

Abstract
Although the human neurotropic polyomavirus, JC virus (JCV), was isolated almost a half century ago, understanding the molecular mechanisms governing its biology remains highly elusive. JCV infects oligodendrocytes and astrocytes in the central nervous system (CNS) and causes a fatal brain disease known as progressive multifocal leukoencephalopathy (PML) in immunocompromised individuals including AIDS. It has a small circular DNA genome (~ 5 kb) and generates two primary transcripts from its early and late coding regions, producing several predicted alternatively spliced products mainly by cis-splicing. Here, we report the discovery and characterization of two novel open reading frames (ORF1 and ORF2) associated with JCV late transcripts, generated by an unusual splicing process called trans-splicing. These ORFs result from (i) the trans-splicing of two different lengths of the 5′-short coding region of VP1 between the coding regions of agnoprotein and VP2 after replacing the intron located between these two coding regions, and (ii) frame-shifts occurring within the VP2 coding sequences terminated by a stop codon. ORF1 and ORF2 are capable of encoding 58 and 72 aa long proteins respectively and are expressed in infected cells and PML patients. Each ORF protein shares a common coding region with VP1 and has a unique coding sequence of their own. When the expression of the unique coding regions of ORFs is blocked by a stop codon insertion in the viral background, the mutant virus replicates less efficiently when compared to wild-type, suggesting that the newly discovered ORFs play critical roles in the JCV life cycle.
Keywords: Progressive multifocal leukoencephalopathy, ORF, VP1, VP2, cis- and trans-splicing, polyomavirus, JCV, SV40, BKV, merkel cell carcinoma virus, DNA replication, transcription, RNA splicing
INTRODUCTION
RNA splicing is a highly regulated and complex nuclear processing event in eukaryotic gene expression where various combinations of exons from pre-mRNA molecules are joined together to generate multiple mRNAs encoding different protein isoforms. In a majority of splicing events, this process takes place within a single RNA precursor molecule called cis-splicing (Adami and Babiss, 1991; Gattoni et al., 1991; van Santen and Spritz, 1986). There are numerous reports demonstrating that exon joining can also take place between two independent pre-mRNA species. This mechanism was first observed in Trypanosome brucei (Murphy et al., 1986; Sutton and Boothroyd, 1986) and later in the nematodes (C. elegans) (Krause and Hirsh, 1987) and was designated as trans-splicing. In both organisms, a species-specific non-coding spliced leader RNA (SL RNA) with a singular 5′ splice donor site was detected to be spliced to various 3′ splice acceptor sites on separate pre-mRNA molecules. Soon after, trans-splicing of SL RNA was also detected in Schistosoma-Mansoni (Rajkovic et al., 1990), Euglena (Tessier et al., 1991) and tunicates (Vandenberghe et al., 2001). Later, another type of trans-splicing event, which mimics cis-splicing but differs from SL RNA splicing, was also found in mammalian cells during the mid-90s (Eul et al., 1995; Eul et al., 1996).
The characteristics of a transcript that lead to trans-splicing are unknown but it usually occurs when cellular and viral pre-mRNAs contain cryptic splice sites which are not accessible to regular cis-splicing (Eul et al., 1995). A prime example of this phenomenon was observed in the formation of a super-transforming phenotype of SV40 large T antigen, where it was found that the 5′ splice donor site of the SV40 large T antigen carries a single point mutation which apparently inhibits a conventional cis-splicing event but favors an alternative trans-splicing reaction via a cryptic 5′ slice donor site (Eul and Patzel, 2013). This splicing event created a 47 amino acid duplication in the retinoblastoma protein (pRb) binding domain of SV40 large T antigen leading to a ~100 kDa rather than a 94 kDa protein. Another evidence for trans-splicing between two different RNAs was provided by Caudevilla et al. who demonstrated that HIV-1 Nef pre-mRNA was involved in a heterologous trans-splicing event with SV40 large T antigen and with several other cellular RNAs, including Alu, transposon gene families and PTPκ (Caudevilla et al., 2001; Green, 1986). The other example of naturally occurring trans-splicing between viral and host pre-mRNA was reported by Kikumori et al., who detected the trans-splicing of adenovirus RNA to several host pre-mRNAs, including β-actin and glyceraldehyde-3-phosphate dehydrogenase transcripts (Kikumori et al., 2002).
Viruses are obligatory intracellular parasites and heavily rely on the regulatory mechanisms provided by host cells for splicing and propagation. JC virus (JCV) is a neurotropic human polyomavirus that infects glial cells including oligodendrocytes and astrocytes in the central nervous system (CNS) and causes a fatal brain disease known as PML in immunocompromised individuals including AIDS and cancer patients (Berger, 1992; Berger, 2000; Berger, 2007). JCV has a small double-stranded circular DNA genome and its late coding regions generate a single polycistronic message that is known to undergo cis-splicing to produce transcripts that are capable of encoding a regulatory protein (agnoprotein) and the structural capsid proteins (VP1, VP2 and VP3) (Coric et al., 2014; Frisque et al., 1984; Saribas et al., 2013; Saribas et al., 2014; Saylor and Venkatesan, 2016; Shishido-Hara et al., 2000). These splicing events primarily produce two major splice products by removing the intron 1 which is located between agnoprotein and VP2 coding regions (a major splice product 1, M1) as well as that of intron 2 which is located between the agnoprotein and VP1 coding regions (another major splice product 2, M2) (Saribas et al., 2013; Shishido-Hara et al., 2000) followed by rejoining of the exons.
In this study, we report the discovery and characterization of two new open reading frames (ORF1 and ORF2) found in the JCV late transcripts, which were first observed during the mutational analysis of the JCV agnoprotein dimerization domain by RT-PCR. These novel ORFs result from two consecutive events: (i) an unusual splicing process known as trans-splicing, where fragment exchange occurs between two different JCV late transcripts followed by (ii) a frame-shift occurring after splicing within the VP2 coding region. More specifically, two different lengths of the short 5′-coding region of VP1 are inserted in place of the intron 1 located between the agnoprotein and VP2 coding regions followed by a frame shift that occurs within the VP2 coding region. These two events create ORF1 and OFR2, which are predicted to encode a 58 and a 72 aa long protein respectively. Both proteins share a common region with the N-terminus of VP1 and have unique carboxy terminal regions of their own. Initial characterization of the ORF transcripts by RT-PCR utilizing total RNA isolated from both the infected cells and the PML brain tissue samples has confirmed that both ORFs are transcribed in vitro and in vivo. Subsequent protein detection studies for the ORF1 protein using a new polyclonal antibody raised against the unique coding region of this protein has revealed that it is expressed both in the infected cells and PML patients. Furthermore, immunocytochemistry studies in the absence of a viral infection cycle not only showed that ORFs localize to both cytoplasmic and nuclear compartments of cells but also a significant level of ORF2 is localized to the nucleus. In contrast, examination of the PML brain samples by immunohistochemistry showed a more exclusive nuclear localization of ORF1, implicating that the viral infection cycle promotes the nuclear localization of this protein and suggests a nuclear role for it. A specific mutation made on ORFs where the expression of the unique regions of both ORF1 and ORF2 is blocked by a stop codon insertion resulted in phenotypes which replicate less efficiently than WT, suggesting critical roles for ORFs during the JCV life cycle.
MATERIALS AND METHODS
Cell lines
SVG-A is a human cell line established by transforming primary human fetal glial cells with an origin-defective SV40 mutant (49). These cells do not express the SV40 late proteins (VP1, VP2, VP3 and Agno) but they do express the early proteins including SV40 LT-Ag (Saribas et al., 2014; Sariyer et al., 2006). HEK293T is a human embryonic kidney epithelial cell line (ATCC no. CRL-326). U-87MG is a glioblastoma cell line (ATCC no. HTB-14). All cell lines were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) (Life Technologies, Waltham, MA, catalog no. 31600-034) supplemented with 10% heat inactivated fetal bovine serum (FBS) and antibiotics [penicillin-streptomycin (100 μg/ml), ciprofloxacin (10 μg/ml)]. Cells were maintained at 37°C in a humidified atmosphere and supplemented with 7 % CO2.
Plasmids
pBluescript KS (+)-JCV Mad-1 WT plasmid was previously described (Saribas et al., 2013). Single point mutations at position G1586 (Mad-1 numbering, GenBank: NC_001699) were made at the cryptic donor sites located within the VP1 coding region using the QuikChange™ site–directed mutagenesis kit (Agilent, Santa Clara, CA, catalog no. 200521) along with appropriate synthetic oligonucleotides. The resulting mutant viruses were designated as Bluescript KS (+)-JCV Mad-1 (G1586A) and Bluescript KS (+)-JCV Mad-1 (G1586C). In addition, a stop codon (TAA) was introduced in the JCV Mad-1 genome after position T524 (Mad-1 numbering) in order to block the expression of the unique C-terminus region of ORF1 and ORF2 by using the QuikChange™ site–directed mutagenesis kit with appropriate primers. The resulting plasmid was designated as Bluescript KS (+)-JCV Mad-1 (T524-Stop). The coding sequences of ORF1, ORF1M (Ile23Met) and ORF2 were subcloned into a pCGT7 vector (Saribas et al., 2017) at XbaI/BamHI sites, where ORFs were tagged with a T7-tag sequence (MASMTGGQQMG) at their N-termini. The resulting expression plasmids were designated as pCGT7-JCV ORF1, pCGT7-JCV ORF1M (Ile23Met) and pCGT7-JCV ORF2.
Transfection and transfection/infection
HEK293T, SVG-A and U-87MG cells were plated onto tissue culture plates (60 mm in diameter, Becton Dickinson, Franklin lakes, NJ, catalog no. 353002) (1 × 106 cells/plate) in triplicate the day before transfection. Cells were washed with growth medium (DMEM), re-fed with fresh medium and grown for 3 h at 37°C. Each culture plate was then transfected with the following expression plasmids separately: pCGT7-JCV ORF1, pCGT7-JCV ORF1M (Ile23Met) and pCGT7-JCV ORF2 (8 μg each) using lipofectamine™ 3000 transfection reagent according to the Manufacturer’s recommendations (ThermoFisher, catalog no. L3000001). At 48 h posttransfection, whole-cell extracts were prepared as described below. SVG-A cells (2 × 106 cells/T75 cm2 flask/8 μg DNA) were also separately transfected with the following plasmid constructs: Bluescript-JCV Mad-1 WT, Bluescript KS (+)-JCV Mad-1 (T524-Stop), Bluescript KS (+)-JCV Mad-1 (G1586C) and Bluescript KS (+)-JCV Mad-1 (G1586A) using lipofectamine™ 3000 transfection reagent according to the Manufacturer’s recommendations. Note that each plasmid was digested with BamHI to liberate the viral genome from the vector [Bluescript KS (+)] prior to transfection. At the indicated time points described in each figure legend, whole-cell extracts and viral DNA were prepared.
Raising a polyclonal antibody against the unique coding region of ORF1
A peptide corresponding to the C-terminal unique region (42-VHGCRTCTFGGPSCYCF-58) of ORF1 was synthesized commercially by Southern Biotech (www.southernbiotech.com). A rabbit polyclonal antibody was raised commercially against this peptide by Lampire Biological Laboratories (www.lampire.com), which was designated as anti-ORF1 (Ab#66) antibody and recognizes the C-terminal unique coding region of ORF1.
