

Abstract
Neuronal gap junctional protein connexin 36 (Cx36) contributes to neuronal death following a range of acute brain insults such as ischemia, traumatic brain injury and epilepsy. Whether Cx36 contributes to neuronal death and pathological outcomes in chronic neurodegenerative diseases, such as amyotrophic lateral sclerosis (ALS), is not known. We show here that the expression of Cx36 is significantly decreased in lumbar segments of the spinal cord of both human ALS subjects and SOD1G93A mice as compared to healthy human and wild-type mouse controls, respectively. In purified neuronal cultures prepared from the spinal cord of wild-type mice, knockdown of Cx36 reduces neuronal death caused by overexpression of the mutant human SOD1-G93A protein. Taken together, these data suggest a possible contribution of Cx36 to ALS pathogenesis. A perspective for the use of blockers of Cx36 gap junction channels for ALS therapy is discussed.
Keywords: Gap junctions, connexin 36, amyotrophic lateral sclerosis, spinal cord, neurodegenerative diseases
Introduction
In the mammalian central nervous system, direct intercellular communication between neighboring neurons occurs through electrical synapses (gap junctions; GJ). Using animal models of acute neuronal injury such as brain ischemia, traumatic brain injury, epilepsy and glutamate-mediated excitotoxicity, we have shown previously that genetic and/or pharmacological blockade of neuronal, connexin 36 (Cx36)-containing GJs is unambiguously neuroprotective [1–4]. In the ischemic mouse cortex in vivo and in vitro, we also demonstrated a transient increase (at 2–3 hrs post-ischemia) with the following profound decrease (at 24 hrs post-ischemia) in the expression of Cx36 [3]. The delayed Cx36 downregulation overlapped with massive post-ischemic death of neurons. Altogether, our data suggested a critical role for Cx36 in neuronal death following acute brain injury. A modified model of glutamate-mediated excitotoxicity, where neuronal GJs are intimately involved, has been proposed [5].
Amyotrophic lateral sclerosis (ALS), is a progressive neurodegenerative disease characterized by preferential degeneration of upper and lower motor neurons, gradual decline of muscle strength and death within 3–5 years after the first symptoms [6]. Mutations of the superoxide dismutase 1 (SOD1) gene are frequently associated with the familial form of ALS [7]. Multiple mechanisms for the contribution of SOD1 variants in ALS-related motor neuron degeneration have been suggested [8,9]. However, whether as in case of the acute brain injury, Cx36 plays a role in neuronal death during ALS and other chronic neurodegenerative diseases, is not known. In the present study, using purified neuronal cultures prepared from the wild-type (WT) mouse spinal cord, we determined whether genetic knockdown of Cx36 reduces neuronal death caused by overexpression of the human SOD1-G93A mutant protein. Using spinal cord samples from ALS human subjects and SOD1G93A mice (a commonly used animal model of familial ALS), we also tested the hypothesis that the expression of Cx36 is decreased at the late stages of ALS, i.e., at the time when neuronal degeneration is observed [6,10–13].
Materials and methods
Postmortem human samples of the lumbar spinal cord were obtained from the NIH NeuroBioBank (see Acknowledgements). SOD1G93A mice (on the C57BL/6 background) were originally obtained from the Jackson Laboratory (stock # 004435). The use of animal subjects in these experiments was approved by the University of Kansas Medical Center Animal Care and Use Committee. The experiments were carried out in accordance with the National Institute of Health Guide for the Care and Use of Laboratory. All studies were conducted blindly.
Western blot experiments were performed using the approaches and antibodies as described in detail [14]. In addition, rabbit anti-Cx45 primary antibody was obtained from Invitrogen (Carlsbad, CA, USA; cat.# 407000). Band optical density was determined using a direct detection with Quantity One software (Bio-Rad, USA). Optical density signals were normalized relative to tubulin and normalized values were compared to controls (set at 1.0). Tubulin levels per unit of total protein did not vary significantly among samples. The following postmortem samples of the lumbar spinal cord were utilized from healthy human subjects and those diagnosed with ALS (gender/years of age): controls - M/60, M/74, F/54; ALS - M/60, M/78, F/54. In addition, three WT mice (C57BL/6) and three SOD1G93A mice were used (gender/days of age): WT - M/172, M/148, M/148; SOD1G93A - M/139, M/150, M/150. The ALS mice were sacrificed when they were unable to right themselves within 30 s when placed on their sides and, thus, were at the end stage of disease. The mice were genotyped using qPCR based protocol to confirm the transgene copy number.
