

Abstract
Traditional cell culture and animal models utilized for preclinical drug screening have led to high attrition rates of drug candidates in clinical trials due to their low predictive power for human response. Alternative models using human cells to build in vitro biomimetics of the human body with physiologically relevant organ-organ interactions hold great potential to act as “human surrogates” and provide more accurate prediction of drug effects in humans. This review is a comprehensive investigation into the development of tissue-engineered human cell-based microscale multi-organ models, or multi-organ microphysiological systems for drug testing. The evolution from traditional models to macro- and microscale multi-organ systems are discussed in regards to the rationale for recent global efforts in multi-organ microphysiological systems. Current advances in integrating cell culture and on-chip analytical technologies, as well as proof-of-concept applications for these multi-organ microsystems are discussed. Major challenges for the field, such as reproducibility and physiological relevance, are discussed with comparisons of the strengths and weaknesses of various systems to solve these challenges. Conclusions focus on the current development stage of multi-organ microphysiological systems and new trends in the field.
Keywords: microphysiological systems, body-on-a-chip, drug development, functional readouts, PBPK
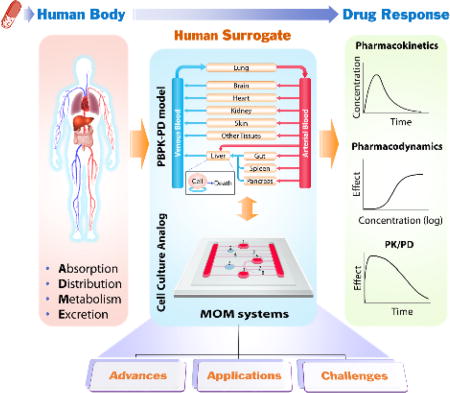
Multi-organ microphysiological (MOM) systems are in vitro microscale cell culture analog of the human body (Body-on-a-chip). Tissue engineered single organ models are interconnected to reproduce complex multi-organ interactions. Such biomimetics of the human body can potentially serve as “human surrogates” to simulate human responses to drugs, providing pharmacokinetic and pharmacodynamic information.
1. Introduction
The current paradigm of pharmaceutical innovation has rendered drug development a lengthy and increasingly expensive process. It takes an average of more than 10 years to develop and bring a safe and effective new treatment to bedside.[1] The average cost for a new drug to reach market has been more than doubling every decade for the last forty years,[1] and has recently estimated to be $2.6 billion per drug.[2] The escalating R&D cost burden is inevitably passed on to patients, and partially contributes to the spiraling price of new drugs.[3] The impetus behind this rising R&D cost is the high drug attrition rate during clinical trials, the most expensive stage in drug development. Around 90% (95% for oncology) of drug candidates selected through preclinical studies fail clinical development mainly due to unexpected safety issues or lack of efficacy.[4,5] New strategies for preclinical screening that can better predict clinical outcomes are an urgent need to expedite drug development and to do so at sustainable prices.
1.1. Current models
Current preclinical drug screening relies heavily on in vitro cell culture models and animal models to project drug responses in humans. While conventional two-dimensional (2D) cell culture models can be easily established in automation-friendly, multi-well microtiter plates and can facilitate high throughput screening (HTS), the screening accuracy is limited. These simplified 2D models in many cases do not capture the in vivo three-dimensional (3D) cell microenvironment and tissue structure that support authentic cell and organ functions. Most importantly, they fail to reproduce complex multi-organ interactions and dynamic drug process that take place in a living animal. Animal models, on the other hand, can provide valuable information on in vivo drug process and responses. Yet, they not only are expensive and time-consuming, raise ethical issues, but have shown disappointingly low predictive power for human drug responses due to cross-species discrepancies between human and experimental animals.[4,6]
1.2. Emerging alternative models
In recognizing the problems with current cell and animal models, the field of drug development, including regulatory agencies, pharmaceutical, and chemical companies, is actively searching for new methodologies, aiming to develop a realistic “human surrogate” that will reduce animal usage and offer more accurate and cost-effective prediction of human responses. Ideally, a human surrogate should recapitulate the dynamic in vivo drug process of absorption, distribution, metabolism and excretion (ADME), reproduce the pharmacokinetics (PK) and pharmacodynamics (PD), and ultimately project the time course of pharmacological response (PK/PD) to guide drug candidate screening and dose selection for clinical trials (Figure 1). The goal for the human surrogate development for drug screening is not to build a perfect mini-human or to replicate a human body, but simply to provide a better predictive model than animal models. Increasing the success of clinical trials from 10% to even 20–30% would reduce drug development costs tremendously.
Figure 1.

Schematic diagram illustrating how in vitro cell culture analog (CCA) combined with PBPK-PD modeling can serve as human surrogates and contribute to improving the accuracy and efficiency of preclinical prediction of drug response in human. Drugs entering the human body undergo a dynamic process of drug absorption, distribution, metabolism and excretion (ADME), and elicit various responses. PBPK-PD models and CCAs are mathematical and physical representation of the human body, respectively. They model the human body as a series of interconnected compartments representing different organs or tissues to simulate drug process and responses. The two types of models are complementary. PBPK models can guide the design of CCAs to improve their physiological relevance, while data from CCAs can be used to reduce the number of adjustable parameters in PBPK models. Comparison of CCA results with PBPK simulation results can lead to improvement or validation of CCA models that aims to reproduce the pharmacokinetics (PK, the time course of drug and metabolite concentrations in the body) and pharmacodynamics (PD, the pharmacological effect of a drug, described as a function of concentration), and ultimately project the time course of pharmacological response (PK/PD).
A promising technology that has gained increasing recognition is a mathematical approach, physiologically based pharmacokinetic (PBPK) modeling. PBPK modeling takes a bottom-up approach that integrates known biological mechanisms with all available compound data.[7] It treats the human body as a series of interconnected compartments representing single organs or tissues with great similarity (Figure 1). The contents within a compartment are considered homogenous. The compartments are reactors, absorbers, or holding tanks, where the concentration of drugs or metabolites can be described by differential equations. Such mathematical models can reflect organ-organ interactions and the dynamic exposure of tissues to drugs, allowing one to predict the pharmacokinetics for a given dose scenario. PBPK models combined with PD models can be potent tools in predicting dynamic drug effects under normal and disease conditions. A rodent PBPK model with five tissue compartments (“lung”, “fat”, “kidney”, “liver”, and “other tissues”) built to describe naphthalene uptake and metabolism in vivo, solely based on the then-current knowledge of naphthalene circulation and biotransformation, as well as physiological and in vitro kinetic parameters, accurately predicted naphthalene responses in animals under various routes of administration, pretreatments and doses.[8] The value of PBPK modeling for predicting human drug responses, however, has not been fully exploited, mainly due to the limited quantitative knowledge of human cell metabolism and a large body of unknown biological mechanisms. Most current PBPK models are either empirical descriptions or more mechanistic with a great number of adjustable parameters, which diminish their predictive power.[7]
In contrast to PBPK modeling’s mathematical representation of the human body, cell culture based multi-compartmental, multi-cellular bioreactors have been proposed as the physical analog of a living system to incorporate organ-organ interactions into in vitro drug testing.[9,10] An early proof-of-concept experiment was conducted in a three-chamber system using naphthalene as a model drug.[11] Simply by interconnecting all cell culture flasks/bottles and providing continuous medium circulation among them, a cell culture based multi-organ model (also called cell culture analog, CCA) with “liver”, “lung”, and “other tissues” chambers recapitulated the toxic effects of naphthalene on lung cells due to the formation of cytotoxic naphthalene oxide in the liver chamber and its circulation to the lung chamber.[11] Such interconnected multi-compartmental CCA models are powerful tools for studying remote actions of compounds with reactive metabolites[11] and other soluble ligand mediated crosstalks among tissues[12], which are usually missing in the traditional cell culture models. Connected cultures of hepatocytes, endothelial cells and adipocytes in the multi-compartment bioreactors (MCB) under circulating perfusion exhibited modified glucose and lipid metabolic profiles as compared to static individual cultures.[12–14] Intestinal and liver cell co-culture in consecutively perfused bioreactors mimicked the intestinal absorption and first-pass liver metabolism of the prodrug phenacetin,[15,16] and indicated synergistic intestinal and liver metabolism in response to paracetamol.[17] The CCA models also offer great flexibility for focusing on specific subsets of organs, creating “knockout models” or changing the cell number or cell metabolic/functional states of specific organ chambers to pursue mechanistic interrogation or mimic pathological conditions, which are usually complicated to achieve with living animals.
Human CCA models hold the potential to simulate human drug responses including those from drug actions of unknown mechanisms that cannot be predicted by PBPK modeling. They are also powerful platforms to generate quantitative knowledge of human cell metabolism and help reduce the number of adjustable parameters in a PBPK model. Meanwhile, PBPK models of the human body can guide the design of CCA models, thus making them more physiologically relevant, and help extrapolate the results from CCA models for clinical trials. Combining the strength of PBPK modeling and human CCA models can be a potent approach to develop realistic human surrogates for drug development.
1.3. New opportunities for developing multi-organ microphysiological systems
The explosive growth in the fields of stem cell technology, 2D and 3D cell culture, microfabrication, and microfluidics since the beginning of the 21st century has brought unprecedented opportunities for developing in vitro microscale biomimetics of the human body as a promising “human surrogate” for the next generation drug screening. The success in generating human induced pluripotent stem cells (hiPSCs) from adult somatic cells[18–20] and differentiating them in vitro into a variety of specific organ cell types, including neurons,[21–23] cardiomyocytes,[24,25] hepatocytes,[26] pancreatic cells,[27] lung and airway epithelial cells,[28] brain microvascular endothelial cells[29] and intestinal cells,[30] offers new possible cell sources for building in vitro human tissue biomimetics. Compared to cell lines, human iPSC derived cells exhibit phenotypes closer to their in vivo counterparts, and the available cell number is potentially unlimited, in contrast to primary cells. Such techniques also allow creation of patient and disease specific cells for personalized drug screening models.[31,32] With the advent of novel biomaterials that mimic native cell microenvironment[33,34] and 3D biofabrication technologies[35,36] that can create cell spheroids or clusters with 3D organization, in vitro re-creation and maintenance of functional organ mimetics has become realistic. Several miniaturized organotypic models of human organs, including the brain,[37] heart,[38] kidney,[39] and liver,[40] have been developed and demonstrated with some organ level functions. Development of micro/nanofabrication technologies and biological microelectromechanical systems (BioMEMS) allows creation of miniaturized bioreactors with interface feature size close to the cell emulating the native cell niche, microarchitectures mimicking tissue structures, as well as in situ microbiosensors.[41] Recent development in microfluidics enables precise microscale fluid control and allows the creation of desired fluidic connections among microscale organ models in mimicking in vivo blood circulation.[42,43] With all these advances in technologies, new global efforts have been mounted to build the next generation drug testing tool, namely, tissue-engineered human cell-based microscale multi-organ models. These multi-organ microphysiological systems (MOMs), also known as microCCA[44,45] or body-on-a-chip (BOC),[41,46–48] are gaining momentum in recent years and hold great potential to better identify drug candidates for clinical trials.
1.4. Focus of the current review
There have been many comprehensive reviews on the design, construction and applications of tissue engineered microphysiological systems (MPS) for single organs or specific diseases.[41,49–59] This review will focus on microscale multi-organ systems with total fluid and tissue volume smaller than 1/10,000 of the human body (~6.5 mL).[60] We will review the current available multi-organ integration platforms, on-chip analytical technologies, and the proof-of-concept applications in drug testing and disease modeling. We will also evaluate different models in terms of their strength and weakness, and focus on discussing several critical challenges that the field is facing and possible strategies as well as future directions.
2. Advances in multi-organ integration platforms
To establish organ-organ interactions in vitro, fluidic connections among organ models are often required. Based on how various organ models are fluidically interconnected, the current integration platforms can be categorized into four major types (Figure 2): i) static microscale platforms, ii) single-pass microfluidic platforms, iii) pump-driven recirculating microfluidic platforms, and iv) pumpless recirculating microfluidic platforms. We will discuss each type of platform by focusing on their approaches to integration, representative models, and their advantages vs. disadvantages. More detailed applications of different platforms will be discussed in Section 4. Challenges that are common for all platforms will be discussed in Section 5.
Figure 2.

Integration platforms for recreating organ-organ interactions in vitro. (A) Static microscale platform presented in four major forms: a) transwell platform; b) microtunnel platform; c) micropattern platform; d) wells-in-a-well platform. (B) Single-pass microfluidic platform connects all organ modules in series in one fluid route (route 1), or with additional routes (e.g., route 2) if barrier tissues are involved. (C) Pump-driven recirculating platform interconnects organ modules in serial and/or parallel in a closed-loop circuit (loop 1). Separate fluidic pools or loops (e.g. loop 2) are needed for barrier tissues. (D) Pumpless recirculating platforms utilize gravity-driven flow and a rocking motion to drive fluid through the organ module network, in which organ modules are connected in serial and/or parallel. On, organ module; Pn, pump; Rn, reservoir; Cn, medium collector; Dn, debubbler. n represents the index of a specific module.
2.1. Static microscale platforms
Static microscale platforms achieve organ-organ interactions mainly through direct physical contact among tissues and/or passive diffusion of soluble ligands, cell metabolites or cellular components via a common medium connecting all organ compartments. Four major forms of static multi-organ MPS are presented below and their advantages and disadvantages are discussed (Figure 2A).
2.1.1. Transwell platform
The transwell platform is a long-established static system for multi-compartmental, multi-cellular co-culture. It was first developed by Dr. Stephen Boyden for leukocyte migration analysis in 1960s.[61] In such a system, a transwell, which is a cylindrical insert with a thin porous polymeric membrane bottom, is placed in a traditional cell culture well, dividing the well into an upper and a lower compartments (Figure 2A (a)).[62,63] It compartmentalizes different organ models while allowing inter-organ medium exchange, cellular contact and even cell migration through micropores of various dimensions. The transwell system accommodates up to three different organs that can be easily analyzed and retrieved individually. The platform is particularly useful for systems involving barrier tissues. With open access to both compartments, drug absorption through a barrier tissue (such as the intestinal wall, the skin, vasculature, and the blood brain barrier) constructed on the porous membrane can be easily evaluated by monitoring the concentrations of test drugs or their metabolites in both donor and receiving compartments over time.
2.1.2. Microtunnel platform
In contrast to a transwell’s vertical connection through micropores, the microtunnel platform creates horizontal ties among organ chambers with microfabricated fluid tunnels (Figure 2A (b)). Inter-organ medium exchange through the microtunnels by diffusion is generally not efficient due to the great length and small cross-sectional area of the fluid path. The resulting biochemical gradient established across the microtunnels, however, can be effective in guiding cell migration or directional growth of cellular projections (such as axons and neurites) with their cell bodies restrained within the organ chambers. The microtunnel platform is therefore often used to create connections between the neural system and other organs, such as muscle[64] or tumors[65].
2.1.3. Micropattern platform
The micropattern platform creates multi-organ co-culture in a single compartment with different organ cells spatially separated using cell micropatterning techniques (Figure 2A (c)).[66] Different types of cells are often selectively attached to or removed from 2D culture substrate with regional (patterned) surface modification that tunes cell attachment,[67,68] or encapsulated in different hydrogels patterned in 3D configuration.[69] Different organ cell interactions are mainly mediated by diffusion through the overlying medium, and sometimes also by physical contact with neighboring cells. The cell micropatterning allows easy allocation of different organ cells for optical interrogation. Selective retrieval of live cells is also possible for further cell/molecular analysis.[70] Yet the spatial constraints based on the differential properties of initially patterned surface or scaffold can gradually lose effectiveness after cells produce their own extracellular matrix and modify their surroundings. Analysis of individual organs in a long-term multi-organ co-culture could be challenging using this platform.
2.1.4. Wells-within-a-well platform
The wells-in-a-well concept modifies the micropattern platform by using physical barriers instead of surface or 3D micropatterning to separate various organ cells (Figure 2A (d)). The integrated discrete multiple organ cell culture (IdMOC) system developed by Li and coworkers represents the first commercial use of the “wells-within-a-well” platform for multi-organ co-culture.[71] The IdMOC plates consist of large co-culture wells with each containing multiple small wells. Each small inner well serves as an isolated culture compartment for individual organs. Different organs can be cultured in organ-specific medium till they are all ready for co-culture. Multi-organ co-culture is initiated by filling up the large co-culture wells above all inner wells with a common medium. Crosstalk among organ compartments is driven by diffusion of soluble cell metabolites through the overlying medium. The IdMOC plates support multi-organ co-culture of up to 6 different organ models. The plates were adapted from conventional multiwell culture plate format and can be easily compatible with automated liquid handling and imaging systems to achieve high-throughput multi-organ drug testing.
2.1.5. Summary
In general, static microscale multi-organ integration platforms are easy to construct and operate, and can recreate basic organ-organ connections in vitro. There are usually no moving parts or forced convection of fluids in the system. The platforms are either commercially available (the transwells and the IdMOC plates) or require minimal microfabrication. The organ compartments are often open for direct access where macroscale cell culture and liquid handling techniques can easily be applied. The horizontal arrangement of multiple organ compartments in most static microsystems, including microtunnel, micropattern and wells-within-a-well models, facilitates optical interrogation, while the transwell platform supports multi-organ models with barrier tissues. The organ-organ interactions in the static systems rely on diffusion and/or physical contact that are mostly established based on biochemical gradients among organ compartments.
Any disturbance to the gradients in the system could lead to outcome variations or unrepeatable results. More importantly, inter-organ communication through the common medium is less controllable in terms of specificity, directionality and extent, unlike the in vivo circulation system with specific perfusion order and rate for different organs.
2.2. Single-pass microfluidic platforms
Single-pass microfluidic platforms integrate different organ models by applying open-loop sequential medium perfusion through all organ units in a single fluid route, or multiple separate ones if barrier tissues are included (Figure 2B). The organ-organ interactions in such systems are mostly unidirectional. Drugs and drug metabolites from upstream organ models are carried along and can act on the downstream organ units while there is no feedback route to the upstream organs. This lack of feedback eliminates the detection of a potentially important route of inter-organ interactions.
Multi-organ microfluidic platforms often take modular approaches to integration. van Midwoud et al. constructed a single-pass multi-organ platform made of polydimethylsiloxane (PDMS) through serial tubing connection of multiple microfluidic biochips previously designed for continuous organ slice perfusion (Figure 3A).[72,73] Each biochip unit adopted a transwell-like structure, where a polycarbonate porous membrane was integrated to divide cylindric microchambers into upper and lower chambers and support organ slices. The medium was perfused using a syringe pump through the porous membrane, around the organ slice, and then passed to the downstream biochip to convey unidirectional organ interactions. Loskill et al. proposed a configurable multi-organ platform, µOrgano, which used Lego®-like plug-and-play connectors for rapid and reliable transformation between discrete single-organ mode and integrated multi-organ mode (Figure 3B).[74] This modular platform decouples the single-organ culture platforms from the multi-organ integration platform, and thus can reduce the complexity of multiple organoid preparation. The demonstration of the above microfluidic integration platforms has so far been limited to single-pass, single-route perfusion for parenchymal tissues (or barrier tissues without consideration of their barrier function).
Figure 3.

