibia length was determined using a digital micrometer after dissection and removal of all soft tissue. The heart to tibia length ratio was computed as heart weight in grams divided by tibia length in cm.
Exercise tolerance test
Given that exercise intolerance is often the first clinical manifestation of diastolic dysfunction, a maximal exercise tolerance test (time to exhaustion during a standardized exercise protocol) was performed to differentiate functional disparities between SHR and WKY strains based on estrogen status. A total of four rats in each group were familiarized with the customized rodent treadmill (EP-131, Insight, São Paulo, Brazil) by walking at a speed of 20 cm/s, 10 min/d, and 5% grade for 1 week prior to testing. Each exercise test was performed after at least 1 day of rest. The protocol for the exercise tolerance evaluation consisted of 3 min at 12 m/min, with 1.2 m/min increases in speed every 3 min until the rats reached exhaustion. Exhaustion was determined when the rats stopped keeping pace with the treadmill and remained at the lower end of the treadmill for more than 5 s, despite gentle nudging by an investigator (DGC) who was blinded to the study groups. The total distance travelled (meters) was used as a measure of exercise capacity.
Echocardiographic evaluation and measurements
For the echocardiogram, rats were anesthetized with an isoflurane 1.5%/oxygen mixture using a nose cone during spontaneous ventilation. Using the Vevo 770 high-frequency ultrasound system coupled with a micro visualization scan head of 30 MHz (VisualSonics, Toronto, Canada), animals were imaged in a shallow left lateral decubitus position while lying on a warmed mat. M-mode 2-dimensional images were obtained from the parasternal short axis view. LV internal dimensions in systole (LVESD) and diastole (LVEDD) and LV wall thicknesses in diastole were measured, and left ventricular ejection fraction (LVEF), fractional shortening (FS), posterior wall thickness (PWT), relative wall thickness (RWT), and LV mass were calculated, as described previously.24,32
Pulsed-Doppler and tissue Doppler imaging of peak early transmitral filling (Emax) and septal mitral annular (e′) velocities, respectively, were obtained from the left parasternal apical four-chamber view. The ratio of early peak transmitral early filling-to-mitral annular descent, or E/e′, was used as an index of filling pressure. An investigator who was masked to the study groups (JS) carried out measurements and calculations off-line using the Vevo 770 Protocol-Based Measurements software. All measured and calculated systolic and diastolic indices are represented as the average of at least five consecutive cardiac cycles to minimize beat-to-beat variability.
Invasive hemodynamic assessment
At 10 weeks after OVX/sham surgery, 10 SHR (n = 5 intact and 5 OVX) and 9 WKY (n = 5 intact and 4 OVX) rats were anesthetized with sodium pentobarbital (50 mg/kg; intraperitoneal) and prepared for terminal arterial blood and LV pressure measurements. This anesthetic was selected to allow for carotid artery catheterization without the hindrance of volatile anesthesia delivery by mask and because pentobarbital has been shown to induce similar effects on cardiac diastolic function, blood pressure and heart rate as isoflurane.34,35 A catheter (PE-50) was introduced into the right carotid artery and connected to a pressure transducer (MLT884, ADInstruments, Inc., Colorado Springs, CO, USA) that measured systolic, diastolic, and mean arterial blood pressure. Subsequently, the catheter was advanced into the left ventricle to record intracavitary pressures including LV systolic pressure and end diastolic pressure (LVEDP). LV contraction and relaxation rates were assessed by positive and negative dP/dt, respectively. Data were recorded on a polygraph (Powerlab, ADInstruments, Inc.) using Lab Chart software (Version 7.0, ADInstruments, Inc.).
Interstitial collagen deposition
Collagen deposition within hearts of 9 SHRs (n = 4 intact and 5 OVX) and 9 WKYs (n = 5 intact and 4 OVX) was evaluated using picrosirius red-stained, 4 μm, 4% paraformaldehyde-fixed, paraffin-embedded sections, as previously described.25,26 Images were captured using an Olympus BX51 Fluorescent Microscope (Spach Optics Inc., Rochester, NY, USA). The ratio of collagen-stained pixels to unstained pixels in 10 fields/heart was quantified using NIH ImageJ software (http://rsbweb.nigh.gov/ij/).
Real-time PCR
Real-time PCR was used to quantify mRNA expression for specific genes in LV tissue. For detailed methodology, please refer to the Supplementary material online. With regard to cardiac chymase, we amplified rat chymase 1, which is also known as mast cell protease 5 (rMCP-5). The NCBI RefSeq number for rat chymase 1 for primer generation is NM_013092.1. The agarose gel electrophoresis of the PCR product for chymase 1 revealed a specific band corresponding to the expected 66 base-pair size (Figure 1). To further confirm the specificity of PCR for rat chymase 1, we isolated the PCR product from the agarose gel using the QIAquick Gel Extraction Kit (QIAGEN Inc., Germantown, MD, USA), and determined the nucleotide sequence with assistance from Eton Bioscience, Inc. (San Diego, CA, USA) (data not shown).
Figure 1.
Agarose gel electrophoresis of PCR products for chymase 1 gene showing a 66 base-pair (bp) fragment from representative WKY and SHR heart tissue. M = DNA marker.
PCR: polymerase chain reaction; SHR: spontaneously hypertensive rat; WKY: Wistar-Kyoto rat.
Immunoblot analysis
LV tissue homogenates were separated by SDS-PAGE and transferred onto membranes, as previously described.23 Immunoblots were probed using antibodies for AT1R (1:250; Alomone Labs, Jerusalem, Israel) and the Ang-(1-7) receptor, Mas (1:5000; Alomone Labs, Ltd, Jerusalem, Israel). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 1:5000; Cell Signaling, Danvers, MA, USA) was used as a loading control. The bands were digitized using MCID image analysis software (Imaging Research, Inc., Ontario, Canada). Each band was expressed in arbitrary units and normalized to its own GAPDH.
LV myocardial ACE, ACE2, and chymase activities
Tissue enzyme activities were determined by high-performance liquid chromatography as previously described.14 Radiolabeled (125I) rat Ang-(1-12) was used as a substrate for the measurement of chymase enzymatic activity. 125I-Ang I was employed as the substrate for ACE activity and 125I-Ang II was used as the substrate for the measurement of ACE2 activity.
Statistical analyses
All values are expressed as mean ± standard error of the mean (SEM). A two-way analysis of variance was used to determine the effects of strain (SHR versus WKY), estrogen status (OVX versus intact) and an interaction (strain × estrogen status), followed by Tukey’s multiple-comparisons test. Simple linear regression analysis was used to determine the relationships between cardiac RAS enzymatic activities and LV diastolic function, characterized by E/e′. Data were analyzed using the software GraphPad Prism Version 6 (GraphPad Software, Inc., La Jolla, CA, USA). A p-value <0.05 was considered statistically significant.
Results
Ovariectomy increases body weight, cardiac mass, and upregulates cardiac estrogen receptor gene expression
At the end of the 10-week protocol, prior to sacrifice, WKY rats were heavier than SHRs and, consistent with our previous studies,24–26 the removal of ovaries increased body weight in both strains (Table 1). Estrogen depletion led to significant increases in whole heart weight, irrespective of strain. Uterine weights were greater in gonadal-intact rats compared with their OVX counterparts (estrogen effect: F (1,17) = 65.49, p = 0.0001) confirming the efficacy of surgical bilateral ovariectomy.36 While there were no differences in exercise capacity between strains (n = 4 rats/group), OVX rats showed a tendency for reduced exercise tolerance (estrogen effect: F (1, 12) = 4.039, p = 0.067), as estrogen status contributed to 19% of the total variance in meters travelled on the treadmill.
Table 1.
