

Abstract
The PDGF signaling pathway plays important roles in development and progression of human cancers. In this study, we aimed to identify genetic variants of the PDGF pathway genes associated with pancreatic cancer risk in European populations by using three published GWAS datasets, which consisted of 9,381 cases and 7,719 controls. The expression quantitative trait loci (eQTL) analysis was also performed by using data from the 1000 Genomes, TCGA and GTEx projects. As a result, we identified two potential susceptibility loci (rs5757573 and rs6001516) of PDGFB associated with pancreatic cancer risk [odds ratio (OR) = 1.10, 95% confidence interval (CI) = 1.05–1.16, and P = 4.70×10−5 for the rs5757573 C allele and 1.21, 1.11–1.32, and 2.01×10−5 for the rs6001516 T allele]. Haplotype analysis revealed that the C-T haplotype carriers had a significantly increased risk of pancreatic cancer than those carrying the T-C haplotype (OR = 1.23, 95% CI = 1.12–1.34, P =5.00×10−6). The multivariate regression model incorporating the number of unfavorable genotypes (NUGs) with age and sex showed that carriers with 1–2 NUGs, particularly among 60–70 age group or males, had an increased risk of pancreatic cancer, compared with those without NUG. Further, the eQTL analysis revealed that both loci were correlated with a decreased mRNA expression level of PDGFB in lymphoblastoid cell lines and pancreatic tumor tissues (P = 0.015 and 0.071, respectively). Our results suggest that genetic variants in PDGFB may play a role in susceptibility to pancreatic cancer. Further population and functional validations of our findings are warranted.
Keywords: single nucleotide polymorphism, PDGF, pancreatic cancer susceptibility, pathway analysis, genome-wide association study
Introduction
Pancreatic cancer is an aggressive malignant disease with a very poor prognosis. In the United States, pancreatic cancer is the fourth leading cause of cancer-related death, and approximate 53,070 people will be diagnosed with pancreatic cancer and 41,780 will die of this disease in 20161. Although most cancers already have a much improved survival, advances in diagnosis and treatment for pancreatic cancers have been slow, with a five-year relative survival of only 8%1. This is mainly because pancreatic cancer is rarely detected at an early stage, and the etiology of pancreatic cancer is still not very clear2–4. As a consequence, there is an urgent need to construct a successful pancreatic cancer risk assessment model to identify people who are at high risk of pancreatic cancer, which can help us advance our understanding of pancreatic carcinogenesis and disease progression and identify susceptible individuals for prevention and early detection to reduce the incidence and mortality of pancreatic cancer.
Age is a key risk factor for pancreatic cancer with the median age at diagnosis of pancreatic cancer of 72 years old5. Other environmental risk factors include male sex, diabetes, cigarette smoking and obesity6. While genetic basis for the majority of familial clustering of pancreatic cancer cases has yet to be explained clearly, several rare, moderately or highly penetrant mutations in some important pancreatic cancer genes have been identified, such as CFTR, BRCA2, PALB2, PRSS1, SPINK1, STK11 and DNA mis-match repair genes7. Some common variants associated with pancreatic cancer risk have also been identified in recent genome-wide association studies (GWASs)8–14. However, the overwhelming majority of the single-nucleotide polymorphisms (SNPs) identified by GWASs have been located in introns or intergenic regions, which do not have clear biological functions. SNPs in genes involved in specific biological pathways that may have important roles in the development and progression of pancreatic cancer need to be further explored. Several candidate gene/pathway-based association studies have been performed and identified multiple SNPs in genes associated with the risk of pancreatic cancer by using the published GWAS dataset 15–20. In the present study, we performed a candidate gene/pathway-based analysis using three GWAS datasets to identify potential susceptibility loci associated with pancreatic cancer risk. This strategy uses a limited number of SNPs based on their gene functions in a specific biological pathway, which significantly decreases dimension or multiple testing of genotyping data.
The platelet-derived growth factor (PDGF) signaling pathway plays important roles in the development and progression of human cancers, because these genes regulate the processes of cell proliferation, apoptosis, migration, invasion and metastasis21–23. The mechanism of the PDGF signaling is to activate important cancer-associated signaling, such as the RAS/PI3K/ERK/AKT signaling, to stimulate DNA synthesis24, 25. Studies have demonstrated a link between the PDGF signaling pathway and pancreatic cancer. For example, a high PDGFR-B expression level correlates with a poor disease-free survival in pancreatic cancer patients, which has been proposed as a possible target for attenuating metastasis in the p53 mutant tumors26. While overexpression of PDGF-BB was found to be associated with a decreased pancreatic cancer growth by increasing tumor pericyte content27, a microarray-based gene expression profiling revealed that the PDGF signaling pathway was differently expressed in pancreatic cancer cell line SW1990, compared with two control cell lines, the HPDE6c7 and PANC-1 cells 28.
Based on these findings, we hypothesized that genetic variants of the PDGF signaling pathway genes are associated with risk of pancreatic cancer. To test this hypothesis, we conducted a comprehensive meta-analysis of genetic variants in genes of the PDGF signaling pathway using previously published GWAS datasets from the PanScan (the Pancreatic Cancer Cohort Consortium, and the Pancreatic Cancer Case-Control Consortium) and Pancreatic Cancer Case Control Association Study. We also explored potential correlations of the identified SNPs with mRNA expression levels.
Methods and Materials
Study subjects
The subjects in this case-control study were from two published GWASs: the PanScan study (phs000206.v5.p3) and the Pancreatic Cancer Case Control Association Study (dbGaP #:phs000648.v1.p1). The PanScan GWAS has three phases, including PanScan I, II and III (1,921 cases and 2,016 controls in PanScan I; 1754 cases, 1889 controls in PanScan II; 1538 cases, 0 controls in PanScan III)8, 9, 13. Because there were no controls in PanScan III, we merged the PanScan II and PanScan III into one dataset PanScan II/III. The other Pancreatic Cancer Case Control Association Study was drawn from the Pancreatic Cancer Case-Control consortium (PanC4) and included case-control studies from the United States, Europe and Australia (4168 cases and 3814 controls)12, 29, 30. Subjects of European ancestry in two GWAS studies were selected in the analysis. As a result, these three GWAS datasets included a total of 15,423 individuals (8,477 cases and 6,946 controls) for the final analysis (Supplementary Table 1). A written informed consent was obtained from study participants. All methods were performed in accordance with the relevant guidelines and regulations for each of participating institutions, and the present study followed the study protocols approved by Duke University Health System Institutional Review Board.
Gene and SNP selection
The keyword “PDGF” was searched in Molecular Signatures Database (MSigDB) (http://www.broadinstitute.org/gsea/msigdb/index.jsp)31, and the resultant 129 related autosomal chromosomes genes involving in the PDGF signaling pathway from of BIOCARTA and REACTOME were included for further analysis (Supplementary Table 2).