Preparation of whole cell extracts, Western blotting and immunoprecipitation/Western blotting
Whole-cell extracts were prepared from cells (SVG-A and HEK293T) transfected with pCGT7-JCV ORF1 or pCGT7-JCV ORF1M (Ile23Met) or pCGT7-JCV ORF2 plasmids as described previously (Coric et al., 2014; Sariyer et al., 2011). Briefly, cells (transfected/untransfected) were lysed in lysis buffer [50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA and 1.0 % NP-40 in the presence of protease inhibitors (Sigma, St. Louis, MO, catalog no. P8340)] on a rocking platform at 4°C for 30 min (2 × 106 cells/1000 μl lysis buffer). The cell lysate was then cleared by centrifugation at 17,000 × g for 10 min at 4°C and stored at − 80°C until use. For Western blot analysis, 40 μg of protein was resolved on a SDS-15% PAGE, transferred onto a nitrocellulose membrane with 0.2 μm pore size (Bio Rad, catalog no 162-0097) for 30 min at 250 mA. Membranes were first blocked with 5 % dry milk solution prepared in TBST (50 mM Tris-HCL, pH 7.4, 150 mM NaCl, 0.01 % Tween 20) buffer for 2h at room temperature. Membranes were then probed with the primary mouse monoclonal anti-T7 (Novagen, catalog no. 69522) and secondary goat anti-mouse IRDye 680LT (LI-COR, Lincoln, NE, catalog no. 926-68070) antibodies. Finally, membranes were washed twice with 1 × PBS and scanned using Odyssey® CLx Infrared Imaging System (LI-COR) to detect the protein of interest. For immunoprecipitation (IP) of ORF1, whole-cell extracts (500 μg protein/1000 μl volume/sample) were prepared from SVG-A cells, untransfected or transfected/infected with JCV Mad-1 WT genome at 15th day posttransfection/infection and were subjected to immunoprecipitation (IP) using anti-ORF1 (Ab#66) antibody (5 μl/sample) in lysis buffer and incubated for 16 h at 4°C on a rotating platform. Samples were also incubated for an additional 4 h with 20 μl protein G Sepharose 4 Fast Flow resin (GE Healthcare, Chicago, IL, catalog no. 17-0618-01) at 4°C on a rotating platform. Immune complexes were then washed three times in lysis buffer (1000 μl) by centrifugation and resolved on a SDS-15% PAGE, transferred onto a nitrocellulose membrane with 0.2 μm pore size (Bio Rad, Hercules, CA, catalog no 162-0097) for 30 min at 250 mA. Membranes were blocked with dry milk as described above and probed with primary rabbit polyclonal anti-ORF1 antibody (Ab#66) (1:1000 dilution) for overnight and washed three times with TBST buffer, followed by incubation with secondary goat anti-rabbit IRDye 800CW (LI-COR, catalog no. 925-32211) antibody (1:10.000 dilution) prepared in TBST for 45 min. Finally, membranes were scanned using LI-COR imaging system to detect the protein of interest. Whole-cell extracts (40 μg/lane) prepared from SVG-A cells, (untransfected or transfected/infected with appropriate JCV WT or its mutant genomes) were also analyzed for detection of JCV VP1 protein expression by Western blotting using mouse monoclonal anti-VP1 (pAB597) and secondary goat anti-mouse IRDye 680LT antibodies. Finally, membranes were scanned using LI-COR imaging system to detect VP1. Each blot was also probed with anti-GAPDH antibody (Abcam, catalog no. ab8245) for a loading control.
Indirect immunofluorescence microscopy
Indirect immunofluorescence microscopy studies were performed as previously described (Sadowska et al., 2003; Sariyer et al., 2011). Briefly, SVG-A cells (2 × 106 cells/T75 cm2 flask) were transfected separately with the following plasmids: pCGT7-JCV ORF1, pCGT7-JCV ORF1M (Ile23Met) and pCGT7-JCV ORF2 (8 μg each) using lipofectamine™ 3000 transfection reagent according to the manufacturer recommendations. At 24 h posttransfection, cells were trypsinized, reseeded at subconfluency on polylysine-coated glass chamber slides (Nunc, catalog no. 154461) and incubated for an additional 24h. The next day, the cells were washed twice with 1 × PBS, fixed in cold acetone and incubated with 5% bovine serum albumin in PBS for 2 h. Chamber slides were then incubated with anti-T7 mouse monoclonal antibody (1:200 dilution) (Novagen, catalog no. 69522) overnight. Cells were washed three times with TBST buffer for 10 min intervals and subsequently incubated either with a fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse secondary antibody (Novus, catalog no. NB720-F) for 45 min. Cells were then finally washed with TBST buffer three times for 10 min each, incubated with DAPI (ThermoFisher, 4′,6-Diamidino-2-Phenylindole, Dihydrochloride, catalog no. D1306) (300 ng/ml) to stain the nucleus, mounted using “ProLong® Gold Antifade” mounting medium (Life Technologies, catalog no. P36934) and dried overnight. Slides were then examined under a fluorescence microscope (Leica, DMI-6000B, objective: HCX PL APD 40×/1.25 oil, LAS AF operating software) for visualization of the protein of interest. SVG-A cells transfected with pCGT7-JCV ORF1 plasmid were also processed for ICC using anti-ORF1 polyclonal antibody (Ab#66) (1:200 dilution) as described under figure 7 legend.
Figure 7. Analysis of the ORF1 protein expression in PML brain tissue samples by immunohistochemistry (IHC).
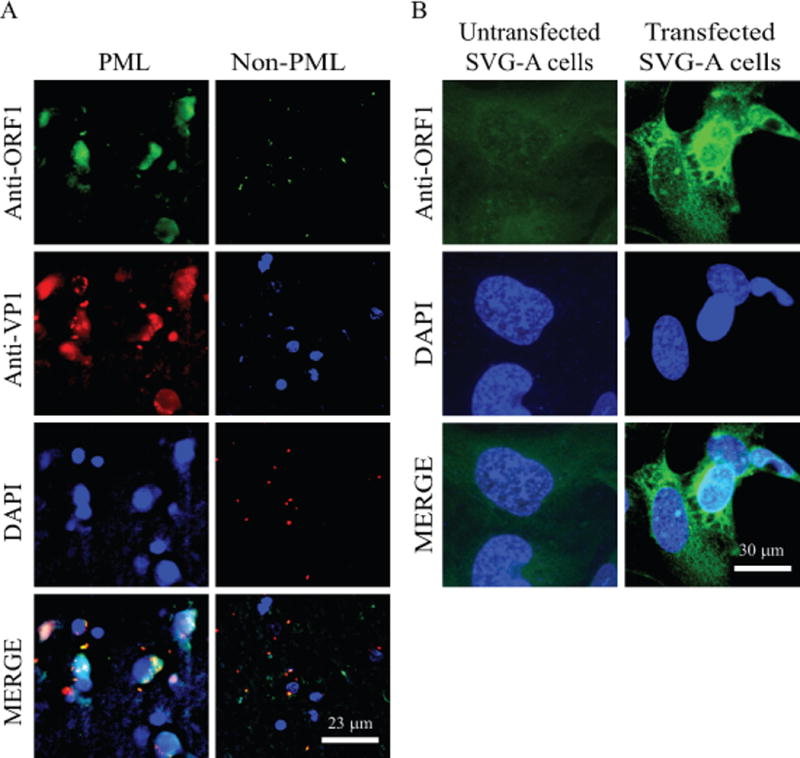
(A) Detection of the ORF1 expression in PML brain tissue sections by IHC. The formalin-fixed, paraffin-embedded and sectioned non-PML and PML brain tissue specimens were provided as “ready to stain” on slides by the NNTC. Samples were first probed with a combination of primary anti-ORF1 (Ab#66) rabbit polyclonal (1:200 dilution) and anti-VP1 (pAB597) monoclonal antibodies overnight. The following day, after washing the samples with PBS, they were incubated with Rhodamine-conjugated goat anti-mouse plus fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit secondary antibodies. Finally, slides were stained DAPI, mounted and examined under a fluorescence microscope as described under figure legend 5B. Scale bar: 23 μm. (B) Detection of ORF1 expression in SVG-A cells using Ab#66 antibody as a control. SVG-A cells were transfected with pCGT7-ORF1 expression plasmid on the glass-chamber slides and cells were then processed for ICC at 24 h posttransfection using primary anti-ORF1 (Ab#66) rabbit polyclonal (1:200 dilution) and secondary fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit antibodies to detect the stable expression of ORF1 protein; and examined under a fluorescence microscope as described under figure legend 5B. Scale bar: 30 μm.
Extraction of JCV DNA from PML brain tissue samples
Brain tissue samples [PML positive (PML+HIV-1 positive) and normal control (non-PML+HIV-1 positive] were obtained from the National NeuroAIDS Tissue Consortium (NNTC, www.nntc.org). Each tissue sample (50-75 mg) was homogenized in 3 ml Qiagen buffer P1 (Qiagen, Germantown, MD, catalog. 19051) using a glass-Teflon homogenizer. Subsequently, samples were lysed in 3 ml of Qiagen lysis buffer P2 (catalog no. 19052) by thoroughly mixing and incubating at room temperature for 10 min. Samples were then neutralized with 3 ml of Qiagen buffer P3 (Qiagen catalog no. 19053) and incubated on ice for 15 min. Cell debris was removed by centrifugation (15,000 rpm, Sorval RC6+ centrifuge, F21-8 × 50y rotor). Cleared lysate was subjected to isopropanol precipitations (sample/isopropanol, 1:1 ratio) by centrifugation (15,000 rpm, Sorval RC6+ centrifuge, F21-8 × 50y rotor) at + 4°C for 30 min. Pelleted samples were resuspended in 500 μl dH2O, extracted with phenol/chloroform and precipitated with ethanol in the presence of tRNA (10 μg/sample) by centrifugation (Eppendorf 5424, 15,000 rpm) at + 4°C for 30 min. Sample pellets were air-dried, resuspended in DNase and RNase free dH2O (50 μl) and stored at −80°C until they were used for PCR amplification.
RNA isolation from PML brain tissue samples and cell culture cells
PML positive and Non-PML brain tissue samples were obtained from the NNTC. Each sample (50-100 mg) was homogenized in 3 ml of Trizol reagent (ThermoFisher Scientific, catalog no. 15596018) using a glass-teflon homogenizer. Subsequently, total RNA was isolated employing the RNeasy lipid tissue mini kit (Qiagen, catalog no. 74804) following the protocols provided by the manufacturer. Total RNA was also isolated from untransfected (SVG-A alone) or transfected/infected (SVG-A cells transfected with JCV genome) (2.2 × 107 cells) cells at specific time points as indicated in the respective figure legends using the RNeasy® lipid tissue mini kit according to the Manufacturer’s recommendations.
RT-PCR and DNA sequencing
RT-PCR amplification was carried out using SuperScript™ III one step RT-PCR system (Invitrogen, catalog no. 12574026) using 250 ng total RNA and appropriate primers (see figure legends) according to the manufacturer’s recommendations. Total RNA was isolated from SVG-A cells transfected/infected separately with Bluescript-JCV Mad-1 WT, Bluescript KS (+)-JCV Mad-1 (G1586C), Bluescript KS (+)-JCV Mad-1 (G1586A) and Bluescript KS (+)-JCV Mad-1 (T524-Stop) plasmids and used in RT-PCR reactions. Each reaction was first incubated at 55°C for 30 min to synthesize cDNA and subsequently amplified by PCR using appropriate primers (see figure legends) as follows: Denaturation: 94°C for 2 min (1 cycle), amplification: 94°C for 15 sec., 55°C for 30 sec., 68°C for 1 min (40 cycles), extension cycle: 68°C for 5 min. (1 cycle), and maintenance: 4°C (1 cycle). Specific PCR reactions were carried out as described under each figure legend. Specific RT-PCR products corresponding to unexpected (ORF1), unspliced and spliced fragments were subcloned into pcDNA3.1 (+) at HindIII and BamHI sites and commercially sequenced (Genewiz,http://www.genewiz.com) as indicated as related figure legends. RT-PCR amplification was also performed to amplify GAPDH cDNA as a control for RT-PCR reactions using the following primers: GAPDH-Forward: 5′-TTCTCCCCATTCCGTCTTCC-3′, GAPDH-Reverse: 5′-GTACATGGTATTCACCACCC-3′. The PCR conditions for GAPDH amplification were the same as described for JCV transcripts above. Note that RNA samples were treated with RNAse-free DNAse I (New England Biolabs, catalog no. M0303S) before samples were used in RT-PCR reactions.
Replication assays
Replication assays were carried out as previously described (Sariyer et al., 2011). Briefly, the plasmid constructs [Bluescript KS (+)-JCV Mad-1 WT, Bluescript KS (+)-JCV Mad-1 (G1586C), Bluescript KS (+)-JCV Mad-1 (G1586A) and Bluescript KS (+)-JCV Mad-1 (T524-Stop) were digested with BamHI to liberate the viral genome from the vector, Bluescript KS (+). SVG-A cells (2 × 106 cells/T75 cm2 flask) were then separately transfected/infected with each plasmid viral DNA (8 μg DNA) using lipofectamine™ 3000 transfection reagent. After overnight incubation, cells were transferred into T175cm2 flasks and fed with DMEM (35 ml) supplemented with 10% FBS and 1% ampicillin/streptomycin. The half-volume of the media was replenished every three days posttransfection. At the indicated time points, low-molecular-weight DNA containing both input and replicated viral DNA was isolated using the alkaline lysis method where samples were lysed in Qiagen P2 buffer (catalog no. 19052) followed by isopropanol precipitation. Samples were then extracted with phenol/chloroform extraction method, precipitated with ethanol and resuspended in 100 μl of dH2O. One fourth of each DNA sample was finally digested with BamHI and DpnI enzymes, resolved on a 1% agarose gel and analyzed by Southern blotting as described previously (Saribas et al., 2011).