Physical characteristics, exercise performance, and cardiac estrogen receptor gene expression.
| SHR Mean ± SEM |
WKY Mean ± SEM |
Strain p-value |
E2 p-value |
Strain × E2 p-value |
|
|---|---|---|---|---|---|
| Body Weight (g) | 0.0228 | 0.0001 | 0.471 | ||
| Intact | 194.5 ± 11.3 | 212.4 ± 7.2 | |||
| OVX | 248.7 ± 9.5* | 281.5 ± 10.5† | |||
| Heart Weight (g) | 0.736 | 0.01 | 0.376 | ||
| Intact | 0.84 ± 0.07 | 0.90 ± 0.05 | |||
| OVX | 1.04 ± 0.03 | 1.01 ± 0.03 | |||
| Tibial Length (cm) | 0.013 | 0.003 | 0.013 | ||
| Intact | 3.37 ± 0.02 | 3.37 ± 0.04 | |||
| OVX | 3.40 ± 0.04 | 3.60 ± 0.04† | |||
| HW/TL (mg/cm) | 0.871 | 0.029 | 0.147 | ||
| Intact | 2.47 ± 0.21 | 2.67 ± 0.13 | |||
| OVX | 3.05 ± 0.09 | 2.80 ± 0.08 | |||
| Uterine Weight (g) | 0.266 | 0.0001 | 0.252 | ||
| Intact | 1.16 ± 0.16 | 0.90 ± 0.13 | |||
| OVX | 0.11 ± 0.02‡ | 0.12 ± 0.01† | |||
| Exercise Capacity (meters) | 0.155 | 0.067 | 0.131 | ||
| Intact | 1,487 ± 94 | 1,464 ± 362 | |||
| OVX | 697 ± 217 | 1380 ± 46 | |||
| ERβ mRNA | 0.078 | 0.0009 | 0.038 | ||
| Intact | 1.27 ± 0.10 | 0.75 ± 0.09 | |||
| OVX | 1.49 ± 0.11 | 1.54 ± 0.22† | |||
| GPER mRNA | 0.117 | 0.0001 | 0.003 | ||
| Intact | 0.80 ± 0.09 | 0.61 ± 0.12 | |||
| OVX | 0.99 ± 0.08 | 1.54 ± 0.15‡ |
Treadmill exercise capacity was measured in SHR and WKY rats at experiment week 8. Body and tissue weights and tibia length, and estrogen receptor gene expression were obtained 10 weeks after OVX or sham (gonads left intact) procedures. The groups consisted of intact-SHRs (n = 6), OVX-SHRs (n = 6), intact-WKY (n = 5), OVX-WKY (n = 4). Using a two-way ANOVA, significant differences in functional, structural, and hemodynamic indices, with respect to strain, estrogen (E2) status (intact versus OVX), and strain × E2 status, were determined. The p-values are presented on the right of the table. For convenience, significant differences are indicated in bold. Post-hoc analysis using Tukey’s multiple comparisons indicate significant differences within-strain for estrogen status.
p < .01 OVX-SHRs versus intact-SHRs;
p < .01 OVX-WKY versus intact-WKY;
p < .001 OVX-SHRs versus intact-SHRs.
ANOVA: analysis of variance; ERβ: estrogen receptor beta; GPER: G-protein coupled estrogen receptor; HW/TL: heart weight-to-tibia length ratio; OVX: ovariectomy; SHR: spontaneously hypertensive rat; WKY: Wistar-Kyoto.
To understand the potential consequences of estrogen loss on cardiac estrogen receptors, we performed mRNA quantitative analysis of nuclear estrogen receptors (ERs), ER-α and ER-β, and the G-protein-coupled membrane receptor (GPER), also called GPR30 in LV tissue. Although both ER-β and GPER were detected in the female heart, expression levels of ER-α were very low. The loss of estrogens significantly increased ER-β gene expression (estrogen effect: F (1,17) = 16, p = 0.0009), particularly in the WKY rats (interaction effect: F (1,17) = 12.38, p = 0.038). While GPER mRNA expression levels were comparable among gonadal-intact rats of both strains at baseline, OVX led to an approximate 250% increase in GPER expression in WKY rats but only a 20% increase in SHRs (interaction effect: F (1,17) = 12, p = 0.003) (Table 1).
Effects of estrogen status on cardiac dimensions, LV function, and hemodynamics
The effects of estrogen status and rat strain on LV structure, systolic function, and hemodynamics are shown in Table 2. As expected, there were significant strain effects with respect to LV morphology, with greater LV end systolic dimensions, PWT and RWT, and LV mass in hypertensive SHRs versus normotensive WKY rats. While OVX led to significant increases in LV dimensions, PWT, and LV mass in both strains, the effect of OVX on LVESD and LVEDD was most pronounced in the WKY rats (interaction effect on LVESD: F (1,17) = 5.70, p = 0.029; and LVEDD: F (1,17) = 7.92, p = 0.012). Systolic function, as defined by LVEF and FS, was not altered by either strain or estrogen status. Doppler indices of diastolic function, however, revealed important differences between SHRs versus WKY rats and OVX versus gonadal-intact rats (Figure 2). Specifically, e′ was lower and E/e′ was higher in SHR compared with WKY (Strain effect on e′: F (1,17) = 17.14, p = 0.0008; and E/e′: F (1,17) = 5.24, p =0.035). While estrogen loss adversely affected both of these indices of diastolic function (estrogen effect on e′: F (1,17) = 26.66, p < 0.0001; and E/e′: F (1,17) = 14.37, p = 0.002), the influence of OVX was again most profound in WKY rats (WKY-intact versus WKY-OVX for e′: p < 0.001, and for E/e′: p < 0.05, after post-hoc multiple comparisons). The strain × estrogen effect, or interaction, was significant only for e′ (F (1,17) = 5.071, p = 0.039). Heart rates under isoflurane were significantly lower in OVX rats, irrespective of strain (estrogen effect: F (1,17) = 7.67, p = 0.013).
Table 2.
Echocardiographic Indices and Invasive Hemodynamics.
| SHR mean ± SEM |
WKY mean ± SEM |
Strain p-value |
E2 p-value |
Strain × E2 p-value |
|
|---|---|---|---|---|---|
| Left ventricular morphology | |||||
| LVEDD (cm) | 0.152 | 0.003 | 0.012 | ||
| Intact | 0.696 ± 0.026 | 0.584 ± 0.041 | |||
| OVX | 0.714 ± 0.012 | 0.749 ± 0.011* | |||
| LVESD (cm) | 0.022 | 0.0001 | 0.029 | ||
| Intact | 0.453 ± 0.012 | 0.382 ± 0.016 | |||
| OVX | 0.509 ± 0.012† | 0.507 ± 0.019‡ | |||
| PWTed (cm) | 0.004 | 0.024 | 0.502 | ||
| Intact | 0.176 ± 0.011 | 0.128 ± 0.018 | |||
| OVX | 0.197 ± 0.008 | 0.164 ± 0.009 | |||
| AWTed (cm) | 0.747 | 0.847 | 0.64 | ||
| Intact | 0.154 ± 0.009 | 0.155 ± 0.014 | |||
| OVX | 0.161 ± 0.007 | 0.115 ± 0.005 | |||
| RWT | 0.022 | 0.29 | 0.527 | ||
| Intact | 0.494 ± 0.035 | 0.422 ± 0.075 | |||
| OVX | 0.558 ± 0.019 | 0.439 ± 0.021 | |||
| Systolic function | |||||
| LVEF (%) | 0.196 | 0.567 | 0.098 | ||
| Intact | 62 ± 3 | 71 ± 3 | |||
| OVX | 66 ± 1 | 65 ± 3 | |||
| FS% | 0.558 | 0.183 | 0.453 | ||
| Intact | 34 ± 3 | 34 ± 3 | |||
| OVX | 29 ± 2 | 32 ± 2 | |||
| Heart Rate (bpm) | 0.979 | 0.013 | 0.156 | ||
| Intact | 302 ± 13 | 324 ± 24 | |||
| OVX | 283 ± 7 | 262 ± 10 | |||
| Invasive hemodynamics | |||||
| Systolic arterial pressure (mmHg) | 0.0006 | 0.137 | 0.754 | ||
| Intact | 131 ± 6 | 94 ± 4 | |||
| OVX | 148 ± 12 | 106 ± 10 | |||
| Diastolic arterial pressure (mmHg) | 0.093 | 0.177 | 0.939 | ||
| Intact | 80 ± 13 | 59 ± 5 | |||
| OVX | 96 ± 14 | 76 ± 8 | |||
| Mean arterial pressure (mmHg) | 0.021 | 0.129 | 0.915 | ||
| Intact | 100 ± 12 | 72 ± 5 | |||
| OVX | 120 ± 14 | 89 ± 10 | |||
| LV end systolic pressure (mmHg) | 0.015 | 0.243 | 0.349 | ||
| Intact | 130 ± 5 | 114 ± 6 | |||
| OVX | 150 ± 11 | 116 ± 9 | |||
| LV end diastolic pressure (mmHg) | 0.016 | 0.002 | 0.364 | ||
| Intact | 6.29 ± 0.84 | 1.88 ± 0.26 | |||
| OVX | 9.57 ± 1.24 | 7.42 ± 2.18§ | |||
| dP/dt max (mmHg/s) | 0.021 | 0.597 | 0.938 | ||
| Intact | 7333 ± 1539 | 4778 ± 350 | |||
| OVX | 6736 ± 680 | 4333 ± 882 | |||
| dP/dt min (mmHg/s) | 0.025 | 0.77 | 0.989 | ||
| Intact | (-)5376 ± 1207 | (-)3386 ± 241 | |||
| OVX | (-)5150 ± 419 | (-)3137 ± 608 | |||
Echocardiograms and invasive hemodynamics were measured in SHRs and WKY rats at experiment weeks 9 and 10, respectively. The groups consisted of intact-SHRs (n=6), OVX-SHRs (n=6), intact-WKY rats (n=5), and OVX-WKY rats (n=4). Using a two-way ANOVA, significant differences in functional, structural, and hemodynamic indices, with respect to strain, estrogen (E2) status (intact versus OVX), and strain × E2 status, were determined. The p-values are presented on the right of the table. For convenience, significant differences are indicated in bold. Post-hoc analysis using Tukey’s multiple comparisons indicate significant differences within-strain for estrogen status.