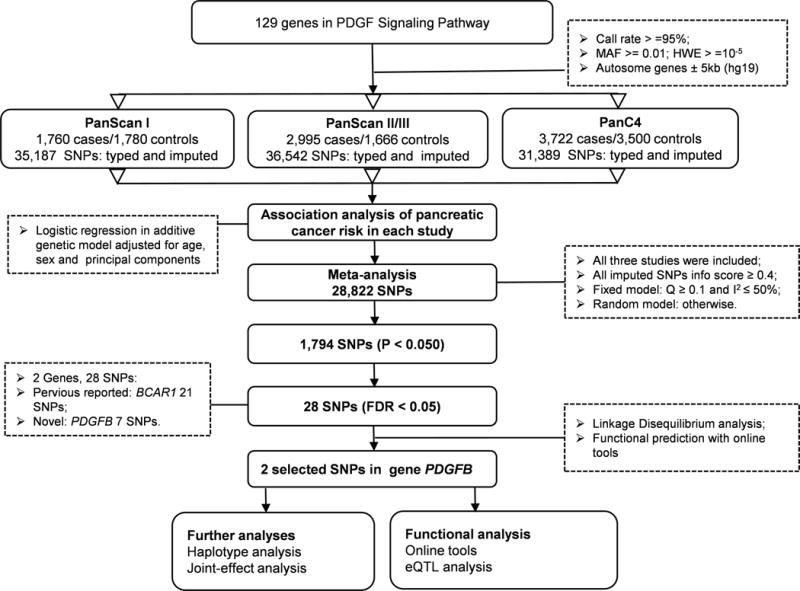
For these GWAS datasets, genotyping was performed using Illumina HumanHap550v3.0, Human610_Quadv1_B, HumanOmniExpress-12v1.0 and HumanOmniExpressExome-8v1. Genotyped SNPs located in these genes and their ± 500-kb flanking regions were extracted for imputation, which was conducted by using IMPUTE2 software with the reference panel from the 1000 Genomes (phase 1 release V3)32. Imputed SNPs with an information score ≥ 0.4 were qualified for further analysis. After quality control, there were 35187, 35142 and 31389 SNPs within 5kb up- and down-streams of genes in the PDGF signaling pathway from populations of the PanScan I, PanScan II/III and panC4 studies, respectively. The final meta-analysis contained 28,822 SNPs with the following inclusion criteria: a call rate ≥ 95%, minor allele frequency (MAF) ≥ 1%, and Hardy-Weinberg Equilibrium (HWE) exact P value ≥ 1×10−5 (Figure 1, Supplementary Figure 1).
Figure 1.
Statistical analysis
SNP association analysis was conducted first in a single locus analysis by using a logistic regression model with adjustment for age, sex and the top principal components in the genotyping data. For each SNP, an odds ratio (OR) and its 95% confidence interval (95% CI) were estimated by unconditional logistic regression analysis of case/control groups with adjustment for age and principal components using PLINK33. With the inverse variance method, a meta-analysis was further employed on the results of a log-additive model of 28,822 SNPs with Stata software (v12, State College, Texas, US). Cochran’s Q statistics and I2 were used to assess the heterogeneity (Q ≤ 0.10 and I2 ≤ 50%)34. The analysis adopted fixed-effects models, if no heterogeneity; otherwise random-effects models were used.
The false discovery rate (FDR) approach with a cut-off value of 0.05 was applied to control for multiple testing and to reduce the probability of false-positive findings35. The association between each SNP and pancreatic cancer risk was assessed by using an additive genetic model. In the combined risk genotype analysis, the multivariable stepwise logistic regression model was carried out to select the independent and significant SNPs, and the number of unfavorable genotypes (NUGs) of the significant SNPs was subsequently used to assess the classification performance of the model. All the individuals were divided into two groups: a low-risk group (0 NUGs) and a high-risk group (1–2 NUGs). Meanwhile, Haploview v4.2 was used to generate the Manhattan plots and LD plots. LocusZoom (http://locuszoom.sph.umich.edu/locuszoom/) was employed to construct the regional association plots by using European populations from the 1000 Genomes Project. The correlations between SNPs and corresponding mRNA expression levels were calculated by using a general linear regression model. Statistical analysis was carried out by R (version 3.3.1), SAS (version 9.4; SAS Institute, Cary, NC, USA) and PLINK (version 1.07), if not specified otherwise.
SNP-mRNA expression correlation analysis
Four online tools, i.e., F-SNP36 (http://compbio.cs.queensu.ca/F-SNP/), SNPinfo37 (http://snpinfo.niehs.nih.gov/), RegulomeDB 38 (http://regulomedb.org/) and HaploReg39 (http://www.broadinstitute.org/mammals/haploreg/haploreg.php) were used to predict the potential functions of the significant SNPs. The expression quantitative trait loci (eQTL) analysis was performed to estimate the associations between the SNPs and the mRNA expression levels of the corresponding gene by using the mRNA expression data from the lymphoblastoid cells of 373 Europeans available in the 1000 Genomes Project40 and 127 Europeans available in The Cancer Genome Atlas (TCGA) (https://tcga-data.nci.nih.gov/tcga/)41. The eQTL result of the online database genotype-tissue expression project (GTEx) was also taken into account in this analysis42. Comparisons of the targeted gene mRNA expression levels between tumor and adjacent normal tissues were performed in the Oncomine™ database (https://www.oncomine.org/)43.
Result
Single locus analysis
The workflow of the analysis is shown in Figure 1. Firstly, the associations between common SNPs (MAF ≥ 0.05) and pancreatic cancer risk for each of the three populations of European ancestry were estimated by using unconditional logistic regression analysis. The SNP number in the PDGF pathway genes was 35187, 36542 and 31389 for PanScan I, PanScan II/III and PanC4, respectively. Single locus analysis revealed that there were 1969, 2277 and 1575 SNPs with a nominal P < 0.05, respectively (Supplementary Figure 2). Secondly, a total of 28822 SNPs were included in a meta-analysis of the three populations, of which 1794 SNPs were associated with pancreatic cancer risk at P < 0.05 in an additive model and 28 SNPs on BCAR1 and PDGFB passed multiple testing corrections with FDR < 0.05 (Figure 2A and Table 1). Although the chromosome region (16q23.1) where BCAR1 is located has been previously reported by a GWAS13, the PDGFB gene located at 22q13.1 with seven SNPs (rs130651, rs6001516, rs35235663, rs56180415, rs6001512, rs71319025 and rs5757573) is a novel finding, for which we performed further in-silico analysis.
Figure 2.