PCR amplification of JCV DNA from PML and non-PML brain samples
Two microliters of the viral DNA isolated from each PML and Non-PML brain tissue sample were subjected to PCR amplification using Fail Safe™ DNA polymerase mix (Epicentre, Madison, WI, PCR-pre mix F, catalog no. SF99060) according to the Manufacturer’s recommendations. The PCR primers used in each of the PCR reactions are specified in the respective figure legends. PCR conditions were as follows: Denaturation: 94°C, 5 min. 1 cycle; amplification: 94°C, 1 min; 60°C, 1 min, 72°C, 2 min, 42 cycles; extension: 72°C, 10 min, 1 cycle. Amplified gene products were analyzed by agarose gel electrophoresis.
Immunohistochemistry on PML brain tissue samples
Formalin-fixed, paraffin-embedded Non-PML and PML patient brain tissue sections were provided by the NNTC as “ready to stain” (5 μm thickness and mounted on salinized slides). Sections were then processed for immunohistochemistry as previously described (Almqvist et al., 2002; Imam et al., 1995). Briefly, sections were deparaffinized by first incubating them in an oven at 60°C for 40 min and treating them with Xylene for 30 min three times. Subsequently, sections were passed through various concentrations of ethanol solutions for hydration (100% for 3 min, two times; 90% for 3 min, two times; 70% for 3 min, two times; dH2O for 3 min, two times). They were then treated with citrate buffer, (0.1 M, pH 6.0) for 30 min and chilled at 4°C for 20 min and washed with PBS three times for 5 min. Samples were blocked with 5% serum prepared in PBS for 2 h and incubated with primary antibodies overnight [a combination of anti-ORF1 (Ab#66) rabbit polyclonal (1:200 dilution) and anti-VP1 (pAB597, a gift from Dr. Walter Atwood, Brown University) monoclonal antibodies]. Next day, samples were rinsed with PBS three times for 10 min intervals and incubated with secondary Rhodamine-conjugated goat anti-mouse (Novus, catalog no. NB120-6786) plus fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit secondary (Novus, Littleton, CO, catalog no. NB7182) antibodies and subsequently washed with PBS. Samples were then incubated with DAPI to stain nuclei for 10 min and washed with PBS three times. Coverslips were finally mounted on the slides using “ProLong® Gold Antifade” mounting medium and were examined under the fluorescence Leica (DMI-6000B) microscope (objective: HCX PL APD 40×/1.25 oil, LAS AF operating software).
RESULTS
Discovery of a novel open reading frame (ORF) associated with JCV late transcripts
Cis-alternative splicing of pre-mRNA is a mechanism by which various open reading frames (ORFs) are generated from the same transcript. Each splice product is then translated into the various isoforms of a protein. Viruses, including JCV, utilize this elegant process to encode proteins that are required to complete their life cycle. This is a highly regulated process in eukaryotic cells and is mediated by the specialized RNA-protein complexes called spliceosomes (Hastings and Krainer, 2001). The JCV late region generates two major transcripts, M1 and M2, after cis-splicing (Fig. 1A and 1B) and these transcripts together encode regulatory agnoprotein and structural capsid proteins (VP1, VP2 and VP3) (Bollag et al., 2006; Frisque et al., 1984).
Figure 1. RT-PCR analysis of JCV late transcripts revealed an unexpected splice product.
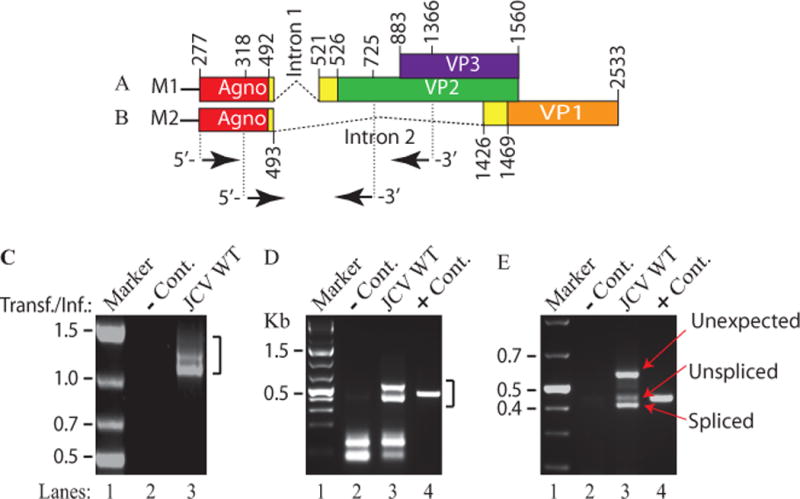
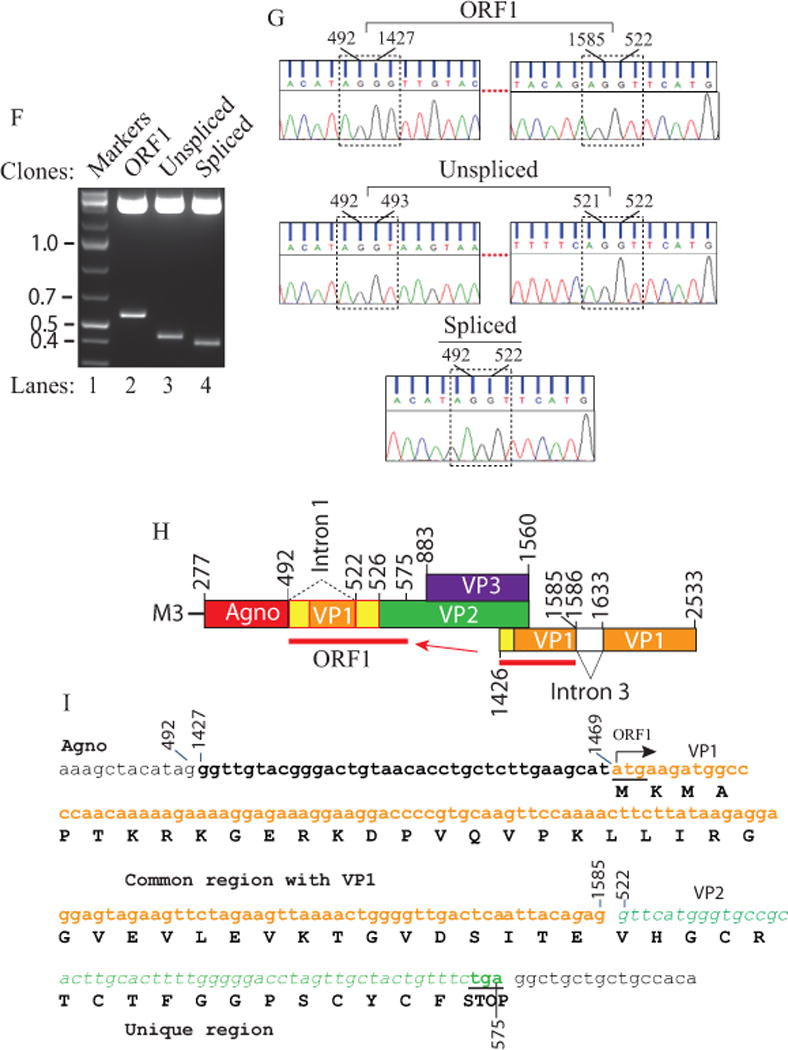
(A) Schematic representation of the JCV Mad-1 first major late transcript (M1) prior to splicing. The position of intron 1 between nucleotides 493 and 521 (JCV Mad-1 numbering) is indicated. (B) Schematic presentation of second major JCV Mad-1 late transcript (M2) prior to splicing. The position of intron 2 between nucleotides 493 and 1426 is indicated. (C) Agarose gel analysis of the RT-PCR products from the M1 transcript with specific primers (5′-JCV Mad-1 277-303 bp region and 3′-(JCV Mad-1, 1366-1346 bp). Total RNA was isolated from SVG-A cells transfected/infected with JCV Mad-1 WT at 15th day posttransfection/infection, subjected to RT-PCR reaction as described in Materials and Methods and analyzed on an agarose gel (1.5%). A bracket points the unresolved RT-PCR products. Transf./Inf.: Transfection/Infection. Total RNA from the untransfected SVG-A cells was also subjected to RT-PCR using the same primers as a negative control (− Cont.). (D) Re-amplification of the RT-PCR products from panel C by PCR using internal primers. The RT-PCR products from the panel C were re-amplified using the following internal primers: 5′-Primer: JCV Mad-1 (318-338) and 3′-primer: JCV Mad-1 (725-701) and resolved on an agarose gel (3 %). In lane 4, PCR-amplified JCV Mad-1 sequence was used as a positive control (+ Cont.) using the same internal primers. A bracket points to the unresolved bands. (E) Further resolution of the unresolved bands shown on panel D and subcloning them for sequencing. The resolved bands, labeled as “unexpected, unspliced and spliced” bands were gel-purified using QIAquick® gel extraction kit (Qiagen, catalog no. 28704), digested with HindIII and BamHI restriction enzymes and subcloned into the pcDNA3.1 (+) vector at HindIII and BamHI sites. Finally, the clones were sequenced commercially (Genewiz,http://www.genewiz.com). (F) Analysis of the cloned fragments by restriction enzyme digestion. The clones were digested with HindIII and BamHI enzymes and analyzed on a 3% agarose gel. (G) Partial representation of the DNA sequencing data of the cloned fragments at the splice junctions. The splice junctions were encased with dashed lines and the nucleotides were numbered according to the JCV Mad-1 strain numbering. (H) Schematic representation of a trans-spliced product (159 bp) of the 5′-end of the VP1 [Mad-1 (1426-1585) region] in place of “Intron 1” and creation of the ORF1 open reading frame. (I) Representation of the ORF1 sequence (159 bp) inserted in place of Intron 1. After insertion, a frame shift occurs within VP2, which is terminated with stop codon at nucleotide position 575. The common region of ORF1 (41 aa long) with VP1 is colored orange and the unique region is colored green (17 aa long).
We have previously reported that JCV agnoprotein forms highly stable dimer/oligomers through its major α-helix domain (Coric et al., 2014; Saribas et al., 2013; Saribas et al., 2011; Saribas et al., 2016). During the characterization of these higher order structures, we also analyzed the viral late transcripts related to the dimerization domain mutants of agnoprotein by RT-PCR (Saribas et al., 2013). These RT-PCR studies suggested that amplified products may contain additional novel bands (Fig. 1C, lane 3) but could not be resolved well on this gel due to their relatively longer length (> 1000 bp). To obtain more evidence for this possibility, we re-amplified the RT-PCR products using internal primers and resolved them on a high percentage agarose gel (3%) (Fig. 1D). Further resolution of the RT-PCR products by gel electrophoresis indeed revealed an additional band migrating slower than expected (unspliced and spliced) gene products of the M1 major transcript (Fig. 1A) of the JCV late gene (unexpected) (Fig. 1E,, lane 3). Each of these discrete bands was then subcloned into pcDNA3.1 (+) vector (Fig. 1F) and commercially sequenced (Fig. 1G and S1). This DNA sequencing data revealed that the “spliced product” underwent a predicted splicing process as we previously demonstrated with respect to the splicing of the intron 1 (Saribas et al., 2013). The “unspliced product”, as expected, resulted from an unspliced form of the intron 1. Surprisingly, we observed that an additional “unexpected band” resulted from a homologous trans-splicing event, where a short 5′-coding region VP1 gene originating from a cryptic splice site at nucleotide G1586 (Fig. 1H and 1G) was inserted in between agnoprotein and VP2 coding regions after replacement of the intron 1 as depicted in Fig. 1H, creating a novel ORF for JCV late transcripts. This trans-insertional fragment consists of a combination of the 5′-untranslated region of VP1 plus a short 5′-coding sequence of VP1 gene (encoding a 42 aa long peptide). After insertion, a frame shift occurs within the coding region of VP2, which is in-frame with VP1 insertional sequences, creating an additional 16 aa peptide and terminated with a stop codon within the VP2 coding region (Fig. 1I). In summary, the new insertional sequence, ORF1, has the capacity to encode a 58 amino acid long protein (Fig. 1I).