Representative single-pass MOM systems. (A) A liver-intestine MOM based on serially connected biochips designed for tissue slice perfusion. Reproduced with permission.[73] Copyright 2010, Royal Society of Chemistry. (B) µOrgano, a modular multi-organ platform using Lego®-like plug&play connectors for inter-organ connection. Adapted under the terms of the CC-BY-4.0 license.[74] Copyright 2015, Loskill et al. (C) An intestine-liver-tumor MOM with both systemic and intestinal perfusion. Reproduced with permission.[75] Copyright 2010, American Chemical Society. (D) A functionally coupled microphysiological system with multiple organ modules constructed at physically distant laboratories and connected through medium transfer in a physiological order. Adapted under the terms of the CC-BY-4.0 license.[77] Copyright 2017, Vernetti et al.
Integration of functional barrier tissues into multi-organ MPS requires multiple separate microphysiological fluid pools or routes. Imura et al. constructed an intestine-liver-tumor microsystem with both apical and basal side perfusion of the intestine tissue driven by two syringe pumps (Figure 3C).[75] A porous membrane was sandwiched between two microfluidic channels made in PDMS to support the intestine tissue culture at the interface of the dual fluid circuits. This enabled quantitative evaluation of intestine absorption through apical drug administration and both-side sampling. The systemic perfusion through “intestine” (basal), “liver” and “tumor” chambers also allowed testing of liver metabolism and the anti-cancer effects of drugs after intestinal absorption and metabolism. Imura and coworkers extended the platform by adding infusion inlets along the apical microchannel before passing through the intestine chamber. Such modification enabled sequential mixing of drug samples with infused artificial gastric juice, neutralization buffer, and artificial intestinal juice, and thus allows additional testing on the stability of drugs throughout the digestion process.[76]
Aside from the physical integration of various organ models, a special form of the single-pass MOMs using “functional coupling” has been demonstrated through a multi-laboratory collaboration (Figure 3D).[77] Individual organ units were constructed separately in four physically distant laboratories. Organ-organ connections were established by transporting medium effluent from one organ module to the next in a physiological order. A liver-muscle model and a four-organ model of intestine, liver, blood brain barrier/neurovascular unit, and kidney were built in such form as proof-of-concept microsystems to model drug ADME process. The integration strategy of “functional coupling” could potentially combine the expertise from different laboratories and include the most sophisticated single organ modules into a multi-organ system. The physical decoupling allowed to construct individual organ models on different microfluidic platforms and culture them in their optimized medium at organ-specific perfusion rate. Yet the results would not be in real-time or dynamic. The extra intermediate steps to connect distant organ modules could bring additional administrative and logistic costs and raise a major concern about to which extent the inter-organ communication can be preserved and recreated after transportation. Cell metabolites and other soluble ligands that could act on downstream organ modules may have lost or compromised their bioactivity during the prolonged process, especially those reactive intermediates. Overall, functional coupling could be a practical strategy for constructing pilot multi-organ models to gain valuable insights for future designs before a universal platform and a common medium are developed to physically integrate all organ modules. Yet it would be difficult to adopt for industrial drug screening in its current format.
In summary, the single-pass microfluidic platforms for multi-organ integration feature open-loop sequential fluid transfer across organ modules through connecting tubing, microfluidic channels, or transportation. The controlled fluid flow allows more reliable mass transfer of cell metabolites among organ models than that in systems solely depend on diffusion. The continuous perfusion of fresh medium and drug sample solutions gives constant nutrient supply, waste removal and a steady dose of drug. Such multi-organ models could be useful in providing a qualitative evaluation of drug pharmacodynamics. Yet their ability to simulate in vivo pharmacokinetics and drug effects is largely limited due to the oversimplified series perfusion architecture and the lack of reciprocal organ-organ interactions.
2.3. Recirculating microfluidic platforms
Recirculating multi-organ platforms connect individual organ compartments with circulating culture media that mimic the blood flow in the body (Figure 2C–D). Such closed-loop perfusion facilitates reciprocal organ interactions that are usually missing in the single-pass perfusion systems. Cell metabolites released from one organ chamber are distributed by circulation and can act on other organ units until clearance. The closed-loop medium circulation also allows simulation of the dynamic exposure of organ cells to drugs and drug metabolites over time, producing valuable pharmacokinetic information.
2.3.1. Pump-driven recirculating MOMs for parenchymal tissues
In 2004, the Shuler group, which had previously demonstrated the CCA models in macroscale, combined the CCA concept with microfabrication technologies to develop a three-compartment microscale CCA (µCCA) model based on a 2.5 cm × 2.5 cm silicon chip with etched microchannels and microchambers.[78] The medium was recirculated through the chip using a peristaltic pump. The group extended this silicon chip based platform to four-chamber models to integrate more parenchymal organs,[44,79,80] and later introduced 3D cell culture in hydrogels into the µCCA platform to better mimic the 3D structure of parenchymal tissues (Figure 4A).[81] These µCCA models were designed based on PBPK models, which incorporated better physiological relevance into the systems. The dimensions of the on-chip microfluidic channels and microchambers were specifically devised and coupled with the pump flow rate based on proportional scaling of the organ volumes and perfusion rates in human. Such systems maintained in vivo organ size ratios and physiological fluid residence time in the corresponding organ chambers. The silicon base for these early models enabled creation of miniaturized fluidic channels and organ chambers, which required hundreds to thousands fold fewer cells and media than the macroscale counterpart,[11] and provided near-physiological level liquid-to-tissue ratios in organ compartments.[78] It also allowed integration of microscale features for local fluidic control, such as using micropillar arrays for rapid and even flow distribution in organ chambers.[78]
Figure 4.

Representative pump-driven recirculating MOM systems demonstrated for parenchymal tissues (A–D) and with barrier tissues (E–F). (A) A liver-marrow-tumor 3-organ µCCA with 3D hydrogel cell culture. Adapted and reproduced with permission.[81] Copyright 2009, Royal Society of Chemistry. (B) A 4-organ MOM with compartmentalized microenvironment. Reproduced with permission.[83] Copyright 2009, Royal Society of Chemistry. (C) A modular MOM system integrated with a flow-controlling breadboard, an on-chip bubble trap, and multiple biosensors. Adapted from Zhang et al.[84] Copyright 2017, Zhang et al. (D) A liver-tumor MOM based on open channels and hanging drop arrays. Reproduced with permission.[85] Copyright 2014, Nature Publishing Group. (E) A 5-compartment, liver-GI 2-organ MOM. Photograph of the system reproduced with permission.[87] Copyright 2009, John Wiley and Sons. (F) A dual-layer, stirrer-based micropump driven liver-intestine-lung MOM.[90] Photo provided by courtesy of Dr. Teruo Fujii. G) Transwell compatible, pneumatic pump driven MOM systems. i) A liver-skin MPS; Reproduced with permission.[92] Copyright 2013, Royal Society of Chemistry. ii) A liver-neurosphereo MPS; Reproduced with permission.[94] Copyright 2015, Elsevier. iii) A four-organ MOM with systemic and excretory circuits. Adapted under the terms of the CC-BY-3.0 license.[42] Copyright 2015, Royal Society of Chemistry. H) A multilayer MOM mimicking the ADME process of oral drugs.1) Caco-2 cell layer; 2) Human umbilical vein endothelial cells (HUVEC) (grey) and hepatocyte (green) layers; 3) Hepatocyte (green) and HUVEC (grey) layers; 4) MCF-7 cell layer; 5) Lung, heart, adipose tissues. Adapted under the terms of the CC-BY-4.0 license.[95] Copyright 2016, Nature Publishing Group. (I) An integrated MOM platform driven by electromagnetically actuated micropumps. Reproduced with permission.[96] Copyright 2016, Royal Society of Chemistry.
In contrast to the serial connection in all single-pass platforms to achieve inter-organ communications, the perfusion through different organ models in a recirculating MOM can be serial,[82] parallel,[80] or combined. The four-compartment µCCA of “lung”, “liver”, “fat”, and “other tissues” developed by Viravaidya et al. adopted a perfusion architecture mimicking the blood circulation.[79] The “liver”, “fat”, and “other tissues” chambers were connected in parallel, and together connected in series with the “lung” chamber, a medium reservoir and a peristaltic pump. A similar perfusion network was used for a 4-organ recirculating MOM of “fat”, “kidney”, “liver” and “lung”, presented by Zhang et al. (Figure 4B).[83] The volumes and the perfusion rates in this 4-organ system, however, were not customized for each organ compartment, leaving the system less physiologically relevant despite the in vivo-like connections among organs. The system integrated four micropillar arrays to confine hydrogel immobilized 3D cell cultures within the organ chambers, and allow peri-chamber perfusion through microchannels defined by the micropillars (Figure 4B (iii)). Such micropillar arrays combined with the hydrogel encapsulation approach protected cells from direct exposure to shear stress from medium perfusion.
To construct more sophisticated recirculation architecture for multi-organ systems, a fluid breadboard based modular approach has recently been proposed.[84] The automated flow control breadboard made of PDMS was integrated with on-chip microchannels and program controllable pneumatic valves and could be used to manage fluid circulation among organ models, on-chip analytical modules, bubble traps, and reservoirs (Figure 4C). The platform was demonstrated with a two-organ model of “liver” and “heart” connected in series. It holds the potential of integrating multiple organ modules and handling complexed fluid network with features of in vivo-like perfusion architecture and organ-specific perfusion rates.
A unique, open channel based multi-organ microfluidic platform has also been developed.[85] Frey et al. transformed an array of hanging drops, commonly used for the 3D culture of cell spheroids or microtissues, into multiple parallel multi-organ microsystems using a multi-channel peristaltic pump and a reconfigurable microfluidic network interconnected through open channels between neighboring hanging drops (Figure 4D). By filling and establishing perfusion through one dimension of the 2D microchannel network, cells from different organs were loaded and cultured in columns of the hanging drop network. Multi-organ units were then created in rows by switching the perfusion to the second dimension of the network. Such a platform supports the 3D culture and analysis of both individual organ units and multi-organ sets, and can flexibly switch between the two modes. The multiple parallel multi-organ sets on chip also allow simultaneous testing of several drugs or drugs at different doses. Yazdi et al. further made the system more compact with built-in pneumatic chambers for flow control.[86] The hanging drop array based design can potentially create an integrated, high throughput platform for recirculating MOM systems. These open-channel based systems usually do not have the problem of bubble formation that occurs in most closed channel microfluidic systems, yet they face a major issue of medium evaporation at rates varying with the surrounding environment. Additional compensating flow is often required. In addition, the current proof-of-concept multi-organ system adopted the simplest serial fluidic network architecture to connect all the different organ chambers, which were perfused at the same flow rate. It is not straightforward whether such a platform can be extended to incorporate in vivo-like circulation structure with parallel branches and organ-specific perfusion rates or accommodate functional barrier tissues.
2.3.2. Pump-driven recirculating MOMs with functional barrier tissues
Barrier and parenchymal organ models are usually constructed on different platforms. Unlike parenchymal organ models established in confined microchambers or hanging drops, models of barrier tissues, such as intestine, lung and skin, are often constructed on microporous membranes that separate the apical and basal space of the barrier. Incorporating barrier tissues into multi-organ microfluidic systems often involves integration of the different platforms for barrier and non-barrier tissues.
Mahler et al. took a simple modular approach for integration by keeping barrier and parenchymal organ models on their own platforms and interlinking them with tubing (Figure 4E).[87] They created a 5-compartment MOM by combining a previously developed transwell-based gastrointestinal (GI) tract model[88] and a four-chamber silicon µCCA. The system provided systemic and intestine apical dual circulation and can be used to model the absorption, metabolism and toxicity of oral drugs and supplements.[87,89] Such modular multi-organ systems are usually less compact than single-chip integrated systems, yet they require minimum adaptation for both barrier and parenchymal organ models.
Kimura et al. proposed a single-chip, dual-layer design for barrier tissue integration (Figure 4F). They constructed a three-organ recirculating microsystem of “liver”, “small intestine” and “lung”.[90] The barrier tissue (small intestine) chamber and the apical fluid circuit were created in the upper PDMS layer, while the parenchymal tissue chambers (liver, and lung, treated as a non-barrier tissue) and the systemic (basolateral) circuit were built in the lower layer. A microporous membrane was sandwiched in between as the interface of the two circuits. To better reproduce the interactions between small intestine and liver, the system preserved many physiological parameters for both organs, such as organ size ratio and perfusion rate, as well as systemic perfusion structure. The built-in stirrer-based micropumps also made the system compact and self-contained (no fluid or gas loops required outside of the device[91]).
Wagner et al. from the Marx group utilized transwell inserts to host barrier tissues and developed a multi-compartment microfluidic platform that could accommodate transwell inserts and provide recirculating perfusion with an on-chip pneumatic pump (Figure 4G (i)).[92] A proof-of-concept system with two organ compartments was demonstrated with liver microtissues and skin biopsy. With a transwell insert installed in the skin compartment, this microfluidic device established the air-liquid interface required for skin tissue culture and supported stable co-culture of the liver and skin tissues for up to 28 d. The group later used the same platform to build liver-skin, liver-neurosphere, and liver-intestine microsystems with 3D cultured liver microtissues, skin biopsies, neurospheres, and intestine constructs (Figure 4G (ii)).[93,94] They endothelialized the microfluidic channels in the liver-skin model to mimic the in vivo vasculature, and observed interactions between endothelial cells and the liver microtissues. These co-cultures were stable and maintained on chip for up to 14 d. The same group further developed a four-organ MOM system of “intestine”, “liver”, “skin”, and “kidney” on a modified device with two recirculating fluid circuits (Figure 4 G (iii)).[42] The chip included a second microfluidic channel layer for the excretory circuit and a sandwiched microporous membrane between the two channel layers to support kidney proximal tubule epithelial cell culture. The microsystem maintained cell viability and tissue structure for 28 d and established a stable gradient of glucose among the three fluid pools representing the intestine lumen, the blood circulation and the kidney excretory circuit. Such a platform can potentially be used to simulate the ADME profile of drug candidates. Overall, these transwell compatible microfluidic systems provide a reliable platform for long-term co-culture of multiple organ models including barrier tissues, and thus can potentially support drug testing with repeated doses.
An et al. proposed a unique vertical stacking structure for constructing MOM systems (Figure 4H).[95] The presented microsystem included cell and tissue models representing intestine, vasculature, liver, heart, lung and adipose tissues, a dialysis membrane representing the kidney, and three recirculating fluid loops representing intestinal lumen, systemic circulation and kidney excretory circuit. Four polycarbonate microporous membranes and the dialysis membrane were sandwiched between six vertically stacked PDMS microchannel or microchamber layers to support barrier and non-barrier tissue culture and serve as permeable interface between microchannels and microchambers. It should be noted that, different from the microfluidic MOMs previously discussed, the transfer of cell metabolites among different organ models in this system depended mainly on diffusion driven by the gradients established between the three fluid circuits. The system was used to simulate the ADME process and toxicity of several oral drugs. The microsystem established interconnections among multiple organ models, yet it had little physiological resemblance in terms of the circulation structure, organ volume and perfusion rate. The testing results were more qualitative than quantitative and should be interpreted with appropriate caution.
More integrative platforms are emerging.[96–99] They usually consist of an organ module layer, a fluidic layer, and a flow control layer embedded with arrays of pneumatic or electrically actuated micropumps (Figure 4I). These integrative MOM boards often provide flexibility of fluid circuit configuration and sophisticated onboard fluid control. They are also featured with scalability and portability attractive for industrial adoption. Coppeta et al. demonstrated both isolated individual organ recirculation and inter-organ recirculation for multiple pairs of airway-liver cultures on a single board, which were maintained with function for 2 weeks.[96] It should be noted, though, that these highly integrative MOM boards have limited flexibility to customize organ chambers for physiologically relevant representation of different organs. Fluid reconfiguration and control also often requires excessive fluid in the system which could dilute drugs and metabolites in the system.
2.3.3. Pumpless recirculating microfluidic platforms
Unlike above discussed recirculating multi-organ platforms that all rely on active fluid pumping to move medium among organ models, Sung et al. proposed a novel microfluidic platform that combined passive gravity-driven flow and a rocking motion to recirculate medium on chip.[100] A proof-of-concept model of three organ compartments representing liver, tumor and bone marrow was created on a 4 cm × 4 cm chip (Figure 5A). All fluidic components, including three organ microchambers, two reservoirs, and interconnecting microchannels, were built on chip with no external fluid or gas loops. The medium recirculation was achieved by positioning the entire chip on a titling rocker platform to create gravity-induced flow through the on-chip fluidic network and switching the tiling direction periodically. Such pumpless microfluidic platform provides reciprocating circulation of medium rather than a closed-loop unidirectional perfusion as in vivo. Yet the difference in the recirculation mode causes little deviation in pharmacokinetic profiles of drugs.[100] The resulting bidirectional flow did not show adverse effects on the metabolism of the liver tissue reconstructed from primary cells.[101] The pumpless platform was further applied to construct a four-organ MOM with cardiac, muscle, neuronal and liver modules with functional readouts (Figure 5B).[43] This recirculating platform maintained the co-culture viable and functional over a 14 d period.
Figure 5.