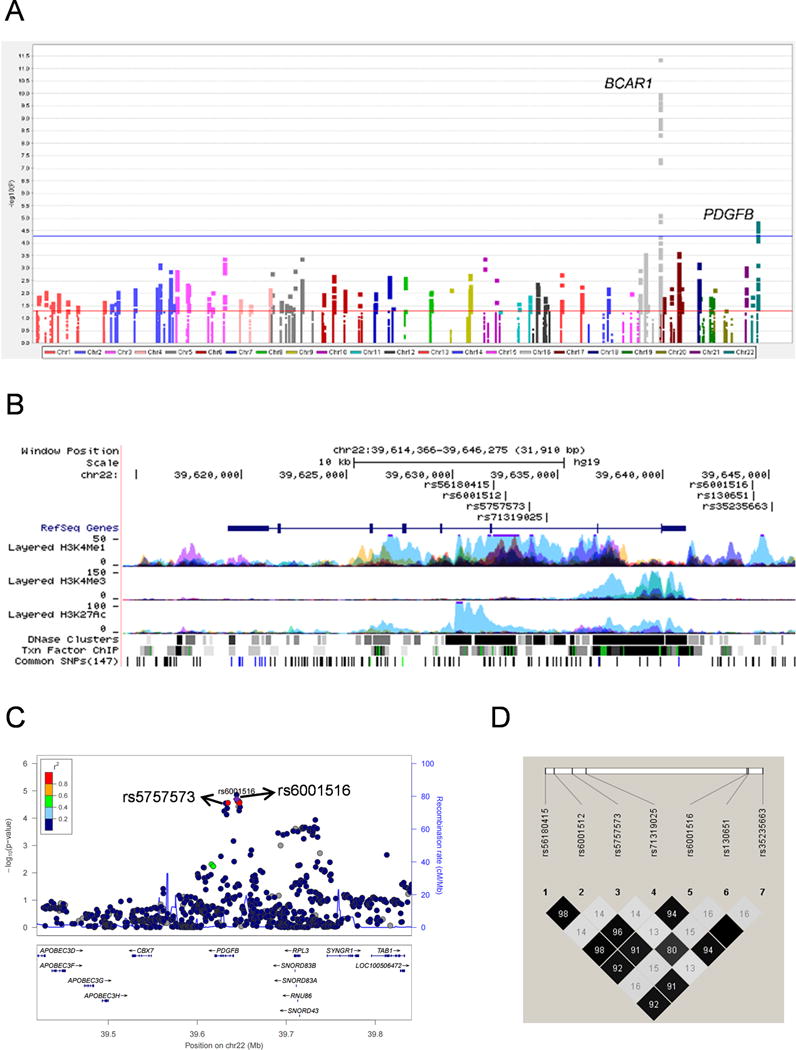
Table 1.
Associations between 28 SNPs in the PDGF signaling pathway and pancreatic cancer risk with FDR < 0.05
| SNP | Gene | Chr. | Position | Allelea | EAF1b | EAF2b | EAF3b | OR (95%CI) c | Pd | FDR |
|---|---|---|---|---|---|---|---|---|---|---|
| rs72802395 | BCAR1 | 16 | 75286484 | G/A | 0.057 | 0.066 | 0.077 | 1.36 (1.25–1.48) | 4.06E-12 | 1.17E-07 |
| rs73605136 | BCAR1 | 16 | 75299967 | C/T | 0.100 | 0.103 | 0.101 | 1.27 (1.18–1.37) | 1.04E-10 | 9.99E-07 |
| rs7185352 | BCAR1 | 16 | 75300248 | G/C | 0.099 | 0.103 | 0.101 | 1.27 (1.18–1.37) | 1.02E-10 | 9.99E-07 |
| rs73605139 | BCAR1 | 16 | 75300038 | C/T | 0.100 | 0.103 | 0.101 | 1.27 (1.18–1.36) | 1.39E-10 | 1.00E-06 |
| rs8046145 | BCAR1 | 16 | 75295021 | G/C | 0.100 | 0.103 | 0.105 | 1.26 (1.17–1.36) | 2.24E-10 | 1.29E-06 |
| rs13337397 | BCAR1 | 16 | 75295639 | C/A | 0.101 | 0.103 | 0.104 | 1.26 (1.17–1.35) | 3.95E-10 | 1.63E-06 |
| rs8063014 | BCAR1 | 16 | 75297673 | G/A | 0.100 | 0.103 | 0.104 | 1.26 (1.17–1.35) | 3.46E-10 | 1.63E-06 |
| rs3971235 | BCAR1 | 16 | 75279982 | A/ACC | 0.099 | 0.103 | 0.105 | 1.25 (1.16–1.35) | 1.03E-09 | 2.84E-06 |
| rs28439846 | BCAR1 | 16 | 75280378 | G/A | 0.099 | 0.103 | 0.105 | 1.25 (1.17–1.35) | 9.80E-10 | 2.84E-06 |
| rs28595463 | BCAR1 | 16 | 75281235 | C/T | 0.099 | 0.103 | 0.105 | 1.25 (1.16–1.34) | 1.08E-09 | 2.84E-06 |
| rs60879082 | BCAR1 | 16 | 75283065 | G/A | 0.099 | 0.103 | 0.105 | 1.25 (1.16–1.35) | 9.88E-10 | 2.84E-06 |
| rs3743614 | BCAR1 | 16 | 75280958 | C/T | 0.100 | 0.104 | 0.105 | 1.25 (1.16–1.34) | 1.22E-09 | 2.94E-06 |
| rs142593735 | BCAR1 | 16 | 75277446 | AAGAC/A | 0.099 | 0.104 | 0.105 | 1.25 (1.16–1.34) | 1.80E-09 | 3.98E-06 |
| rs3826110 | BCAR1 | 16 | 75277780 | G/A | 0.100 | 0.104 | 0.105 | 1.25 (1.16–1.34) | 1.94E-09 | 3.99E-06 |
| rs8048529 | BCAR1 | 16 | 75275630 | C/T | 0.099 | 0.104 | 0.105 | 1.25 (1.16–1.34) | 2.17E-09 | 4.18E-06 |
| rs13337017 | BCAR1 | 16 | 75272367 | C/T | 0.099 | 0.104 | 0.105 | 1.24 (1.16–1.33) | 4.18E-09 | 7.53E-06 |
| rs28690217 | BCAR1 | 16 | 75260329 | G/C | 0.097 | 0.102 | 0.104 | 1.23 (1.14–1.32) | 4.00E-08 | 6.79E-05 |
| rs2287990 | BCAR1 | 16 | 75258617 | C/T | 0.096 | 0.101 | 0.104 | 1.23 (1.14–1.32) | 4.48E-08 | 7.18E-05 |
| rs13331385 | BCAR1 | 16 | 75259218 | T/A | 0.097 | 0.102 | 0.104 | 1.22 (1.14–1.32) | 5.17E-08 | 7.84E-05 |
| rs1035539 | BCAR1 | 16 | 75276775 | A/G | 0.315 | 0.334 | 0.338 | 1.12 (1.06–1.17) | 7.03E-06 | 0.010 |
| rs11645191 | BCAR1 | 16 | 75274980 | G/A | 0.311 | 0.329 | 0.335 | 1.11 (1.06–1.17) | 1.17E-05 | 0.016 |
| rs130651 | PDGFB | 22 | 39644273 | A/G | 0.327 | 0.324 | 0.329 | 1.12 (1.06–1.17) | 1.39E-05 | 0.018 |
| rs6001516 | PDGFB | 22 | 39644203 | C/T | 0.065 | 0.071 | 0.073 | 1.21 (1.11–1.32) | 2.01E-05 | 0.025 |
| rs35235663 | PDGFB | 22 | 39645139 | G/A | 0.065 | 0.071 | 0.072 | 1.21 (1.11–1.32) | 2.41E-05 | 0.029 |
| rs56180415 | PDGFB | 22 | 39631963 | G/T | 0.064 | 0.070 | 0.073 | 1.20 (1.10–1.31) | 2.86E-05 | 0.031 |
| rs6001512 | PDGFB | 22 | 39632523 | G/A | 0.064 | 0.070 | 0.073 | 1.20 (1.10–1.31) | 2.90E-05 | 0.031 |
| rs71319025 | PDGFB | 22 | 39634444 | C/A | 0.064 | 0.071 | 0.073 | 1.20 (1.10–1.31) | 2.74E-05 | 0.031 |
| rs5757573 | PDGFB | 22 | 39633622 | T/C | 0.367 | 0.363 | 0.369 | 1.10 (1.05–1.16) | 4.70E-05 | 0.048 |
SNP, single nucleotide polymorphism; PDGF, platelet-derived growth factor; FDR, false discovery rate; Chr, chromosome; EAF, effect allele frequency; OR, odds ratio.