ORF1 is expressed in cells infected with JCV
We next investigated whether ORF1 is translated into a protein in cells infected with JCV. For this purpose, first, a rabbit polyclonal antibody (Ab#66) was raised against the predicted carboxy-terminus unique region of ORF1as indicated in Fig. 2A. Then, the expression of ORF1 in infected cells was first assessed by a direct Western blotting with no success due to the low levels of ORF1 expressed in infected cells. Its expression was then examined by immunoprecipitation (IP) followed by Western blotting (WB) as demonstrated in Fig. 2B. For this purpose, whole-cell extracts prepared from SVG-A cells either untransfected or transfected/infected with JCV Mad-1 genome were subjected to IP followed by WB analysis using anti-ORF1 antibody, as described in the legend for Fig. 2. IP/WB results demonstrated the detection of a distinct protein band, migrating at the level of expected size, which clearly suggests that the ORF1 message is translated into a protein during the JCV replication cycle. Note that we use the term ‘transfection/infection” to explain the case where the viral DNA is transfected into the cells and, upon transfection, the viral infection cycle starts. In addition to IP/WB assay, the specificity of the anti-ORF1 antibody was also tested when the ORF1 protein was expressed in cells in relatively high levels by transfection and detected by immunocytochemistry as demonstrated in Fig. 7B.
Figure 2. Detection of ORF1 expression in JCV infected cells by IP/Western.
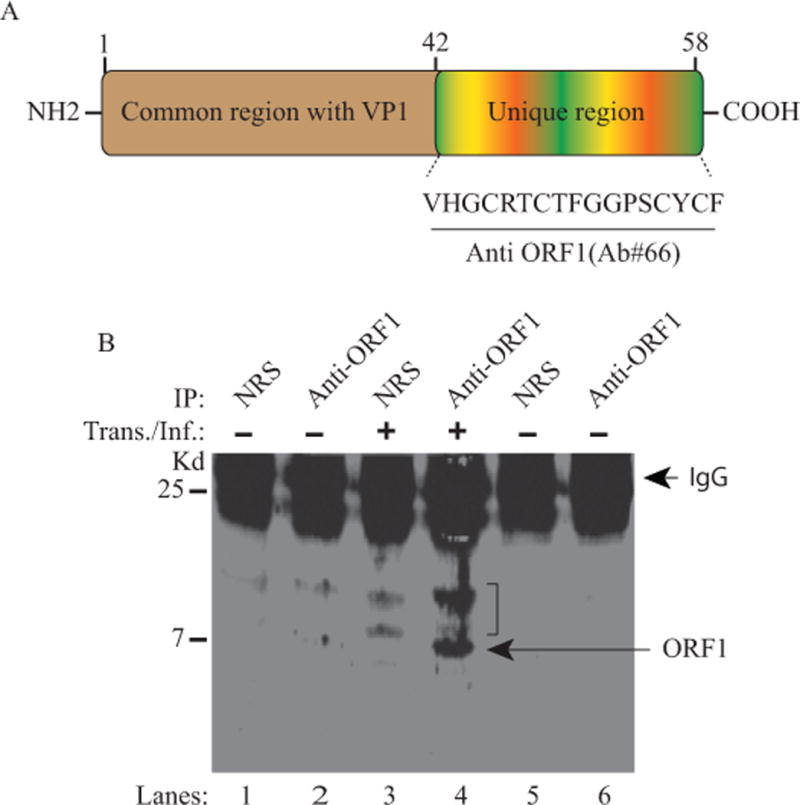
(A) Schematic representation of ORF1 protein (58 aa long). The N-terminus (aa 1-41) of ORF1 overlaps with the N-terminus of VP1 (aa 1-41) but the C-terminus of the protein is unique (aa 42-58). An anti- ORF1 polyclonal antibody was raised in a rabbit against the unique region of ORF1 and is designated as Ab#66. (B) Detection of ORF1 expression in SVG-A cells infected with JCV Mad-1 strain by immunoprecipitation followed by Western blot (IP/WB) as described in Material and Methods using Ab#66 antibody. Membranes were probed with primary anti-ORF1 (Ab#66) and secondary (goat anti-rabbit IRDye 800CW) antibodies and analyzed on an Odyssey CLx imaging system. In lanes 5 and 6, normal rabbit serum (NRS) and Ab#66 antibody respectively were immunoprecipitated and loaded as additional controls in order to demonstrate whether there is any contribution to the non-specific detection of the bands observed on lanes 1 and 2 by the degradation of the antibodies. IP: immunoprecipitation, Kd.: Kilodalton. IgG: Immunoglobulin light chain. A bracket indicates the non-specific bands detected by the primary antibody.
Examination of the PML brain tissue samples for detection of the ORF1 message
Expression of ORF1 in infected cells at the RNA and protein level strongly suggested a possibility that this protein would be expressed in PML patients as well. To explore this possibility, postmortem PML and non-PML brain tissue samples were initially obtained from the Manhattan HIV Brain Bank and processed for preparation of the total RNA and JCV DNA. RT-PCR studies using total RNA and specific PCR primers for detection of ORF1 showed a similar three-banding pattern as observed for the infected tissue culture cells (compare Fig. 3A, lane 3 to Fig 1E, lane 3). In parallel, we did not detect such amplified bands for the RNA sample isolated from a non-PML patient (Fig. 3A, lane 2), indicating the specificity of the ORF1-like expression only in PML patients (designated by hatched and filled arrow heads). The identity of these bands was further explored by PCR amplification using internal primers followed by DNA sequencing as described below. Moreover, the detection of the JCV genome in the same PML sample but not in the non-PML brain tissue sample was demonstrated by PCR amplification to further support the specificity of the amplification of RT-PCR in the PML patient sample (Fig. 3B, lane 2).
Figure 3. Detection of the ORF1-like expression in a PML brain tissue sample by RT-PCR.
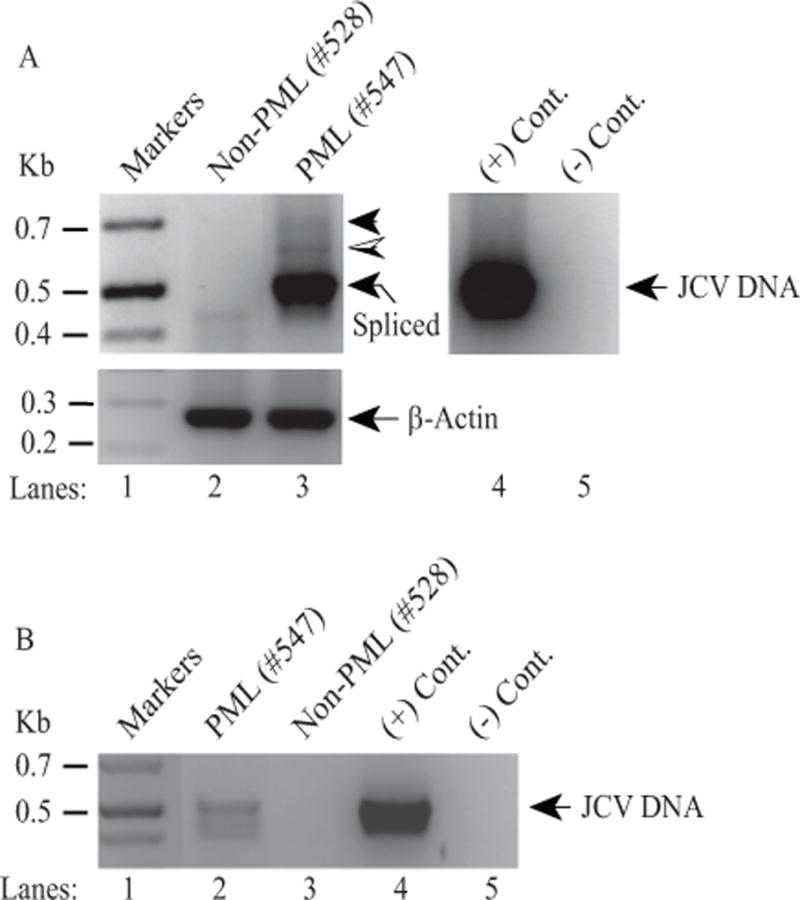
(A) (Upper panel) Detection of a three-banding pattern in a PML patient tissue samples by RT-PCR. Initially, PML and non-PML tissue samples were obtained from the Manhattan HIV Brain Bank and total RNA isolated from those samples was subjected to one step RT-PCR as described in Materials and Methods using JCV 5′-primer (277-302) and JCV 3′-primer (780-752) (Mad-1 numbering). A new internal 3′-primer was used in this RT-PCR to amplify relatively short fragments and therefore resolve them well on the agarose gels. The hatched and filled arrow heads indicate the possible expression of ORF1 and perhaps additional ORFs respectively. The labeled arrow points to the expected spliced product. In lane 4, JCV Mad-1 DNA was used as positive control (+ Cont.) and in lane 5, water was used as a negative control (− Cont.) in PCR reactions. (Lower panel) Total RNA was also subjected to RT-PCR using human actin primers as a control: Forward primer: Human actin (354-375) 5′-CTACAATGAGCTGCGTGTGGC-3′ and Reverse primer: Human actin (624-603) 5′-CAGGTCCAGACGCAGGATGGC-3′. (B) Detection of JCV DNA in PML tissue samples. In parallel to the panel A, JCV DNA isolated from the same PML and non-PML brain tissue samples were also amplified by PCR as described in Materials and Methods, using the same primers described for panel A.
PML patients express additional novel ORFs
To further validate the detection of the ORF1 expression both in the JCV infected cells in tissue culture and in the PML brain tissue samples, total RNA from the same PML and non-PML patients were further subjected to RT-PCR amplification using the internal primers as described in the legend to Fig. 4. Analysis of the amplified products on an agarose gel followed by DNA sequencing confirmed the detection of ORF1 expression not only in infected cells but also in PML brain tissue sample (Fig. 4B, lanes 3 and 4 respectively). As expected, non-PML patient RNA did not produce ORF1 band (Fig. 4B, lane 5). Agarose gel analysis of the RT-PCR products also showed the detection of an additional DNA band running slower than the ORF1 band (lane 4). DNA sequencing analysis of this band led us to discover an additional novel trans-spliced product of JCV from a PML patient inserted into the same region as ORF1, which is capable of creating another open reading frame, called ORF2, which originates from another cryptic splice site positioned at nucleotide G1644 (Fig. 4A). A similar banding pattern for the RNA isolated from the infected cells in vitro is also barely detectable. That is, the infected tissue culture cells also express this trans-spliced ORF2 at low levels (Fig. 4B, lane 3). The translation of the sequencing data revealed that ORF2 protein shares its first 42 aa sequence with ORF1 at the amino acid level as it does with VP1 and has an additional 18 amino acid stretch, again originating from the VP1 coding region (totaling 60 aa from VP1) (Fig. 4C). Furthermore, a frame-shift occurring within the VP2 coding region resulted in additional unique 12 amino acids. In summary, the ORF2 message is predicted to encode a 72 aa long protein (Fig. 4C). Furthermore, a detailed analysis of the cloned RT-PCR products of the same PML patient by DNA sequencing revealed that one of the ORF1 clones has a mutation at residue 23 [isoleucine to methionine (Ile23Met)]. This mutant is designated as “ORF1 mutant” (ORF1M). It is not clear whether it is an isolated case or if PML patients can have additional such variations as well. Additionally, what are the consequences of this mutation for the viral life cycle? Addressing these types of questions are beyond the scope of our current work. In addition, one may consider that the detection of ORF1 and ORF2 by RT-PCR may be due to artifacts forming during the PCR amplification. This possibility has been extensively investigated by our group using different sets of PCR primers and RNA preparations either from the infected cells or PML positive brain tissue samples. In each case, we obtained the same consistent results, which strongly argue against such a possibility. Moreover, RNase protection assays may provide a more direct form of evidence for the detection of the ORF transcripts and eliminate all such lingering questions. However, running such assays appear to not be feasible for ORF1 and ORF2 at all because of the following two reasons: (i) ORF1 and ORF2 share common regions with VP1 and VP2 coding regions and (ii) the VP1 and VP2 transcripts are highly dominant in the JCV late transcript population. As a result, RNA probes prepared for the detection of ORF1 and ORF2 transcripts will primarily hybridize with VP1 and VP2 transcripts and consequently interfere with the detection of ORF1 and ORF2 transcripts.