Representative pumpless recirculating MOM systems. Self-contained, integrated MOMs that support co-culture of 2 to 13 organs have been demonstrated on pumpless microfluidic platforms. (A) A liver-tumor-marrow three-organ MOM. Reproduced with permission.[100] Copyright 2010, Royal Society of Chemistry. (B) A palm-sized 4-organ MOM system with integrated electrical activity and contractile force sensors for non-invasive functional monitoring.[43] (C) A modular microsystem that approximates unidirectional perfusion and supports the co-culture of GI tract epithelium and 3D primary liver construct with in situ TEER measurement capacity.[104] (D) A whole-body microphysiological system that considers explicitly thirteen organs.[105]
To accommodate barrier tissues, such as the intestine and endothelial layers, which can be effected by bidirectional flows, a “step chamber” has been introduced to minimize the bidirectional shear stress in the pumpless systems.[102,103] Passive valves were also used to approximate unidirectional perfusion in the organ chambers (Figure 5C).[104]
The Shuler group later developed a whole-body model based on the pumpless microfluidic platform, which considered explicitly thirteen organs (Figure 5D).[105] This body-on-a-chip device was designed based on a PBPK model. Organ chamber volumes and perfusion rates were scaled down proportionally to keep in vivo organ size ratios and physiological fluid resident time. To accommodate barrier tissues, they incorporated microporous membranes and adopted a layered design to separate barrier and non-barrier tissue cultures. A proof-of-concept demonstration was performed with 3D cultured cells from five different organs. The microfluidic platform maintained the co-cultures at high viability and liver functionality over a seven-day period. Their work demonstrated the feasibility of constructing and operating a physiologically relevant whole-body microphysiological system (with a large number of organ compartments) for a sustained period with the capability of measuring cellular functions.
Overall, the pumpless recirculating MOM systems are highly integrated, self-contained, and relatively easy and cost-effective to construct and maintain. With all fluidic components (microchambers, microchannels and reservoirs) built on chip requiring no external fluid or gas loops, these microsystems can be compact and stackable,[91] and thus hold great potential for relatively high throughput application in pharmaceutical industries. The gravity-driven passive flow system keeps the MOM systems free of air bubbles, which commonly occurs in a closed-channel recirculating systems and affects or interrupts their operation. The pumpless feature makes the MOMs easy to set up and maintain and can support reliable, long-term operation. The open reservoir design also provides easy access for liquid sampling. Current pumpless MOMs are all based on PBPK models that incorporate human physiological parameters. By designing microfluidic channel dimension and the tilting angle, desired organ-specific perfusion rates can be achieved. A price for this highly-integrated fluid system is losing some flexibility of flow network reconfiguration. Once the pumpless device is fabricated, the ratio of flow through different channels has been fixed, although the total flow rate can be adjusted by changing the tilting angle. The reservoirs required for reciprocal recirculation also increase the total volume of the fluid system resulting in higher systemic liquid-to-tissue ratio.
2.3.4. Summary
Recirculating multi-organ microfluidic platforms allow re-creation of the reciprocal organ-organ interactions that are missing in single-pass platforms. Different organ modules can be connected in parallel or in more complex circuits while still achieving organ-organ interactions. Without being limited to serial circuits, it is possible to create in vivo-like circulation architecture and provide differential perfusion over various organ modules. However, most of current recirculating MOMs are still at the stage of establishing reciprocal connections among organ modules. Many still used the simplest single serial circuit to connect all modules. PBPK model based µCCAs and the pumpless MOMs show greater physiological relevance by incorporating physiological parameters into designs of the microchambers, microchannels and the perfusion networks.
3. Advances in on-chip analytical technologies for MOMs
To construct reliable multi-organ MPS and perform drug testing, it is crucial to maintain desired cell microenvironment (such as pH, temperature, and oxygen level) and constantly assess cell responses in terms of viability, metabolism, and functionality. Integration of on-chip analytical technologies into MOMs often enables in situ non-invasive real-time monitoring of those important systemic and cellular parameters. Online biosensors and bioanalytical assays using electrical, electromechanical, electrochemical, and optical signals to extract physical, chemical, and biological information from in vitro culture systems have been developed and initially demonstrated in single-organ models. We will focus on major on-chip analytical technologies that have currently been integrated into MOMs.
3.1. Monitoring cellular microenvironment
Important physicochemical factors such as pH, dissolved oxygen, and temperature, as well as specific biochemical factors strongly affect cell function, behavior and growth.[106] In vitro cell culture aims to reproduce to some extent the complex in vivo microenvironment by controlling these physicochemical and biochemical parameters.[107] Monitoring of these parameters is crucial in MOM systems to provide reproducible conditions during in vitro culture.
Oxygen is a key element in the behavior, function and energy metabolism of cells.[108] Maintaining desired levels of dissolved oxygen is essential to provide optimal culture conditions. Approaches using electrochemical and luminescent optical sensors have been developed for dissolved oxygen measurement. Optical methods are usually preferred in body-on-a-chip applications for their non-invasive nature and good signal stability over extended periods of time.[109] Fluorescence quenching of oxygen sensitive dyes is often utilized for oxygen sensing.[110] Sin et al. integrated a dissolved oxygen fluorometric sensor in a 3-chamber µCCA, which detected the fluorescence quenching of a ruthenium complex upon collision with oxygen molecules.[78] The ruthenium complex was immobilized in resin based particles and further encapsulated into a PDMS patch integrated in the µCCA chip. The patch was excited by a blue silicon LED, and the fluorescent emission was measured with a photodiode module. This in situ real-time monitoring approach indicated that the µCCA chip achieved adequate oxygen levels for culture. Zhang et al. incorporated an optical multi-analyte sensing module for in-line monitoring of pH and dissolved oxygen into a multi-organ microsystem.[84] The oxygen sensing used a similar setup as described above (Figure 6A).[111] The pH sensor utilized a silicon photodiode to quantify optical absorbance of the culture medium containing a pH-sensitive dye phenol red (Figure 6B). These sensors combined with a temperature probe allowed monitoring three key aspects of the microenvironment of the multi-organ platform (Figure 6C–D).
Figure 6.

Working principle of dissolved oxygen and pH measurement using optical sensors. (A) Measurement of dissolved oxygen is based on the luminescent quenching of an oxygen sensitive dye. A blue LED is used to excite the dye and a photodiode measures the changes in luminescent intensity. Reproduced with permission.[111] Copyright 2016, AIP Publishing. (B) Measurement of pH is based on the detection of light absorbed by phenol red containing cell culture media. Reproduced with permission.[111] Copyright 2016, AIP Publishing. (C) Schematic diagram showing the pH, dissolved oxygen and temperature sensors within a microfluidic module. Adapted from Zhang et al., 2017.[84] Copyright 2017, Zhang et al. (D) Photograph of the oxygen and pH sensing module. Reproduced with permission.[111] Copyright 2016, AIP Publishing.
3.2. Evaluating cell viability, morphology and functionality
Application of MOM systems requires effective readouts to assess the effects of a specific challenge. Cell viability and morphological analyses are the most commonly used approaches for evaluating cell response. Applications for drug bioavailability and efficacy testing often require more in-depth information on the functional state of cells and organs. Functional measurements of organ-specific parameters, such as cell metabolism, contractile force, electrical activity, and barrier permeability, produce a more sensitive evaluation of drug responses and provide insight into the underlying mechanisms.[91] Ideally, on-chip analytical technologies for integration into MOMs should provide non-invasive evaluation of these parameters to minimize any potential alteration of the system and allow live-cell monitoring throughout the culture period.
3.2.1. Cell morphology and viability
Imaging techniques such as light and fluorescence microscopy are a key tool in modern cell biology. A simple approach to monitor cell morphology in multi-organ systems is by developing optical transparent MOM systems with a size scale that allows visual inspection using standard optical microscopy. Materials such as PDMS, poly(methyl methacrylate) (PMMA), and glass with high optical transparency have been widely used in multi-organ systems.[112] Combined with automated imaging systems and cell viability indicators, transparent MOMs allow real-time in situ high-throughput analysis of cell morphology and viability. For example, Oh et al. developed a high-throughput approach to monitor long-term growth of liver (HepG2/C3A) and cancer (MES-SA) cells in multiple µCCA chips using a portable epi-fluorescence based optical microscope system that fitted into a standard cell culture incubator to image a large number of cells simultaneously.[113]
3.2.2. Cell metabolism
Cell metabolites secreted into the culture medium are often used to assess cell status during in vitro culture. They are useful functional markers for evaluating cell and organ response to physical or chemical challenges. Detection and quantification of these biomarkers with miniaturized on-chip biosensors using minimal sample volumes are desired in MOM systems. Much effort has been devoted to adapting macroscale biosensors for on-chip applications. Zhang et al. integrated electrochemical immunobiosensors in a liver-heart MOM system for continuous monitoring of the liver biomarkers albumin and glutathione S-transferase α (GST-α), and the cardiac specific marker creatine kinase (CK-MB) (Figure 7A).[84] The electrochemical sensors used biotinylated antibodies attached to the functionalized surface of microfabricated gold electrodes for antigen detection. The immunobiosensor module could also be regenerated using a cleaning mechanism (Figure 7A ii). This approach allows for continuous monitoring of cell secreted biomarkers through extended periods of time. Misun et al. incorporated in situ biosensing electrodes for monitoring lactate secretion and glucose consumption into a previously developed open channel, hanging drop array based MOM platform (Figure 7B).[85,114] The chip design allowed for plug-and-play usage of the biosensing electrodes, which could improve operation as the sensors could be calibrated off-chip and inserted before use or when needed during the culture period.
Figure 7.

On-chip biosensors integrated in MOM systems for cell metabolite detection. (A) Electrochemical immune biosensor. Adapted from Zhang et al., 2017.[84] Copyright 2017, Zhang et al. i) Photograph of a microelectrode set and SEM image of the working electrode surface. ii) Schematic showing the functionalization and regeneration process of the electrode. SAM, self-assembled monolayer; SPV, streptavidin. (B) Schematics of the microfluidic hanging-drop network and the plug-in biosensor chip. Adapted under the terms of the CC-BY-4.0 license.[114] Copyright 2016, Misun et al. i) 3D exploded view of the device and the plug-in biosensors. ii) 2D schematic of the hanging drop chip. The dashed lines represent the slot where the biosensor electrode is inserted. iii) Schematic and working principle of the enzyme-based biosensor. A hydrogel containing either glucose oxidase (GOx) or lactate oxidase (LOx) transforms glucose or lactate to produce hydrogen peroxide, which is electrochemically detected on the Pt surface at 0.65 V vs. Ag/AgCl.
3.2.3. Contractile force
Skeletal, cardiac and smooth muscle tissues are specialized for contraction, generating contractile forces. Functional readouts on muscle contractility are often desired when assessing a compound’s toxicity and efficacy on muscle tissues due to their superior sensitivity.[115] Traditional in vitro measurement approaches for muscle contractility often involve invasive and complex setups that are difficult to apply to microscale systems.[116,117] Novel miniaturized biosensors combined with non-invasive recording systems are being developed and incorporated into microphysiological systems.[118]
Adoption of silicone microcantilevers, originally developed for microelectromechanical systems (MEMS), has enabled on-chip biomolecular recognition[119] and force sensing[115] at micro- and nanoscale. Mechanical forces generated from myocytes cultured on the cantilever surface are transferred to the cantilever beam causing bending of the silicone cantilever. Based on this, the Hickman group developed a microcantilever array chip (Figure 8A) and an optical recording system (Figure 9A) to measure contractile force from cardiac or skeletal muscle cells.[43,120–126] The microcantilever chips with 32 parallel microcantilever beams were microfabricated from silicon-on-insulator wafers using photolithography (Figure 8B), and were surface modified to facilitate the anchorage of muscle cells.[126] Bending of the microcantilever beam due to muscle tissue contraction was non-invasively detected through a photodetector that measured changes in the deflection of a laser beam directed at the tip of the cantilever (Figure 9A–B). The contractile force was then calculated through the Stoney equation considering the mechanical properties of the cantilever and the geometry of the system.[123,124] Recently, Oleaga et al. has incorporated this technology into a pumpless four-organ MOM system (Figure 5B), and demonstrated and validated the use of biomechanical readouts on muscle tissues for drug testing in a multi-organ setting.[43] The system integrated two silicon microcantilever array chips as the on-chip biosensors to analyze changes in contractility of either human stem cell (hSC)-derived cardiomyocytes or skeletal muscle cells in response to drugs. This microcantilever based approach allows comprehensive measurement of peak force, time to fatigue and other characteristics that are difficult to quantify in in vitro systems.
Figure 8.

Design and fabrication of microcantilever chips for contractile force measurement. (A) Photograph of a silicon cantilever chip (1 cm × 1 cm). The chip contains 32 microcantilevers. Arrows indicate the position of cantilever 1, 16, 17 and 32. CL: cantilever. Reproduced with permission.[126] Copyright 2017, Springer Science+Business Media New York. (B) Schematics of the top and bottom photomasks for fabricating 24 cantilever chips on a single 4-inch wafer.
Figure 9.

Cardiac functional analysis with human cells using a microelectrode array (MEA) and microcantilever based system. (A) Diagram of the cantilever-based force measurement system. (B) Top: Phase contrast micrograph and immunocytochemistry micrograph of aligned human cardiomyocytes on the microcantilever chip. Bottom: Example traces of deflection and torsional force produced by the myocyte contractions. (C) Schematics of the patterning process on MEA. (D) Top left: Phase contrast micrograph of MEA with patterned human-derived cardiomyocytes. Top right: Immunostaining micrograph showing that human derived cells differentiated to cardiomyocytes. Bottom: Rhythm generation recorded by the MEA electrodes. Scale: The inter-electrode distance on the MEA is 200 µm. Reproduced with permission.[125] Copyright 2015, Elsevier.
Compliant micropillar based approaches have also been used for contractile force measurement.[127,128] Uzel et al. utilized PDMS micropillars to assess the force generation of muscle strips in a neuromuscular junction (NMJ) model (Figure 10).[128] Motoneurons derived from photosensitized mouse embryonic stem cells (mESCs) were optically excited to fire action potentials and further trigger muscle contraction. The contraction of a muscle strip anchored to a pair of PDMS micropillars was quantified by imaging-based automated tracking of the muscle strip deformation and micropillar deflection, and was converted to the contractile force based on the stress-strain characteristics of PDMS. The integration of these force sensors allowed for a non-invasive and quantitative characterization of muscle contraction due to neural excitation and NMJ function.
Figure 10.