Reference allele/effect allele.
EAF1 was EAF in PanScan I controls; EAF2 was EAF in PanScan II/III controls; EAF3 was EAF in PanC4 controls.
Fixed effect models were used when no heterogeneity was found between studies (Q test P > 0.10 and I2 < 50.0%); otherwise, random effect models were used.
Meta-analysis of the three studies.
The results of the seven PDGFB SNPs in each of GWAS datasets and the final meta-analysis are summarized in Table 2 and Supplementary Figure 3. All SNPs showed a low heterogeneity among these three GWAS datasets (all Q-test P > 0.300 and I2 < 20.0, Table 2).
Table 2.
Associations between 7 SNPs in the PDGFB gene and pancreatic cancer risk in three GWAS datasets.
| SNP | Location | Allelea | PanScan I | PanScan II/III | PanC4 | Heterogeneity c | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||||||
| EAF | OR (95% CI)b | Pc | EAF | OR (95% CI)b | Pc | EAF | OR (95% CI)b | Pc | Q | I2 | |||
| rs130651 | 5′-Upstream | A/G | 0.327 | 1.11 (1.00–1.22) | 0.053 | 0.324 | 1.11 (1.01–1.22) | 0.029 | 0.329 | 1.13 (1.05–1.21) | 0.001 | 0.947 | 0.0 |
| rs6001516 | 5′-Upstream | C/T | 0.065 | 1.37 (1.14–1.64) | 7.57E-04 | 0.071 | 1.13 (0.96–1.33) | 0.134 | 0.073 | 1.18 (1.05–1.34) | 0.008 | 0.294 | 18.3 |
| rs35235663 | 5′-Upstream | G/A | 0.065 | 1.36 (1.13–1.63) | 8.97E-04 | 0.071 | 1.14 (0.97–1.34) | 0.123 | 0.072 | 1.18 (1.04–1.34) | 0.009 | 0.315 | 13.6 |
| rs56180415 | Intron | G/T | 0.064 | 1.36 (1.14–1.63) | 8.61E-04 | 0.070 | 1.15 (0.97–1.35) | 0.101 | 0.073 | 1.17 (1.03–1.32) | 0.014 | 0.313 | 14.0 |
| rs6001512 | Intron | G/A | 0.064 | 1.35 (1.12–1.62) | 0.001 | 0.070 | 1.15 (0.98–1.36) | 0.090 | 0.073 | 1.17 (1.03–1.33) | 0.013 | 0.380 | 0.0 |
| rs71319025 | Intron | C/A | 0.064 | 1.37 (1.14–1.64) | 7.28E-04 | 0.071 | 1.15 (0.98–1.36) | 0.092 | 0.073 | 1.16 (1.03–1.32) | 0.016 | 0.292 | 18.8 |
| rs5757573 | Intron | T/C | 0.367 | 1.09 (0.99–1.2) | 0.079 | 0.363 | 1.11 (1.02–1.21) | 0.021 | 0.369 | 1.11 (1.03–1.19) | 0.004 | 0.962 | 0.0 |
SNP, single nucleotide polymorphism; GWAS, genome-wide association study; EAF: effect allele frequency in controls; OR: odd ratio; CI: confidence interval;
Referring to “common allele/effect allele”.
Logistic regression analysis was adjusted for sex, age and principal components in each study.
Heterogeneity were defined as Q-test P ≤ 0.100 or I2 ≥ 50.0%.
Linkage disequilibrium (LD) and haplotype analysis
Further LD analysis revealed that of the seven PDGFB SNPs, five (rs6001516, rs35235663, rs56180415, rs6001512 and rs71319025) and another two SNPs (rs130651and rs5757573) shared a high LD, respectively (r2 ≥ 0.80, Figure 2C and 2D). According to online functional prediction and LD analysis, therefore, we selected two proxy PDGFB SNPs (rs5757573 and rs6001516) for further analysis.
Haplotype (ht) analysis was employed to evaluate the combined effect of the two proxy SNPs in PDGFB. There are four different haplotypes [ht1 (T-C), ht2 (C-C), ht3 (T-T), and ht4 (C-T)] at the rs5757573 and rs6001516 positions, but the frequency of ht3 was so low that we cannot provide a valid statistical interpretation. In addition, the results showed that haplotype ht4 including two risk alleles C and T was most significantly associated with an increased risk of pancreatic cancer in PanScan I, PanScan II/III, PanC4 and pooled datasets [OR (95%CI) = 1.38 (1.15–1.65), 1.18 (1.00–1.40), 1.19 (1.05–1.35), and 1.23 (1.12–1.34), respectively; P = 0.001, 0.052, 0.006 and 5E-06, respectively] (Table 3).
Table 3.