Figure 4. Discovery of additional ORFs from PML brain tissue samples.
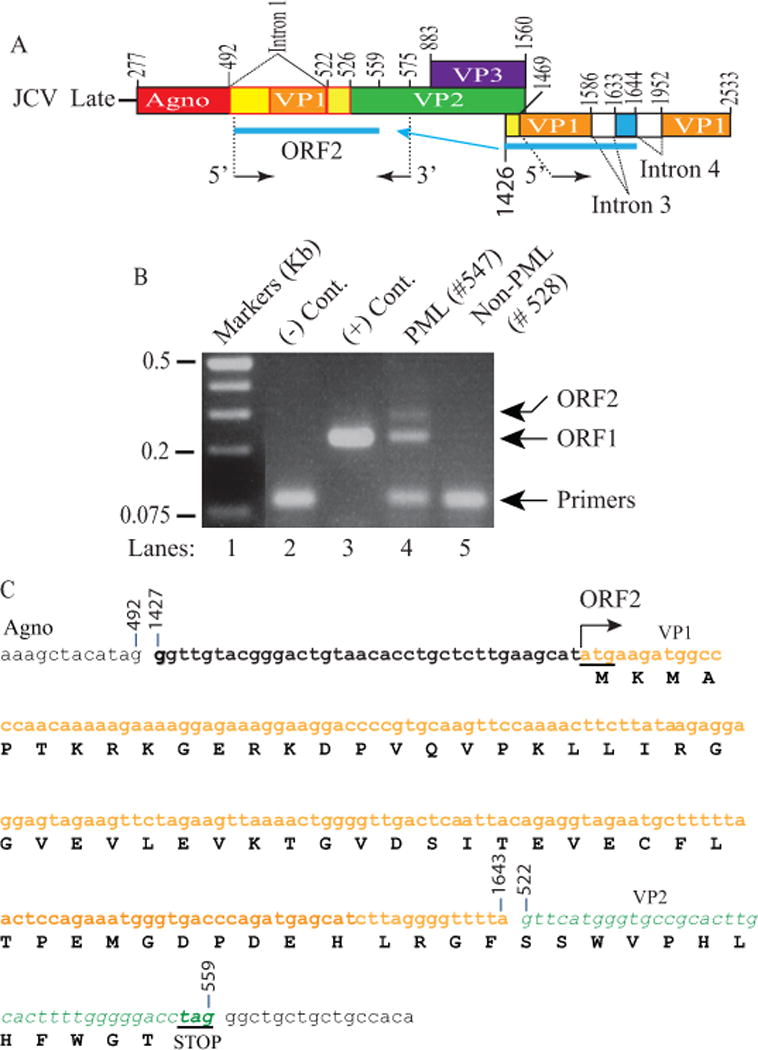
(A) Schematic representation of JCV late transcript containing the ORF2 coding region. (B) RT-PCR amplification of the ORF1 coding region with specific internal primers followed by analysis of the amplified products by agarose gel (3%) electrophoresis. Total RNA isolated from SVG-A cells [uninfected (-Cont., lane 2) and infected with JCV Mad-1 genome, (+ Cont., lane 3)]; and from PML (lane 4) and non-PML (lane 5) patients were subjected to RT-PCR using the following internal primers: 5′-primer: JCV Mad-1 VP1 (1469-1481), 3′-primer: JCV Mad-1 VP2 (575-555). The RT-PCR products were gel-purified and subcloned into XbaI/BamHI sites of the pCGT7 and sequenced. The sequencing data showed that, in addition to ORF1, a longer fragment of VP1 (216 bp) was also inserted in place of Intron 1 in another splice product creating another new ORF designated as a ORF2. Sequencing data also revealed that a mutant form of ORF1 also exists in PML tissue samples, designated ORF1M where Ile23 is mutated to Met (Ile23Met). (C) Schematic representation of a trans-spliced fragment (216 bp) of the 5′-end of the VP1 [Mad-1 (1427-1643) region] in place of “Intron 1” and creation of ORF2 (72 aa). After insertion, a frame shift occurs within VP2, which is terminated with a stop codon at nucleotide position 559. The common region of ORF1 (60 aa long) with VP1 is colored in orange and the unique region is colored in green (12 aa long).
Investigating the expression profiles of ORF1, ORF1M and ORF2 in cell lines
Next, we sought to examine the expression profiles of ORFs in various cells by Western blotting and immunocytochemistry (ICC), because it is already known that not every protein can be easily expressed in tissue culture cells. First, the coding sequences of ORF1, ORF1M and ORF2 were subcloned into pCGT7 expression vector in which they were tagged at their NH2 terminus with T7 tag; and the resulting plasmids were transfected into either epithelial origin, HEK293T, or glial origin, SVG-A cells. At 24 h posttransfection, whole cell extracts were prepared and analyzed by Western blotting using anti-T7 antibody. Western blotting data showed that ORF1, ORF1M and ORF2 are all detectably expressed in these two different cell types.
In parallel, subcellular distribution of each ORF protein was also examined by ICC primarily in SVG-A cells due to the fact that these cells have comparatively larger cell volume than HEK393T cells and therefore these cells provide more detailed microscopic images. Although all three ORF proteins showed a relatively similar cytoplasmic and nuclear distribution property in the transfected cells, ORF2 protein exhibited an additional punctate nuclear accumulation pattern which is distinctly different from than those of ORF1 and ORF1M. These ICC studies suggest that the proteins may have roles in both compartments (cytoplasm and nucleus). Additional ICC distribution studies have also been performed using another cell type, U-87MG, which also has a relatively larger cell volume than HEK293T cells. These studies also showed that ORFs have similar distribution properties as observed for SVG-A cells, suggesting that these three proteins do not exhibit a cell-type specific subcellular distribution pattern (data not shown).
Detection of ORF1 and ORF2 transcripts in RNA samples isolated from additional PML patients
To further validate the expression of ORF1 and ORF2 transcripts in PML patient brains, we have obtained additional PML and non-PML brain tissue samples from the National NeuroAIDS Tissue Consortium (NNTC) for RNA, JCV DNA isolation and immunohistochemistry (IHC) studies. Total RNA from brain samples was then subjected to the RT-PCR as described under Materials and Methods. RT-PCR data showed that ORF1 transcript is clearly detected in all PML tissue samples (Fig. 6A, lanes 6–9). Only one patient sample showed undetectable levels of ORF2, which may be due to the poor quality of the RNA isolated and consequent weak amplification by RT-PCR (Fig. 6A, lane 7). As expected, none of the non-PML patients expressed either of the ORFs (lanes 2–5), confirming the specificity of the RT-PCR amplification. In parallel, the detection of the JCV viral DNA in PML and non-PML patients was also examined. As expected, viral DNA was readily detected by PCR in all PML patients, but not in non-PML normal brain controls thus correlating with our findings from the RNA analysis of the ORF1 and ORF2 expression in PML patients (Fig. 6B, lanes 6–9).
Figure 6. Detection of ORF1 and ORF2 transcripts in additional PML patients.

(A) Additional PML and Non-PML tissue samples were obtained from the National NeuroAIDS Tissue Consortium (NNTC) and total RNA isolated from these samples were subjected to RT-PCR amplification using the following primers: 5′-primer (1469-1489) and 3′-primer (780-755) as described in Materials and Methods. RT-PCR products were then analyzed by agarose gel (3%) electrophoresis. The position of the primers used in RT-PCR is indicated by an arrow head. Total RNA isolated from the PML and non-PML patients was also subject to RT-PCR amplification of the GAPDH cDNA as a control. (B) Detection of JCV DNA in PML brain tissue samples. In parallel to the RT-PCR assays in panel A, JCV DNA was also isolated from the PML and non-PML brain tissue samples and was PCR-amplified using 5′-primer (277-302) and 3′-primer (1366-1346) and analyzed by agarose gel (1%) electrophoresis as described in Materials and Methods. An arrow head points to the position of the primers used in the PCR reaction. In lane 10, JCV Mad-1 DNA was also PCR amplified as a positive control (+ Cont.).
The expression of ORF1 in PML tissue sections was also examined by immunohistochemistry (IHC). Since we raised an antibody (Ab#66) against the unique carboxyl terminus of ORF1, ICC was first performed using this antibody allowing the detection of ORF1 protein expression in transfected cells (Fig. 7B), which confirmed the specificity of this antibody against ORF1. Paraffin-embedded and sectioned tissue samples from the non-PML and PML patients were obtained as “ready to stain” slides from the NNTC. Slides were then incubated with a combination of primary and secondary antibodies for detection of ORF1 expression as described in Materials and Methods. Examination of the slides under a fluorescence microscope revealed that the ORF1 expression is readily detectable in PML but not in non-PML tissue samples and it is localized mostly to the nucleus (Fig. 7A), which is in contrast with our findings from its cellular distribution studies from the transfected tissue culture cells (Fig. 5B, 7B), where we found that it is mostly localized to both the cytoplasmic and nuclear compartments of the transfected cells, suggesting that the infection conditions influence its predominant localization to the nucleus. As a positive control for the expression of a JCV protein in brain tissue sections, we have demonstrated the co-detection of JCV VP1, which is a nuclear protein (White MK, 2016), in the same serial section of the PML tissue sample (Fig. 7A). It is also noteworthy that the tissue sections were prepared from the matched PML (#6064) and non-PML (#1059) patient samples from which RNA and DNA samples were originally isolated for RT-PCR and PCR amplifications respectively. As mentioned above, the specificity of anti-ORF1 antibody was also tested in transfected cells before it was used on IHC samples (Fig. 7B). Moreover, additional PML tissue sections were also examined to detect ORF1 expression using the same antibody and we obtained the similar results as shown in Fig. 6A, further confirming the expression of ORF1 in PML brain tissue samples (data not shown).
Figure 5. Stable expression analysis of ORF1, ORF1M and ORF2 in different cell lines by Western blotting and immunocytochemistry (ICC).
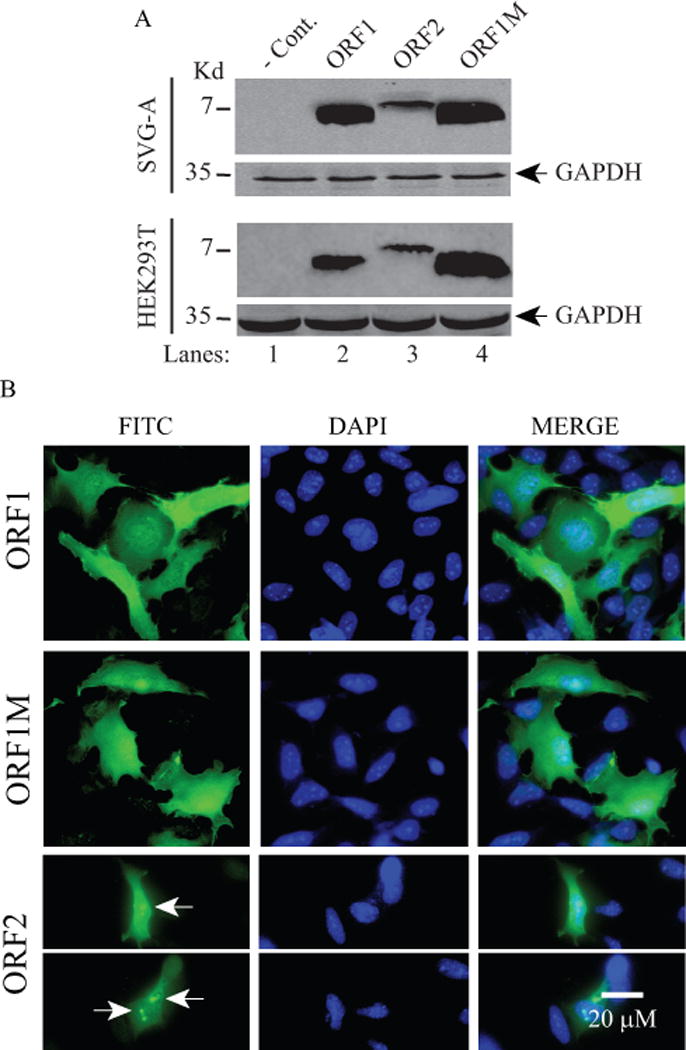
(A) Analysis of ORF1, ORF1M and ORF2 expression in SVG-A and HEK293T cells by Western blotting. These two cell lines were transfected with either vector alone (pCGT7) or the expression plasmids (pCGT7-ORF1 or ORF1M, pCGT7-ORF1M or pCGT7-ORF2). At 48 h posttransfection, whole-cell extracts (WCE) were prepared and analyzed by Western blot using primary mouse anti-T7 monoclonal and secondary goat anti-mouse IRDye 680LT antibodies as described in Materials and Methods. Each blot was also probed with anti-GAPDH antibody for control. Since the anti-ORF1 antibody (Ab#66) only detects ORF1 but not ORF2, therefore it was not used in these Western blots and ICC assays. In lane 1, WCE prepared from the untransfected (transfected with only the vector) cells were loaded as a negative control (− Cont.) (B) Analysis of the cellular distribution of ORF1, ORF1M and ORF2 proteins in SVG-A cells by ICC. In parallel to panel A, these three proteins were also examined by ICC on SVG-A cells using primary mouse anti-T7 monoclonal and secondary FITC-conjugated goat anti-mouse antibodies. Cells were also incubated with DAPI to stain the nucleus and examined under a fluorescence microscope as described in Materials and Methods. Arrows point to a more intense punctate localization patterns of ORF2 in the nucleus. Scale bar: 20 μM.