p < 0.01 OVX-WKY versus intact-WKY;
p < 0.05 OVX-SHR versus intact-SHRs;
p < 0.001 OVX-WKY versus intact-WKY;
p < 0.05 OVX-WKY versus intact-WKY.
ANOVA: analysis of variance; AWTed: anterior wall thickness end diastole; bpm: beats per minute; dP/dt max: maximum derivative of change in LV systolic pressure over time; dP/dt min: minimum derivative of change of LV diastolic pressure over time; FS: fractional shortening; LV: left ventricular; LVEDD: left ventricular end diastolic dimension; LVEF: left ventricular ejection fraction; LVESD: left ventricular end systolic dimension; OVX: ovariectomy; PWTed: posterior wall thickness end diastole; RWT: relative wall thickness; SEM: standard error of the mean; SHR: spontaneously hypertensive rat; WKY: Wistar-Kyoto rats;
Figure 2.

Early mitral annular velocity (e′) and early transmitral filling velocity-to-mitral annular velocity ratio (E/e′) in SHRs and WKY rats. Data indicate differences in (a) septal e′ and (b) E/e′ with respect to strain and estrogen status (intact versus OVX). Values are reported as means ± SEM; SHRs, n = 6/group. WKY rats, n = 4–5/group. Black bars represent gonadal-intact rats; open bars represent OVX rats. Data were analyzed using two-way analysis of variance for strain and estrogen status (intact versus OVX) effects. Overall, SHRs had reduced ventricular lusitropy (e′) and higher filling pressures (E/e′) compared with WKY rats (strain effect on e′: F (1,17) = 17.14, p = 0.0008; and E/e′: F (1,17) = 5.24, p =0.035, respectively) *p < 0.05 and ***p < 0.001 indicate significance between strains. Even though estrogen loss reduced e′ and increased E/e′ in both strains (estrogen effect on e′: F (1,17) = 26.66, p < 0.0001; and E/e′: F (1,17) = 14.37, p = 0.002, respectively), the effects were most pronounced in the WKY rats, particularly e′ (interaction effect on e′: F (1,17) = 5.071, p = 0.039; and E/e′: F (1,17) = 1.622, not significant). Post-hoc analysis using Tukey’s multiple comparisons indicate significant differences only within the WKY strain for these diastolic functional parameters, with respect to estrogen status.
#p < 0.05 and ###p < 0.001.
OVX: ovariectomy; SEM: standard error of the mean; SHR: spontaneously hypertensive rat; WKY: Wistar-Kyoto rat.
Systemic and LV pressures and the rate of LV pressure development (dP/dtmax) and relaxation (dP/dtmin) were significantly higher in SHR versus WKY rats, independent of estrogen status (Table 2). While LVEDP also showed significant strain effects (F (1, 15) = 7.37, p = 0.016), this surrogate of left atrial pressure was affected by estrogen status (F (1, 15) = 13.32, p = 0.002). Estrogen loss increased LVEDP most markedly in the WKY strain (sham versus OVX, p < 0.05), which may be reflective of the greater increases in left ventricular dimensions in this group after OVX compared with SHRs (Table 2).
Effects of estrogen status on cardiac interstitial collagen deposition
Quantitative histological analysis of cardiac collagen deposition in the left ventricle from SHRs and WKY-intact and OVX rats are presented in Figure 3. Corresponding to their greater RWT, SHRs had a modest but significant increase in interstitial collagen deposition compared with WKY rats (strain effect: F (1,14) = 4.66, p < 0.05), irrespective of estrogen status.
Figure 3.
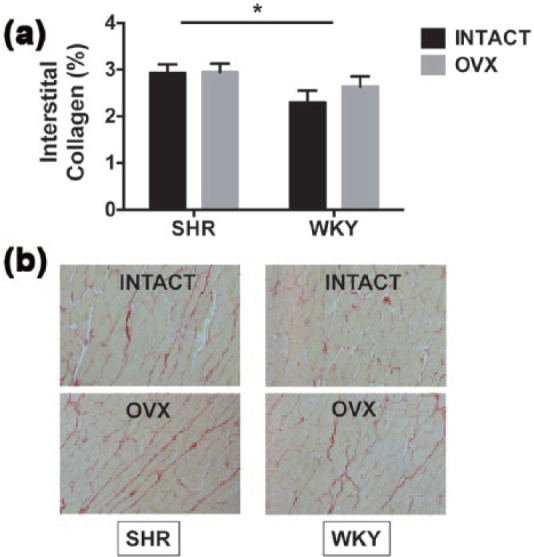
Cardiac interstitial collagen deposition in SHRs and WKY female rats. (a) Quantification of cardiac interstitial collagen from histological analysis. Data indicate differences in % interstitial collagen deposition with respect to strain (SHR versus WKY) and estrogen status (intact versus OVX). Values are reported as means ± SEM; SHR, n = 4 intact and 5 OVX; WKY, n = 5 intact and 4 OVX. Black bars represent gonadal-intact rats; grey bars represent OVX rats. SHR hearts exhibited more collagen deposition than WKY hearts, irrespective of estrogen status (strain effect). There were no estrogen or interaction effects. *p < 0.05 (strain effect). (b) Representative images of LV free wall interstitial collagen deposition were taken at 20× magnification. The picrosirius red-stained slides show modest increases in collagen deposition in hypertensive (SHR) hearts when compared with normotensive (WKY) hearts.
OVX: ovariectomy; SEM: standard error of the mean; SHR: spontaneously hypertensive rat; WKY: Wistar-Kyoto rat.