Schematics of the neuromuscular MOM using integrated compliant micropillars to measure force generation from muscle strips. (A) Two-dimensional scheme of the chip design. B) Three-dimensional view showing the coculture of neurospheres and hydrogel embedded muscle bundles attached to the micropillars. C) Exploded view of the system showing the position of the micropillar layer. Adapted under the terms of the CC BY-NC 4.0 license.[128] Copyright 2016, Uzel et al.
3.2.4. Electrical activity
Measurement of the electrical activity provides information on the functionality of electrical conductive tissues, such as cardiac, muscle and nervous tissues. Techniques such as patch clamp and microelectrode arrays (MEA) have been developed to study the electrical activity of conductive tissues at the micro and meso scale.[129,130] While the patch clamp has greatly extended our knowledge of the electrophysiology of those tissues, it is an invasive, time-consuming, labor-intensive and skill-demanding technique that is difficult to integrate into MOM systems.[131] MEA based technologies, on the other hand, provide a non-invasive and high-throughput solution for monitoring the electrical activity of conductive tissues in microphysiological systems.[132,133]
MEAs allow detection of field potentials generated by cells in close proximity to substrate-integrated electrodes.[132,134,135] Based on that, the Hickman group designed a custom MEA (cMEA) chip for in vitro recording of the electrical activity of cardiac cells first demonstrated in a single organ system (Figure 9C–D).[125,136] With cells directly cultured on the MEA surface, this approach also allows to monitor cell networks and study signal propagation. Combined with cell patterning techniques, MEA recording can provide in-depth functional information, such as QT intervals, conduction velocity, in addition to beat frequency.[125,136,137] The cMEA chips have recently been incorporated into a pumpless multi-organ system to provide non-invasive functional readouts of cardiac and neuronal electrical activity (Figure 5B).[43] Integration of MEA and microcantilever chips into a same MOM system, enabling electrical and mechanical readouts, allows for comprehensive functional evaluation as well as mechanistic interrogation of organ response to challenges by compounds and drugs.
3.2.5. Barrier function
Barrier tissues, such as skin, GI tract and blood brain barrier, play an important role in regulating drug bioavailability and distribution in the body. Establishing and maintaining barrier functions in those tissue models in vitro are critical for multi-organ systems to reproduce in vivo pharmacokinetics. Transepithelial/transendothelial electrical resistance (TEER) is a commonly used indicator for barrier integrity. Non-invasive measurement of TEER can be conducted with electrodes on both sides of the cellular layer to measure the electrical resistance across the barrier.[138,139] This approach has been applied using microfabricated on-chip thin-film electrodes to monitor the barrier integrity of in vitro microfluidic models of intestine, lung alveolar epithelium, and blood brain barrier.[140,141] Circular Ag/AgCl pellet electrodes have also been integrated into organ chambers on a pumpless platform to measure the barrier properties of a blood-brain barrier model based on co-culture of brain microvascular endothelial cells and astrocytes cells.[103] The in-line electrode system provided comparable readouts to the commonly used chopstick electrode system designed for transwells. The same setup has recently been incorporated into a GI-liver model to monitor the barrier function of the GI tract cell layer over a 14 d culture period.
3.3. Summary
The continual development of MOM systems will require incorporating biosensor technology to monitor cell microenvironment and functionality in real time, non-invasively to move beyond acute evaluations of drugs and toxins to chronic assessment. Some effort has been done in incorporating non-invasive technologies that allow in situ monitoring of different organ modules but this work is still in its infancy and will be a major hurdle to overcome to enable more mainstream use of this technology. Several approaches have been used either by incorporating the biosensors inside the main platform system or by developing the biosensors as separate modules as indicated in the previous section and while these systems are functional their incorporation into multi-organ systems remains a challenge.
4. Applications for multi-organ microphysiological systems
Main applications of MOM systems are toward toxicity testing of compounds and industrial chemicals, drug efficacy evaluation, as well as modeling of diseases that involve multiple organs. NCATS at NIH and DARPA recently funded a $150,000,000 program for grants in this important area. Many proof-of-concept applications have been demonstrated on different platforms.
4.1. Toxicity assay and drug efficacy testing
One of the main areas of application of MOM systems is for the pharmaceutical, chemical and cosmetic industries to assess the potential toxicological hazard and effects of diverse compounds.[142,143] With multiple interconnected organ models, MOM systems hold the potential to simulate the dynamic in vivo ADME process of diverse substances,[144] and predict compound toxicity and drug efficacy. Systems with functional readouts could at the same time provide long-term evaluation of compounds that can currently only be done in animals.
4.1.1. Hepatic metabolism and Bioactivity
Liver models are the most commonly included module in a MOM system due to the essential role of liver in drug metabolism. Various compounds can undergo biotransformation in the liver that modifies their bioactivity. Prodrugs are transformed to active forms through enzymatic activation, while some toxins can be detoxified by hepatocytes. Therefore, a great number of MOM systems that includes the liver model have been developed to test the effects of drug and drug metabolites.
Metabolism-dependent, organ-specific toxicity
Dual-organ MOM systems combining models of the liver and another organ, such as heart, lung, and kidney, are used to test organ-specific toxicity of drugs and drug metabolites in addition to hepatotoxicity.
Cardiotoxicity is one of the main reasons of drug attrition during preclinical and clinical studies as well as post-approval withdraw.[145] Zhang et al. developed a liver-heart MOM system on the fluid breadboard based modular platform with on-chip electrochemical immunobiosensors and a mini-microscope to evaluate the side effects of an anti-cancer prodrug, capecitabine, and an anti-inflammatory drug, acetaminophen.[84] Capecitabine induced enhanced cell death in heart organoids (prepared from human iPSC-derived cardiomyocytes) in the present of liver organoids (prepared from human primary hepatocytes), likely due to the enzymatic bioactivation of capecitabine by hepatocytes into cardiotoxic 5-fluorouracil (5-FU). In contrast, 2 d exposure to acetaminophen resulted in dose-dependent hepatoxicity indicated by increased GST-α secretion and decreased albumin production and cell viability, and only slight changes in cardiac CK-MB secretion and beating rate. This demonstrates that a live-heart MOM system can be a useful tool to detect metabolism-dependent cardiotoxicity and hepatotoxicity of drug candidates.
Nephrotoxicity is another common adverse drug effect that could lead to significant clinical syndromes, such as acute kidney injury. Biotransformation of xenobiotics through the liver into more water-soluble compounds favors their disposal through the kidneys.[146,147] The kidney’s blood filtration process extracts these xenobiotic compounds and metabolites from the blood stream, which are concentrated in the kidney microenvironment.[148] This combined with constantly high levels of renal blood flow makes the kidney especially poised as a target for toxicity. Liver-kidney MOMs have been applied to investigate liver metabolism-dependent nephrotoxicity. Choucha-Snouber et al. established a liver-kidney system on a pump-driven recirculating MOM platform to test the nephrotoxicity of an anti-cancer prodrug ifosfamide.[82] Kidney cells (MDCK) proliferation was significantly inhibited in response to ifosfamide exposure in liver-kidney models constructed with HepaRG hepatocytes, but not with HepG2/C3A cells. This is mainly due to the much higher expression levels of cytochrome P450 (CYP)3A4 and CYP2B6 in HepaRG cells, which are involved in biotransformation of ifosfamide into nephrotoxic chloroacetaldehyde.
Other organ models, such as the lung and skin, have also been integrated with the liver models for toxicological studies. A liver-lung µCCA developed by Viravaidya et al. demonstrated liver metabolism dependent naphthalene toxicity towards lung epithelial cells.[44] A liver-fibroblast model built on the IdMOC platform showed that the presence of hepatocytes modified the viability of fibroblasts in response to 4-aminophenol and cyclophosphamide treatment through diffusible metabolites after liver detoxification or activation of the drugs,[149] while the cytotoxic effects of aflatoxin B1 were only limited to the liver compartment after the metabolic activation with no toxicant diffused to outside of the liver cells.[150] Liver-skin models using HepaRG hepatocyte spheroids and skin biopsies were built on the transwell compatible, pneumatic micropump-driven recirculating MOM platform, and used to study chronic toxicity of repeated dosing of troglitazone for up to 9 days.[92,93] The study showed that repeated exposure to troglitazone induced hepatotoxicity with an increase of LDH activity and CYP3A4 in a dose dependent manner.
Efficacy and toxicity of anti-cancer drugs
Oncology drug development has the lowest clinical success rate among all therapeutic areas, with only 5.1% of candidates entering Phase I eventually get FDA approval.[4] Clinical safety concerns on serious adverse events and lack of efficacy are two primary causes of failure in clinical trials.[151] To assess the efficacy as well as toxicity of an anti-cancer treatment in vitro, MOM systems that integrate models of the liver, tumor, and other organs have been developed.
Efficacy of anti-cancer drugs and drug metabolites can simply be evaluated using dual liver-cancer models. Frey et al. constructed a liver-colon cancer model with HCT-116 colon cancer spheroids and rat liver cells using the hanging drop array-based platform, and demonstrated the anti-cancer effects of 5-FU and liver-bioactivated cyclophosphamide (CP), indicated by inhibited cell growth of HCT-116 cells.[85] Lee et al. developed a liver-tumor model with HepG2 and HeLa cells on the pumpless recirculating platform to investigate the anti-cancer activity of a flavonoid, luteolin, which was found metabolized by liver cells into more potent tumor-killing metabolites in a concentration dependent manner.[152] This dual organ system also revealed significantly weaker anti-cancer activity than that predicted by a 96-well plate study, likely due to the simultaneous liver metabolism and tumor growth inhibition in the MOM system, versus the sequential presence of those two activities using the multiwell plate platform.
More organ models, in addition to the liver and cancer, have been integrated into MOM systems to simultaneously evaluate the efficacy and toxicity of anti-cancer therapeutics. Two heart-liver cancer MOM systems have been developed to test a chemotherapeutic drug, doxorubicin.[84,99] They were constructed on either the fluid breadboard based platform or an integrative pneumatic pump-driven recirculating platform, using HepG2/C3A hepatocellular carcinoma cells and primary or iPSC-derived human cardiomyocytes. Both systems captured the anti-cancer effects of doxorubicin indicated by decreased albumin secretion, cell proliferation and viability of the liver cancer cells. These models also detected significant cardiotoxicity from doxorubicin treatment, likely due to the cardiotoxic liver metabolite doxorubicinol. Sung et al. designed a liver-colon cancer-marrow three-organ µCCA to study the drug effects of 5-FU, its oral prodrug, Tegafur, and a combination of Tegafur with uracil.[81] The µCCA were built with 3D hydrogel cultures of colon cancer cells (HCT-116), liver (HepG2/C3A) and myeloblast (Kasumi-1) cells, and demonstrated the anti-cancer effects of 5-FU and liver-bioactivated Tegafur and their cytotoxicity towards myeloblasts. Administration of Tegafur with uracil to the system enhanced the anti-cancer effects by inhibiting 5-FU degradation. Sung et al. later reinvented the 3-organ system using a pumpless platform, and combined with a PK-PD model to study the dose responses to 5-FU from all three cell types.[100]
With increased number of organs incorporated into the basic liver-cancer model, MOM systems can also be used to evaluate the differential multi-organ toxicity beyond the anti-cancer effect of cancer therapeutic drugs. Li et al. constructed a 6-organ MOM on the wells-within-a-well IdMOC platform using cocultures of MCF-7 breast cancer cells and primary human cells from five major organs, including liver (hepatocytes), kidney (renal proximal tubule cells), lung (small airway epithelial cells), central nervous system (astrocytes) and vasculature (aortic endothelial cells).[71] This multi-organ coculture system demonstrated the anti-cancer effect of tamoxifen as well as its differential cytotoxicity on primary cells from various organs.
Multidrug resistant (MDR) cancers are especially challenging to treat. While current MDR modulators all come with various side effects, combining chemotherapeutics with a mixture of MDR modulators with different side effects may generate useful treatment strategies. In a proof-of-concept study, Tatosian et al. used a 4-chamber µCCA with liver (HepG2/C3A), bone marrow (MEG-01), uterine cancer (MES-SA), and MDR uterine cancer (MES-SA/DX-5) to test drug combinations that selectively attack MDR cancer cells.[80] The study identified a combination of a chemotherapeutic, doxorubicin and two MDR modulators, cyclosporine and nicardipine that provided optimal efficacy with acceptable side effects.
Chronic drug toxicity and efficacy testing with functional readouts
Chronic testing of drug efficacy and side-effects are often needed in preclinical screening, while most of current MOM systems developed have been focused on acute studies, due to the challenge of long-term maintenance of multi-organ co-cultures and many destructive, end-point assays they use. Increasing the lifespan of multi-organ systems is critical to enable the feasibility of performing repeated dose and chronic studies of xenobiotics. MOM systems that support long-term multi-organ co-cultures, and provide non-invasive functional readouts are desired for chronic applications. In a landmark investigation, Oleaga et al. used a four-organ MOM to test the effects of doxorubicin, atorvastatin, valproic acid, acetaminophen and a control compound N-acetyl-m-aminophenol in human cardiac (iPSC-derived cardiomyocytes), liver (HepG2/C3A), skeletal muscle (human skeletal muscle) and neurons (human motoneurons) in a recirculating serum-free medium.[43] The co-cultures were maintained for up to 14 days. Results obtained upon drug exposure in the multi-organ system showed a good correlation with results observed in the literature. Doxorubicin at 5uM induced hepatotoxicity, cardiotoxicity and a decrease in cardiac and skeletal muscle functionality while atorvastatin showed myotoxicity, neurotoxicity, hepatotoxicity and reduction in muscle functionality. Exposure to valproic acid at 2 mM affected the liver compartment with a reduction in viability and urea production while showing neuroprotective effects. Exposure to an overdose concentration of 5 mM acetaminophen decreased liver viability while preserving cardiac viability and functionality. The multi-organ platform was later extended by the Hickman group for up to 28 days in vitro, which maintained organ function, and demonstrated the potential of this platform for further chronic studies.
4.1.2. GI absorption and metabolism of oral drugs
Oral drug delivery is the most commonly used route of drug administration due to its convenience and low cost. Orally delivered drugs need to go through the GI system, where they are selectively absorbed and metabolized before entering the hepatic portal vein. GI absorption and the subsequent liver metabolism, also called first-pass metabolism, play an important role in determining the bioavailability of an oral drug and its bioactive metabolites in the systemic blood circulation. GI tract models are, therefore, often integrated with liver models into a MOM system to simulate this process and to predict oral drug bioavailability.[42,62,63,73,75,76,87,89,90,93,102,104]
Choi et al. established a two-organ models of the GI-liver on the transwell platform to evaluate the bioavailability and first-pass metabolism of benzo[a]pyrene (B[a]P).[62] The system, built with Caco-2 (GI) and HepG2 (liver) cells, revealed low level of cytotoxicity of B[a]P treatment despite that B[a]P could be biotransformed into toxic metabolites by both GI and liver cells. This was due to the low bioavailability of B[a]P and its metabolites, which was transported back to the apical side by Caco-2 cells. Lau et al. used a similar transwell based model of the GI (Caco-2) and liver (human primary hepatocytes) to test the bioavailability of 24 compounds.[63] The concentration versus time curve (AUC) of parent compounds over a 3 h period correlated reasonably well with their in vivo oral bioavailability. These static microscale models demonstrated the feasibility of using GI-liver MOM systems to predict oral drug bioavailability.
Microfluidic MOM systems involving the GI and liver models have also been applied to investigate the intestinal absorption and first-pass metabolism of ingested substances. A GI-liver µCCA model constructed with HepG2/C3A liver cells and cocultures of Caco-2 and mucin-producing TH29-MTX cells was challenged with carboxylated polystyrene nanoparticles, in modeling for inert, negatively charged nanoparticles, at a high dose of possible daily human consumption.[89] The study showed a relatively low permeability of nanoparticles across the GI barrier. Yet those single nanoparticles and small aggregates, a small portion that passed through the barrier, induced aspartate aminotransferase (AST) release, indicating liver cell injury.
In addition to its barrier function, the role of GI tract in drug metabolism has also gain increasing attention. A single-pass GI-liver model using freshly isolated rat intestine and liver slices developed by van Midwoud et al. highlighted intestine’s metabolic function in drug process.[73] The perfusion of chenodeoxycholic acid (a primary bile acid) containing medium through the intestine biochip to the liver biochip resulted in enhanced down-regulation of cytochrome P450 7A1 in the liver in response to the bile acid, due to the bile acid induced expression of fibroblast growth factor 15 (FGF15) in the intestine chamber and the transport of FGF15 to the liver chamber. In another GI-liver model developed on the pumpless platform by Choe et al., the coculture of Caco-2 (GI) and HepG2 (liver) cells was maintained for up to four weeks and demonstrated synergistic enhancement of metabolic activity in both organ models and modified Caco-2 absorption under flow.[102] The model yielded a more realistic metabolic profile of apigenin, a flavonoid, with the presence of both organ models.[102]
Microfluidic MOM systems have integrated GI-liver models with other organ models to assess the bioavailability, metabolism and bioactivity of ingested substances in the same system. Using a liver-GI-lung three-organ recirculating MOM system (Figure 4F), Kimura et al. tested the effects of the orally administered anti-cancer drugs epirubicin and cyclophosphamide.[90] Administration of epirubicin through the apical side of the intestine barrier showed limited efficacy of killing liver cancer cells compared to systemic administration, due to reduced bioavailability of EPI. In contrast, a large portion of cyclophosphamide dose passed through the intestine barrier and was bioactivated to anti-cancer metabolites targeting the liver cancer cells while maintaining lung cell viability. Imura et al. developed a single-pass three-organ microchip integrating GI, liver, and breast cancer models and characterized the intestinal absorption, hepatic metabolism and bioactivity towards cancer cells for a variety of ingested substances, including the anti-cancer drugs cyclophosphamide, epirubicin, and the estrogen-like compounds 17- estradiol, as well as soy isoflavone.[75]. Their results on cyclophosphamide and epirubicin were similar to those from Kimura et al.’s study. The bioavailability of estrogen like compounds was not affected by the intestinal barrier and their application increased breast cancer cell viability. Further studies using this platform incorporated synthetic digestive juices to mimic the effects of digestion and test for substance stability during digestion.[76]
4.1.3. Blood-brain barrier penetration
The blood-brain barrier (BBB) consists a dynamic physical and metabolic barrier strictly regulating the molecular exchange between the blood and the brain. It also limits the access to the central neural system (CNS) for therapeutic delivery. A large proportion of drug candidates for CNS disorders, such as brain cancer, stroke, and Alzheimer’s disease, failed due to inadequate brain penetration.[153] Several in vitro microphysiological models of the BBB have been developed for the permeability testing of CNS drugs and potential brain toxins.[103,141,154] Recently, a neurovascular model, consisting of human brain microvascular endothelial cells, iPSC derived human neurons, pericytes and astrocytes, was incorporated into a MOM system with the intestine, liver and kidney models through functional coupling.[77] The system was used to investigate the BBB permeability for terfenadine, trimethylamine, vitamin D3 and their intestine-liver metabolites. Terfenadine was metabolized by intestinal and liver cells into fexofenadine, which did not cross the BBB model. On the contrary, vitamin D3 and its metabolites all penetrated the barrier. The study also discovered that trimethylamine-N-oxide (TMAO), the liver metabolite of trimethylamine (TMA, a microbiome metabolite), was able to penetrate the BBB. This model demonstrated the use of BBB models in a multi-organ setting for permeability testing for drugs and drug metabolites.
4.1.4. Bioaccumulation
The dynamic process of bioaccumulation plays an important role in defining the pharmacokinetics and drug effects in vivo. Adipose tissue, one of the largest organs in the human body, is a major site of bioaccumulation for many compounds, especially lipophilic compounds. It acts as a reservoir that modulates the levels of those compounds in the body over time. Despite all these, adipose tissue and bioaccumulation are often underrepresented in MOM systems. To date, only four MOMs have considered adipose tissue with two of them used for drug testing.[79,83,95,105] In a multilayer MOM system, An et al., compared bioaccumulation of propranolol and pentobarbital in the lung, heart and fat tissue models, and showed preferred deposition of pentobarbital in the fat chamber, and propranolol deposition across all three organ with difference in concentrations.[95] A 4-compartment µCCA model of “lung” (L2), “liver” (HepG2/C3A), “fat” (differentiated 3T3-L1 adipocytes), and “other tissues” developed by Viravaidya et al. further demonstrated the role of bioaccumulation in regulating drug effects.[79] This microsystem reproduced liver metabolism dependent toxicity of naphthalene, which was further modulation by the bioaccumulation in adipose tissues. The presence of the fat chamber significantly decreased naphthalene and naphthoquinone induced lung cell injury due to GSH depletion. The system, coupled with PBPK models, provided a basis to understand mechanistically why mice are much more sensitive to nepheline than rats.