Associations between haplotypes of the two potentially functional SNPs (rs5757573 and rs6001516) and pancreatic cancer risk
| Haplotypea | Group
|
OR (95% CI)b | Pb | |
|---|---|---|---|---|
| Case (%) | Control (%) | |||
| PanScan I | ||||
| T-C | 2165 (61.5) | 2253 (63.3) | 1.00 | |
| C-C | 1061 (30.1) | 1081 (30.4) | 1.02 (0.92–1.13) | 0.707 |
| C-T | 294 (8.4) | 223 (6.3) | 1.38 (1.15–1.65) | 0.001 |
| T-Td | 0 (0) | 1 (0) | – | – |
| PanScan II/III | ||||
| T-C | 3679 (61.4) | 2122 (63.7) | 1.00 | |
| C-C | 1841 (30.7) | 980 (29.4) | 1.1 (0.99–1.19) | 0.099 |
| C-T | 469 (7.8) | 229 (6.9) | 1.18 (1.0–1.4) | 0.052 |
| T-Td | 1 (0.03) | 1 (0.03) | 0.6 (0.04–10.4) | 0.756 |
| PanC4 | ||||
| T-C | 4525 (60.8) | 4417 (63.1) | 1.00 | |
| C-C | 2305 (31) | 2083 (29.8) | 1.09 (1.01–1.17) | 0.028 |
| C-T | 608 (8.2) | 497 (7.1) | 1.19 (1.05–1.35) | 0.006 |
| T-Td | 4 (0.1) | 1 (0) | 3.84 (0.43–34.14) | 0.229 |
| Pooled datasetd | ||||
| T-C | 10369 (61.2) | 8792 (63.3) | 1.00 | |
| C-C | 5207 (30.7 | 4144 (29.8) | 1.07 (1.02–1.12) | 0.009 |
| C-T | 1371 (8.1) | 949 (6.8) | 1.23 (1.12–1.34) | 5E-06 |
| T-Td | 5 (0.03) | 3 (0.02) | 1.44 (0.34–6.05) | 0.623 |
OR, odds ratio; CI. Confidence interval; Dom, dominance genetic model; Rec, recessive genetic model;
Referring to rs5757573 allele- rs6001516 allele;
Adjusted for age, sex and data source;
The dataset merged all the population;
There are too few cases to prove statistically significant.
Joint-effect analysis
We also assessed the joint effect of the two identified SNPs in the presence of age and sex in a multivariate stepwise logistic regression model, in which the data source was also taken into consideration in the pooled dataset of PanScan and PanC4 studies. As a result, the two independent SNPs remained significantly associated with pancreatic cancer risk (Supplementary Table 4). The results showed that rs5757573 T/C was significantly associated with pancreatic cancer risk in all the genetic models, so was rs6001516 C/T but not in the recessive genetic model. In an additive model, the associations between these two SNPs and cancer risk indeed had a linear trend as the frequency of the minor allele increased (trend test: P = 0.0001 and P = 3.3E–05, respectively, Supplementary Table 5).
Consistent with previous results, individuals with genotypes of rs5757573 CT+CC and rs6001516 TC+TT had an increased risk of pancreatic cancer, compared with those with the wild-type genotype of each SNP in the pooled dataset (P = 0.0004 and P = 3.1E–05, respectively, Supplementary Table 5). Using a dominant model, we combined risk genotypes of rs5757573 CT+CC and rs6001516 TC+TT into a single variable as NUGs. The trend test indicated a significant association between increased NUGs and the risk of pancreatic cancer (P = 5E–06, Supplementary Table 6). Since the differences in age groups and gender are statistically significant in each dataset (Supplementary Table 1) and advancing age is a known risk factor for pancreatic cancer, we first performed subgroups (<60, 60–70, >70) analysis by age and found that the risk associated with NUG was more evident in 60–70 age group (OR = 1.19, 95% CI = 1.07–1.33, P = 0.002, Supplementary Table 7). In the stratified analysis by sex, we found that the risk associated with NUG was more evident in males (OR = 1.16, 95% CI = 1.06–1.27, P = 0.001, Supplementary Table 7). However, no interaction and heterogeneity were observed between these strata (P > 0.05, Supplementary Table 7).
Correlation analysis
Potential influence of the seven PDGFB SNPs on mRNA expression levels was preliminarily inferred through the online tools (Supplementary Table 3). Three SNPs (rs130651, rs6001516 and rs35235663) are located in the 5′ upstream region of PDGFB, and the other four SNPs are located in the intronic regions, but all of these seven SNPs are located in the enhancer region of histone H3 mono methyl K4 (H3k4me1) that marks active/poised enhancers and in a DNase hypersensitive site representing open and active chromatins (Figure 2B). Moreover, we used the Genotype-Tissue Expression Project (GTEx) database (http://www.gtexportal.org/home/) to perform eQTL and showed that in the whole blood samples, the rs5757573 C allele was associated down-regulated mRNA expression levels of PDGFB (P = 5.8E–06) in an additive model (Supplementary Figure 4F).
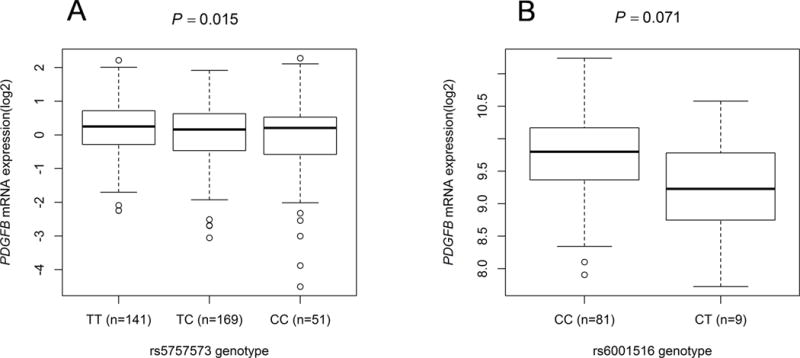
To substantiate the associations between the identified SNPs and pancreatic cancer risk, we also evaluated correlations between SNPs and mRNA expression levels of the corresponding genes in normal lymphoblastoid cell lines from 373 Europeans from the 1000 Genomes Project and in pancreatic tissues from 127 Europeans in the TCGA-PAAD Project. However, we failed to impute the genotype of rs5757573 based on the current quality control in the population of the TCGA-PAAD Project. As tested by Student t-test or linear regression analysis of the logarithm transformed expression values (log2) using the data of lymphoblastoid cell lines in the 1000 Genomes Project, the rs5757573 C allele was significantly associated with lower levels of PDGFB mRNA expression (P = 0.015 in an additive model [Figure 3A] and P = 0.050 and 0.039 in dominant and recessive models, respectively [Supplementary Figure 4D and 4E]) and the rs6001516 T allele had a marginal significant association with a decreased mRNA expression level of PDGFB in pancreatic tumor tissues from the TCGA (P = 0.071, Figure 3B). However, the eQTL results for SNP rs6001516 were non-significant in lymphoblastoid cell lines in either genetic models (Supplementary Figures 4A, 4B and 4C). In addition, we queried the eQTL results in the GTEx database and found that SNP rs5757573 also had a significant correlation with a decreased level of PDGFB mRNA expression in normal pancreatic tissues (P = 0.037, Supplementary Table 8), which is consistent with the results in whole blood cells from GTEx (Supplementary Figure 4F) and lymphoblastoid cell lines from the 1000 Genomes project (Figure 3A).
Figure 3.
Discussion
To investigate whether common germline variants in genes of the PDGF signaling pathway contribute to pancreatic cancer risk, we performed association analysis by using the data from three GWAS datasets: PanScan I, II / III from PanScan study and PanC4 from Pancreatic Cancer Case Control Association Study. Through meta-analysis, we identified two potential susceptibility loci for pancreatic cancer risk, which are located in PDGFB at 22q13.1. We further showed that both variants were independently or jointly associated with an increased pancreatic cancer risk, especially in the group with age of 60–70 and males. Further eQTL analysis revealed that those two SNPs might influence the mRNA expression levels of PDGFB.