JCV switches to ORF2 expression when ORF1 splicing is blocked by mutagenesis
We next investigated the impact of the new ORFs on the viral life cycle by mutagenesis. We made point mutations within one of the cryptic splice donor sites of ORF1 positioned at nucleotide 1586 (JCV Mad-1 numbering) where guanine (G) was mutated to either adenine (A) or cytosine (C) so that ORF1 expression could be completely blocked. Wild-type and mutant viral genomes were then transfected/infected into SVG-A cells and total RNA was isolated at both 5th and 15th day posttransfection/infection time points to amplify the messages for ORF1 and ORF2 by RT-PCR using specific primers as shown in Fig. 8A. Note that, JCV has a relatively long life cycle compared to other polyomaviruses such as SV40 and it takes 6 or 7 days to complete its first round of the viral life cycle in SVG-A cells. In order to better understand the effect of these mutations, we therefore chose to analyze first two life cycles of the virus rather than the first life cycle only. As expected, we have detected the expression of ORF1 (predominant) and ORF2 messages (barely detectable) for WT at both 5th and 15th day posttransfection/infection (Fig. 8B, lanes 3 and 6). Interestingly, when ORF1 expression is blocked by mutations, both mutant viruses then switched to express ORF2 message only (Fig. 8B, lanes 4, 5, 7 and 8). Although these mutants expressed ORF2 transcripts at the comparable levels with WT during the first round of replication (5th day data points), they showed a differential expression pattern during the second round of replication cycle (15th day data point). That is, while the G1586A mutant maintained ORF2 expression level (Fig. 8B, lane 7) as observed for the first round of replication cycle (Fig. 8B, lane 5), the expression level of the G1586C mutant, on the other hand, significantly decreased (Fig. 8B, lane 8) compared to that of G1586A mutant (lane 7) during the second round of replication.
Figure 8. Analysis of the ORF1 splice-deficient mutants.
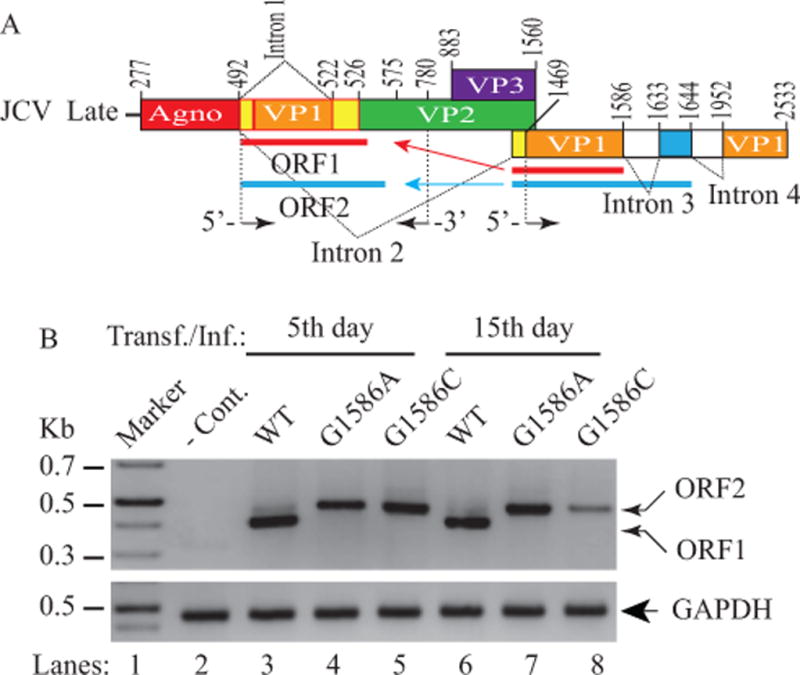
(A) Schematic representation of JCV Mad-1 late transcript illustrating the trans-splicing of the small fragments from the 5′-VP1 in place of Intron 1, which results in the formation of the ORF1 and ORF2 open reading frames. The G nucleotide at a cryptic splice donor site positioned at the nucleotide 1586 was mutated to either A or C as described in Materials and Methods. (B) RT-PCR analysis of the JCV late transcripts isolated from SVG-A cells transfected/infected with either JCV Mad-1 WT or its ORF1 splice-deficient mutants. Cells were transfected/infected with the following plasmid constructs: Bluescript KS (+)-JCV Mad-1 WT, Bluescript KS (+)-JCV Mad-1 (G1586A) and Bluescript KS (+)-JCV Mad-1 (G1586C) as described in Materials and Methods. At the indicated time points, total RNA was isolated from those cells and was subjected to RT-PCR amplification using the following primers: 5′-primer (1469-1489) and 3′-primer (780-755) as described in Materials and Methods. In lane 1, total RNA from untransfected cells was also used in RT-PCR reactions using the same primers as a negative control (− Cont.) and amplified products were then analyzed on a 3% agarose gel. The total RNA was also subjected to RT-PCR for amplification of GAPDH RNA as described for Fig. 6A.
Effect of ORF1 splice-deficient mutants on JCV replication
We next examined the viral replication efficiency of these mutants compared to WT by DpnI/Southern blot assays (Saribas et al., 2011; Saribas et al., 2012). JCV Mad-1 WT and mutant JCV genomes were individually transfected/infected into SVG-A cells and low molecular weight DNA was isolated at 5th and 15th day posttransfection/infection as described in Materials and Methods. In order to distinguish the newly-replicated viral DNA from the transfected DNA (bacterially produced and methylated), DNA samples were digested with DpnI enzyme (Hirt, 1967). In addition, to obtain a single banded pattern on Southern blots, DNA samples were also digested with BamHI, which cuts JCV genome once and thereby linearizes the circular genome. DNA samples were then analyzed by Southern blot using a probe prepared from JCV Mad-1 genome as a template. As shown in Fig. 9A and 9B, the replication efficiency of each mutant slightly decreased compared to that of WT by the 5th day data point (Fig. 9A, lanes 3 and 4; Fig. 9B). G1586A mutant also replicated almost as efficiently as WT by the 15th day posttransfection/infection (Fig. 9A, lane 7; Fig. 9B). However, the replication levels of the G1586C mutant drastically decreased by the 15th day (Fig. 8B, lane 8, Fig. 9B). Consistent with these findings, the expression levels of VP1 was also found to be below the detection levels (Fig. 9C, lane 8) in our experimental conditions. These negative results suggested the following possibilities: One possibility is the G1586C mutation could have deleterious effects on the stability of transcripts or their translation. Another possibility is that the G1586C mutation (Val42Leu) perhaps has a more dramatic effect on VP1 structure (conformation) than that of the G1586A mutation (Val42Ile) has, such that VP1 can no longer fulfill its functional activities during the viral life cycle. In order to avoid such lingering questions, we independently introduced a specific stop codon mutation between the unique and common regions of the ORF1 and ORF2 so that the unique regions of both proteins are not expressed during the viral life cycle. Additionally, this mutation does not affect either any of the splicing sites of the JCV late transcripts or their expression at the protein level as indicated (Fig 10A). In other words, if the ORF proteins have impact on the viral replication cycle, this mutant should manifest its effects as explained below.
Figure 9. Analysis of the replication properties of the ORF1 splice-deficient mutants.
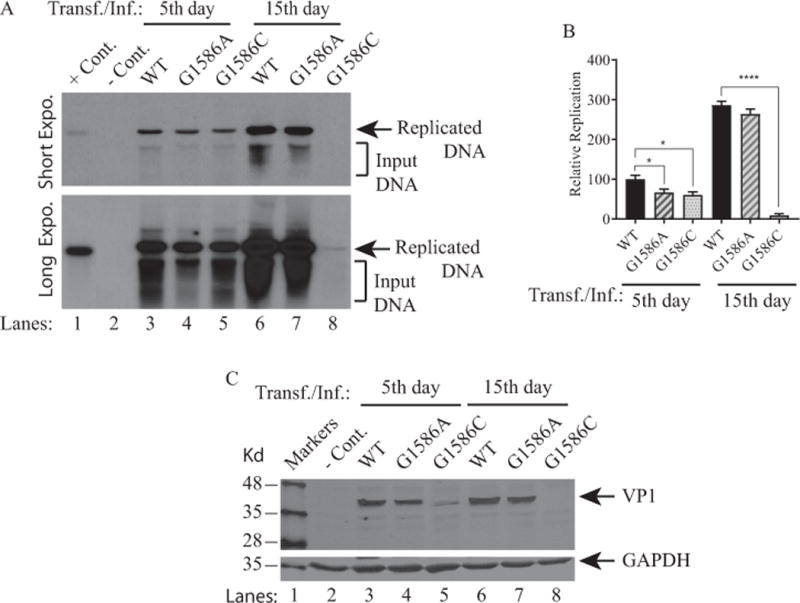
(A) SVG-A cells were either untransfected (− Cont.) or transfected/infected with Bluescript KS (+)-JCV Mad-1 WT or Bluescript KS (+) JCV Mad-1 (G1586A) or Bluescript KS (+)-JCV Mad-1 (G1586C) as described in Materials and Methods. At the indicated time points, low-molecular-weight DNA containing both input and replicated viral DNA was isolated and analyzed by Southern blot as described in Materials and Methods. This assay was repeated more than three times with independent assays and a representative data is shown here with short and long exposure (Expo.) times. In lane 1, 2 ng of JCV Mad-1 DNA was linearized by BamHI digestion and loaded as a positive control (+ Cont.). In lane 2, low-molecular-weight DNA was also isolated from the untransfected SVG-A cells, subjected to enzyme digestion as the other experimental samples and loaded as a negative control (− Cont.). (B) Quantitative analysis of the results. The band intensities for the replicated DNA on Southern blots were determined by using an Image J program (https://imagej.nih.gov/ij/); and statistically analyzed and graphed by using GraphPad Prism 7 program (https://www.graphpad.com/scientific-software/prism/). One way ANOVA was used to determine the statistical differences between WT and mutants. **** P< 0.0001 and * P< 0.0001 indicate the statistical differences between WT and mutants with respect to their replication efficiency as indicated. (C) Analysis of VP1 expression by Western blot for the ORF1 mutants. In parallel to the replication assays described in panel A, whole-cell extracts (WCE) prepared from untransfected and transfected/infected cells were resolved on a SDS-10% PAGE and blotted on a nitrocellulose membrane. The membrane was then probed with a primary monoclonal anti-VP1 (pAB597) (Saribas et al., 2013) and secondary IRDye 680 goat anti-mouse antibodies; and scanned on an Odyssey CLx imaging system. In lane 2, WCE from the untransfected cells was loaded as a negative control (− Cont.). The Western blot was also probed with anti-GAPDH antibody to demonstrate equal loading.
Figure 10. Analysis of the splicing efficiency of the truncated forms of the ORF1 and ORF2 mutants by RT-PCR.