4.1.5. Renal drug clearance
Other than being metabolized by the liver, many drugs are cleared unchanged from the body through elimination by the kidney. Impaired renal function could affect drug elimination and alter a drug’s PK and PD, which often requires changes in the clinical dosing. To better predict drug effects in the body, several reductionist models of the kidney have been incorporated in MOM systems to simulate this process to some extent. In a vertically stacked multi-organ model (Figure 4H), An et al. used a dialysis membrane to mimic the excretion of pentobarbital, thiopentone and propranolol in the kidney.[95] In a functionally coupled MOM model (Figure 3D), the 3D kidney model constructed from proximal tubule endothelial cells and human umbilical vein endothelial cells (HUVECs) demonstrated kidney elimination of TMA and its metabolite TMAO, as well as fexofenadine, the intestinal and liver metabolite of terfenadine.[77]
4.2. Human disease modeling
Another important area of application of MOM systems is to model complex human diseases that involve multi-organ interactions to improve understanding, diagnosis, prevention, and therapeutic treatment of the disease. Proof-of-concept models have been established in the areas of cancer metastasis and neuromuscular communication.
Cancer cell metastasis is most commonly modeled using multi-organ microsystems.[155] Metastatic cancer cells detach from the primary tumor site, transmigrate across the endothelial-basement membrane barrier into the bloodstream (i.e. intravasation), circulate, and extravasate from the vasculature to distant organ sites.[156] In vitro modeling of this process is an important approach to interrogate the underlying mechanisms of the metastatic cascade and inspire novel therapeutic strategies for cancer treatment.[157] Zervantonakis et al. developed a 3D microfluidic model for tumor cell intravasation that involves interactions among tumor cells, endothelial cells, and macrophages.[158] The system allowed for real-time monitoring and quantification of the dynamics of tumor cell intravasation. This model revealed that macrophage secreted TNF-α increased endothelial permeability, tumor-endothelial cell interactions and tumor cell intravasation rate. Riahi et al. presented a 3D microfluidic system to model extravasation of circulating tumor cells into metastasis target organs releasing CXCL12 ligands.[159,160] In addition to their interactions with the vasculature (intravasation, circulation, and extravasation) during metastasis, cancer cell interactions with the local organ microenvironment have also been modeled and investigated with MOM systems. Xu et al. designed a multi-organ single-pass microfluidic system to study the mechanisms involved in lung cancer metastasis to three distant organs, including the brain (astrocytes), bone (osteoblast cells) and liver (hepatocytes).[155] The model showed that lung cancer cells co-cultured with bronchial epithelial cells gained invasive phenotype, migrated through the bronchial epithelial cell layer, and reached and affected the remote target organs. To study the local microenvironment responsible for breast cancer progression and metastasis, Choi et al. developed a compartmentalized 3D microfluidic model of breast ductal carcinoma in situ (DCIS) with co-cultures of mammary ductal epithelial cells, mammary fibroblasts, and incorporated breast cancer spheroids.[161] This model can potentially facilitate mechanistic interrogation on malignant progression of DCIS in early-stage breast cancer. Recently, peri-neural invasion of cancer cells has also been modeled using a neuron-cancer microtunnel co-culture system.[65] The model recapitulated the reciprocal interactions between neurons and cancer cells that led to the neurite growth along microchannels and the cancer cell migration along neurites. Cancer cells with higher levels of clinical peri-neural invasion (prostate and pancreatic cancer cells vs. breast cancer cells) showed greater peri-neurite migration on chip. The model can be a useful tool for studying the dynamics and mechanisms of peri-neural cancer invasion and screening for compounds inhibiting the progression.
Besides cancer cell metastasis, multi-organ microsystems have also been applied to model intercellular communications between neurons and muscle cells, which can be altered in neurodegenerative diseases. The Hickman lab has developed rat[162] as well as human[163,164] based systems, which indicated functional innervation. Zahavi et al. developed a two-compartment microtunnel system for mouse motoneuron-muscle co-culture. The model recreated functional excitation and contraction coupling between motoneurons and muscles through motor axon extension across the microtunnels and neuromuscular junction (NMJ) formation.[64] Using this model, the study revealed the role of glial-derived neurotrophic factor (GDNF) in promoting muscle innervation, axon growth, and soma survival.
The application of MOMs in disease modeling is currently demonstrated in limited areas, yet they hold great potential in modeling human multi-organ diseases, such as metabolic syndrome and type 2 diabetes mellitus. They can be especially useful for elucidating mechanisms and developing therapies of rare diseases that affect multiple organ systems, such as Churg-Strauss syndrome and POEMS syndrome, where the drug development is extremely challenging due to the limited number of available human subjects.
5. Challenges
To be validated and qualified as next generation in vitro models and tools for preclinical drug screening, a multi-organ microphysiological system shall reproducibly generate a result that has “specific interpretation and application in drug development”.[165] Multi-organ microphysiological systems (MOMs), or body-on-a-chips (BOCs), have advanced significantly by integrating new technologies in the platforms, such as stem cell biology, biomaterials, microelectronics and microfluidics. Yet the field is still facing critical challenges to provide reliable and cost-effective solutions to construct integrated MOMs with reproducibility and physiological relevance, and at the same time, in a relatively high throughput format to enable widespread industrial adoption. In this section, we will focus on three major challenges and discuss promising strategies.
5.1. Blood surrogate and controlled culture environment
Human blood circulated throughout the body is in constant exchange with interstitial fluid surrounding the tissue cells. It provides continuous supply of oxygen and nutrients (such as amino acids, electrolytes, glucose, and fatty acids), removes metabolite wastes (such as carbon dioxide, urea, and lactic acid), and mediates inter-organ communication. It is not trivial to develop a common culture medium as a blood surrogate that can maintain the viability and function of various organ models in vitro, and approximate the interaction of blood components with drugs. Lack of effective blood surrogate often hinders the development and demonstration of integrated multi-organ systems.[74,77]
In vitro cell culture media are usually optimized specifically for the cell type in culture to best maintain their viability and phenotypic cellular function. The formula can thus vary significantly with cell types. In addition, the increased use of human stem-cell derived cells for constructing in vitro human organ mimetics often requires different specialized media for different differentiation stages to induce or maintain a specific phenotype. To coordinate such diverse needs for various cell types at different stages, modular approaches that physically decouple single organ model preparation and multi-organ co-culture are often used to reduce the challenge of developing a universal medium for all modeled organs. Organ mimetics constructed from human primary cells or stem-cell derived cells are more resilient to environmental changes after they mature, and are more likely to maintain their phenotypic function to a degree in response to culture medium switch. It is possible to use basal media without any cell-type specific supplements to maintain short-term (up to 24 h) co-culture of multiple separately prepared organ models for acute drug studies.[71] For chronic drug studies, several different strategies have been developed aiming to provide reliable long-term multi-organ co-culture. Many multi-organ systems with 2 to 4 organ compartments including the liver simply chose liver cell culture media as the blood surrogate to assure hepatic functions under co-culture, due to the pivotal role of liver metabolism in drug process.[42,63,85,92,93,102,104] Some chose to include all specialized media for individual organs involved and mixed them in equal ratio as the blood surrogate to meet the needs of all cell types to a certain extent.[83,84] Both strategies have maintained viable multi-organ (up to 4 organs) co-cultures for several days to up to four weeks. Yet these strategies may not be able to meet challenges to maintain high levels of organ functions with increased number of organ units in the system. Some growth factors can have contradictory effects on different organ cells. Zhang et al. incorporated controlled release of TGF-β1 in the lung chamber to promote lung cell function while minimizing the inhibitory effects of TGF-β1 on hepatic functions.[83] Such compartmentalized release of organ-specific growth factors can be a useful approach to balance the differential needs from various organs. Similarly, organ-specific extracellular matrix proteins (collagen, fibronectin, Matrigel, fibrin etc.) are often used to better mimic in vivo cellular microenvironment. These compartmentalized modifications are important routes to supplement the systemic blood surrogate circulation.
In addition to maintaining viable and functional multi-organ co-cultures in vitro, the ideal blood surrogate need to be fully chemically defined to create reliable multi-organ models and generate reproducible results for drug testing. Most current multi-organ models (> 90%) have human or animal sera supplemented in the culture medium to provide a wide range of growth factors, hormones, vitamins, amino acids, fatty acids and trace elements that promote cell growth or maintain cell phenotypes.[42,44,62,78–80,82,84,90,92,93,95,105] The composition of human or animal-sourced sera can vary significantly from batch to batch. Chemically well-defined, serum-free formula of culture media are thus desired to improve system reliability and reproducibility. Based on analysis of serum composition, in 1995, the Hickman group developed a serum-free formula for hippocampal neuron cell culture by replacing serum supplement with known chemical components providing similar support for cell growth and maintenance.[166] The formula were further adapted to support reliable long-term culture of skeletal myotubes,[167] cardiomyocytes,[125] and motoneurons,[22,168] as well as dual culture of motoneurons with sensory neurons to build the stretch reflex arc,[169–171] or with oligodendrocyte progenitor cells to model myelination.[172] This laid a foundation for introducing serum-free cell culture media into the multi-organ systems. Recently, Oleaga et al. demonstrated in a four-organ MOM, in which serum-free culture medium maintained hepatic function, cardiac and neuronal electrical activity, and cardiac and skeletal muscle contraction capacity.[43] This was the first system to incorporate a serum-free recirculating medium in a multi-organ system. Development of blood surrogate with full chemical definition is the first step towards reliable and reproducible MOMs.
The functional groups presented on the surface of 2D culture substrate or 3D extracellular matrix further define cellular microenvironment and can greatly affect cell adhesion and phenotype. Surface modification techniques can often be used to precisely control the surface properties for cell adhesion and alignment.[173–175] Human or animal sourced extracellular matrices, such as human placental collagen and Matrigel, are frequently used for surface coating or cell encapsulation to mimic native cellular niche. Yet they also contribute as a source of variations that affect the reproducibility of organ models. Bioinspired synthetic biomaterials with clear chemical definition, such as PGmatrix (19 amino acid residues inspired by spider silk and the trans-membrane segment of human muscle L-type calcium channel),[176] are emerging as extracellular matrix mimics, and could potentially help further standardize cell microenvironment and improve the reproducibility of MOM systems.
5.2. Drug adsorption and absorption to platform materials
Adsorption and absorption of drugs and their metabolites to platform materials can skew the PK and PD profiles recreated in the MOM systems. To obtain accurate and reproducible results on PK/PD parameters, it is crucial to minimize and thoroughly characterize the amount of drug adsorption and absorption onto the platform materials. Strategies to reduce drug adsorption and absorption include selecting proper platform materials and surface modification techniques to achieve low absorptive and adsorptive properties for the testing drugs and minimizing the exposed surface area to absorptive and adsorptive materials.
Common platform materials can be quite different in adsorptive and absorptive capacity for specific drugs. While stainless-steel, glass and Teflon were found virtually inert to naphthalene, polypropylene, high density polyethylene and acetal showed high adsorption and absorption of naphthalene.[11] None of commonly used tubing (Tygon, Pharmed, C-flex, Norprene and Viton) were found inert to naphthalene.[11] Therefore, it is important to select proper materials to construct the fluid route for the testing drugs. It should be noted that most of current MOMs are demonstrated in PDMS due to its low cost and easy fabrication. PDMS surface is highly hydrophobic and can attract significant adsorption and absorption of hydrophobic drugs and their metabolites.[177] PDMS surface modification,[178] lipophilic coating,[179] or pinhole-free parylene coating[180] can be applied to modify its adsorptive and absorptive properties. Alternatively, thermoplastic materials, including PMMA, polystyrene, polycarbonate, and cyclic olefin copolymer (COC), are promising platform materials that can achieve relatively low drug adsorption and absorption with proper surface modification,[177] have shown great biocompatibility and optical and mechanical properties, and are cost-effective and compatible with industrial manufacturing. Those thermal plastics are thus more likely to be adopted as the platform materials by the pharmaceutical industry. Minimizing fluid-exposed surface area can also reduce drug adsorption and absorption in the system. The pumpless platform[43,100,102,104,105,152] and recirculating MOMs driven by on-chip micropumps[42,90,92–94] inherently benefit from the elimination of external fluid tubing with significant reduction of exposed surface area. It should be noted that even after all the above optimization steps, a thorough characterization on the adsorption and absorption of testing drugs onto the platform is still necessary to appropriately interpret the PK/PD results generated by MOMs. Such characterization testing is missing currently from many published MOMs.
5.3. Physiological relevance
The physiological relevance of MOM systems is of the essence for them to reproduce in vivo pharmacokinetics and pharmacodynamics and provide meaningful information for clinical trials. Lack of physiological relevance can make it difficult to interpret their data and greatly affect their eligibility as an in vitro tool for predicting drug process and effects. Most of current MOMs, except for PBPK model based systems (such as µCCAs,[11,44,78–81,87] pumpless recirculating MOMs,[43,100,102,104,105,152] and the stirrer-based micropump driven liver-intestine MOM[90]), have primarily focused on maintaining viable organ models and establishing interconnections among organs, yet have incorporated little consideration of physiological relevance of the inter-organ interactions into the design. Designs to maintain physiological ratios of organ size and incorporate sophisticated fluid circuits mimicking the blood circulation with organ specific perfusion rates can be the first steps towards physiologically relevant MOMs.
A more challenging aim for the in vitro MOM models is to establish physiologically realistic ratios of liquid to cell volumes at both local and systemic levels. Most biochemical reactions and cell signaling are concentration dependent. A large liquid-to-cell ratio leading to over dilution of drug metabolites and soluble ligands that mediate inter-organ interactions could result in incorrect PK/PD data, mechanistically different drug responses, or failure to detect certain drug effects through organ-organ interactions. We summarize in Table 1 the systemic and organ-specific liquid-to-cell ratios, as well as metabolic burden measured by liquid per hepatocyte for currently published MOM models, and make a quantitative comparison of those parameters with macroscale models and the human body. While in general, both systemic and organ-specific liquid-to-cell ratios, and the metabolic burden (liquid per hepatocyte) in microfluidic MOMs are smaller than those in macroscale and static microscale models, they are often tens to thousands-fold larger than the human physiological values (Figure 11). Notably, the adoption of microfabricated microfluidic platforms (including single-pass and recirculating ones) has dramatically brought down the median organ-specific liquid-to-cell ratios (~100 fold less vs. macroscale models, and ~15 fold less vs. static microscale models, Figure 11A), yet not so much for the systemic liquid-to-cell ratios (3~4 fold less, Figure 11B). While macroscale and static microscale models have high levels of both systemic and organ-specific liquid-to-cell ratios, microfluidic MOM models show differential liquid distribution at local and systemic levels depending on how the platforms construct organ chambers and build interconnections among different organ modules. Silicon chip based µCCA using extremely shallow cell chamber (20 – 30 µm) with relatively large growth area achieved lowest (near-physiological) organ-specific liquid-to-cell ratios (Figure 11A, red circle α).[44,78,79,87] Organ chambers filled with tissue biopsies[92,93] or 3D constructs built on scaffolds[104] or hydrogels[105] with high-density cell incorporation also showed low organ-specific liquid-to-cell ratio. Organ modules with in situ analytical components providing valuable organ-specific functional analysis, such as electrical or mechanical activity monitoring, often require integration of BioMEMS devices and 2D cell culture configuration, and thus usually have relatively large liquid-to-cell ratio.[43] Strategies that separate metabolic and functional representation of specific organ units in the system may help balance functional information acquisition and unfavorable drug and metabolite dilution.
Table 1.
Quantitative comparison on liquid-to-cell volume ratios and metabolic burden between the human body and in vitro systems.
| Biological System | Organ-specific liquid-to-cell ratio (v/v)a) |
Systemic liquid-to-cell ratio (v/v)a) |
Liquid per hepatocyte [nL]a) |
Reference |
|---|---|---|---|---|
| Human Body | 0.1 ~ 1.5b) | 0.3 | 0.06 | Graf et al.[186] |
| Macroscale physiological system | ||||
| 3-compartment CCA | Liver: 640; Lung: 12500 | 1800 | 10.7 | Sweeney et al.[11] |
| MCB | 76280 | 375 | Vozzi et al.[12] | |
| MCB | Liver: 14000; Fat: 110 | 330 | 120 | Vinci et al.[13] |
| MCB | Liver: 4200; Fat: 200 | 950 | 60 | Iori et al.[14] |
| Static microscale platform | ||||
| Transwell | Liver: 262; GI: 148 | 220 | 1.0 | Choi et al.[62] |
| Transwell | Liver:203; GI:302 | 217 | 1.2 | Lau et al.[63] |
| IdMOC | Liver: 81; Kidney: 1914; Lung: 2279; CNS: 924; Tumor: 502 | 3816 | 25 | Li et al.[71] |
| IdMOC | Liver: 233; fibroblast: 3941 | 1834 | 9.5 | Li et al.[150] |
| IdMOC | Liver: 233; fibroblast: 2815 | 2152 | 11.4 | Cole et al.[149] |
| Single-pass microphysiological system | ||||
| Liver: 19; GI: 4 | van Midwoud et al.[73] | |||
| Liver: 10; GI: 32; Tumor: 150 | Imura et al.[75,76] | |||
| Pump-driven recirculating microphysiological system | ||||
| 3-compartment µCCA | Liver: 1; Lung:31 | 708 | 4.0 | Sin et al.[78] |
| 4-compartment µCCA | Liver: 2; Lung: 25; | 857 | 2.5 | Viravaidya et al.[44,79] |
| 4-compartment µCCA | Liver:10; Marrow: 7; Tumor:7 | 474 | 3.4 | Tatosia et al.[80] |
| 3-compartment µCCA | 280 | 2.0 | Sung et al.[81] | |
| Liver: 18; Lung: 23; Kidney: 23; Fat: 1 | 29350 | 1852 | Zhang et al.[83] | |
| Liver: 22; Kidney: 20 | 1115 | 6.7 | Choucha-Snouber et al.[82] | |
| Liver: 17; Heart: 16 | 550 | 5.6 | Zhang et al.[84] | |
| Hanging drop network | Liver: 1454; Tumor: 14000 | 4520 | 26 | Frey et al.[85] |
| µCCA with barrier tissues | Liver: 1; GI: 23 | 390 | 1.3 | Mahler et al.[87] |
| Liver: 7; GI: 16 | 63 | 3.6 | Esch et al.[89] | |
| Liver: 69; Skin: 7 | 14 | 0.4 | Wagner et al.[92] | |
| Liver: 44; Skin: 8; EC: 620 | 15 | 0.5 | Maschmeyer et al.[93] | |
| Liver: 44; Intestine: 110 | 102 | 0.5 | Maschmeyer et al.[93] | |
| Liver: 139; Intestine: 28; Skin: 20; Kidney 2856 | 52 | 1.7 | Maschmeyer et al.[42] | |
| Pumpless recirculating microphysiological system | ||||
| Liver: 98; Bone marrow: 70; Tumor: 70 | 1094 | 7.5 | Sung et al.[100] | |
| Liver: 8; GI: 14 | 223 | 2.4 | Esch et al.[104] | |
| Liver: 139; Heart: 55; Brain: 614; Skeletal Muscle 2500 | 350 | 8.0 | Oleaga et al.[43] | |
| 14-chamber whole-body MOM | Liver: 8; GI: 12; Lung: 39; Bone marrow: 5; Kidney: 23 | 334 | 3.0 | Miller et al.[105] |
| Liver: 33; GI: 500 | 842 | 3.2 | Choe et al.[102] | |
| Liver: 17; Tumor:250 | 1310 | 4.0 | Lee et al.[152] | |
Liquid-to-cell ratios and liquid per hepatocyte were estimated based on published MOM literature and following physiological parameters: (A) human body volume, 65L;[60] (B) extracellular fluid volume, 15L; (C) total hepatocyte cell number, 2.41×1011;[60] (D) average cell volumes: average mammalian cells, 4000 um3;[60] hepatocyte, 4900 um3;[60] HepG2 cell, 2850 um3;[187] mouse hepatocyte, 5560 um3;[188] lung epithelial cell, 1764 um3;[189] kidney proximal tubular cell, 2100 um3;[190] cardiomyocyte, 15600 um3;[191] adipocyte, 3×105 um3;[192] astrocyte (soma and process): 4349 um3;[193] 3T3 cell: 2040 um3;[194] L2 cell: 382 um3;[195] MDCK cell, 1480 um3;[196] (E) mature Caco-2 monolayer thickness, 30 um;[197] and (F) cell doubling time: MCF7, 24 h; HAEC 28.6 h; HepG2, 48 h; HK-2, 96 h; MEG-01, 48 h;
Organ-specific liquid-to-cell ratios for the human body are listed in the supporting information Table S1.
Figure 11.