The PDGF signaling pathway plays an important role in the occurrence or/and development of pancreatic cancer. A series of studies have progressively revealed the complexity of the PDGF signaling network, among which the core factors are the PDGFs and their tyrosine kinase receptors (PDGFRs)44. The PDGF isoforms are composed of four different polypeptide chains encoded by four different genes, namely PDGFA, PDGFB, PDGFC and PDGFD, corresponding to PDGF-A, PDGF-B, PDGF-C and PDGF-D. The four PDGF chains assemble into disulphide-bonded dimers via homo- or heterodimerization, and five different dimeric isoforms have been reported so far (PDGF-AA, PDGF-AB, PDGF-BB, PDGF-CC and PDGF-DD), which exert the effects by binding to, and activating, two protein tyrosine kinase receptors, alpha and beta45.
In the previous studies on the effect of the PDGF signaling pathway in pancreatic cancer, the high PDGFRβ expression levels correlate with a poor disease-free survival in pancreatic cancer patients with a gain-of-function activity of the mutant p53 that promotes invasion and metastasis26. However, one study investigated the mRNA expression in 13 human pancreatic tumors and found that although PDGF-A was expressed by nearly all of the specimens, 6 of 13 expressed low levels of PDGF-B46. Another recent study showed that the overexpression of PDGF-BB, which encoded by PDGFB, was found to inhibit tumor growth in human pancreatic cancer cells by increasing tumor pericyte content27. By using the online Oncomine database, we also found that the PDGFB expression levels in tumor tissue were lower than that in normal tissue in two studies from European populations. These reports and the Oncomine data suggest that PDGFB may act as a tumor suppressor in pancreatic cancer.
In the present study, we found that the PDGFB rs5757573 C and PDGFB rs6001516 T alleles were associated with an increased risk of pancreatic cancer possibly by decreasing the mRNA expression levels of PDGFB in Europeans populations. It has been reported that SNP rs5757573 is correlated with mean arterial pressure in a study exploring the relationship between SNPs in inflammatory genes and vascular stiffness47. Other SNPs in PDGFB were also reported to be associated with primary biliary cirrhosis, inflammatory bowel disease and multiple myeloma48–50. These results indicated that genetic variants in PDGFB had effects on inflammatory reaction, specifically involved in chronic inflammatory diseases. Unresolved chronic inflammation is implicated in all stages of cancer development, and an inflammatory tumor microenvironment is considered a hallmark of cancer, including pancreatic cancer51. During pathophysiological processes (e.g., inflammation, fibrosis and tumor growth), pericytes can be activated by a combination of growth factors, especially PDGF-B, which can recruit pericytes into tumor blood vessels by the PDGF-B/PDGFRβ signaling52. Increasing pericytes would inhibit the growth of endothelial cells 53. Taken together, genetic variants in PDGFB may lead to a decrease in pericytes by down-regulating the expression of PDGF-B in the stage of precancerous inflammation to provide a feasible tumor microenvironment for pancreatic cancer cells, then finally leading to an increase in the risk of pancreatic cancer. However, additional mechanistic studies are required to investigate the biological mechanisms underlying the observed associations.
There are some limitations in the present study. Firstly, we had no access to family history and others clinical data in publically available datasets, which may have an impact on pancreatic cancer risk. Also, there was no information about any treatment or survival in the phenotype data, and thus we were unable to neither adjust for these covariates in the risk assessment model, nor assess their effects on clinical prognosis. Secondly, there was no control data in the PanScan III GWAS, which might have led the merged PanScan II/III GWAS datasets heterogeneous. Thirdly, we were limited to evaluate whether a particular SNP had the biological function by only using the available online tools and eQTL analysis. More functional investigations are warranted to provide direct functional evidence to support our findings and to enable us consider which genetic variant in PDGFB may have played an important role in constructing the pancreatic cancer risk assessment model. Lastly, except for SNPs nearby BCAR1 (a reported locus), no other SNPs could passed the Bonferroni correction for multiple testing (0.05/35187 SNPs = 1.4×10−6), even after we used the Meff method to calculate the number of independent SNPs (0.05/7061 independent SNP= 7.08×10−6) 54, 55. Although we applied FDR to control for false positive findings and in-silico functional evidence has also been provided, further independent studies are required to replicate our findings.
In conclusion, our present study analyzed the associations between genetic variants in PDGF signaling pathway genes and pancreatic cancer risk in European populations. We identified two SNPs in PDGFB (rs5757573 T>C and rs6001516 C>T) that were associated with an increased risk of pancreatic cancer. Our results suggested that two identified SNPs in PDGFB may play a role in susceptibility to pancreatic cancer. Further population and functional validations of our findings are warranted.
Supplementary Material
Acknowledgments
As Duke Cancer Institute members, QW acknowledge support from the Duke Cancer Institute as part of the P30 Cancer Center Support Grant (Grant ID: NIH CA014236). QW was also supported by a start-up fund from Duke Cancer Institute, Duke University Medical Center.
The authors thank all the participants of the PanScan GWAS Study and Pancreatic Cancer Case Control Association Study. The authors would also like to acknowledge dbGaP repository for providing the cancer genotyping dataset.
PanScan
The PanScan project was funded in whole or in part with federal funds from the National Cancer Institute (NCI), US National Institutes of Health (NIH) under contract number HHSN261200800001E. Additional support was received from NIH/NCI K07 CA140790, the American Society of Clinical Oncology Conquer Cancer Foundation, the Howard Hughes Medical Institute, the Lustgarten Foundation, the Robert T. and Judith B. Hale Fund for Pancreatic Cancer Research and Promises for Purple. A full list of acknowledgments for each participating study is provided in the Supplementary Note of the manuscript with PubMed ID: 25086665. The dbGaP accession number for this study used in this manuscript is phs000206.v5.p3.
PanC4
The patients and controls for this study were derived from the following PANC4 studies: Johns Hopkins National Familial Pancreas Tumor Registry, Mayo Clinic Biospecimen Resource for Pancreas Research, Ontario Pancreas Cancer Study (OPCS), Yale University, MD Anderson Case Control Study, Queensland Pancreatic Cancer Study, University of California San Francisco Molecular Epidemiology of Pancreatic Cancer Study, International Agency of Cancer Research and Memorial Sloan Kettering Cancer Center. This work is supported by NCI R01CA154823 Genotyping services were provided by the Center for Inherited Disease Research (CIDR). CIDR is fully funded through a federal contract from the National Institutes of Health to The Johns Hopkins University, contract number HHSN2682011000111. The dbGaP accession number for this study used in this manuscript is phs000648.v1.p1.