To knockdown Cx36, we used a lentivirus expressing Cx36-targeted small hairpin RNA (shRNA-Cx36) that we designed previously (shRNA1-Cx36 from [4]). A lentivirus expressing shRNA directed against Cypridina luciferase (shRNA-LUC; [4]) was used as a control. A multiplicity of transduction of 20 for both lentiviral vectors was employed. Suppression of Cx36 protein by shRNA-Cx36 was confirmed using western blots (see Results).
An expression plasmid for (Myc-DDK-tagged)-human soluble SOD1 was purchased from Origene, Inc. (Rockville, MD, USA; cat.# RC200725; Accession number NM_000454.4). The codon for glycine at position 93 was mutated to a codon for alanine using site-directed mutagenesis (QuikChange II kit; Agilient, Inc., Santa Clara, CA, USA). The SOD1 cDNA in both the purchased plasmid and the G93A mutation were fully sequenced to confirm identity. An empty vector (VECT) was used as a transfection control. WT mouse spinal cord cultures were transfected with Lipofectamine 2000 (Life Technologies, Inc., Carlsbad, CA) and plasmid DNA (2 μg plasmid, 2 μl Lipofectamine 2000 combined in a total volume of 200 μl media, per well in 24-well plates containing primary mouse neurons cultured as outlined below). To confirm the expression of the SOD1 and SOD1-G93A mutant we employed western blots and an anti-FLAG antibody (see Results).
Purified neuronal cultures containing ~95% neurons were prepared (as we described [14]) from the spinal cord of embryonic day 18–19 WT mice. On day in vitro 3 (DIV3), the cultures were singly transduced with shRNA-LUC or shRNA-Cx36 (as we described [4]). On DIV4, the cultures also were singly transfected with the control plasmid or plasmids inducing human SOD1 or SOD1-G93A mutant. On DIV10, methyl thiazolyl tetrazolium (MTT) assay was conducted. The MTT tests were designed to specifically analyze the death of neurons as we described previously [3].
Data were analyzed using the two-tailed unpaired Student’s t-test or ANOVA with post hoc Tukey and InStat software (GraphPad Software, San Diego, CA, USA). Data are reported as mean ± SE for the number of samples indicated.
Results
We tested whether the expression of connexins, and particularly Cx36, was altered during the late stages of ALS. Western blot experiments were conducted in postmortem samples of the lumbar spinal cord obtained from the healthy human subjects and diagnosed ALS patients (see Materials and methods). We observed a significant downregulation of the expression of neuronal Cx36 protein in the ALS spinal cord (Fig. 1A). In contrast, the expression of non-neuronal Cx43, that is found in astrocytes [15] and activated microglia [16], was increased during ALS (Fig. 1B). Cx45, that also may be expressed by neurons [17], was detected in neither control nor ALS conditions (Fig. 1C). Essentially identical results were obtained in the spinal cord samples utilized from WT and SOD1G93A mice (Fig. 1D–F). Together, these findings indicate that neuronal Cx36 expression is decreased in the spinal cord at the late stages of ALS, while astroglial Cx43 is upregulated.
Fig. 1.