Effects of ovariectomy on cardiac RAS, neurohormonal and calcium regulatory genes and AT1R and Mas proteins
Gene expression levels of RAS components and their corresponding protein expression levels with respect to strain and estrogen status are presented in Table 3. Myocardial tissue from both WKY and SHR expressed gene transcripts for chymase, ACE, ACE2, and angiotensinogen, as well as AT1R and Mas receptor. mRNA levels of renin and AT2R, however, were either not detected or were very low in the adult female heart, which is consistent with previous reports by others.37,38 While the loss of estrogens was associated with an overall increase in cardiac chymase gene expression, the response, again, was most marked in the WKY strain (strain effect: F (1,15) = 24.38, p = 0.0001). Specifically, OVX led to an approximately 200% increase in cardiac chymase expression in WKY rats but only about a 60% increase in SHR (interaction: F (1,15) = 16.08, p = 0.0011; Table 3).
Table 3.
RAS, neurohormonal, and calcium regulatory-related genes and AT1R and Mas proteins.
| SHR mean ± SEM |
WKY mean ± SEM |
Strain p-value |
E2 p-value |
Strain × E2 p-value |
|
|---|---|---|---|---|---|
| Genes | |||||
| Chymase mRNA | 0.0002 | 0.0001 | 0.0011 | ||
| Intact | 0.47 ± 0.06 | 0.58 ± 0.16 | |||
| OVX | 0.72 ± 0.10 | 1.78 ± 0.13* | |||
| ACE mRNA | 0.05 | 0.024 | 0.613 | ||
| Intact | 0.82 ± 0.16 | 0.42 ± 0.05 | |||
| OVX | 1.12 ± 0.19 | 0.88 ± 0.08 | |||
| ACE2 mRNA | 0.51 | 0.57 | 0.48 | ||
| Intact | 0.5 ± 0.08 | 0.49 ± 0.22 | |||
| OVX | 0.47 ± 0.12 | 0.72 ± 0.32 | |||
| Angiotensinogen mRNA | 0.104 | 0.012 | 0.896 | ||
| Intact | 0.71 ± 0.17 | 0.42 ± 0.05 | |||
| OVX | 1.12 ± 0.19 | 0.88 ± 0.08 | |||
| AT1R mRNA | 0.031 | 0.331 | 0.273 | ||
| Intact | 1.34 ± 0.10 | 1.56 ± 0.23 | |||
| OVX | 1.32 ± 0.10 | 1.94 ± 0.29 | |||
| Mas mRNA | 0.0001 | 0.0001 | 0.0007 | ||
| Intact | 1.05 ± 0.08 | 1.73 ± 0.32 | |||
| OVX | 1.28 ± 0.12 | 3.98 ± 0.46* | |||
| ANF mRNA | 0.513 | 0.573 | 0.484 | ||
| Intact | 0.50 ± 0.08 | 0.49 ± 0.22 | |||
| OVX | 0.47 ± 0.12 | 0.72 ± 0.32 | |||
| BNP mRNA | 0.22 | 0.015 | 0.21 | ||
| Intact | 0.63 ± 0.15 | 0.32 ± 0.10 | |||
| OVX | 0.79 ± 0.12 | 0.80 ± 0.06 | |||
| NOX2 mRNA | 0.037 | 0.006 | 0.047 | ||
| Intact | 0.91 ± 0.03 | 0.92 ± 0.08 | |||
| OVX | 0.98 ± 0.07 | 1.30 ± 0.13† | |||
| SERCA2 mRNA | 0.0001 | 0.973 | 0.63 | ||
| Intact | 1.65 ± 0.11 | 0.85 ± 0.19 | |||
| OVX | 1.59 ± 0.13 | 0.93 ± 0.09 | |||
| PLB mRNA | 0.0001 | 0.006 | 0.638 | ||
| Intact | 1.21 ± 0.14 | 0.36 ± 0.06 | |||
| OVX | 1.65 ± 0.14 | 0.69 ± 0.08 | |||
| PLB/SERCA2 mRNA | 0.74 ± 0.08 | 0.47 ± 0.05 | 0.0004 | 0.0002 | 0.881 |
| Intact OVX | 1.40 ± 0.04‡ | 0.75 ± 0.06† | |||
| Proteins | |||||
| AT1R | 0.0001 | 0.0002 | 0.0814 | ||
| Intact | 3.12 ± 0.28 | 1.84 ± 0.11 | |||
| OVX | 1.95 ± 0.08§ | 1.34 ± 0.07 | |||
| Mas | 0.0005 | 0.006 | 0.053 | ||
| Intact | 2.43 ± 0.16 | 1.65 ± 0.05 | |||
| OVX | 1.79 ± 0.12∥ | 1.52 ± 0.08 | |||
Cardiac gene transcripts and protein expression were measured in SHR and WKY hearts 10 weeks after OVX or sham procedures (gonads intact). The groups consisted of intact-SHR (n = 6), OVX-SHR (n = 5), intact-WKY (n = 5), OVX-WKY (n = 4). Using a two-way ANOVA, significant differences in cardiac gene and protein expression, with respect to strain, estrogen (E2) status (intact versus OVX), and strain x E2 status, were determined. The p-values are presented on the right of the table. For convenience, significant differences are indicated in bold. Post-hoc analysis using Tukey’s multiple comparisons indicate significant differences within-strain for estrogen status.
p < 0.0001 OVX-WKY versus intact-WKY;
p < 0.05 OVX-WKY versus intact-WKY;
p < 0.01 OVX-SHRs versus intact-SHR;
p < 0.001 OVX-SHRs versus intact-SHRs;
p < 0.01 OVX-SHRs versus intact-SHRs.
ACE: angiotensin converting enzyme; ACE2: angiotensin converting enzyme 2; AT1R: angiotensin receptor 1; ANF: atrial natriuretic factor; BNP: brain natriuretic peptide; NOX2: NADPH oxidase 2; OVX: ovariectomy; PLB: unphosphorylated phospholamban; RAS: renin–angiotensin system; SEM: standard error of the mean; SERCA2: sarcoplasmic-endoplasmic calcium ATPase 2; SHR: spontaneously hypertensive rats; WKY: Wistar-Kyoto rats.
ACE mRNA levels were nearly double in SHR versus WKY (strain effect: p = 0.05), even though levels of the transcripts after estrogen loss increased comparably in both strains (estrogen effect: p = 0.024). However, unlike chymase gene expression, the strain × estrogen effect (interaction) for ACE mRNA was not significant. By contrast, ACE2 gene expression was not affected by strain or estrogen status. Even though angiotensinogen expression was not overtly different between strains, OVX caused markedly higher levels in both SHR and WKY rats (estrogen effect: F (1,16) = 8.068, p = 0.012). AT1 receptor gene expression was significantly higher in WKY versus SHR hearts (strain effect: F (1,17) = 5.560, p = 0.031), independent of estrogen status. Gene levels of the Mas receptor were also higher in WKY rats versus SHRs (strain effect: F (1,17) = 47.77, p = 0.0001), and this differential expression between strains was most pronounced with the loss of estrogens (interaction effect: F (1,17) = 17.16, p = 0.0007). Specifically, Mas receptor levels were 2.3-times higher in OVX-WKY rats when compared with their gonadal-intact littermates (p < 0.0001), whereas estrogen status accounted for only a 20% difference in Mas receptor mRNA expression in the SHR strain.
Protein expression levels of AT1R and Mas receptor normalized to GAPDH are shown in Table 3. AT1R and Mas receptors protein were nearly 75% higher in gonadal-intact SHR versus their normotensive WKY counterparts. Estrogen loss reduced expression of both receptors (estrogen effect on AT1R: F (1,17) = 21.46, p = 0.0002; and Mas: F (1,17) = 9.736, p = 0.006), particularly in the SHR strain (AT1R: intact-SHR versus OVX-SHR, p < 0.001; Mas: intact-SHRs versus OVX-SHRs, p < 0.01). Even so, the interaction effect (strain × estrogen status) did not achieve statistical significance for either receptor (Table 3).
