

Abstract
A model that predicts retention for peptides using a HALO® penta-HILIC column and gradient elution was created. Coefficients for each amino acid were derived using linear regression analysis and these coefficients can be summed to predict the retention of peptides. This model has a high correlation between experimental and predicted retention times (0.946), which is on par with previous RP and HILIC models. External validation of the model was performed using a set of H. pylori samples on the same LC-MS system used to create the model, and the deviation from actual to predicted times was low. Apart from amino acid composition, length and location of amino acid residues on a peptide were examined and two site-specific corrections for hydrophobic residues at the N-terminus as well as hydrophobic residues one spot over from the N-terminus were created.
Keywords: Peptide, Retention, Hydrophilic Liquid Interaction Chromatography, Amino Acid, Retention Time, Prediction Model
The use of hydrophilic interaction liquid chromatography (HILIC) columns has grown tremendously due to the various types of columns available as well as their ability to separate polar analytes. Although reversed-phase (RP) chromatography is the method of choice for proteomic experiments, HILIC is able to separate peptides that are not retained on RP columns, or those that may exhibit inadequate selectivity differences. These two complimentary chromatographic techniques have even been paired as a two dimensional approach for more complex separations [1–3].
Standard proteomic experiments have long used chromatography coupled to mass spectrometry for analysis. In these experiments, peptides are identified by their mass-to-charge (m/z) ratio and fragmentation data, which usually involves database searching. While this technique is very common, researchers may have trouble identifying multiple peptides with the same m/z ratio in which fragmentation data is insufficient in identification. To this end, chromatography can be used to further the identification process, as retention times of peptides are related to their amino acid sequences. By predicting what the retention would be, peptides can quickly be identified by their m/z ratio as well as their retention time, and peptides with the same mass but different sequences can be identified separately due to differing retention times. This can decrease the time spent in identification as well as increase the confidence of identifications [4,5] Targeted approaches can also benefit from this, as the time of analysis spent looking for specific peptides can be shortened.
O’Hare and Nice were the first researchers to notice that peptide retention was directly related to amino acid composition, and this discovery in 1979 opened the door for models that were able to predict the retention of peptides [1,2,4–17]. Almost all of these models have been made using RP as the means of separation, but there have been several HILIC models that have been made recently [1,2,4,17]. These models derive coefficients for each amino acid, describing their hydrophilic or hydrophobic behavior. When summed together, the coefficients can accurately predict elution position for a particular column, operated under defined conditions. The processes to create these models can range from using linear regression analysis to substituting amino acids on a synthetic peptide, and can even include sequence corrections, size corrections, or various modifications [4,7,9,10–13,17]. Even though most of the prediction models have been created for RP columns, the number of HILIC models has increased as the types of HILIC columns available have increased throughout the years. The first HILIC peptide prediction model was created by Yoshida in 1998 on an TSK Amide-80 column, and then Gilar et. al. created coefficients for three HILIC columns with different stationary phases: bare silica, bridge-ethyl hybrid silica, and an amide modified bridge-ethyl hybrid silica [1,2]. All of these models have very high correlation between experimental and actual retention times of peptides (in the range of 0.92–0.97), however they have also shown that the amino acid coefficients can change with different HILIC stationary phases and are also dependent on operating conditions such as pH, which affect ionization, thus polarity, of amino acyl side chains. Due to this concern, new peptide retention models need to be made for new HILIC stationary phases, operated under specific conditions, such as temperature, gradient profile, and mobile phase components.
In this paper, we have created a HILIC peptide retention prediction model using 297 peptides from various proteomic samples for a HALO® penta-HILIC column. Coefficients for each amino acid have been derived using linear regression analysis and the correlation is very high (0.94553), indicating the agreement between predicted and actual retention times. We also introduce a site-specific correction for peptides with hydrophobic amino acids at the N-terminus, criteria for peptides selection, and retention expression in glucose units (GU) so that the model can be ran on any LC-MS system. This useful model will be able to increase protein confidence and reduce the time spent in identification by predicting the retention of peptides.
EXPERIMENTAL SECTION
1.1 Protein Digestion
Human IgGs were separated from human serum (Sigma-Aldrich, St. Louis, MO, USA) using a HiTrap™ Protein G column (General Electric Company, Fairfield, CT, USA). Myoglobin, transferrin, concanavalin A, fetuin, cytochrome C, lysozyme, ribonuclease B, carbonic anhydride, and dextran were purchased from Sigma-Aldrich (St. Louis, MO, USA). Bovine serum albumin was purchased from Waters (Milford, MA, USA). These proteins as well as yeast proteins, mosquito cuticular proteins, and H. pylori proteins were reduced using 10-mM DTT and then alkylated using 55-mM IDA, which were both purchased from Sigma Aldrich (St. Louis, MO, USA). Sequencing-grade trypsin or chymotrypsin purchased from Promega (San Luis Obispo, CA, USA) was added (50:1, w/w, protein/trypsin) and samples were incubated overnight.