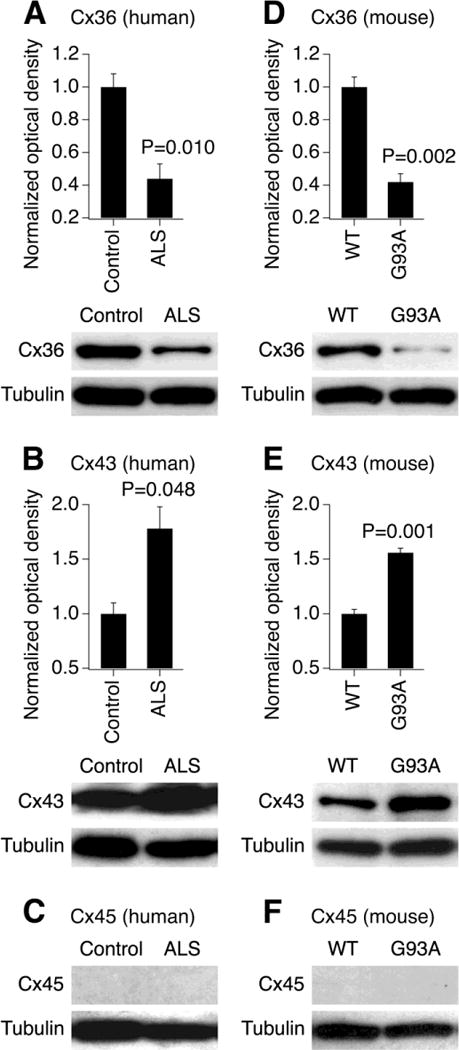
Expression of neuronal Cx36 decreases and glial Cx43 increases in ALS conditions. Representative blots and statistical data from western blot experiments in the lumbar spinal cord samples obtained from human subjects (A–C) and mice (D–F) are shown. The analysis was done for the expression of Cx36 (A, D), Cx43 (B, E) and Cx45 (C, F). The expression of Cx45 was not detected. In all graphs, optical density signals are normalized relative to tubulin and compared to the control (healthy human subjects or WT mice). Statistical analysis: two-tailed unpaired Student’s t-test relative to the control; n = 3 per group in all groups; data are shown as mean ± SE. G93A, SOD1G93A mice.
Previous studies have shown that transfection of cultured rat spinal cord motor neurons with a plasmid inducing SOD1-G93A mutant protein triggers mitochondrial fragmentation and neuronal death [9]. However, this does not occur in cultures transfected with a plasmid inducing WT SOD1 [9]. Since we have compelling evidence that Cx36 GJ coupling between cortical neurons contributes to neuronal death following acute injury [4], we wanted to determine if this held true for neuronal death associated with SOD1-G93A expression. Purified neuronal cultures were prepared from WT mouse embryonic spinal cord. On DIV3, the cultures were singly transduced with shRNA-LUC or shRNA-Cx36 lentiviral vectors. On DIV4, the cultures also were singly transfected with the empty vector (VECT) or plasmids expressing human SOD1 or the SOD1-G93A mutant. On DIV10, western blots were performed to confirm the suppression of Cx36 by shRNA-Cx36 (Fig. 2B) and the induction of WT or mutant SOD1 by the plasmids (Fig. 2C). In addition, MTT assay was performed to analyze neuronal death (Fig. 2A). As expected, the cultures transfected with the empty vector did not show anti-FLAG reactivity (Fig. 2C). The expression of FLAG was slightly lower in cultures transfected with the G93A mutant as compared to the WT SOD1 (Fig. 2C). However, in control conditions (shRNA-LUC transduction), the former induced neuronal death while the latter did not (Fig. 2A). In conditions of Cx36 knockdown (shRNA-Cx36 transduction; Fig. 2B), the G93A-mediated neuronal death did not occur (Fig. 2A). This suggests a protective role of Cx36 blockade in ALS-related death of neurons.
Fig. 2.
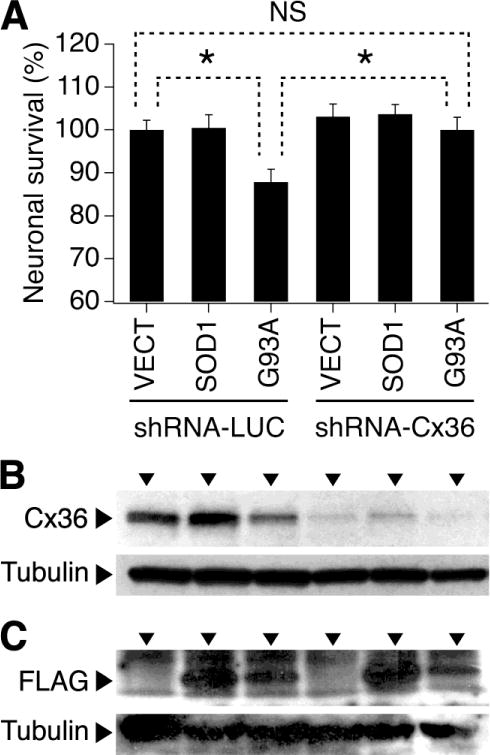
Knockdown of Cx36 prevents neuronal death caused by overexpression of the SOD1-G93A mutant protein. Statistical data from MTT assay (A) and representative images from western blot experiments (B, C) in WT mouse purified neuronal spinal cord cultures are shown. The cultures were transduced with shRNA-LUC or shRNA-Cx36 and also were transfected with the empty vector (VECT) or plasmids inducing human SOD1 or G93A mutant. shRNA-Cx36 effectively suppressed Cx36 protein expression (B). Cultures transfected with the SOD1 and G93A mutant demonstrated the FLAG reactivity (C). Statistical analysis in A: ANOVA with post hoc Tukey; n = 8 per group in all groups; *P<0.05; NS, non-significant.