Since important direct and indirect effects of estrogens on the maintenance of cardiac structure and function during health and disease include modulation of inflammation, cardiac myocyte survival, reactive oxygen species production, and intracellular calcium homeostasis,39 LV expression of genes associated with myocardial injury, oxidative stress, and sarcoplasmic reticulum calcium, namely brain natriuretic peptide (BNP), atrial natriuretic factor (ANF), NADPH oxidase 2 (NOX2), sarcoplasmic-endoplasmic calcium ATPase 2 (SERCA2) and phospholamban, were also measured (Table 3). BNP mRNA levels were increased in OVX rats, irrespective of strain, while ANF was not influenced by either of these factors. NOX2, one of the isoforms of NADPH oxidase, which is involved in the formation of reactive oxygen species, was also increased by the loss of estrogens, particularly in WKY rats (p < 0.05 for intact-WKY versus OVX-WKY). SERCA2 gene levels were nearly two-fold higher in SHRs versus WKY rats (strain effect: F (1,17) = 28.01, p = 0.0001), independent of estrogen status. While mRNA levels of phospholamban were also higher in SHRs versus WKY rats (strain effect: F (1,17) = 55.41, p = 0.0001), the loss of estrogens increased this important inhibitory protein of SERCA2 in both strains (estrogen effect: F (1,17) = 9.84, p = 0.006). Strain and estrogenic differences were also apparent in the ratio unphosphorylated phospholamban (PLB):SERCA2 (strain effect: F (1,17) = 19.49, p = 0.004; estrogen effect: F (1,17) = 21.53, p = 0.002), suggesting the potential for more calcium handling impairments in SHRs versus WKY rats at basal ovarian hormone states, with augmentation of this adverse effect after OVX, independent of strain.
Influence of estrogen loss and strain on cardiac RAS enzymatic activities
As we reported recently in male SHR hearts,14 chymase, as opposed to ACE activity predominates in female hearts; cardiac chymase activity was 10–15-fold higher than cardiac ACE activity, irrespective of strain. Corresponding to the increased chymase mRNA expression following estrogen loss (Table 3), the activity of chymase was also increased after OVX (estrogen effect: F (1,15) = 15.41, p = 0.001), particularly in WKY rats (p < 0.05) (Figure 4). Interestingly, the metabolism of Ang II by ACE2 showed both strain (F (1,17) = 5.661, p = 0.029) and estrogen status effects (F (1,17) = 9.100, p = 0.008). Specifically, ACE2 activity was 25% higher in gonadal-intact WKY versus gonadal-intact SHRs and the loss of ovarian estrogens markedly reduced the activity of ACE2 by nearly 30% in the normotensive WKY heart (OVX-WKY versus intact-WKY: p < 0.05), compared with a 13% decrease in hypertensive SHR hearts. There were no interaction effects (strain × estrogen status) among the cardiac RAS enzymatic activities.
Figure 4.

Myocardial chymase, ACE, and ACE2 activities in SHR and WKY rats. Data indicate differences in (a) Chymase, (b) ACE, and (c) ACE2 enzymatic activities with respect to strain and estrogen status (intact versus OVX). Values are reported as means ± SEM; SHR, n = 5–6/group. WKY rats, n = 4–5/group. Black bars represent gonadal-intact rats; open bars represent OVX rats. Data were analyzed using two-way analysis of variance for strain and estrogen status (intact versus OVX). Main effects for estrogen status consist of: chymase activity F(1,15) = 15.41, p = 0.0013; ACE activity (F(1,16) = 4.204, p = 0.057; ACE2 activity (F(1,17) = 9.100, p = 0.0078. *p < 0.05 indicates significance between strains for ACE2 activity. Post-hoc analysis using Tukey’s multiple comparisons indicate significant difference within-strain for estrogen status #p < 0.05.
ACE: angiotensin converting enzyme; OVX: ovariectomy; RAS: renin–angiotensin system; SEM: standard error of the mean; SHR: spontaneously hypertensive rat; WKY: Wistar-Kyoto rat.
Relationship between diastolic function and cardiac RAS metabolism
Regression plots of cardiac RAS enzymatic activities and diastolic function, defined by E/e′, in WKY and SHR hearts are shown in Figure 5. In the WKY strain, worsening diastolic function, as indicated by elevated filling pressures, was significantly related to chymase activity, as 77% of the variation in E/e′ could be explained by the variation in cardiac chymase activity. There was also a strong trend for an association between elevated filling pressures and a reduction in cardiac ACE2 activity (p = 0.055), as 66% of the variance in E/e′ could be inversely attributed to ACE2 (the Ang-(1-7)-forming enzyme). Even though no overt differences in cardiac RAS gene expression or enzymatic activity were observed between intact and OVX-SHRs, diastolic function in the SHR strain was inversely related to cardiac ACE2 activity (p = 0.007).
Figure 5.
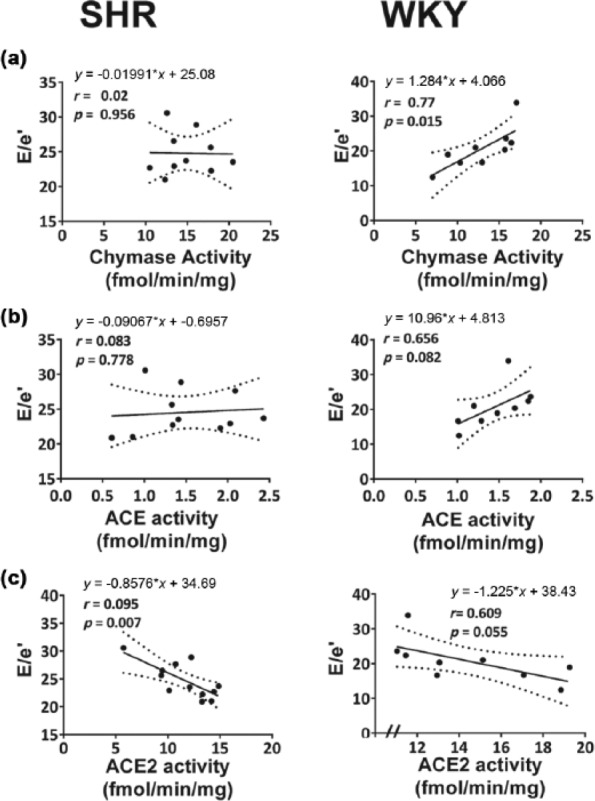
Relationship between cardiac RAS enzymatic activities and diastolic function, as defined by E/e′, in SHR and WKY rat hearts. Linear relationships (solid line) and 95% confidence intervals (dotted line) between (a) chymase activity and E/e′, (b) ACE activity and E/e′, and (c) ACE2 activity and E/e′ among SHRs (left panel) and WKY rats (right panel) are shown with regression equations and correlation coefficients, r. In SHRs, worsening diastolic function, indicated by elevations in E/e′, was inversely related to ACE2 activity (left panel (c)), while ACE and chymase activities were not significantly linked to diastolic dysfunction in this strain. In WKY rats, worsening diastolic function, or increases in E/e′, were positively linked to increasing cardiac chymase and ACE activities, respectively (right panel (a) and (b), and inversely related to ACE2 activity (right panel (c)).
ACE: angiotensin converting enzyme; RAS: renin–angiotensin system; SHR: spontaneously hypertensive rat; WKY: Wistar-Kyoto rat.
Discussion
In this comprehensive study of estrogen as a modulator of cardiac function and biomarkers of RAS activity and inflammation, we report for the first time a robust differential response of OVX between normotensive WKY and SHR. Overall, estrogen appears to exert a more significant regulatory role upon biochemical components of RAS expression and activity in WKY rats when compared with hypertensive SHR counterparts. These hormonal and signaling differences in cardiac tissue components between the two strains in response to loss of estrogen are associated with worsening of diastolic function as reflected by increases in LV filling pressure and reductions in myocardial relaxation. Importantly, this pathobiologic response in WKY rats was directly linked to increases in gene expression and enzymatic activity of cardiac chymase, and a modest but significant reduction in cardiac ACE2 activity. While estrogen loss had no overt influence on the SHR cardiac RAS markers, worsening diastolic function in this strain was associated with reductions in cardiac ACE2 activity. These differential effects of estrogen depletion were independent of blood pressure differences but associated with important changes in markers of oxidative stress. In documenting an inverse effect of estrogen depletion on the opposing arms of the RAS composed of the Ang II-forming chymase-ACE axis and the Ang II-degrading ACE2 axis, the findings reveal a critical role of sex steroids in modulating the lusitropic and structural components of the cardiac phenotype in the presence and absence of hypertension.