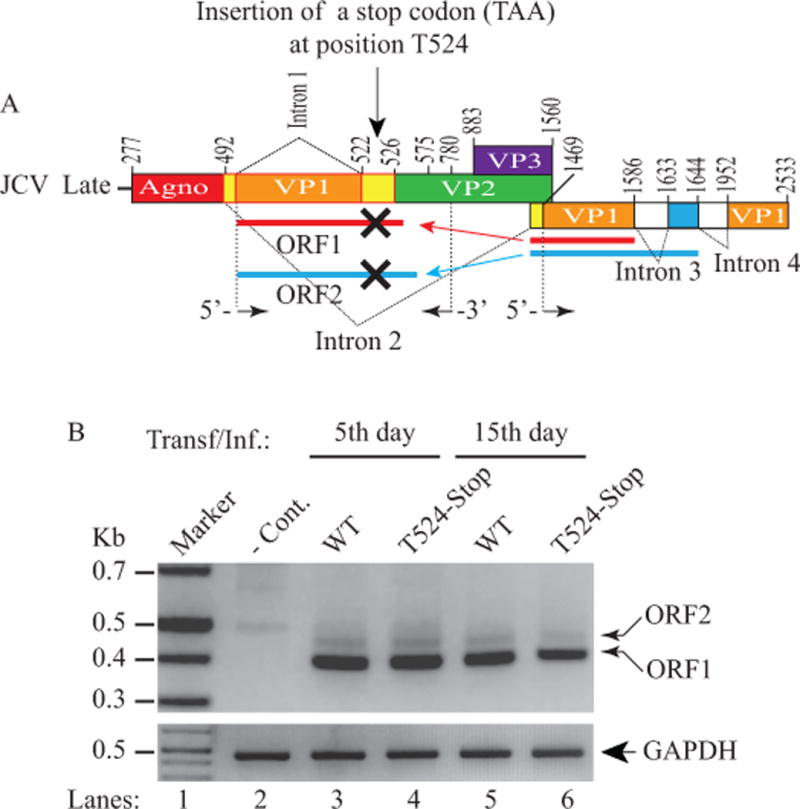
(A) Schematic representation of a JCV Mad-1 late transcript illustrating the insertion of a stop codon (TAA) at position after T524 (GTT524 TAA C525ATGG) to block the expression of the unique amino acid regions of both ORF1 and ORF2 proteins. (B) RT-PCR analysis of the JCV late transcripts isolated from SVG-A cells either untransfected (− Cont.) or transfected/infected with JCV Mad-1 WT or Mad-1 T524 stop mutant using the following primers: 5′-primer (1469-1489) and 3′-primer (780-755) as described in Materials and Methods. The total RNA was also subjected to RT-PCR for amplification of GAPDH RNA as described for Fig. 6A. Amplified products were then analyzed on a 3% agarose gel.
Effect of truncated forms of ORF1 and ORF2 on JCV life cycle
In order to further investigate the impact of ORF1 and ORF2 on the JCV life cycle, as explained above, we created a simultaneously truncated form of ORF proteins (ORF1 Δ42-58 and ORF2 Δ60-72) in the viral background by introducing a stop codon (TAA) after nucleotide position T524 (Fig. 10A, Fig. 10B) so that only the common (originating from VP1) but not the unique regions of ORFs (ORF1 and ORF2) are allowed to be expressed. The fundamental unique features of this mutation are that it does not alter any of the splice sites or the coding regions of JCV, including ORF1 and ORF2. This stop codon mutation also allows trans-splicing of ORF1 and ORF2 messages in place of intron 1 as usual, as described previously. As a result, if there were to be an impact of ORF1 and ORF2 on viral DNA replication, this mutation would manifest it. The message levels of ORF1 and ORF2 resulting from this mutant were first assessed by RT-PCR. Results showed indistinguishable message levels from that of WT by the 5th day data point for both ORF1 and ORF2 (Fig 10A, lanes 3 and 4). However, the level of the ORF1 message substantially decreased by the 15th day compared to that of the WT (~ 48%, Fig. 10B, lane 6).
Next, the replication efficiency of the truncated mutants was investigated by Southern blot as described in the legend to Fig. 9. Results clearly showed that truncated mutants of ORF1 and ORF2 replicate less efficiently than WT even during the first round of replication (5th day data point) (Fig. 11A, lane 4) but not statistically significant. However, during the second round of replication, (15th day data point), this difference becomes more pronounced (Fig. 11A, lane 6; 11B), (~ 5 fold less), indicating the importance of the unique regions of ORF1 and ORF2 in viral replication. In parallel, the results from the Western blot analysis correlates with our findings from the replication assays (Fig. 11A) in that VP1 protein expression of the stop mutant was found to be significantly diminished compared to that of WT by 15th day data point (Fig. 11C, lane 6). Taken together, ORF1 and ORF2 appear to play critical roles during the JCV replication cycle.
Figure 11. Analysis of the replication efficiency of the truncated forms of the ORF1 and ORF2 mutants.
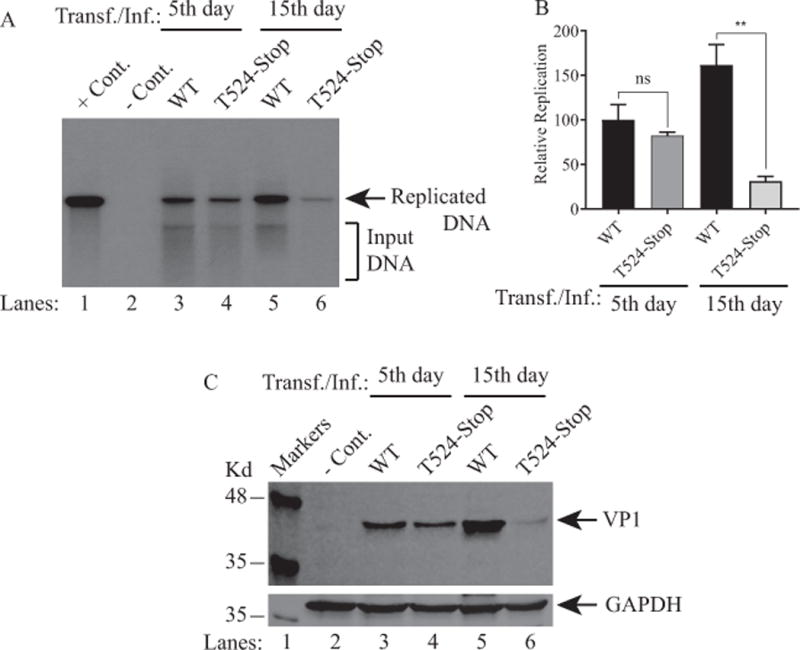
(A) SVG-A cells were either untransfected (− Cont.) or transfected/infected with Bluescript KS (+)-JCV Mad-1 WT or Bluescript KS (+) JCV Mad-1 (T524-Stop) mutant. At the indicated time points, the low-molecular-weight DNA containing both input and the replicated viral DNA was isolated and analyzed by Southern blotting as described under figure legend 9A. This assay was repeated more than three times with independent assays. A representative figure is shown here. In lane 1, 2 ng of JCV Mad-1 DNA digested and linearized by BamHI was loaded as a positive control (+ Cont.). (B) Quantitative analysis of the results. The band intensities for the replicated DNA on Southern blots were quantified and graphed as described for figure legend 9B. One way ANOVA was used to determine the statistical differences between WT and mutants. ** P< 0.0042 indicates the statistical difference between WT and the mutants with respect to their replication efficiency as indicated. NS: indicates no statistical difference. (C) Analysis of VP1 expression for both truncated mutants by Western blotting as described for Fig. 9C.
DISCUSSION
Trans-splicing is an alternative form of splicing which occurs between two independent pre-mRNA transcripts. In this study, we report the discovery of two novel trans-splicing products of the JCV late transcripts, which occur when two overlapping but different lengths of 5′ region of VP1 are spliced out and inserted in place of intron 1, located between the coding regions of agnoprotein and VP2. The exact mechanism of this event is currently unknown, but it is clear that there are fragment exchanges between two independent JCV late transcripts in order for trans-splicing to occur. The insertion products are 159 bp and 216 bp long for ORF1 and ORF2 respectively. Upon insertion, frame shifts occur within the VP2 coding region of JCV, creating new open reading frames that are capable of encoding 58 and 72 aa long proteins, designated as ORF1 and ORF2, respectively. These observed trans-splicing events occur not only in vitro tissue culture infection systems but also in JCV-infected brains of PML patients. It should be noted here that these newly discovered ORFs do not result from either the degradation products of the late transcripts or from the artificial rearrangements that may occur during the RT-PCR reactions. These two questions were satisfactorily addressed by using several different combinations of RT-PCR primers and using different batches of RNA preparations from the infected cells in vitro and from the PML patient samples in vivo. These studies undoubtedly reveal the fact that the ORFs are genuine splicing products of the late transcripts. Data from the current study also showed that JCV predominantly expresses ORF1 trans-spliced product over ORF2 regardless of whether it results from an in vitro or in vivo infection cases. We next investigated whether ORFs are associated with protein expression by Western blot and immunohistochemistry. To perform these detection assays, a polyclonal antibody was first raised against the unique region of ORF1 (Fig. 2A). Initial detection studies by a direct Western blot analysis were unable to provide satisfactory results from the infected cells due to the low level of expression of ORF1. However, immunoprecipitation assays clearly demonstrated that ORF1 is indeed expressed during the viral infection cycle (Fig. 2B). The expression properties of the ORF proteins in the absence of an infection cycle were also shown. For this, the coding sequences of the ORFs (ORF1, ORF1M, and ORF2) were subcloned under a strong CMV promoter and tagged with T7-tag at their N-terminus (pCGT7 vector), because the T7-tag is a strong antigenic epitope commonly used to label proteins; and their expression was then monitored in tissue culture by Western blotting (Fig. 5A) and immunocytochemistry (Fig. 5B). They were clearly found to be detectable by both techniques. In order to directly demonstrate the specificity of the newly raised anti-ORF1 antibody, additional immunocytochemistry assays were also performed using the same T7-tagged ORF1 expression plasmid where ORF1 protein was found to be detectable by the new anti-ORF1 antibody (Fig. 7B).
It was expected that ORF1, ORF2 and ORFM proteins display a nuclear distribution pattern in vitro transfection assays due to the fact that all contain the nuclear localization signal of VP1 (MAPT K5R6K7 GEK8K9D) in their N-terminus sequences. On the contrary, they were distributed both to cytoplasmic and nuclear compartments of the transfected cells (Fig. 5B). It was noticed that in addition to being distributed to both the cytoplasm and nucleus, ORF2 also consistently showed a distinct punctate distribution pattern in the nucleus (Fig. 5B). However, intracellular distribution pattern of ORF proteins, at least, that of ORF1 showed deviations from this pattern in PML patient samples. For example, while ORF1 is detected at the cytoplasmic compartment of the infected cells in vitro (Fig. 5B), it was mostly found to be localized to the nucleus in the PML patient tissue samples in vivo (Fig. 7A). This may be explained, in part, by a previously reported case by Shishido-Hara et al (Shishido-Hara et al., 2000), who addressed a similar question as to why ORF proteins do not exclusively localize to nucleus in vitro cases, even if they have the same nuclear localization signal as VP1 has. Shishido-Hara et al., demonstrated that the putative nuclear localization signal of JCV VP1 (MAPT K5R6K7 GEK8K9D) is indeed a weak nuclear localization signal compared to that of SV40 (MADTKRKGSCPGAAPKKPKE) and VP1 requires the expression of VP2 and VP3 proteins for its efficient nuclear localization, otherwise it is distributed to both cytoplasm and nucleus in cells. SV40 VP1, on the other hand, does not have such a requirement (Shishido-Hara et al., 2000), suggesting that although ORF1, ORF2 and ORF1M all have the complete JCV VP1 localization signal in their sequence, these proteins appear to behave similar to JCV VP1 as previously reported (Shishido-Hara et al., 2000) in the absence of VP2 and VP3 expression.