Discussion
Protective effects of GJ blockade
Our previous work demonstrated a transient increase in the expression of Cx36 shortly (at 2–3 hrs) after cortical ischemia in vivo and in vitro and also in cell culture models of traumatic brain injury and epilepsy [3]. This increase was induced by activation of group II metabotropic glutamate receptors. In all our studies employing acute brain injury models or glutamate-mediated excitotoxicity in vivo and in vitro, genetic or pharmacological blockade of Cx36 GJs or inhibition of the mechanisms that control the post-injury upregulation of Cx36 had significant neuroprotective effects [1–4,18]. These studies implicated a critical role for Cx36 in neuronal death following acute brain injury. Based on this work, we proposed a modified model of glutamate-mediated excitotoxicity, where GJs play the central role in regulation of the extent of the secondary neuronal death following injury [5]. We also suggested that, independently of the nature of the primary neuronal death, the amount of the secondary neuronal death will be significantly extended as a result of the contribution of neuronal GJs.
To our knowledge, a single report has been published [19] that addressed the role of GJs/hemichannels in ALS. The study showed that a non-selective GJ hemichannel blocker, INI-0602, significantly suppressed neuronal loss in the spinal cord and extended survival in two transgenic mouse models of ALS, SOD1G93A and SOD1G37R [19]. In the present study with the use of purified neuronal spinal cord cultures, we demonstrate that knockdown of neuronal Cx36 reduces death of neurons caused by overexpression of the SOD1-G93A mutant protein. As in case of acute ischemic and excitotoxic neuronal death [4], the contribution of Cx36 may potentially be via inter-neuronal GJs and, perhaps, propagation of some “death signals” from neurons dying from overexpression of the G93A mutant to other, unaffected or less-affected neurons. However, the possibility remains for the contribution of Cx36 via hemichannels or channel-independent mechanisms, as was shown previously for non-neuronal connexins with the use of various models of acute cell death (reviewed in [20]). Future studies will address this issue.
Downregulation of Cx36 at late stages of neuronal injury
Previous study with the use of in vivo and in vitro models of acute cortical ischemia [3] also showed the delayed decrease in Cx36 expression. Cx36 was reduced below the background levels, this was observed 24 hrs post-ischemia and occurred at the time when massive ischemic and excitotoxic neuronal death occurs [1,3]. The delayed downregulation of Cx36 presumably reflected the primary (i.e., ischemia-induced) and secondary (i.e., glutamate receptor-dependent) death of neurons expressing Cx36. Degeneration of spinal motor neurons also occurs in both human subjects and mouse models at late stages of ALS [6,10–13]. In the present study, we observed the reduced expression of Cx36 in the spinal cord at the late stages of ALS in humans and mice. We speculate that this finding reflects a decrease in the number of Cx36-expressing neurons as part of the overall neuronal loss at the terminal stages of ALS. Some of this neuronal loss presumably is also mediated by Cx36 GJs (see the previous section). Future experiments will determine the proportion of neurons expressing and not expressing Cx36 that die during ALS.
We also observed the upregulation of glial Cx43. This apparently reflects “reactive gliosis” that occurs during ALS [10,12,13]. We do not have an information on whether or not the postmortem human samples where collected from the ALS patients who have had the SOD1-G93A mutation. However, because the human and mouse results were similar, the possibility exists that the decrease in Cx36 and increase in Cx43 expression may be general phenomena at the terminal stages of ALS, regardless the ALS etiology.