The development of systolic hypertension after the menopause is an independent risk factor for diastolic dysfunction and its progression to HFpEF.17 As diastolic dysfunction can also emerge independent of pre-existing hypertension,40–42 even among postmenopausal women,29 we compared the role of endogenous estrogens in the maintenance of cardiac morphology and function in SHR and its genetic, normotensive counterpart, the WKY. Given that Ang II contributes to the development of diastolic dysfunction via direct and indirect hemodynamic, tissue, and cellular effects43 (we also examined the changes in cardiac RAS components in response to estrogen status). Thus, the intact versus OVX/SHRs and WKY experimental paradigm provides a preclinical translational window to aid in the understanding of the underpinnings of diastolic dysfunction after ovarian estrogen loss in the context of normal blood pressure and pre-existing hypertension. Another particular advantage to using the SHR model to identify the link between estrogen loss and the cardiac RAS is that the SHR has a heightened Ang II receptor type 1 (AT1R) system,44 as opposed to elevated basal levels of tissue Ang II, that underlie the pathophysiology of hypertension in the mRen2.Lewis rat.45
While no overt effects of estrogen deficiency on systemic blood pressure were observed in either strain, consistent with previous reports by others,46,47 estrogen loss led to maladaptive changes in e′, E/e′, and LVEDP only in WKY rats. These data indicate that pre-existing hypertension has a ceiling effect on the already impaired diastolic function in the hypertensive strain. Compared with gonadal-intact WKY rats, SHRs displayed an elevated PLB:SERCA2 ratio, signifying impaired calcium reuptake at basal hormonal states. However, since the magnitude of changes in the calcium regulatory genes after estrogen loss were similar in SHRs and WKY rats, dysregulation of the intracardiac calcium signaling pathway is not likely to be a causal factor in the loss of diastolic function in the normotensive WKY rats. Moreover, it does not appear that cardiac collagen deposition contributed to the worsening of diastolic function in WKY rats, as estrogen status did not overtly impact this marker of remodeling. Indeed, cardiac collagen is one of the main factors related to diastolic function through effects on passive elastic properties of the left ventricle. Whether age plays a role in this process remains speculative. We recently showed no significant interstitial remodeling following OVX in normotensive Brown Norway × Fischer 344 (BNF344) rats at mid-life, while estrogen loss late-in-life in this strain augmented collagen deposition.36 Taken together, these findings suggest the presence of a very strong tonic action of estrogen on the cellular mechanisms that account for myocardial relaxation in normotensive WKY rats, an interpretation that agrees with the greater, probably compensatory, expression of ER beta and GPER gene transcripts in ovariectomized WKY. In keeping with this interpretation, Witnich et al.48 found lower expression of ER alpha and beta in SHR hearts compared with WKY rats after estrogen loss.
Although androgens and cardiac androgen receptors were not measured in the present study, they have been linked to cardiac hypertrophy and hypertension in SHRs.49 Fortepaini et al.46 showed that removal of the ovaries from cycling SHRs protects from the development of exacerbated hypertension when compared with older SHRs that have ceased from cycling. These authors claim that the balance between androgens and estrogens may be more importantly involved in the development of hypertension, and presumably hypertensive heart disease, than either sex steroid alone in this hypertensive strain. Since our rats underwent ovariectomy while ovaries were still functioning, any role that tissue-specific androgen metabolism might have had on cardiac structure or function was likely negligible.
Our data are consistent with other studies that have linked the effects of estrogen loss on diastolic function to a change in the cardiac RAS. In the renin-overexpressing mRen2.Lewis rat, we found that the surgical loss of ovarian estrogens at a young age correlated with upsurges in cardiac Ang II expression, cardiac chymase, and mast cell number, and that chronic E2 replacement attenuated these effects,22 as well as an OVX-related diastolic dysfunctional phenotype.24 Li et al.50 recently showed that 17β-estradiol inhibited mast cell chymase release to prevent adverse remodeling in OVX rats subjected to pressure overload by transverse aortic constriction; however, other components of the intracardiac RAS were not assessed in their study. We now show that OVX led to an approximate 200% increase in cardiac chymase gene expression in WKY rats, whereas gene expression of chymase increased by only 60% in OVX-SHRs. A similar pattern was also seen for chymase activity. Chymase, a serine protease found in mast cell granules and non-cardiac myocytes,51 is released into the cardiac interstitium following injury or inflammation. Chymase acts within the primary ACE-independent pathway of Ang II formation, and also functions to activate TGF-beta and other promoters of extracellular matrix degradation, thereby playing a role in tissue remodeling.15,16 Whether the exaggerated expression and activity of chymase in WKY rats after OVX contributed to the greater impairment in diastolic function, or are merely a reflection of an intermediate state prior to more overt HF remains unclear. Nonetheless, it is important to note that we showed that cardiac chymase enzymatic activity, as opposed to cardiac ACE activity, predominates in the female. Cardiac chymase-mediated Ang II formation from Ang-(1-12) was found to be 10–15-times higher than ACE-mediated Ang II formation from Ang I, corroborating our recently reported findings in the male SHR heart.14
Chymase might be activated in early transition states, from compensated hypertrophy to HF, where the processes of tissue remodeling are most activated. For example, in the 2K1C hypertensive heart,52 chymase was markedly activated 32 weeks after clipping, coinciding with the development of cardiac fibrosis. Analogous to this concept, and similar to the relatively unchanged chymase profile among SHRs after OVX, chymase failed to up-regulate in the late stages of HF.53 Whether steady-state chronic conditions, such as long-standing hypertension and end-stage HF, are immune to exacerbations from cardiac chymase remains speculative.
In addition to chymase- and ACE-mediated generation of Ang II that results in adverse remodeling and cardiac dysfunction, decreased ACE2 can shift the balance in the local cardiac RAS to the Ang II/AT1R axis, also leading to disease progression. In mice with ACE2 gene deletion54 and in hypertensive rats chronically treated with an ACE2 antagonist,55 impairments in cardiac function and structure were linked to increases in circulating and cardiac Ang II levels. Upregulation of ACE2 has been seen in pathologic states such as hypertension and HF,56–58 suggesting its role in counter-regulating the development or progression of cardiac disease. In the present study, ACE2 deactivation showed a robust correlation to the diastolic dysfunctional phenotype, defined by E/e′, of the SHR and a modest relationship to worsening filling pressures in the WKY. Whether this was due to the loss of estrogen-mediated modulation of ACE2,59 or a positive feedback mechanism in the RAS whereby Ang II facilitates the loss of its negative regulator ACE2,60 is not known. Ishiyama et al.61 first reported that Ang II may negatively regulate cardiac expression of ACE2. Given that ACE2 and the Ang-(1-7) receptor Mas are also localized in cardiomyocytes, cardiac fibroblasts, and endothelial cells,62 it will be important to determine the exact cell type and role of ACE2-mediated Ang-(1-7) formation from Ang II in the maintenance of cardiac structure and function in the female heart.
Conclusion
The female adult OVX WKY rat is an estrogen-sensitive rodent model that can be used to provide insight into the potential mechanisms that underlie subtle cardiac changes that might occur after the loss of ovarian estrogens in the absence of hypertension. Because the rapid decline in estrogen secondary to OVX in the adult rodent does not mimic the decline that gradually occurs during the many years of the human menopause phase, extrapolation of findings to pathologic conditions that occur late-in-life should be interpreted with caution. Nonetheless, diastolic dysfunction in normotensive WKY rats after estrogen loss is primarily associated with marked increases in both cardiac chymase gene expression and chymase-mediated Ang II formation from Ang-(1-12), and modest reductions in cardiac ACE2 activity. Deactivation of cardiac ACE2 activity was also linked to modest changes in the already present diastolic dysfunctional phenotype of the SHR. Indeed, while our knowledge of the mechanisms that underlie sex hormone related-differences in heart disease is still developing, increasing information regarding the specific RAS components, metabolic pathways, and cell–cell interactions, in conjunction with estrogens and their receptors, will be important in order to realize the promise of personalized medicine.