In order to understand the role of ORFs in the JCV replication cycle, two mutations were made at the specific splice donor site of ORF1 at position G1586 to block the trans-splicing of ORF1. Guanine nucleotide at position 1586 was mutation to adenine (A) or cytosine (C), generating two mutants (G1586A and G1586C). Results showed that, as expected, both mutants lost the expression of their ORF1 messages (Fig. 8B), but surprisingly switched to the expression of the ORF2 only (Fig. 8B) and replicated relatively well compared to WT during the first round of the viral replication cycle (Fig. 9A, Fig. 9B). However, both mutants showed differential replication efficiency during the second round of viral replication cycle. That is, the replication efficiency of G1586A mutant remained essentially unaltered compared to WT, but that of G1586C mutant almost completely abolished (Fig. 9A, 9B). One of the possible explanations for this finding is that perhaps G1586C mutation (Val42Leu) compared to that of G1586A (Val42Ile) on VP1, although both amino acid changes are considered to be relatively conservative, may introduce dramatic structural changes on VP1 protein, which could then be translated into the loss of function in various aspects of VP1 during the viral infection cycle, including but not limited to, its cytoplasmic/nuclear transport, capsid assembly and interaction with VP2/VP3 etc. Such functional losses by VP1 could lead to drastic effects on the viral replication cycle.
In order to eliminate such difficult questions, we have introduced a stop codon mutation (TAA) at a position after nucleotide thymidine 524 (T524) (Mad-1 numbering), which allows the expression of a truncated version of each ORF from the viral background (Fig. 10A), but does not affect any of the splice sites of JCV early and late transcripts nor the expression of the JCV early and late proteins (large T antigen, small T protein, T′-proteins, VP1, VP2, VP3 and agnoprotein). The functional consequences of this insertion were then examined by means of the replication assays. Data revealed that JCV virus with this stop codon replicated slightly less efficiently compared to WT during the first round of replication (5th day data point). However, the replication efficiency of this mutant virus dramatically decreased during the second round of replication (15th day of data point) (~ 5 fold less replication than WT) (Fig. 11A and 11B), suggesting that the ORFs play significant roles in the efficient completion of JCV life cycle.
The discovery of these novel ORFs and partial understanding of their function during the viral replication cycle prompted us to explore the existence of additional splice variants of JCV late transcripts and therefore the formation of the additional open reading frames. Our initial evaluation of such splice variants are encouraging and suggests that small viruses, such as JCV, utilizes uncommon trans-splicing mechanisms to amplify their genomic capacity by creating new open frames in order to have a highly efficient replication cycle. We have just begun to characterize the biological functions of such ORFs.
Supplementary Material
Acknowledgments
This work was made possible by grants awarded to M.S. [(RO1NS090949) by the NIH and by the Temple University Drug Discovery Initiative (161398)]. PML and non-PML patient brain tissue samples were provided by the National NeuroAIDS Tissue Consortium (NNTC, www.nntc.org). We specifically thank Dr. Susan Morgello (Manhattan HIV Brain Bank) for providing initial PML and Non-PML tissue samples as well as helping us obtaining additional brain tissue samples from NNTC.
Footnotes
CONFLICT OF INTEREST
Authors declare no conflict of interest.
References
- Adami G, Babiss LE. DNA template effect on RNA splicing: two copies of the same gene in the same nucleus are processed differently. EMBO J. 1991;10(11):3457–3465. doi: 10.1002/j.1460-2075.1991.tb04910.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almqvist PM, Mah R, Lendahl U, Jacobsson B, Hendson G. Immunohistochemical detection of nestin in pediatric brain tumors. The journal of histochemistry and cytochemistry: official journal of the Histochemistry Society. 2002;50(2):147–158. doi: 10.1177/002215540205000203. [DOI] [PubMed] [Google Scholar]
- Berger JR. PML in AIDS. Neurology. 1992;42(9):1845–1846. doi: 10.1212/wnl.42.9.1845-b. [DOI] [PubMed] [Google Scholar]
- Berger JR. Progressive Multifocal Leukoencephalopathy. Curr Treat Options Neurol. 2000;2(4):361–368. doi: 10.1007/s11940-000-0053-7. [DOI] [PubMed] [Google Scholar]
- Berger JR. Progressive multifocal leukoencephalopathy. Curr Neurol Neurosci Rep. 2007;7(6):461–469. doi: 10.1007/s11910-007-0072-9. [DOI] [PubMed] [Google Scholar]
- Bollag B, Kilpatrick LH, Tyagarajan SK, Tevethia MJ, Frisque RJ. JC virus T′135, T′136 and T′165 proteins interact with cellular p107 and p130 in vivo and influence viral transformation potential. J Neurovirol. 2006;12(6):428–442. doi: 10.1080/13550280601009553. [DOI] [PubMed] [Google Scholar]
- Caudevilla C, Da Silva-Azevedo L, Berg B, Guhl E, Graessmann M, Graessmann A. Heterologous HIV-nef mRNA trans-splicing: a new principle how mammalian cells generate hybrid mRNA and protein molecules. Febs Letters. 2001;507(3):269–279. doi: 10.1016/s0014-5793(01)02957-x. [DOI] [PubMed] [Google Scholar]
- Coric P, Saribas AS, Abou-Gharbia M, Childers W, White MK, Bouaziz S, Safak M. Nuclear magnetic resonance structure revealed that the human polyomavirus JC virus agnoprotein contains an alpha-helix encompassing the Leu/Ile/Phe-rich domain. Journal of virology. 2014;88(12):6556–6575. doi: 10.1128/JVI.00146-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eul J, Graessmann M, Graessmann A. Experimental evidence for RNA trans-splicing in mammalian cells. Embo J. 1995;14(13):3226–3235. doi: 10.1002/j.1460-2075.1995.tb07325.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eul J, Graessmann M, Graessmann A. Trans-splicing and alternative-tandem-cis-splicing: two ways by which mammalian cells generate a truncated SV40 T-antigen. Nucleic Acids Res. 1996;24(9):1653–1661. doi: 10.1093/nar/24.9.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eul J, Patzel V. Homologous SV40 RNA trans-splicing A new mechanism for diversification of viral sequences and phenotypes. Rna Biology. 2013;10(11):1689–1699. doi: 10.4161/rna.26707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisque RJ, Bream GL, Cannella MT. Human polyomavirus JC virus genome. J Virol. 1984;51(2):458–469. doi: 10.1128/jvi.51.2.458-469.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattoni R, Chebli K, Himmelspach M, Stevenin J. Modulation of alternative splicing of adenoviral E1A transcripts: factors involved in the early-to-late transition. Genes Dev. 1991;5(10):1847–1858. doi: 10.1101/gad.5.10.1847. [DOI] [PubMed] [Google Scholar]
- Green MR. Pre-Messenger-Rna Splicing. Annu Rev Genet. 1986;20:671–708. doi: 10.1146/annurev.ge.20.120186.003323. [DOI] [PubMed] [Google Scholar]
- Hastings ML, Krainer AR. Pre-mRNA splicing in the new millennium. Curr Opin Cell Biol. 2001;13(3):302–309. doi: 10.1016/s0955-0674(00)00212-x. [DOI] [PubMed] [Google Scholar]
- Hirt B. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol. 1967;26(2):365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- Imam SA, Young L, Chaiwun B, Taylor CR. Comparison of two microwave based antigen-retrieval solutions in unmasking epitopes in formalin-fixed tissue for immunostaining. Anticancer research. 1995;15(4):1153–1158. [PubMed] [Google Scholar]
- Kikumori T, Cote GJ, Gagel RF. Naturally occurring heterologous trans-splicing of adenovirus RNA with host cellular transcripts during infection. Febs Letters. 2002;522(1–3):41–46. doi: 10.1016/s0014-5793(02)02878-8. [DOI] [PubMed] [Google Scholar]
- Krause M, Hirsh D. A trans-spliced leader sequence on actin mRNA in C. elegans. Cell. 1987;49(6):753–761. doi: 10.1016/0092-8674(87)90613-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy WJ, Watkins KP, Agabian N. Identification of a novel Y branch structure as an intermediate in trypanosome mRNA processing: evidence for trans splicing. Cell. 1986;47(4):517–525. doi: 10.1016/0092-8674(86)90616-1. [DOI] [PubMed] [Google Scholar]
- Rajkovic A, Davis RE, Simonsen JN, Rottman FM. A Spliced Leader Is Present on a Subset of Messenger-Rnas from the Human Parasite Schistosoma-Mansoni. P Natl Acad Sci USA. 1990;87(22):8879–8883. doi: 10.1073/pnas.87.22.8879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowska B, Barrucco R, Khalili K, Safak M. Regulation of human polyomavirus JC virus gene transcription by AP-1 in glial cells. J Virol. 2003;77(1):665–672. doi: 10.1128/JVI.77.1.665-672.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saribas AS, White MK, Safak M. Structure-based release analysis of the JC virus agnoprotein regions: A role for the hydrophilic surface of the major alpha helix domain in release. J Cell Physiol. 2017 doi: 10.1002/jcp.26106. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saribas AS, Abou-Gharbia M, Childers W, Sariyer IK, White MK, Safak M. Essential roles of Leu/Ile/Phe-rich domain of JC virus agnoprotein in dimer/oligomer formation, protein stability and splicing of viral transcripts. Virology. 2013;443(1):161–176. doi: 10.1016/j.virol.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saribas AS, Arachea BT, White MK, Viola RE, Safak M. Human polyomavirus JC small regulatory agnoprotein forms highly stable dimers and oligomers: implications for their roles in agnoprotein function. Virology. 2011;420(1):51–65. doi: 10.1016/j.virol.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saribas AS, Coric P, Hamazaspyan A, Davis W, Axman R, White MK, Abou-Gharbia M, Childers W, Condra JH, Bouaziz S, Safak M. Emerging From the Unknown: Structural and Functional Features of Agnoprotein of Polyomaviruses. Journal of cellular physiology. 2016;231(10):2115–2127. doi: 10.1002/jcp.25329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saribas AS, Mun S, Johnson J, El-Hajmoussa M, White MK, Safak M. Human polyoma JC virus minor capsid proteins, VP2 and VP3, enhance large T antigen binding to the origin of viral DNA replication: evidence for their involvement in regulation of the viral DNA replication. Virology. 2014;449:1–16. doi: 10.1016/j.virol.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saribas AS, White MK, Safak M. JC virus agnoprotein enhances large T antigen binding to the origin of viral DNA replication: Evidence for its involvement in viral DNA replication. Virology. 2012;433(1):12–26. doi: 10.1016/j.virol.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sariyer IK, Akan I, Palermo V, Gordon J, Khalili K, Safak M. Phosphorylation mutants of JC virus agnoprotein are unable to sustain the viral infection cycle. J Virol. 2006;80(8):3893–3903. doi: 10.1128/JVI.80.8.3893-3903.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sariyer IK, Saribas AS, White MK, Safak M. Infection by agnoprotein-negative mutants of polyomavirus JC and SV40 results in the release of virions that are mostly deficient in DNA content. Virol J. 2011;8:255. doi: 10.1186/1743-422X-8-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saylor D, Venkatesan A. Progressive Multifocal Leukoencephalopathy in HIV-Uninfected Individuals. Curr Infect Dis Rep. 2016;18(11):33. doi: 10.1007/s11908-016-0543-8. [DOI] [PubMed] [Google Scholar]
- Shishido-Hara Y, Hara Y, Larson T, Yasui K, Nagashima K, Stoner GL. Analysis of capsid formation of human polyomavirus JC (Tokyo-1 strain) by a eukaryotic expression system: splicing of late RNAs, translation and nuclear transport of major capsid protein VP1, and capsid assembly. J Virol. 2000;74(4):1840–1853. doi: 10.1128/jvi.74.4.1840-1853.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton RE, Boothroyd JC. Evidence for trans splicing in trypanosomes. Cell. 1986;47(4):527–535. doi: 10.1016/0092-8674(86)90617-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier LH, Keller M, Chan RL, Fournier R, Weil JH, Imbault P. Short leader sequences may be transferred from small RNAs to pre-mature mRNAs by trans-splicing in Euglena. EMBO J. 1991;10(9):2621–2625. doi: 10.1002/j.1460-2075.1991.tb07804.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Santen VL, Spritz RA. Alternative splicing of SV40 early pre-mRNA in vitro. Nucleic Acids Res. 1986;14(24):9911–9926. doi: 10.1093/nar/14.24.9911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberghe AE, Meedel TH, Hastings KE. mRNA 5′-leader trans-splicing in the chordates. Genes Dev. 2001;15(3):294–303. doi: 10.1101/gad.865401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MK, S M. In: Molecular biology of JC virus and the human demyelinating disease, progressive multifocal encephalopathy. Reiss CS, editor. New York: Springer; 2016. pp. 75–110. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.