Conclusions
Altogether, our data suggest a possibility for contribution of Cx36 to ALS progression and pathogenesis. Currently, a non-selective blocker of GJs, carbenoxolone, is in preparation for clinical trials for neuroprotection in Huntington’s disease. The anti-malarial agent mefloquine (Lariam®), which is a potent blocker of Cx36 GJs [1,21], provides substantial neuroprotection during acute brain injury [1,2], but also has been patented for neuroprotection in a neurodegenerative disease. This suggests a possibility for the use of neuronal GJ blockade, and the blockade of Cx36 GJs particularly, for ALS therapy.
Highlights.
A role for neuronal connexin 36 in the pathogenesis of ALS is proposed
The mechanism relies on contribution of connexin 36 to ALS-related neuronal death
A perspective for the use of connexin 36 blockade for ALS therapy is discussed
Acknowledgments
This research was supported by a Lied basic science award from the University of Kansas Medical Center Research Institute to A.B.B and NIH 1R01NS078214 to H.N. The study also was supported in part by P30 AG035982 and UL1 TR000001. Core support was provided by NIH HD002528. Human spinal cord samples were obtained from the NIH NeuroBioBank’s Brain and Tissue repository at the University of Mariland-Baltimore and the University of Miami.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributors
Conceived and designed the experiments: A.B.B. and J.D.F. Provided materials: H.N. and J.D.F. Performed research: A.B.B., J.V.D. and J.D.F. Analyzed data: A.B.B., J.V.D. and J.D.F. Wrote the paper: A.B.B., H.N. and J.D.F. All authors have approved the final article.
References
- 1.Wang Y, Denisova JV, Kang KS, Fontes JD, Zhu BT, Belousov AB. Neuronal gap junctions are required for NMDA receptor-mediated excitotoxicity: implications in ischemic stroke. J Neurophysiol. 2010;104:3551–3556. doi: 10.1152/jn.00656.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belousov AB, Wang Y, Song JH, Denisova JV, Berman NE, Fontes JD. Neuronal gap junctions play a role in the secondary neuronal death following controlled cortical impact. Neurosci Lett. 2012;524:16–19. doi: 10.1016/j.neulet.2012.06.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Y, Song JH, Denisova JV, Park WM, Fontes JD, Belousov AB. Neuronal gap junction coupling is regulated by glutamate and plays critical role in cell death during neuronal injury. J Neurosci. 2012;32:713–725. doi: 10.1523/JNEUROSCI.3872-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fontes JD, Ramsey J, Polk JM, Koop A, Denisova JV, Belousov AB. Death of Neurons following Injury Requires Conductive Neuronal Gap Junction Channels but Not a Specific Connexin. PLoS One. 2015;10:e0125395. doi: 10.1371/journal.pone.0125395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Belousov AB, Fontes JD. Neuronal gap junctions: making and breaking connections during development and injury. Trends Neurosci. 2013;36:227–236. doi: 10.1016/j.tins.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brooks BR, Sanjak M, Belden D, Juhasz-Poscine K, Waclawik A. Natural history of amyotrophic lateral sclerosis - impairment, disability, handicap. In: Brown RHJ, Meininger V, Swash M, editors. Amyotrophic lateral sclerosis. Dunitz; London: 2000. pp. 31–58. [Google Scholar]
- 7.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 8.Liu R, Narla RK, Kurinov I, Li B, Uckun FM. Increased hydroxyl radical production and apoptosis in PC12 neuron cells expressing the gain-of-function mutant G93A SOD1 gene. Radiat Res. 1999;151:133–141. [PubMed] [Google Scholar]