Supplementary Material
Acknowledgments
We extend sincere appreciation to Marina Lin, MS for her technical contributions to the study. Data were presented in part at the American Heart Associations’ Hypertension 2016 Meeting, September 14–17, 2016, Orlando, FL, USA and at the Annual Meeting of the North American Section of the International Academy of Cardiovascular Sciences, September 22–24, 2016, Sherbrooke, Quebec, Canada.
Footnotes
Declaration of conflicting interest: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The studies described here were funded, in part, by Program Project Grant 2P01 HL-051952 from the National Heart, Lung, and Blood Institute of the National Institutes of Health (to CMF, LG, JV, LJD), AG033727 of the National Institute on Aging of the National Institutes of Health (to LG), and Conselho Nacional de Desenvolvimento Cientifico e Tecnológico (CNPq) (to DG-C), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) (to JdS), and Instituto Nacional de Ciência e Tecnologia de Fármacos e Medicamentos (INCT-INOFAR) (to GZS).
References
- 1. Go AS, Mozaffarian D, Roger VL, et al. Heart disease and stroke statistics-2014 update: a report from the American Heart Association. Circulation 2014; 129: e28–e292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bybee KA, Stevens TL. Matters of the heart: cardiovascular disease in US women. Mo Med 2013; 110: 65–70. [PMC free article] [PubMed] [Google Scholar]
- 3. Hogg K, Swedberg K, McMurray J. Heart failure with preserved left ventricular systolic function; epidemiology, clinical characteristics, and prognosis. J Am Coll Cardiol 2004; 43: 317–327. [DOI] [PubMed] [Google Scholar]
- 4. Aurigemma GP, Gaasch WH. Clinical practice. Diastolic heart failure. N Engl J Med 2004; 351: 1097–1105. [DOI] [PubMed] [Google Scholar]
- 5. Massie BM, Carson PE, McMurray JJ, et al. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med 2008; 359: 2456–2467. [DOI] [PubMed] [Google Scholar]
- 6. Shah RV, Desai AS, Givertz MM. The effect of renin-angiotensin system inhibitors on mortality and heart failure hospitalization in patients with heart failure and preserved ejection fraction: a systematic review and meta-analysis. J Card Fail 2010; 16: 260–267. [DOI] [PubMed] [Google Scholar]
- 7. Yusuf S, Pfeffer MA, Swedberg K, et al. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet 2003; 362: 777–781. [DOI] [PubMed] [Google Scholar]
- 8. Garg R, Yusuf S. Overview of randomized trials of angiotensin-converting enzyme inhibitors on mortality and morbidity in patients with heart failure. Collaborative Group on ACE Inhibitor Trials. JAMA 1995; 273: 1450–1456. [PubMed] [Google Scholar]
- 9. Ghali JK, Krause-Steinrauf HJ, Adams KF, et al. Gender differences in advanced heart failure: insights from the BEST study. J Am Coll Cardiol 2003; 42: 2128–2134. [DOI] [PubMed] [Google Scholar]
- 10. Shekelle PG, Rich MW, Morton SC, et al. Efficacy of angiotensin-converting enzyme inhibitors and beta-blockers in the management of left ventricular systolic dysfunction according to race, gender, and diabetic status: a meta-analysis of major clinical trials. J Am Coll Cardiol 2003; 41: 1529–1538. [DOI] [PubMed] [Google Scholar]
- 11. Wing LM, Reid CM, Ryan P, et al. A comparison of outcomes with angiotensin-converting–enzyme inhibitors and diuretics for hypertension in the elderly. N Engl J Med 2003; 348: 583–592. [DOI] [PubMed] [Google Scholar]
- 12. Biollaz J, Brunner HR, Gavras I, et al. Antihypertensive therapy with MK 421: angiotensin II–renin relationships to evaluate efficacy of converting enzyme blockade. J Cardiovasc Pharmacol 1982; 4: 966–972. [PubMed] [Google Scholar]
- 13. Ahmad S, Varagic J, Groban L, et al. Angiotensin-(1-12): a chymase-mediated cellular angiotensin II substrate. Curr Hypertens Rep 2014; 16: 429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ahmad S, Varagic J, VonCannon JL, et al. Primacy of cardiac chymase over angiotensin converting enzyme as an angiotensin-(1-12) metabolizing enzyme. Biochem Biophys Res Commun 2016; 478: 559–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ferrario CM. Cardiac remodelling and RAS inhibition. Ther Adv Cardiovasc Dis 2016; 10: 162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ferrario CM, Ahmad S, Varagic J, et al. Intracrine angiotensin II functions originate from noncanonical pathways in the human heart. Am J Physiol Heart Circ Physiol 2016; 311: H404–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hulley S, Grady D, Bush T, et al. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) research group. JAMA 1998; 280: 605–613. [DOI] [PubMed] [Google Scholar]
- 18. Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. JAMA 2002; 288: 321–333. [DOI] [PubMed] [Google Scholar]
- 19. Owan TE, Hodge DO, Herges RM, et al. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med 2006; 355: 251–259. [DOI] [PubMed] [Google Scholar]
- 20. Regitz-Zagrosek V, Oertelt-Prigione S, Seeland U, et al. Sex and gender differences in myocardial hypertrophy and heart failure. Circ J 2010; 74: 1265–1273. [DOI] [PubMed] [Google Scholar]
- 21. Zhao Z, Wang H, Jessup JA, et al. Role of estrogen in diastolic dysfunction. Am J Physiol Heart Circ Physiol 2014; 306: H628–H640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang H, Jessup JA, Zhao Z, et al. Characterization of the cardiac renin angiotensin system in oophorectomized and estrogen-replete mRen2.Lewis rats. PLoS One 2013; 8: e76992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Groban L, Yamaleyeva LM, Westwood BM, et al. Progressive diastolic dysfunction in the female mRen(2). Lewis rat: influence of salt and ovarian hormones. J Gerontol A Biol Sci Med Sci 2008; 63: 3–11. [DOI] [PubMed] [Google Scholar]
- 24. Jessup JA, Wang H, MacNamara LM, et al. Estrogen therapy, independent of timing, improves cardiac structure and function in oophorectomized mRen2.Lewis rats. Menopause 2013; 20: 860–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jessup JA, Zhang L, Chen AF, et al. Neuronal nitric oxide synthase inhibition improves diastolic function and reduces oxidative stress in ovariectomized mRen2.Lewis rats. Menopause 2011; 18: 698–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jessup JA, Zhang L, Presley TD, et al. Tetrahydrobiopterin restores diastolic function and attenuates superoxide production in ovariectomized mRen2.Lewis rats. Endocrinology 2011; 152: 2428–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang H, Jessup JA, Lin MS, et al. Activation of GPR30 attenuates diastolic dysfunction and left ventricle remodelling in oophorectomized mRen2.Lewis rats. Cardiovasc Res 2012; 94: 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chappell MC, Gallagher PE, Averill DB, et al. Estrogen or the AT1 antagonist olmesartan reverses the development of profound hypertension in the congenic mRen2. Lewis rat. Hypertension 2003; 42: 781–786. [DOI] [PubMed] [Google Scholar]
- 29. Hirokawa M, Daimon M, Lee SL, et al. Early menopause does not influence left ventricular diastolic dysfunction: A clinical observational study in healthy subjects. J Cardiol 2016; 68: 548–553. [DOI] [PubMed] [Google Scholar]