1.2 LC-MS/MS Settings and Instrumentation
Data were acquired using a Finnegan LTQ (Thermo-Fisher, San Jose, CA, USA) and an 1100 Series Capillary LC system (Agilent Technologies, Palo Alto, CA, USA) with an ESI source that used spray tips made in-house. Samples were suspended in 25%H2O, 75% ACN and 0.1% formic acid (Sigma-Aldrich, St. Louis, MO, USA) and 8 μL of each sample was injected into the LC. Peptides were separated using a 200μm × 150 mm HALO® penta-HILIC column packed with 2.7-μm diameter superficially porous particles (Advanced Materials Technology, Wilmington, DE, USA). The separation was carried out at room temperature due to the absence of a column oven on this LC-MS system. The gradient used for each sample was 95–30% ACN over 90 minutes at a 2μL/min flow rate, at ambient laboratory temperature of 20°C. The acetonitrile mobile phase contained 0.1% formic acid (Sigma Aldrich, St. Louis, MO, USA) and the stronger elution solvent (water) contained 50 mM ammonium formate (Thermo-Fisher, San Jose, CA, USA). Acetonitrile was used as the organic solvent due to its compatibility with ESI as well as its low viscosity, which allows for higher flow rates to be used with lower back pressures. The low pH of the mobile phase (around 3–4) was used to enhance the protonation of analytes, which will increase the sensitivity in the mass spectrometer, as well as influence the retention of peptides that contain charged residues. The settings for the mass spectrometer included taking the five most intense ions in positive ionization mode from each full mass spectrum (m/z 400–2000) for fragmentation using collision-induced dissociation, and the resulting MS/MS spectra were recorded.
To make sure that this model would be universal, some of the same digested proteins were run on a 4000 Q Trap (AB Science, Chatham, NJ, USA). Peptides were separated by a 2.1 mm × 15 cm HALO® penta-HILIC column packed with 2.7-μ diameter superficially porous particles using a Nexera UFLC (Shimadzu, Columbia, MD, USA). The gradient used the same solvents described above, with gradient elution of 78–48% ACN over 80 minutes at a 0.4-mL/min flow rate. Spectra were obtained using an ESI source.
1.3 Database Search Parameters
The resulting RAW files were converted using Trans-Proteomic Pipeline (Seattle Proteome Center, Seattle, WA, USA), then the MS/MS spectra of each sample were searched using Mascot (Matrix Scientific, Boston, MA, USA) against corresponding protein databases of theoretical MS/MS spectra. The following parameters were utilized in Mascot: a peptide tolerance of 1000 ppm, a fragment tolerance of 0.6 Da, two max missed cleavages of trypsin, and a fixed modification of carbamidomethyl (C).
1.4 Selection of Peptides for Prediction Model and Post-Run Data Analysis
All peptides that had a higher Mascot score than 10 were considered. Peptide retention times were found by hand from RAW files from the apex of the peaks using Xcalibur software (Thermo-Fisher, San Jose, CA, USA), and resulting MS/MS data were visually inspected to verify the peptide assignments. Chromatographic peaks for each peptide had to have a peak asymmetry value of between 0.25 - 4, and peptides exhibiting peak widths greater than 5.5 minutes were excluded from analysis. Peptides had to be fewer than 15 amino acids in length. Peptide retention times in minutes were converted to glucose units based on dextran samples that were run immediately before. Linear regression analysis using StatPlus (AnalystSoft, Walnut, CA, USA) was used to find the coefficients for each amino acid and 297 peptides were used in this study.
RESULTS AND DISCUSSION
2.1 Amino Acid Coefficients
Different HILIC columns exhibit different selectivites from one another, making the creation of a new model for the penta-HILIC stationary phase a requirement in order to predict peptide retention [2,3]. To this end, linear regression analysis was used to find coefficients for each amino acid, and these results are shown in Table 1. Using Equation 1 shown below, predicted retention times of peptides, RT, can be calculated, where Li is the amount of residue i in the peptide, AAi is the amino acid coefficient of residue i, and b0 is the intercept of the model:
| (1) |
The predicted retention times of the 297 peptides in this model were plotted against their actual times and the derived correlation coefficient is 0.94553, which expresses the minimal differences in deviation between actual and predicted retention times using these amino acid coefficients (Supplementary Figure 1). This value is on the higher end of previous RP and HILIC peptide retention prediction models [1,2,4–17]. A bar graph is shown in Figure 1 that displays the distribution of the experimental-calculated deviations compared to a theoretical Gaussian distribution. This figure shows that the actual and theoretical distributions of the deviations match up very well, with actual data having slightly more instances at lower deviations in general.
Table 1.