- 9.Song W, Song Y, Kincaid B, Bossy B, Bossy-Wetzel E. Mutant SOD1G93A triggers mitochondrial fragmentation in spinal cord motor neurons: neuroprotection by SIRT3 and PGC-1α. Neurobiol Dis. 2013;51:72–81. doi: 10.1016/j.nbd.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoo YE, Ko CP. Treatment with trichostatin A initiated after disease onset delays disease progression and increases survival in a mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2011;231:147–159. doi: 10.1016/j.expneurol.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 11.Ralph GS, Radcliffe PA, Day DM, Carthy JM, Leroux MA, Lee DC, Wong LF, Bilsland LG, Greensmith L, Kingsman SM, Mitrophanous KA, Mazarakis ND, Azzouz M. Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat Med. 2005;11:429–433. doi: 10.1038/nm1205. [DOI] [PubMed] [Google Scholar]
- 12.Jouroukhin Y, Ostritsky R, Assaf Y, Pelled G, Giladi E, Gozes I. NAP (davunetide) modifies disease progression in a mouse model of severe neurodegeneration: protection against impairments in axonal transport. Neurobiol Dis. 2013;56:79–94. doi: 10.1016/j.nbd.2013.04.012. [DOI] [PubMed] [Google Scholar]
- 13.Parone PA, Da Cruz S, Han JS, McAlonis-Downes M, Vetto AP, Lee SK, Tseng E, Cleveland DW. Enhancing mitochondrial calcium buffering capacity reduces aggregation of misfolded SOD1 and motor neuron cell death without extending survival in mouse models of inherited amyotrophic lateral sclerosis. J Neurosci. 2013;33:4657–4671. doi: 10.1523/JNEUROSCI.1119-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park WM, Wang Y, Park S, Denisova JV, Fontes JD, Belousov AB. Interplay of chemical neurotransmitters regulates developmental increase in electrical synapses. J Neurosci. 2011;31:5909–5920. doi: 10.1523/JNEUROSCI.6787-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rash JE, Staines WA, Yasumura T, Patel D, Furman CS, Stelmack GL, Nagy JI. Immunogold evidence that neuronal gap junctions in adult rat brain and spinal cord contain connexin-36 but not connexin-32 or connexin-43. Proc Natl Acad Sci U S A. 2000;97:7573–7578. doi: 10.1073/pnas.97.13.7573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eugenin EA, Eckardt D, Theis M, Willecke K, Bennett MV, Saez JC. Microglia at brain stab wounds express connexin 43 and in vitro form functional gap junctions after treatment with interferon-γ and tumor necrosis factor-α. Proc Natl Acad Sci U S A. 2001;98:4190–4195. doi: 10.1073/pnas.051634298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li X, Kamasawa N, Ciolofan C, Olson CO, Lu S, Davidson KG, Yasumura T, Shigemoto R, Rash JE, Nagy JI. Connexin45-containing neuronal gap junctions in rodent retina also contain connexin36 in both apposing hemiplaques, forming bihomotypic gap junctions, with scaffolding contributed by zonula occludens-1. J Neurosci. 2008;28:9769–9789. doi: 10.1523/JNEUROSCI.2137-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Rivero Vaccari JC, Corriveau RA, Belousov AB. Gap junctions are required for NMDA receptor-dependent cell death in developing neurons. J Neurophysiol. 2007;98:2878–2886. doi: 10.1152/jn.00362.2007. [DOI] [PubMed] [Google Scholar]
- 19.Takeuchi H, Mizoguchi H, Doi Y, Jin S, Noda M, Liang J, Li H, Zhou Y, Mori R, Yasuoka S, Li E, Parajuli B, Kawanokuchi J, Sonobe Y, Sato J, Yamanaka K, Sobue G, Mizuno T, Suzumura A. Blockade of gap junction hemichannel suppresses disease progression in mouse models of amyotrophic lateral sclerosis and Alzheimer’s disease. PLoS One. 2011;6:e21108. doi: 10.1371/journal.pone.0021108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Decrock E, Vinken M, De Vuyst E, Krysko DV, D’Herde K, Vanhaecke T, Vandenabeele P, Rogiers V, Leybaert L. Connexin-related signaling in cell death: to live or let die? Cell Death Differ. 2009;16:524–536. doi: 10.1038/cdd.2008.196. [DOI] [PubMed] [Google Scholar]
- 21.Cruikshank SJ, Hopperstad M, Younger M, Connors BW, Spray DC, Srinivas M. Potent block of Cx36 and Cx50 gap junction channels by mefloquine. Proc Natl Acad Sci U S A. 2004;101:12364–12369. doi: 10.1073/pnas.0402044101. [DOI] [PMC free article] [PubMed] [Google Scholar]