Quantitative comparison of liquid-to-cell ratios and systemic metabolic burden among the human body and various in vitro systems. Organ-specific (A) and systemic (B) liquid-to-cell ratios and metabolic burden measured by liquid per hepatocyte (C) are significantly lower in microfluidics based single-pass and recirculating MOMs than in macroscale and static microscale systems, yet are still mostly higher than the in vivo values. Scatter dots within the red circle α represent data from silicon chip based µCCAs with near-physiological organ-specific liquid-to-cell ratios. Scatter dots within the red circle β and γ represent data mostly from transwell compatible, pneumatic pump driven MOMs. Data are presented as scatter dot plots displaying median with range.
The transwell-based, pneumatic pump-driven MOMs from the Marx group achieved so far the lowest systemic liquid-to-cell ratios (Figure 11B, red circle β) and system metabolic burden (Figure 11C, red circle γ) with moderately high organ-specific liquid-to-cell ratios (Table 1). These closed-loop fluid circuits integrated on chip require minimal amount of liquid for circulation. The organ chambers designed for plug-in of transwell inserts, however, accumulate a significant amount of fluid. With the organ number increased from two to four, the systematic liquid-to-cell ratio almost quadrupled (Table 1). Such platforms would face serious challenges to control systemic liquid-to-cell ratios when more organs or whole body models are involved. µCCA chips with low organ-specific liquid-to-cell ratio yielded moderately high systemic liquid-to-cell ratios (Table 1), mainly due to the large amount of fluid in the reservoirs serving as bubble traps. The systemic ratios for these systems can be significantly improved by replacement with on-chip bubble-trap devices.[181] Similarly, a whole-body MOM constructed by Miller et al. on the pumpless platform showed relatively low organ liquid to cell ratios and moderately high systemic ratio, due to some unfilled organ chambers and excessive medium in the reservoirs to reduce the frequency of manual medium replenishment.[105] With more organ cells present and automated liquid handling capability integrated into the system, the systemic ratio would decrease greatly. Emerging integrative systems with flexible fluid network configuration[85] and sophisticated flow control capability[84,96] often have large systemic liquid to cell ratios. Balancing those desired features with physiologically relevant liquid to cell ratios remains challenging for MOM system development.
5.4. Summary
Current challenges for MOM systems across different platforms are to achieve acceptable reproducibility and physiological relevance. Major aspects to address include developing blood surrogate and controlled culture environment, minimizing and characterizing drug adsorption and absorption to platform materials, as well as incorporating physiological considerations to the system design, A continuing challenge for the MOM systems is the compromise between high-content and high-throughput screening. Trietsch et al. has recently demonstrated in a single-organ chip (Gut chip) the feasibility of providing high content analysis in a relatively high throughput format with 40 parallel intestinal tubes in a single plate (357 total for the study) on the pumpless platform.[182] The current focus on high content is appropriate for development of the technology. We anticipate further efforts to approach higher throughput multi-organ systems as those systems mature.
6. Conclusions and future directions
This field is entering a critical phase with the transition of this emerging technology into practical applications. To date, the emphasis has been mostly on demonstrating the feasibility of the technology with emergence of multiple small companies and their partnership with large pharmaceutical and biotechnology related companies.[59] Increasing numbers of examples should be evident soon where the technology is being used to address critical biological and medical questions, especially in disease models. This will be driven not by demonstration of technology but by answering pressing questions in pharmaceutical development, medicine and toxicology. If the technology addresses these challenges in a powerful and unique way, the utilization of these systems and the emergence of companies will continue to grow. If the technology fails to offer useful, cost-effective guidance, interest will decrease as has been seen with numerous technologies in the past whose capabilities were oversold in the beginning of the field. Thus, the next 3 to 5 years will be critical in determining if this new technology will be adopted widely for drug development and chemical toxicity testing.
While all systems discussed will have potentially profound impacts on drug development, we believe that multi-organ microsystems have unique advantages that allow early determination of both a drug’s efficacy and toxicity. With multiple organs present, potential side effects can be tested for both the parental compound and metabolites on a variety of organs if the system is designed to have a low enough volume (to prevent over dilution of metabolites released into a blood surrogate) and physiologically relevant recirculation times (for compounds with decay times on order of minutes). Such systems are now emerging.
We also believe that human in vitro systems will allow the rapid, early testing of the body’s response to multiple drugs. In some cases, treatment with multiple drugs will be of interest and in other cases one will seek to understand drug to drug interactions. Given the large number of combinations of drugs and the importance of the order of exposure (e.g., drug A before B or vice versa can make a significant difference in response), these human in vitro systems coupled with PBPK models may be able to provide guidance on potential human response.
Some key opportunities for these in vitro human systems will be in regard to predicting individual human response to drugs. Genetic and epigenetic characteristics differ among the human population. A human in vitro system could be constructed using patient derived cells. There are significant challenges to make this approach workable in terms of obtaining sufficient cells to test a wide variety of drugs for both efficacy and toxicity. While iPSC cells from an individual could address this problem, there are issues in terms of time required to develop appropriate model tissues, the degree of maturity of cells, and retention of epigenetic features. Functional improvements in iPS technologies may help address some of these issues. Alternatively, and more practically, would be to develop a range of human surrogates from iPSC cells representing major genetic subgroups. Data from such systems could serve to guide clinical trial design and target therapies to the subgroup most likely to benefit from a treatment option.
The design of novel drug delivery systems is another area that can benefit from the use of body-on-a-chip technologies. Drug delivery systems can improve the safety and efficacy of a therapeutic compound.[83,183,184] One challenge in this field is the translation of an in vitro drug delivery system to a preclinical setting.[185] In this regard, multi-organ systems can be used as a promising alternative for testing and development of drug delivery systems.
Other challenges remain. Although there have been some efforts to include immune systems in these in vitro systems, the resulting models are still rather premature. Another area that is needed, but quite challenging, is to understand the potential effects of a drug on cognitive functions of the brain. This is being addressed with certain advancements in the Peripheral Nervous System but still limited for Central Nervous System effects. Being able to anticipate side effects from the two key functions at an early stage of drug development would be quite useful.
While these human in vitro systems have made great strides in the last five years, the next five years will be critical to transforming this technology into something that will greatly assist drug development process. We are optimistic that these technical challenges will be addressed sufficiently well that we can generate more useful drugs at a significantly reduced cost. Ultimately, we expect that these systems will provide human kind with more useful drugs at a lower cost.
Supplementary Material
Acknowledgments
The preparation of this review has been supported by the National Institutes of Health (R44 TR001326 and R01NS050452).
Biographies