TCGA
The results published here are in whole or part based upon data generated by The Cancer Genome Atlas pilot project established by the NCI and NHGRI. Information about TCGA and the investigators and institutions who constitute The Cancer Genome Atlas (TCGA) Research Network can be found at “http://cancergenome.nih.gov”. The TCGA SNP data analyzed here are requested through dbGAP (accession#: phs000178.v1.p1).
Footnotes
Conflict of interest
The authors disclose no potential conflicts of interest.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Ma J, Siegel R, Jemal A. Pancreatic cancer death rates by race among US men and women, 1970–2009. J Natl Cancer Inst. 2013;105:1694–700. doi: 10.1093/jnci/djt292. [DOI] [PubMed] [Google Scholar]
- 3.Kamisawa T, Wood LD, Itoi T, Takaori K. Pancreatic cancer. Lancet. 2016;388:73–85. doi: 10.1016/S0140-6736(16)00141-0. [DOI] [PubMed] [Google Scholar]
- 4.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–21. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 5.Pandol S, Gukovskaya A, Edderkaoui M, Dawson D, Eibl G, Lugea A. Epidemiology, risk factors, and the promotion of pancreatic cancer: role of the stellate cell. J Gastroenterol Hepatol. 2012;27(Suppl 2):127–34. doi: 10.1111/j.1440-1746.2011.07013.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stolzenberg-Solomon RZ, Amundadottir LT. Epidemiology and Inherited Predisposition for Sporadic Pancreatic Adenocarcinoma. Hematol Oncol Clin North Am. 2015;29:619–40. doi: 10.1016/j.hoc.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klein AP. Genetic susceptibility to pancreatic cancer. Mol Carcinog. 2012;51:14–24. doi: 10.1002/mc.20855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amundadottir L, Kraft P, Stolzenberg-Solomon RZ, Fuchs CS, Petersen GM, Arslan AA, Bueno-de-Mesquita HB, Gross M, Helzlsouer K, Jacobs EJ, LaCroix A, Zheng W, et al. Genome-wide association study identifies variants in the ABO locus associated with susceptibility to pancreatic cancer. Nat Genet. 2009;41:986–90. doi: 10.1038/ng.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petersen GM, Amundadottir L, Fuchs CS, Kraft P, Stolzenberg-Solomon RZ, Jacobs KB, Arslan AA, Bueno-de-Mesquita HB, Gallinger S, Gross M, Helzlsouer K, Holly EA, et al. A genome-wide association study identifies pancreatic cancer susceptibility loci on chromosomes 13q22.1, 1q32.1 and 5p15.33. Nat Genet. 2010;42:224–8. doi: 10.1038/ng.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diergaarde B, Brand R, Lamb J, Cheong SY, Stello K, Barmada MM, Feingold E, Whitcomb DC. Pooling-based genome-wide association study implicates gamma-glutamyltransferase 1 (GGT1) gene in pancreatic carcinogenesis. Pancreatology. 2010;10:194–200. doi: 10.1159/000236023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu C, Miao X, Huang L, Che X, Jiang G, Yu D, Yang X, Cao G, Hu Z, Zhou Y, Zuo C, Wang C, et al. Genome-wide association study identifies five loci associated with susceptibility to pancreatic cancer in Chinese populations. Nat Genet. 2011;44:62–6. doi: 10.1038/ng.1020. [DOI] [PubMed] [Google Scholar]
- 12.Childs EJ, Mocci E, Campa D, Bracci PM, Gallinger S, Goggins M, Li D, Neale RE, Olson SH, Scelo G, Amundadottir LT, Bamlet WR, et al. Common variation at 2p13.3, 3q29, 7p13 and 17q25.1 associated with susceptibility to pancreatic cancer. Nat Genet. 2015;47:911–6. doi: 10.1038/ng.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolpin BM, Rizzato C, Kraft P, Kooperberg C, Petersen GM, Wang Z, Arslan AA, Beane-Freeman L, Bracci PM, Buring J, Canzian F, Duell EJ, et al. Genome-wide association study identifies multiple susceptibility loci for pancreatic cancer. Nat Genet. 2014;46:994–1000. doi: 10.1038/ng.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Low SK, Kuchiba A, Zembutsu H, Saito A, Takahashi A, Kubo M, Daigo Y, Kamatani N, Chiku S, Totsuka H, Ohnami S, Hirose H, et al. Genome-wide association study of pancreatic cancer in Japanese population. PloS one. 2010;5:e11824. doi: 10.1371/journal.pone.0011824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pierce BL, Austin MA, Ahsan H. Association study of type 2 diabetes genetic susceptibility variants and risk of pancreatic cancer: an analysis of PanScan-I data. Cancer causes & control : CCC. 2011;22:877–83. doi: 10.1007/s10552-011-9760-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cotterchio M, Lowcock E, Bider-Canfield Z, Lemire M, Greenwood C, Gallinger S, Hudson T. Association between Variants in Atopy-Related Immunologic Candidate Genes and Pancreatic Cancer Risk. PloS one. 2015;10:e0125273. doi: 10.1371/journal.pone.0125273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arem H, Yu K, Xiong X, Moy K, Freedman ND, Mayne ST, Albanes D, Arslan AA, Austin M, Bamlet WR, Beane-Freeman L, Bracci P, et al. Vitamin D metabolic pathway genes and pancreatic cancer risk. PloS one. 2015;10:e0117574. doi: 10.1371/journal.pone.0117574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang H, Wei P, Duell EJ, Risch HA, Olson SH, Bueno-de-Mesquita HB, Gallinger S, Holly EA, Petersen G, Bracci PM, McWilliams RR, Jenab M, et al. Axonal guidance signaling pathway interacting with smoking in modifying the risk of pancreatic cancer: a gene- and pathway-based interaction analysis of GWAS data. Carcinogenesis. 2014;35:1039–45. doi: 10.1093/carcin/bgu010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Campa D, Pastore M, Gentiluomo M, Talar-Wojnarowska R, Kupcinskas J, Malecka-Panas E, Neoptolemos JP, Niesen W, Vodicka P, Delle Fave G, Bueno-de-Mesquita HB, Gazouli M, et al. Functional single nucleotide polymorphisms within the cyclin-dependent kinase inhibitor 2A/2B region affect pancreatic cancer risk. Oncotarget. 2016;7:57011–20. doi: 10.18632/oncotarget.10935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leenders M, Bhattacharjee S, Vineis P, Stevens V, Bueno-de-Mesquita HB, Shu XO, Amundadottir L, Gross M, Tobias GS, Wactawski-Wende J, Arslan AA, Duell EJ, et al. Polymorphisms in genes related to one-carbon metabolism are not related to pancreatic cancer in PanScan and PanC4. Cancer causes & control : CCC. 2013;24:595–602. doi: 10.1007/s10552-012-0138-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilhelm A, Aldridge V, Haldar D, Naylor AJ, Weston CJ, Hedegaard D, Garg A, Fear J, Reynolds GM, Croft AP, Henderson NC, Buckley CD, et al. CD248/endosialin critically regulates hepatic stellate cell proliferation during chronic liver injury via a PDGF-regulated mechanism. Gut. 2016;65:1175–85. doi: 10.1136/gutjnl-2014-308325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–25. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 23.Hoch RV, Soriano P. Roles of PDGF in animal development. Development. 2003;130:4769–84. doi: 10.1242/dev.00721. [DOI] [PubMed] [Google Scholar]