- 30. Kane GC, Karon BL, Mahoney DW, et al. Progression of left ventricular diastolic dysfunction and risk of heart failure. JAMA 2011; 306: 856–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Andersen NH, Hjerrild BE, Sorensen K, et al. Subclinical left ventricular dysfunction in normotensive women with Turner’s syndrome. Heart 2006; 92: 1516–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jessup JA, Lindsey SH, Wang H, et al. Attenuation of salt-induced cardiac remodeling and diastolic dysfunction by the GPER agonist G-1 in female mRen2.Lewis rats. PLoS One 2010; 5: e15433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim HS, Kang TS, Kang IH, et al. Validation study of OECD rodent uterotrophic assay for the assessment of estrogenic activity in Sprague-Dawley immature female rats. J Toxicol Environ Health A 2005; 68: 2249–2262. [DOI] [PubMed] [Google Scholar]
- 34. Stein AB, Tiwari S, Thomas P, et al. Effects of anesthesia on echocardiographic assessment of left ventricular structure and function in rats. Basic Res Cardiol 2007; 102: 28–41. [DOI] [PubMed] [Google Scholar]
- 35. Zuurbier CJ, Keijzers PJ, Koeman A, et al. Anesthesia’s effects on plasma glucose and insulin and cardiac hexokinase at similar hemodynamics and without major surgical stress in fed rats. Anesth Analg 2008; 106: 135–142. [DOI] [PubMed] [Google Scholar]
- 36. Alencar AK, da Silva JS, Lin M, et al. Effect of Age, Estrogen Status, and Late-Life GPER Activation on Cardiac Structure and Function in the Fischer344xBrown Norway Female Rat. J Gerontol A Biol Sci Med Sci 2017; 72: 152–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Biwer LA, D’Souza KM, Abidali A, et al. Time course of cardiac inflammation during nitric oxide synthase inhibition in SHR: impact of prior transient ACE inhibition. Hypertens Res 2016; 39: 8–18. [DOI] [PubMed] [Google Scholar]
- 38. Everett AD, Fisher A, Tufro-McReddie A, et al. Developmental regulation of angiotensin type 1 and 2 receptor gene expression and heart growth. J Mol Cell Cardiol 1997; 29: 141–148. [DOI] [PubMed] [Google Scholar]
- 39. Murphy E. Estrogen signaling and cardiovascular disease. Circ Res 2011; 109: 687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Boyer JK, Thanigaraj S, Schechtman KB, et al. Prevalence of ventricular diastolic dysfunction in asymptomatic, normotensive patients with diabetes mellitus. Am J Cardiol 2004; 93: 870–875. [DOI] [PubMed] [Google Scholar]
- 41. Fontes-Carvalho R, Mancio J, Marcos A, et al. HIV patients have impaired diastolic function that is not aggravated by anti-retroviral treatment. Cardiovasc Drugs Ther 2015; 29: 31–39. [DOI] [PubMed] [Google Scholar]
- 42. Wu J, Yu SY, Wo D, et al. Risks and predictors of mild diastolic dysfunction among middle-aged and aged women: a population-based cohort study. J Hum Hypertens 2016; 30: 335–340. [DOI] [PubMed] [Google Scholar]
- 43. Sciarretta S, Paneni F, Palano F, et al. Role of the renin-angiotensin-aldosterone system and inflammatory processes in the development and progression of diastolic dysfunction. Clin Sci (Lond) 2009; 116: 467–477. [DOI] [PubMed] [Google Scholar]
- 44. Iyer SN, Lu D, Katovich MJ, et al. Chronic control of high blood pressure in the spontaneously hypertensive rat by delivery of angiotensin type 1 receptor antisense. Proc Ntl Acad Sci USA 1996; 93: 9960–9965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jessup JA, Gallagher PE, Averill DB, et al. Effect of angiotensin II blockade on a new congenic model of hypertension derived from transgenic Ren-2 rats. Am J Physiol Heart Circ Physiol 2006; 291: H2166–H2172. [DOI] [PubMed] [Google Scholar]
- 46. Fortepiani LA, Zhang H, Racusen L, et al. Characterization of an animal model of postmenopausal hypertension in spontaneously hypertensive rats. Hypertension 2003; 41: 640–645. [DOI] [PubMed] [Google Scholar]
- 47. Wallen WJ, Cserti C, Belanger MP, et al. Gender-differences in myocardial adaptation to afterload in normotensive and hypertensive rats. Hypertension 2000; 36: 774–779. [DOI] [PubMed] [Google Scholar]
- 48. Wittnich C, Wallen J, Belanger M. The role of 17beta-estradiol in myocardial hypertrophy in females in the presence and absence of hypertension. Cardiovasc Drugs Ther 2015; 29: 347–353. [DOI] [PubMed] [Google Scholar]
- 49. Thum T, Borlak J. Testosterone, cytochrome P450, and cardiac hypertrophy. FASEB J 2002; 16: 1537–1549. [DOI] [PubMed] [Google Scholar]
- 50. Li J, Jubair S, Janicki JS. Estrogen inhibits mast cell chymase release to prevent pressure overload-induced adverse cardiac remodeling. Hypertension 2015; 65: 328–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fu L, Wei CC, Powell PC, et al. Increased fibroblast chymase production mediates procollagen autophagic digestion in volume overload. J Mol Cell Cardiol 2016; 92: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shiota N, Jin D, Takai S, et al. Chymase is activated in the hamster heart following ventricular fibrosis during the chronic stage of hypertension. FEBS Lett 1997; 406: 301–304. [DOI] [PubMed] [Google Scholar]
- 53. Studer R, Reinecke H, Muller B, et al. Increased angiotensin-I converting enzyme gene expression in the failing human heart. Quantification by competitive RNA polymerase chain reaction. J Clin Invest 1994; 94: 301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Crackower MA, Sarao R, Oudit GY, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002; 417: 822–828. [DOI] [PubMed] [Google Scholar]
- 55. Trask AJ, Groban L, Westwood BM, et al. Inhibition of angiotensin-converting enzyme 2 exacerbates cardiac hypertrophy and fibrosis in Ren-2 hypertensive rats. Am J Hypertens 2010; 23: 687–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Keidar S, Kaplan M, Gamliel-Lazarovich A. ACE2 of the heart: From angiotensin I to angiotensin (1–7). Cardiovasc Res 2007; 73: 463–469. [DOI] [PubMed] [Google Scholar]
- 57. Trask AJ, Averill DB, Ganten D, et al. Primary role of angiotensin-converting enzyme-2 in cardiac production of angiotensin-(1–7) in transgenic Ren-2 hypertensive rats. Am J Physiol Heart Circ Physiol 2007; 292: H3019–H3024. [DOI] [PubMed] [Google Scholar]
- 58. Zisman LS, Keller RS, Weaver B, et al. Increased angiotensin-(1–7)-forming activity in failing human heart ventricles: evidence for upregulation of the angiotensin-converting enzyme Homologue ACE2. Circulation 2003; 108: 1707–1712. [DOI] [PubMed] [Google Scholar]
- 59. Brosnihan KB, Hodgin JB, Smithies O, et al. Tissue-specific regulation of ACE/ACE2 and AT1/AT2 receptor gene expression by oestrogen in apolipoprotein E/oestrogen receptor-alpha knock-out mice. Exp Physiol 2008; 93: 658–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kassiri Z, Zhong J, Guo D, et al. Loss of angiotensin-converting enzyme 2 accelerates maladaptive left ventricular remodeling in response to myocardial infarction. Circ Heart Fail 2009; 2: 446–455. [DOI] [PubMed] [Google Scholar]
- 61. Ishiyama Y, Gallagher PE, Averill DB, et al. Upregulation of angiotensin-converting enzyme 2 after myocardial infarction by blockade of angiotensin II receptors. Hypertension 2004; 43: 970–976. [DOI] [PubMed] [Google Scholar]
- 62. Patel VB, Zhong JC, Grant MB, et al. Role of the ACE2/angiotensin 1–7 axis of the renin-angiotensin system in heart failure. Circ Res 2016; 118: 1313–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.