Ying I. Wang received her undergraduate and Master’s degrees, both in Biomedical Engineering, at Zhejiang University in China. Wang continued her research career at the University of California at Davis, where she received her PhD degree in Biomedical Engineering in 2013. Wang then joined Dr. Michael Shuler’s lab at Cornell University as a postdoctoral research fellow. Her research mainly focuses on the development of organ-on-a-chip and body-on-a-chip microphysiological systems for drug screening.

James J. Hickman is the Founding Director of the NanoScience Technology Center and a Professor of Nanoscience Technology, Chemistry, Biomolecular Science, Material Science and Electrical Engineering at UCF with a Ph.D. from MIT in Chemistry. For the past 25 years, he has studied the interaction of biological species with modified surfaces to create cell-based sensors and cellular integration with MEMS devices. He is interested in hybrid systems for biosensor and biological computation applications and the creation of functional in vitro systems for human body-on-a-chip applications. He is also the founder and Chief Scientist of Hesperos which is focusing on cell-based systems for drug discovery and toxicity.

Michael L. Shuler is Samuel Eckert Professor of Engineering in the School of Biomedical Engineering and the School of Chemical and Biomolecular Engineering at Cornell University. Shuler received both of his degrees in chemical engineering (BS, U. Notre Dame, and PhD, U. Minnesota) and has been a faculty member at Cornell University since 1974. Shuler’s research is focused on biomolecular engineering and includes the development of “Body-on-a-Chip” or microphysiological system for drug testing, development of plant cell cultures for the production of paclitaxel, and construction of computer models of cells relating the physiological function to the genomic structure.
Footnotes
Supporting Information is available from the Wiley Online Library or from the author.
References
- 1.PhRMA. Profile Biopharmaceutical Research Industry. 2015 [Google Scholar]
- 2.DiMasi JA, Grabowski HG, Hansen RW. J Health Econ. 2016;47:20. doi: 10.1016/j.jhealeco.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 3.Tefferi A, Kantarjian H, Rajkumar SV, Baker LH, Abkowitz JL, Adamson JW, Advani RH, Allison J, Antman KH, Bast RC, Jr, Bennett JM, Benz EJ, Jr, Berliner N, Bertino J, Bhatia R, Bhatia S, Bhojwani D, Blanke CD, Bloomfield CD, Bosserman L, Broxmeyer HE, Byrd JC, Cabanillas F, Canellos GP, Chabner BA, Chanan-Khan A, Cheson B, Clarkson B, Cohn SL, Colon-Otero G, Cortes J, Coutre S, Cristofanilli M, Curran WJ, Jr, Daley GQ, DeAngelo DJ, Deeg HJ, Einhorn LH, Erba HP, Esteva FJ, Estey E, Fidler IJ, Foran J, Forman S, Freireich E, Fuchs C, George JN, Gertz MA, Giralt S, Golomb H, Greenberg P, Gutterman J, Handin RI, Hellman S, Hoff PM, Hoffman R, Hong WK, Horowitz M, Hortobagyi GN, Hudis C, Issa JP, Johnson BE, Kantoff PW, Kaushansky K, Khayat D, Khuri FR, Kipps TJ, Kripke M, Kyle RA, Larson RA, Lawrence TS, Levine R, Link MP, Lippman SM, Lonial S, Lyman GH, Markman M, Mendelsohn J, Meropol NJ, Messinger Y, Mulvey TM, O'Brien S, Perez-Soler R, Pollock R, Prchal J, Press O, Radich J, Rai K, Rosenberg SA, Rowe JM, Rugo H, Runowicz CD, Sandmaier BM, Saven A, Schafer AI, Schiffer C, Sekeres MA, Silver RT, Siu LL, Steensma DP, Stewart FM, Stock W, Stone R, Storb R, Strong LC, Tallman MS, Thompson M, Ueno NT, Van Etten RA, Vose JM, Wiernik PH, Winer EP, Younes A, Zelenetz AD, LeMaistre CA. Mayo Clin Proc. 2015;90:996. doi: 10.1016/j.mayocp.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mullard A. Nat Rev Drug Discov. 2016;15:447. doi: 10.1038/nrd.2016.135. [DOI] [PubMed] [Google Scholar]
- 5.Kola I, Landis J. Nat Rev Drug Discov. 2004;3:711. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 6.Eastwood D, Findlay L, Poole S, Bird C, Wadhwa M, Moore M, Burns C, Thorpe R, Stebbings R. Br J Pharmacol. 2010;161:512. doi: 10.1111/j.1476-5381.2010.00922.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peters SA. Physiologically-Based Pharmacokinetic (PBPK) Modeling and Simulations: Principles, Methods, and Applications in the Pharmaceutical Industry. Wiley; Hoboken, N.J.: 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sweeney LM, Shuler ML, Quick DJ, Babish JG. Ann Biomed Eng. 1996;24:305. doi: 10.1007/BF02667357. [DOI] [PubMed] [Google Scholar]
- 9.Sweeney LM. Ph.D. Thesis. Cornell University; Aug, 1993. [Google Scholar]
- 10.Shuler ML, Ghanem A, Quick D, Wong MC, Miller P. Biotechnology and bioengineering. 1996;52:45. doi: 10.1002/(SICI)1097-0290(19961005)52:1<45::AID-BIT5>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 11.Sweeney LM, Shuler ML, Babish JG, Ghanem A. Toxicol In Vitro. 1995;9:307. doi: 10.1016/0887-2333(95)00007-u. [DOI] [PubMed] [Google Scholar]
- 12.Vozzi F, Heinrich JM, Bader A, Ahluwalia AD. Tissue Eng Part A. 2009;15:1291. doi: 10.1089/ten.tea.2008.0066. [DOI] [PubMed] [Google Scholar]
- 13.Vinci B, Murphy E, Iori E, Marescotti MC, Avogaro A, Ahluwalia A. Biotechnol J. 2010;5:618. doi: 10.1002/biot.201000009. [DOI] [PubMed] [Google Scholar]
- 14.Iori E, Vinci B, Murphy E, Marescotti MC, Avogaro A, Ahluwalia A. PLoS One. 2012;7:e34704. doi: 10.1371/journal.pone.0034704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bricks T, Paullier P, Legendre A, Fleury MJ, Zeller P, Merlier F, Anton PM, Leclerc E. Toxicol In Vitro. 2014;28:885. doi: 10.1016/j.tiv.2014.02.005. [DOI] [PubMed] [Google Scholar]
- 16.Bricks T, Hamon J, Fleury MJ, Jellali R, Merlier F, Herpe YE, Seyer A, Regimbeau JM, Bois F, Leclerc E. Biopharm Drug Dispos. 2015;36:275. doi: 10.1002/bdd.1940. [DOI] [PubMed] [Google Scholar]
- 17.Prot JM, Maciel L, Bricks T, Merlier F, Cotton J, Paullier P, Bois FY, Leclerc E. Biotechnology and bioengineering. 2014;111:2027. doi: 10.1002/bit.25232. [DOI] [PubMed] [Google Scholar]
- 18.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Cell. 2007;131:861. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 19.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA. Science (New York, N.Y.) 2007;318:1917. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 20.Lowry WE, Richter L, Yachechko R, Pyle AD, Tchieu J, Sridharan R, Clark AT, Plath K. Proc Natl Acad Sci U S A. 2008;105:2883. doi: 10.1073/pnas.0711983105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karumbayaram S, Novitch BG, Patterson M, Umbach JA, Richter L, Lindgren A, Conway AE, Clark AT, Goldman SA, Plath K, Wiedau-Pazos M, Kornblum HI, Lowry WE. Stem Cells. 2009;27:806. doi: 10.1002/stem.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo X, Johe K, Molnar P, Davis H, Hickman J. J Tissue Eng Regen Med. 2010;4:181. doi: 10.1002/term.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo X, Spradling S, Stancescu M, Lambert S, Hickman JJ. Biomaterials. 2013;34:4418. doi: 10.1016/j.biomaterials.2013.02.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang J, Wilson GF, Soerens AG, Koonce CH, Yu J, Palecek SP, Thomson JA, Kamp TJ. Circ Res. 2009;104:e30. doi: 10.1161/CIRCRESAHA.108.192237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lian X, Hsiao C, Wilson G, Zhu K, Hazeltine LB, Azarin SM, Raval KK, Zhang J, Kamp TJ, Palecek SP. Proc Natl Acad Sci U S A. 2012;109:E1848. doi: 10.1073/pnas.1200250109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Si-Tayeb K, Noto FK, Nagaoka M, Li J, Battle MA, Duris C, North PE, Dalton S, Duncan SA. Hepatology. 2010;51:297. doi: 10.1002/hep.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang D, Jiang W, Liu M, Sui X, Yin X, Chen S, Shi Y, Deng H. Cell Res. 2009;19:429. doi: 10.1038/cr.2009.28. [DOI] [PubMed] [Google Scholar]
- 28.Huang SX, Islam MN, O'Neill J, Hu Z, Yang YG, Chen YW, Mumau M, Green MD, Vunjak-Novakovic G, Bhattacharya J, Snoeck HW. Nat Biotechnol. 2014;32:84. doi: 10.1038/nbt.2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lippmann ES, Azarin SM, Kay JE, Nessler RA, Wilson HK, Al-Ahmad A, Palecek SP, Shusta EV. Nat Biotechnol. 2012;30:783. doi: 10.1038/nbt.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spence JR, Mayhew CN, Rankin SA, Kuhar MF, Vallance JE, Tolle K, Hoskins EE, Kalinichenko VV, Wells SI, Zorn AM, Shroyer NF, Wells JM. Nature. 2011;470:105. doi: 10.1038/nature09691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W, Croft GF, Saphier G, Leibel R, Goland R, Wichterle H, Henderson CE, Eggan K. Science (New York, N.Y.) 2008;321:1218. doi: 10.1126/science.1158799. [DOI] [PubMed] [Google Scholar]
- 32.Maehr R, Chen S, Snitow M, Ludwig T, Yagasaki L, Goland R, Leibel RL, Melton DA. Proc Natl Acad Sci U S A. 2009;106:15768. doi: 10.1073/pnas.0906894106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tibbitt MW, Anseth KS. Biotechnology and bioengineering. 2009;103:655. doi: 10.1002/bit.22361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang YS, Khademhosseini A. Science (New York, N.Y.) 2017:356. doi: 10.1126/science.aaf3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bajaj P, Schweller RM, Khademhosseini A, West JL, Bashir R. Annu Rev Biomed Eng. 2014;16:247. doi: 10.1146/annurev-bioeng-071813-105155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murphy SV, Atala A. Nat Biotechnol. 2014;32:773. doi: 10.1038/nbt.2958. [DOI] [PubMed] [Google Scholar]
- 37.Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA. Nature. 2013;501:373. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang YS, Arneri A, Bersini S, Shin SR, Zhu K, Goli-Malekabadi Z, Aleman J, Colosi C, Busignani F, Dell'Erba V, Bishop C, Shupe T, Demarchi D, Moretti M, Rasponi M, Dokmeci MR, Atala A, Khademhosseini A. Biomaterials. 2016;110:45. doi: 10.1016/j.biomaterials.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Freedman BS, Brooks CR, Lam AQ, Fu H, Morizane R, Agrawal V, Saad AF, Li MK, Hughes MR, Werff RV, Peters DT, Lu J, Baccei A, Siedlecki AM, Valerius MT, Musunuru K, McNagny KM, Steinman TI, Zhou J, Lerou PH, Bonventre JV. Nat Commun. 2015;6:8715. doi: 10.1038/ncomms9715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takebe T, Sekine K, Enomura M, Koike H, Kimura M, Ogaeri T, Zhang RR, Ueno Y, Zheng YW, Koike N, Aoyama S, Adachi Y, Taniguchi H. Nature. 2013;499:481. doi: 10.1038/nature12271. [DOI] [PubMed] [Google Scholar]
- 41.Bhatia SN, Ingber DE. Nat Biotechnol. 2014;32:760. doi: 10.1038/nbt.2989. [DOI] [PubMed] [Google Scholar]
- 42.Maschmeyer I, Lorenz AK, Schimek K, Hasenberg T, Ramme AP, Hubner J, Lindner M, Drewell C, Bauer S, Thomas A, Sambo NS, Sonntag F, Lauster R, Marx U. Lab on a chip. 2015;15:2688. doi: 10.1039/c5lc00392j. [DOI] [PubMed] [Google Scholar]
- 43.Oleaga C, Bernabini C, Smith AS, Srinivasan B, Jackson M, McLamb W, Platt V, Bridges R, Cai Y, Santhanam N, Berry B, Najjar S, Akanda N, Guo X, Martin C, Ekman G, Esch MB, Langer J, Ouedraogo G, Cotovio J, Breton L, Shuler ML, Hickman JJ. Sci Rep. 2016;6:20030. doi: 10.1038/srep20030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Viravaidya K, Sin A, Shuler ML. Biotechnol Prog. 2004;20:316. doi: 10.1021/bp0341996. [DOI] [PubMed] [Google Scholar]
- 45.Esch MB, Sung JH, Shuler ML. J Biotechnol. 2010;148:64. doi: 10.1016/j.jbiotec.2010.02.020. [DOI] [PubMed] [Google Scholar]
- 46.Esch MB, King TL, Shuler ML. Annu Rev Biomed Eng. 2011;13:55. doi: 10.1146/annurev-bioeng-071910-124629. [DOI] [PubMed] [Google Scholar]
- 47.Sung JH, Esch MB, Prot JM, Long CJ, Smith A, Hickman JJ, Shuler ML. Lab on a chip. 2013;13:1201. doi: 10.1039/c3lc41017j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sutherland ML, Fabre KM, Tagle DA. Stem Cell Res Ther. 2013;4(Suppl 1):I1. doi: 10.1186/scrt361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.An F, Qu Y, Liu X, Zhong R, Luo Y. Anal Chem Insights. 2015;10:39. doi: 10.4137/ACI.S28905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Benam KH, Dauth S, Hassell B, Herland A, Jain A, Jang KJ, Karalis K, Kim HJ, MacQueen L, Mahmoodian R, Musah S, Torisawa YS, van der Meer AD, Villenave R, Yadid M, Parker KK, Ingber DE. Annu Rev Pathol. 2015;10:195. doi: 10.1146/annurev-pathol-012414-040418. [DOI] [PubMed] [Google Scholar]
- 51.Caplin JD, Granados NG, James MR, Montazami R, Hashemi N. Adv Healthc Mater. 2015;4:1426. doi: 10.1002/adhm.201500040. [DOI] [PubMed] [Google Scholar]
- 52.Hutson MS, Alexander PG, Allwardt V, Aronoff DM, Bruner-Tran KL, Cliffel DE, Davidson JM, Gough A, Markov DA, McCawley LJ, McKenzie JR, McLean JA, Osteen KG, Pensabene V, Samson PC, Senutovitch NK, Sherrod SD, Shotwell MS, Taylor DL, Tetz LM, Tuan RS, Vernetti LA, Wikswo JP. Applied In Vitro Toxicology. 2016;2:97. [Google Scholar]
- 53.Lee SH, Ha SK, Choi I, Choi N, Park TH, Sung JH. Biotechnol J. 2016;11:746. doi: 10.1002/biot.201500551. [DOI] [PubMed] [Google Scholar]
- 54.Polini A, Prodanov L, Bhise NS, Manoharan V, Dokmeci MR, Khademhosseini A. Expert Opin Drug Dis. 2014;9:335. doi: 10.1517/17460441.2014.886562. [DOI] [PubMed] [Google Scholar]
- 55.Ribas J, Sadeghi H, Manbachi A, Leijten J, Brinegar K, Zhang YS, Ferreira L, Khademhosseini A. Applied In Vitro Toxicology. 2016;2:82. doi: 10.1089/aivt.2016.0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Skardal A, Shupe T, Atala A. Drug Discov Today. 2016;21:1399. doi: 10.1016/j.drudis.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yi Y, Park J, Lim J, Lee CJ, Lee SH. Trends Biotechnol. 2015;33:762. doi: 10.1016/j.tibtech.2015.09.007. [DOI] [PubMed] [Google Scholar]
- 58.Zhang YS, Zhang YN, Zhang W. Drug Discov Today. 2017 [Google Scholar]
- 59.Zhang B, Radisic M. Lab on a chip. 2017;17:2395. doi: 10.1039/c6lc01554a. [DOI] [PubMed] [Google Scholar]
- 60.Bianconi E, Piovesan A, Facchin F, Beraudi A, Casadei R, Frabetti F, Vitale L, Pelleri MC, Tassani S, Piva F, Perez-Amodio S, Strippoli P, Canaider S. Ann Hum Biol. 2013;40:463. doi: 10.3109/03014460.2013.807878. [DOI] [PubMed] [Google Scholar]
- 61.Boyden S. J Exp Med. 1962;115:453. doi: 10.1084/jem.115.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Choi S, Nishikawa M, Sakoda A, Sakai Y. Toxicol In Vitro. 2004;18:393. doi: 10.1016/j.tiv.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 63.Lau YY, Chen YH, Liu TT, Li C, Cui X, White RE, Cheng KC. Drug Metab Dispos. 2004;32:937. [PubMed] [Google Scholar]
- 64.Zahavi EE, Ionescu A, Gluska S, Gradus T, Ben-Yaakov K, Perlson E. J Cell Sci. 2015;128:1241. doi: 10.1242/jcs.167544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lei Y, Li J, Wang N, Yang X, Hamada Y, Li Q, Zheng W, Jiang X. Integrative biology : quantitative biosciences from nano to macro. 2016;8:359. doi: 10.1039/c5ib00309a. [DOI] [PubMed] [Google Scholar]
- 66.Nakanishi J, Takarada T, Yamaguchi K, Maeda M. Anal Sci. 2008;24:67. doi: 10.2116/analsci.24.67. [DOI] [PubMed] [Google Scholar]
- 67.March S, Ramanan V, Trehan K, Ng S, Galstian A, Gural N, Scull MA, Shlomai A, Mota MM, Fleming HE, Khetani SR, Rice CM, Bhatia SN. Nat Protoc. 2015;10:2027. doi: 10.1038/nprot.2015.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhong H, Xuan L, Wang D, Zhou J, Li Y, Jiang Q. RSC Adv. 2017;7:21837. [Google Scholar]
- 69.Sugiura S, Cha JM, Yanagawa F, Zorlutuna P, Bae H, Khademhosseini A. J Tissue Eng Regen Med. 2016;10:690. doi: 10.1002/term.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shin DS, You J, Rahimian A, Vu T, Siltanen C, Ehsanipour A, Stybayeva G, Sutcliffe J, Revzin A. Angew Chem Int Edit. 2014;53:8221. doi: 10.1002/anie.201404323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li AP, Bode C, Sakai Y. Chem Biol Interact. 2004;150:129. doi: 10.1016/j.cbi.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 72.van Midwoud PM, Groothuis GM, Merema MT, Verpoorte E. Biotechnology and bioengineering. 2010;105:184. doi: 10.1002/bit.22516. [DOI] [PubMed] [Google Scholar]
- 73.van Midwoud PM, Merema MT, Verpoorte E, Groothuis GM. Lab on a chip. 2010;10:2778. doi: 10.1039/c0lc00043d. [DOI] [PubMed] [Google Scholar]
- 74.Loskill P, Marcus SG, Mathur A, Reese WM, Healy KE. PLoS One. 2015;10:e0139587. doi: 10.1371/journal.pone.0139587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Imura Y, Sato K, Yoshimura E. Anal Chem. 2010;82:9983. doi: 10.1021/ac100806x. [DOI] [PubMed] [Google Scholar]
- 76.Imura Y, Yoshimura E, Sato K. Anal Sci. 2012;28:197. doi: 10.2116/analsci.28.197. [DOI] [PubMed] [Google Scholar]
- 77.Vernetti L, Gough A, Baetz N, Blutt S, Broughman JR, Brown JA, Foulke-Abel J, Hasan N, In J, Kelly E, Kovbasnjuk O, Repper J, Senutovitch N, Stabb J, Yeung C, Zachos NC, Donowitz M, Estes M, Himmelfarb J, Truskey G, Wikswo JP, Taylor DL. Sci Rep. 2017;7:42296. doi: 10.1038/srep42296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sin A, Chin KC, Jamil MF, Kostov Y, Rao G, Shuler ML. Biotechnol Prog. 2004;20:338. doi: 10.1021/bp034077d. [DOI] [PubMed] [Google Scholar]
- 79.Viravaidya K, Shuler ML. Biotechnol Prog. 2004;20:590. doi: 10.1021/bp034238d. [DOI] [PubMed] [Google Scholar]
- 80.Tatosian DA, Shuler ML. Biotechnology and bioengineering. 2009;103:187. doi: 10.1002/bit.22219. [DOI] [PubMed] [Google Scholar]
- 81.Sung JH, Shuler ML. Lab on a chip. 2009;9:1385. doi: 10.1039/b901377f. [DOI] [PubMed] [Google Scholar]
- 82.Choucha-Snouber L, Aninat C, Grsicom L, Madalinski G, Brochot C, Poleni PE, Razan F, Guillouzo CG, Legallais C, Corlu A, Leclerc E. Biotechnology and bioengineering. 2013;110:597. doi: 10.1002/bit.24707. [DOI] [PubMed] [Google Scholar]
- 83.Zhang C, Zhao Z, Abdul Rahim NA, van Noort D, Yu H. Lab on a chip. 2009;9:3185. doi: 10.1039/b915147h. [DOI] [PubMed] [Google Scholar]
- 84.Zhang YS, Aleman J, Shin SR, Kilic T, Kim D, Mousavi Shaegh SA, Massa S, Riahi R, Chae S, Hu N, Avci H, Zhang W, Silvestri A, Sanati Nezhad A, Manbohi A, De Ferrari F, Polini A, Calzone G, Shaikh N, Alerasool P, Budina E, Kang J, Bhise N, Ribas J, Pourmand A, Skardal A, Shupe T, Bishop CE, Dokmeci MR, Atala A, Khademhosseini A. Proc Natl Acad Sci U S A. 2017;114:E2293. doi: 10.1073/pnas.1612906114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Frey O, Misun PM, Fluri DA, Hengstler JG, Hierlemann A. Nat Commun. 2014;5:4250. doi: 10.1038/ncomms5250. [DOI] [PubMed] [Google Scholar]
- 86.Yazdi SR, Shadmani A, Burgel SC, Misun PM, Hierlemann A, Frey O. Lab on a chip. 2015;15:4138. doi: 10.1039/c5lc01000d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mahler GJ, Esch MB, Glahn RP, Shuler ML. Biotechnology and bioengineering. 2009;104:193. doi: 10.1002/bit.22366. [DOI] [PubMed] [Google Scholar]