- 24.Welsh M, Claesson-Welsh L, Hallberg A, Welsh N, Betsholtz C, Arkhammar P, Nilsson T, Heldin CH, Berggren PO. Coexpression of the platelet-derived growth factor (PDGF) B chain and the PDGF beta receptor in isolated pancreatic islet cells stimulates DNA synthesis. Proc Natl Acad Sci U S A. 1990;87:5807–11. doi: 10.1073/pnas.87.15.5807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baghy K, Horvath Z, Regos E, Kiss K, Schaff Z, Iozzo RV, Kovalszky I. Decorin interferes with platelet-derived growth factor receptor signaling in experimental hepatocarcinogenesis. FEBS J. 2013;280:2150–64. doi: 10.1111/febs.12215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weissmueller S, Manchado E, Saborowski M, Morris JPT, Wagenblast E, Davis CA, Moon SH, Pfister NT, Tschaharganeh DF, Kitzing T, Aust D, Markert EK, et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell. 2014;157:382–94. doi: 10.1016/j.cell.2014.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCarty MF, Somcio RJ, Stoeltzing O, Wey J, Fan F, Liu W, Bucana C, Ellis LM. Overexpression of PDGF-BB decreases colorectal and pancreatic cancer growth by increasing tumor pericyte content. J Clin Invest. 2007;117:2114–22. doi: 10.1172/JCI31334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu Q, Fu R, Yin G, Liu X, Liu Y, Xiang M. Microarray-based gene expression profiling reveals genes and pathways involved in the oncogenic function of REG3A on pancreatic cancer cells. Gene. 2016;578:263–73. doi: 10.1016/j.gene.2015.12.039. [DOI] [PubMed] [Google Scholar]
- 29.Borgida AE, Ashamalla S, Al-Sukhni W, Rothenmund H, Urbach D, Moore M, Cotterchio M, Gallinger S. Management of pancreatic adenocarcinoma in Ontario, Canada: a population-based study using novel case ascertainment. Can J Surg. 2011;54:54–60. doi: 10.1503/cjs.026409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McWilliams RR, Bamlet WR, de Andrade M, Rider DN, Cunningham JM, Petersen GM. Nucleotide excision repair pathway polymorphisms and pancreatic cancer risk: evidence for role of MMS19L. Cancer Epidemiol Biomarkers Prev. 2009;18:1295–302. doi: 10.1158/1055-9965.EPI-08-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–25. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS genetics. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. American journal of human genetics. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. Bmj. 2003;327:557–60. doi: 10.1136/bmj.327.7414.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behavioural brain research. 2001;125:279–84. doi: 10.1016/s0166-4328(01)00297-2. [DOI] [PubMed] [Google Scholar]
- 36.Lee PH, Shatkay H. F-SNP: computationally predicted functional SNPs for disease association studies. Nucleic Acids Res. 2008;36:D820–4. doi: 10.1093/nar/gkm904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu Z, Taylor JA. SNPinfo: integrating GWAS and candidate gene information into functional SNP selection for genetic association studies. Nucleic acids research. 2009;37:W600–5. doi: 10.1093/nar/gkp290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, Karczewski KJ, Park J, Hitz BC, Weng S, Cherry JM, Snyder M. Annotation of functional variation in personal genomes using RegulomeDB. Genome research. 2012;22:1790–7. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40:D930–4. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lappalainen T, Sammeth M, Friedlander MR, t Hoen PA, Monlong J, Rivas MA, Gonzalez-Porta M, Kurbatova N, Griebel T, Ferreira PG, Barann M, Wieland T, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501:506–11. doi: 10.1038/nature12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rodgers K, Network CGAR Comprehensive molecular profiling of lung adenocarcinoma (vol 511, pg 543, 2014) Nature. 2014;514 doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Consortium GT. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–60. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fredriksson L, Li H, Eriksson U. The PDGF family: four gene products form five dimeric isoforms. Cytokine Growth Factor Rev. 2004;15:197–204. doi: 10.1016/j.cytogfr.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 45.Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22:1276–312. doi: 10.1101/gad.1653708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ebert M, Yokoyama M, Friess H, Kobrin MS, Buchler MW, Korc M. Induction of platelet-derived growth factor A and B chains and over-expression of their receptors in human pancreatic cancer. International journal of cancer. 1995;62:529–35. doi: 10.1002/ijc.2910620507. [DOI] [PubMed] [Google Scholar]
- 47.Schnabel R, Larson MG, Dupuis J, Lunetta KL, Lipinska I, Meigs JB, Yin X, Rong J, Vita JA, Newton-Cheh C, Levy D, Keaney JF, Jr, et al. Relations of inflammatory biomarkers and common genetic variants with arterial stiffness and wave reflection. Hypertension. 2008;51:1651–7. doi: 10.1161/HYPERTENSIONAHA.107.105668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cordell HJ, Han Y, Mells GF, Li Y, Hirschfield GM, Greene CS, Xie G, Juran BD, Zhu D, Qian DC, Floyd JA, Morley KI, et al. International genome-wide meta-analysis identifies new primary biliary cirrhosis risk loci and targetable pathogenic pathways. Nat Commun. 2015;6:8019. doi: 10.1038/ncomms9019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, Ripke S, Lee JC, Jostins L, Shah T, Abedian S, Cheon JH, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015;47:979–86. doi: 10.1038/ng.3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Swaminathan B, Thorleifsson G, Joud M, Ali M, Johnsson E, Ajore R, Sulem P, Halvarsson BM, Eyjolfsson G, Haraldsdottir V, Hultman C, Ingelsson E, et al. Variants in ELL2 influencing immunoglobulin levels associate with multiple myeloma. Nat Commun. 2015;6:7213. doi: 10.1038/ncomms8213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pesic M, Greten FR. Inflammation and cancer: tissue regeneration gone awry. Curr Opin Cell Biol. 2016;43:55–61. doi: 10.1016/j.ceb.2016.07.010. [DOI] [PubMed] [Google Scholar]
- 52.Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 53.Darland DC, D’Amore PA. TGF beta is required for the formation of capillary-like structures in three-dimensional cocultures of 10T1/2 and endothelial cells. Angiogenesis. 2001;4:11–20. doi: 10.1023/a:1016611824696. [DOI] [PubMed] [Google Scholar]
- 54.Gao X, Starmer J, Martin ER. A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Genetic epidemiology. 2008;32:361–9. doi: 10.1002/gepi.20310. [DOI] [PubMed] [Google Scholar]
- 55.Gao X. Multiple testing corrections for imputed SNPs. Genetic epidemiology. 2011;35:154–8. doi: 10.1002/gepi.20563. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.