- 88.Mahler GJ, Shuler ML, Glahn RP. J Nutr Biochem. 2009;20:494. doi: 10.1016/j.jnutbio.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 89.Esch MB, Mahler GJ, Stokol T, Shuler ML. Lab on a chip. 2014;14:3081. doi: 10.1039/c4lc00371c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kimura H, Ikeda T, Nakayama H, Sakai Y, Fujii T. J Lab Autom. 2015;20:265. doi: 10.1177/2211068214557812. [DOI] [PubMed] [Google Scholar]
- 91.Wang YI, Oleaga C, Long CJ, Esch MB, McAleer CW, Miller PG, Hickman JJ, Shuler ML. Experimental Biology and Medicine. 2017;0 doi: 10.1177/1535370217694101. 153537021769410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wagner I, Materne EM, Brincker S, Sussbier U, Fradrich C, Busek M, Sonntag F, Sakharov DA, Trushkin EV, Tonevitsky AG, Lauster R, Marx U. Lab on a chip. 2013;13:3538. doi: 10.1039/c3lc50234a. [DOI] [PubMed] [Google Scholar]
- 93.Maschmeyer I, Hasenberg T, Jaenicke A, Lindner M, Lorenz AK, Zech J, Garbe LA, Sonntag F, Hayden P, Ayehunie S, Lauster R, Marx U, Materne EM. Eur J Pharm Biopharm. 2015;95:77. doi: 10.1016/j.ejpb.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Materne EM, Ramme AP, Terrasso AP, Serra M, Alves PM, Brito C, Sakharov DA, Tonevitsky AG, Lauster R, Marx U. J Biotechnol. 2015;205:36. doi: 10.1016/j.jbiotec.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 95.An F, Qu Y, Luo Y, Fang N, Liu Y, Gao Z, Zhao W, Lin B. Sci Rep. 2016;6:25022. doi: 10.1038/srep25022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Coppeta JR, Mescher MJ, Isenberg BC, Spencer AJ, Kim ES, Lever AR, Mulhern TJ, Prantil-Baun R, Comolli JC, Borenstein JT. Lab on a chip. 2016;17:134. doi: 10.1039/c6lc01236a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chen WLK, Edington C, Suter E, Yu J, Velazquez JJ, Velazquez JG, Shockley M, Large EM, Venkataramanan R, Hughes DJ, Stokes CL, Trumper DL, Carrier RL, Cirit M, Griffith LG, Lauffenburger DA. Biotechnology and bioengineering. 2017 doi: 10.1002/bit.26370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tsamandouras N, Chen WLK, Edington CD, Stokes CL, Griffith LG, Cirit M. AAPS J. 2017 doi: 10.1208/s12248-017-0122-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kamei K, Kato Y, Hirai Y, Ito S, Satoh J, Oka A, Tsuchiya T, Chen Y, Tabata O. Rsc Adv. 2017;7:36777. [Google Scholar]
- 100.Sung JH, Kam C, Shuler ML. Lab on a chip. 2010;10:446. doi: 10.1039/b917763a. [DOI] [PubMed] [Google Scholar]
- 101.Esch MB, Prot JM, Wang YI, Miller P, Llamas-Vidales JR, Naughton BA, Applegate DR, Shuler ML. Lab on a chip. 2015;15:2269. doi: 10.1039/c5lc00237k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Choe A, Ha SK, Choi I, Choi N, Sung JH. Biomed Microdevices. 2017;19:4. doi: 10.1007/s10544-017-0242-8. [DOI] [PubMed] [Google Scholar]
- 103.Wang YI, Abaci HE, Shuler ML. Biotechnology and bioengineering. 2017;114:184. doi: 10.1002/bit.26045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Esch MB, Ueno H, Applegate DR, Shuler ML. Lab on a chip. 2016;16:2719. doi: 10.1039/c6lc00461j. [DOI] [PubMed] [Google Scholar]
- 105.Miller PG, Shuler ML. Biotechnology and bioengineering. 2016;113:2213. doi: 10.1002/bit.25989. [DOI] [PubMed] [Google Scholar]
- 106.Bloom AB, Zaman MH. Physiol Genomics. 2014;46:309. doi: 10.1152/physiolgenomics.00170.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Esch MB, Smith AS, Prot JM, Oleaga C, Hickman JJ, Shuler ML. Advanced drug delivery reviews. 2014;69–70:158. doi: 10.1016/j.addr.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Oomen PE, Skolimowski MD, Verpoorte E. Lab on a chip. 2016;16:3394. doi: 10.1039/c6lc00772d. [DOI] [PubMed] [Google Scholar]
- 109.Vollmer AP, Probstein RF, Gilbert R, Thorsen T. Lab on a chip. 2005;5:1059. doi: 10.1039/b508097e. [DOI] [PubMed] [Google Scholar]
- 110.McEvoy AK, McDonagh CM, MacCraith BD. Analyst. 1996;121:785. [Google Scholar]
- 111.Mousavi Shaegh SA, De Ferrari F, Zhang YS, Nabavinia M, Binth Mohammad N, Ryan J, Pourmand A, Laukaitis E, Banan Sadeghian R, Nadhman A, Shin SR, Nezhad AS, Khademhosseini A, Dokmeci MR. Biomicrofluidics. 2016;10:044111. doi: 10.1063/1.4955155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ren K, Zhou J, Wu H. Acc Chem Res. 2013;46:2396. doi: 10.1021/ar300314s. [DOI] [PubMed] [Google Scholar]
- 113.Oh TI, Sung JH, Tatosian DA, Shuler ML, Kim D. Cytometry. Part A : the journal of the International Society for Analytical Cytology. 2007;71:857. doi: 10.1002/cyto.a.20427. [DOI] [PubMed] [Google Scholar]
- 114.Misun PM, Rothe J, Schmid YRF, Hierlemann A, Frey O. Microsyst Nanoeng. 2016;2:16022. doi: 10.1038/micronano.2016.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pirozzi KL, Long CJ, McAleer CW, Smith AS, Hickman JJ. Applied physics letters. 2013;103:83108. doi: 10.1063/1.4817939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Park KH, Brotto L, Lehoang O, Brotto M, Ma J, Zhao X. Journal of visualized experiments : JoVE. 2012:4198. doi: 10.3791/4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Effron MB, Bhatnagar GM, Spurgeon HA, Ruano-Arroyo G, Lakatta EG. Circ Res. 1987;60:238. doi: 10.1161/01.res.60.2.238. [DOI] [PubMed] [Google Scholar]
- 118.Agarwal A, Goss JA, Cho A, McCain ML, Parker KK. Lab on a chip. 2013;13:3599. doi: 10.1039/c3lc50350j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fritz J, Baller MK, Lang HP, Rothuizen H, Vettiger P, Meyer E, Guntherodt H, Gerber C, Gimzewski JK. Science (New York, N.Y.) 2000;288:316. doi: 10.1126/science.288.5464.316. [DOI] [PubMed] [Google Scholar]
- 120.Das M, Gregory CA, Molnar P, Riedel LM, Wilson K, Hickman JJ. Biomaterials. 2006;27:4374. doi: 10.1016/j.biomaterials.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 121.Das M, Wilson K, Molnar P, Hickman JJ. Nat Protoc. 2007;2:1795. doi: 10.1038/nprot.2007.229. [DOI] [PubMed] [Google Scholar]
- 122.Wilson K, Molnar P, Hickman J. Lab on a chip. 2007;7:920. doi: 10.1039/b617939h. [DOI] [PubMed] [Google Scholar]
- 123.Wilson K, Das M, Wahl KJ, Colton RJ, Hickman J. PLoS One. 2010;5:e11042. doi: 10.1371/journal.pone.0011042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Smith AS, Long CJ, McAleer C, Bobbitt N, Srinivasan B, Hickman JJ. Journal of visualized experiments : JoVE. 2014:e51866. doi: 10.3791/51866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Stancescu M, Molnar P, McAleer CW, McLamb W, Long CJ, Oleaga C, Prot JM, Hickman JJ. Biomaterials. 2015;60:20. doi: 10.1016/j.biomaterials.2015.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Oleaga C, Legters G, Bridges LR, Kumanchik L, Martin C, Cai Y, Schnepper M, McAleer CW, Long CJ, Hickman JJ. In: Method Pharmacol Tox. Clements M, Roquemore L, editors. Springer New York; New York, NY: 2017. p. 229. [Google Scholar]
- 127.Vandenburgh H, Shansky J, Benesch-Lee F, Barbata V, Reid J, Thorrez L, Valentini R, Crawford G. Muscle & nerve. 2008;37:438. doi: 10.1002/mus.20931. [DOI] [PubMed] [Google Scholar]
- 128.Uzel SG, Platt RJ, Subramanian V, Pearl TM, Rowlands CJ, Chan V, Boyer LA, So PT, Kamm RD. Sci Adv. 2016;2:e1501429. doi: 10.1126/sciadv.1501429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Offenhausser A, Knoll W. Trends in Biotechnology. 2001;19:62. doi: 10.1016/s0167-7799(00)01544-4. [DOI] [PubMed] [Google Scholar]
- 130.Obien ME, Deligkaris K, Bullmann T, Bakkum DJ, Frey U. Front Neurosci. 2014;8:423. doi: 10.3389/fnins.2014.00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Elkins RC, Davies MR, Brough SJ, Gavaghan DJ, Cui Y, Abi-Gerges N, Mirams GR. Journal of pharmacological and toxicological methods. 2013;68:112. doi: 10.1016/j.vascn.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Natarajan A, Molnar P, Sieverdes K, Jamshidi A, Hickman JJ. Toxicol In Vitro. 2006;20:375. doi: 10.1016/j.tiv.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 133.Varghese K, Molnar P, Das M, Bhargava N, Lambert S, Kindy MS, Hickman JJ. PLOS ONE. 2010;5:e8643. doi: 10.1371/journal.pone.0008643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Meyer T, Boven KH, Gunther E, Fejtl M. Drug safety. 2004;27:763. doi: 10.2165/00002018-200427110-00002. [DOI] [PubMed] [Google Scholar]
- 135.Jung DR, Cuttino DS, Pancrazio JJ, Manos P, Cluster T, Sathanoori RS, Aloi LE, Coulombe MG, Czamaski MA, Borkholder DA, Kovacs GTA, Bey P, Stenger DA, Hickman JJ. J Vac Sci Technol A. 1998;16:1183. [Google Scholar]
- 136.Natarajan A, Stancescu M, Dhir V, Armstrong C, Sommerhage F, Hickman JJ, Molnar P. Biomaterials. 2011;32:4267. doi: 10.1016/j.biomaterials.2010.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Langhammer CG, Kutzing MK, Luo V, Zahn JD, Firestein BL. Biotechnol Prog. 2011;27:891. doi: 10.1002/btpr.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Benson K, Cramer S, Galla HJ. Fluids Barriers CNS. 2013;10:5. doi: 10.1186/2045-8118-10-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Srinivasan B, Kolli AR, Esch MB, Abaci HE, Shuler ML, Hickman JJ. J Lab Autom. 2015;20:107. doi: 10.1177/2211068214561025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Walter FR, Valkai S, Kincses A, Petneházi A, Czeller T, Veszelka S, Ormos P, Deli MA, Dér A. Sensors and Actuators B: Chemical. 2016;222:1209. [Google Scholar]
- 141.Booth R, Kim H. Lab on a chip. 2012;12:1784. doi: 10.1039/c2lc40094d. [DOI] [PubMed] [Google Scholar]
- 142.Igielska-Kalwat J, Goscianska J, Witkowska B, Nowak I. Postepy Dermatol Alergol. 2016;33:163. doi: 10.5114/ada.2016.60607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Parasuraman S. J Pharmacol Pharmacother. 2011;2:74. doi: 10.4103/0976-500X.81895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Abaci HE, Shuler ML. Integrative biology : quantitative biosciences from nano to macro. 2015;7:383. doi: 10.1039/c4ib00292j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cross MJ, Berridge BR, Clements PJ, Cove-Smith L, Force TL, Hoffmann P, Holbrook M, Lyon AR, Mellor HR, Norris AA, Pirmohamed M, Tugwood JD, Sidaway JE, Park BK. Br J Pharmacol. 2015;172:957. doi: 10.1111/bph.12979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Fagerholm U. The Journal of pharmacy and pharmacology. 2007;59:1463. doi: 10.1211/jpp.59.11.0002. [DOI] [PubMed] [Google Scholar]
- 147.Alavijeh MS, Chishty M, Qaiser MZ, Palmer AM. NeuroRx : the journal of the American Society for Experimental NeuroTherapeutics. 2005;2:554. doi: 10.1602/neurorx.2.4.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ferguson MA, Vaidya VS, Bonventre JV. Toxicology. 2008;245:182. doi: 10.1016/j.tox.2007.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cole SD, Madren-Whalley JS, Li AP, Dorsey R, Salem H. J Biomol Screen. 2014;19:1402. doi: 10.1177/1087057114550399. [DOI] [PubMed] [Google Scholar]
- 150.Li AP, Uzgare A, LaForge YS. Chem Biol Interact. 2012;199:1. doi: 10.1016/j.cbi.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 151.Waring MJ, Arrowsmith J, Leach AR, Leeson PD, Mandrell S, Owen RM, Pairaudeau G, Pennie WD, Pickett SD, Wang J, Wallace O, Weir A. Nat Rev Drug Discov. 2015;14:475. doi: 10.1038/nrd4609. [DOI] [PubMed] [Google Scholar]
- 152.Lee H, Kim DS, Ha SK, Choi I, Lee JM, Sung JH. Biotechnology and bioengineering. 2017;114:432. doi: 10.1002/bit.26087. [DOI] [PubMed] [Google Scholar]
- 153.Pardridge WM. NeuroRx : the journal of the American Society for Experimental NeuroTherapeutics. 2005;2:3. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Brown JA, Pensabene V, Markov DA, Allwardt V, Neely MD, Shi M, Britt CM, Hoilett OS, Yang Q, Brewer BM, Samson PC, McCawley LJ, May JM, Webb DJ, Li D, Bowman AB, Reiserer RS, Wikswo JP. Biomicrofluidics. 2015;9:054124. doi: 10.1063/1.4934713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Xu Z, Li E, Guo Z, Yu R, Hao H, Xu Y, Sun Z, Li X, Lyu J, Wang Q. ACS Appl Mater Interfaces. 2016;8:25840. doi: 10.1021/acsami.6b08746. [DOI] [PubMed] [Google Scholar]
- 156.Chiang SP, Cabrera RM, Segall JE. American journal of physiology. Cell physiology. 2016;311:C1. doi: 10.1152/ajpcell.00238.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Chaffer CL, Weinberg RA. Science (New York, N.Y.) 2011;331:1559. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 158.Zervantonakis IK, Hughes-Alford SK, Charest JL, Condeelis JS, Gertler FB, Kamm RD. Proc Natl Acad Sci U S A. 2012;109:13515. doi: 10.1073/pnas.1210182109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Riahi R, Yang YL, Kim H, Jiang L, Wong PK, Zohar Y. Biomicrofluidics. 2014;8:024103. doi: 10.1063/1.4868301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verastegui E, Zlotnik A. Nature. 2001;410:50. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 161.Choi Y, Hyun E, Seo J, Blundell C, Kim HC, Lee E, Lee SH, Moon A, Moon WK, Huh D. Lab on a chip. 2015;15:3350. doi: 10.1039/c5lc00514k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Das M, Rumsey JW, Bhargava N, Stancescu M, Hickman JJ. Biomaterials. 2010;31:4880. doi: 10.1016/j.biomaterials.2010.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Smith AS, Long CJ, Pirozzi K, Hickman JJ. Technology (Singap World Sci) 2013;1:37. doi: 10.1142/S2339547813500015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Guo X, Gonzalez M, Stancescu M, Vandenburgh HH, Hickman JJ. Biomaterials. 2011;32:9602. doi: 10.1016/j.biomaterials.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Fifth AIMBE/NIH Workshop on Validation and Qualification of New In Vitro Tools and Models for the Pre-clinical Drug Discovery Process. Bethesda, MD: 2015. [Google Scholar]
- 166.Schaffner AE, Barker JL, Stenger DA, Hickman JJ. J Neurosci Meth. 1995;62:111. doi: 10.1016/0165-0270(95)00063-1. [DOI] [PubMed] [Google Scholar]
- 167.Smith AS, Long CJ, Pirozzi K, Najjar S, McAleer C, Vandenburgh HH, Hickman JJ. J Biotechnol. 2014;185:15. doi: 10.1016/j.jbiotec.2014.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Berry BJ, Akanda N, Smith AS, Long CJ, Schnepper MT, Guo X, Hickman JJ. Biotechnol Prog. 2015;31:1613. doi: 10.1002/btpr.2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Guo X, Ayala JE, Gonzalez M, Stancescu M, Lambert S, Hickman JJ. Biomaterials. 2012;33:5723. doi: 10.1016/j.biomaterials.2012.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Guo X, Colon A, Akanda N, Spradling S, Stancescu M, Martin C, Hickman JJ. Biomaterials. 2017;122:179. doi: 10.1016/j.biomaterials.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Rumsey JW, Das M, Bhalkikar A, Stancescu M, Hickman JJ. Biomaterials. 2010;31:8218. doi: 10.1016/j.biomaterials.2010.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Davis H, Gonzalez M, Stancescu M, Love R, Hickman JJ, Lambert S. Biomaterials. 2014;35:8840. doi: 10.1016/j.biomaterials.2014.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Spargo BJ, Testoff MA, Nielsen TB, Stenger DA, Hickman JJ, Rudolph AS. Proc Natl Acad Sci U S A. 1994;91:11070. doi: 10.1073/pnas.91.23.11070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wilson KA, Finch CA, Anderson P, Vollmer F, Hickman JJ. Biomaterials. 2012;33:225. doi: 10.1016/j.biomaterials.2011.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ravenscroft MS, Bateman KE, Shaffer KM, Schessler HM, Jung DR, Schneider TW, Montgomery CB, Custer TL, Schaffner AE, Liu QY, Li YX, Barker JL, Hickman JJ. Journal of the American Chemical Society. 1998;120:12169. [Google Scholar]
- 176.Huang HZ, Shi JS, Laskin J, Liu ZY, McVey DS, Sun XZS. Soft Matter. 2011;7:8905. [Google Scholar]
- 177.van Midwoud PM, Janse A, Merema MT, Groothuis GM, Verpoorte E. Anal Chem. 2012;84:3938. doi: 10.1021/ac300771z. [DOI] [PubMed] [Google Scholar]
- 178.Gomez-Sjoberg R, Leyrat AA, Houseman BT, Shokat K, Quake SR. Anal Chem. 2010;82:8954. doi: 10.1021/ac101870s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.van Meer BJ, de Vries H, Firth KS, van Weerd J, Tertoolen LG, Karperien HB, Jonkheijm P, Denning C, AP IJ, Mummery CL. Biochem Biophys Res Commun. 2017;482:323. doi: 10.1016/j.bbrc.2016.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Sasaki H, Onoe H, Osaki T, Kawano R, Takeuchi S. Sensor Actuat B-Chem. 2010;150:478. [Google Scholar]
- 181.Sung JH, Shuler ML. Biomed Microdevices. 2009;11:731. doi: 10.1007/s10544-009-9286-8. [DOI] [PubMed] [Google Scholar]
- 182.Trietsch SJ, Naumovska E, Kurek D, Setyawati MC, Vormann MK, Wilschut KJ, Lanz HL, Nicolas A, Ng CP, Joore J, Kustermann S, Roth A, Hankemeier T, Moisan A, Vulto P. Nat Commun. 2017;8:262. doi: 10.1038/s41467-017-00259-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Rosen H, Abribat T. Nat Rev Drug Discov. 2005;4:381. doi: 10.1038/nrd1721. [DOI] [PubMed] [Google Scholar]
- 184.Park K. J Control Release. 2016;240:2. doi: 10.1016/j.jconrel.2015.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Park K. Mol Pharmaceut. 2016;13:2143. doi: 10.1021/acs.molpharmaceut.6b00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Graf JF, Scholz BJ, Zavodszky MI. J Pharmacokinet Pharmacodyn. 2012;39:37. doi: 10.1007/s10928-011-9229-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Weiss EC, Wehner F, Lemor RM. 2007 [Google Scholar]
- 188.Gray RH, AdamRodwell G, Maris D, Haskins JR, Stoner GD. Toxicol Pathol. 1995;23:644. doi: 10.1177/019262339502300602. [DOI] [PubMed] [Google Scholar]
- 189.Crapo JD, Barry BE, Gehr P, Bachofen M, Weibel ER. Am Rev Respir Dis. 1982;126:332. doi: 10.1164/arrd.1982.126.2.332. [DOI] [PubMed] [Google Scholar]
- 190.Simmons NL. Q J Exp Physiol Cms. 1984;69:83. doi: 10.1113/expphysiol.1984.sp002798. [DOI] [PubMed] [Google Scholar]
- 191.Calvillo L, Latini R, Kajstura J, Leri A, Anversa P, Ghezzi P, Salio M, Cerami A, Brines M. Proc Natl Acad Sci U S A. 2003;100:4802. doi: 10.1073/pnas.0630444100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Leonhardt W, Hanefeld M, Haller H. Int J Obes. 1978;2:33. [PubMed] [Google Scholar]
- 193.Bennett M. Virginia Woolf and Neuropsychiatry. Springer; Netherlands, Dordrecht: 2013. p. 107. [Google Scholar]
- 194.Lang F, Ritter M, Woll E, Weiss H, Haussinger D, Hoflacher J, Maly K, Grunicke H. Pflugers Arch. 1992;420:424. doi: 10.1007/BF00374615. [DOI] [PubMed] [Google Scholar]
- 195.Folkesson HG, Matthay MA, Hasegawa H, Kheradmand F, Verkman AS. P Natl Acad Sci USA. 1994;91:4970. doi: 10.1073/pnas.91.11.4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Barker G, Simmons NL. Q J Exp Physiol. 1981;66:61. doi: 10.1113/expphysiol.1981.sp002529. [DOI] [PubMed] [Google Scholar]
- 197.Baranov M, Ter Beest M, Reinieren-Beeren I, Cambi A, Figdor CG, van den Bogaart G. J Cell Sci. 2014;127:1052. doi: 10.1242/jcs.141226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Baxter LT, Zhu H, Mackensen DG, Butler WF, Jain RK. Cancer Res. 1995;55:4611. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.