Derived coefficients for each amino acid. Red amino acids are hydrophilic, blue amino acids are hydrophobic, and the contribution to retention for green amino acids did not achieve statistical significance.
| Amino Acid | Coefficient |
|---|---|
| Alanine (A) | 0.164 |
| Cysteine (C)* | 0.293 |
| Aspartic Acid (D) | 0.800 |
| Glutamic Acid (E) | 0.719 |
| Phenylalanine (F) | −0.967 |
| Glycine (G) | 0.233 |
| Histidine (H) | 1.564 |
| Isoleucine (I) | −0.615 |
| Lysine (K) | 2.121 |
| Leucine (L) | −0.799 |
| Methionine (M) | −0.337 |
| Asparagine (N) | 0.610 |
| Proline (P) | 0.129 |
| Glutamine (Q) | 0.703 |
| Arginine (R) | 1.828 |
| Serine (S) | 0.334 |
| Threonine (T) | 0.357 |
| Valine (V) | −0.306 |
| Tryptophan (W) | −1.138 |
| Tyrosine (Y) | −0.430 |
| Intercept | 1.535 |
| R-Squared Value | 0.94553 |
Carbamidomethylated cysteine
Figure 1.
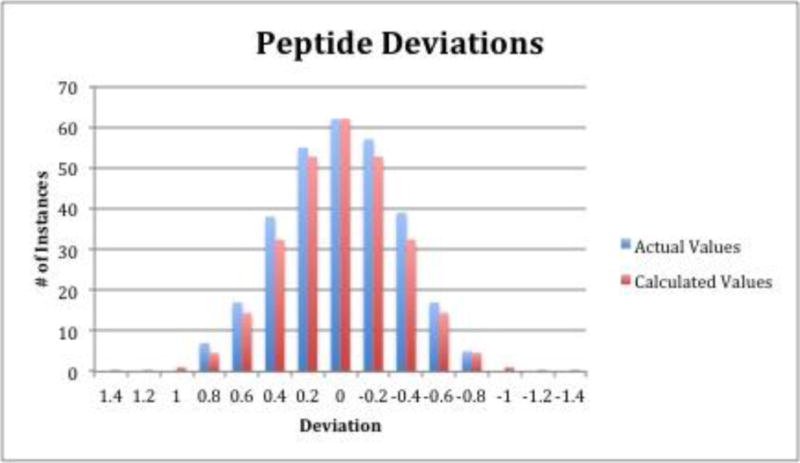
Deviations of actual and theoretical retention times of the 297 peptides used in the study compared against a calculated Gaussian distribution of the data. Each deviation has a range of +/− 0.1 GU, so the value listed at 0 GU would include the range 0.1 to −0.1 GU.
The amino acid residues that have positively charged side chains (arginine, lysine, and histidine) have a positive effect on retention and have the largest effect overall, consistent with other studies [1,2,4,18]. These side-chains interact with the stationary phase to a greater extent and increase the retention of the peptides. Aspartic acid and glutamic acid have negatively charged side chains that also increase retention, but they do not have as great of an effect as the positively charged side chains. This is because the pH of the mobile phase (around 3) is lower than that of the pKa of both residues (3.86 for aspartic acid and 4.07 for glutamic acid), making them neutral and thus interact less strongly with the stationary phase than a charged species. The large, aromatic or aliphatic amino acid residues such as phenylalanine, tryptophan, and tyrosine all decreased the retention of peptides due to the hydrophobic nature of the side chains minimally interacting with the highly polar stationary phase. While the coefficients for these residues are inversely related to reverse phase models, Gilar, et. al. showed that it is not necessarily a linear correlation, and that HILIC and RP can be combined in multidimensional HPLC for more complex separations [2]. Several amino acids that had p-values indicating statistical insignificance for contribution to retention. These amino acids are small (i.e. glycine and alanine) or had both hydrophobic and hydrophilic characteristics (i.e. proline and methionine). It is noted that retention is described with a comparably large intercept value, which may describe the hydrophilic character of both the N and C termini on a peptide (ionization of carboxylic acid amine functional groups), as well as the time it takes for the unretained peptides to travel through the column and reach the MS detector.
All of the coefficients are expressed in glucose units (GU) rather than minutes to permit the model to be used on any LC-MS system. Procainamide-labeled dextran samples were analyzed twice before each sample, averaged, and then the retention time of peptides in minutes was converted to GU based on the logarithmic fit for the dextran samples. Dextran samples elute in order of increasing monosaccharide number, providing a reference for the retention times of peptides. Conversion to time units (minutes) is simple, and an example of that is shown in Figure 2. This approach allows the model to be used regardless of LC-MS system as long as the dextran standard is employed for calibration before the samples. System calibration also permits modifications to the LC-MS system, such as capillary line changes in length or diameter, or instrumental changes, such as inclusion of additional detectors (absorbance or fluorescence). To ensure that dextran is a suitable retention time calibrant, a set of peptide standards were run on different LC-MS systems over the course of a month and the relative retention times of the standards exhibited minimal changes when suitably calibrated.
Figure 2.
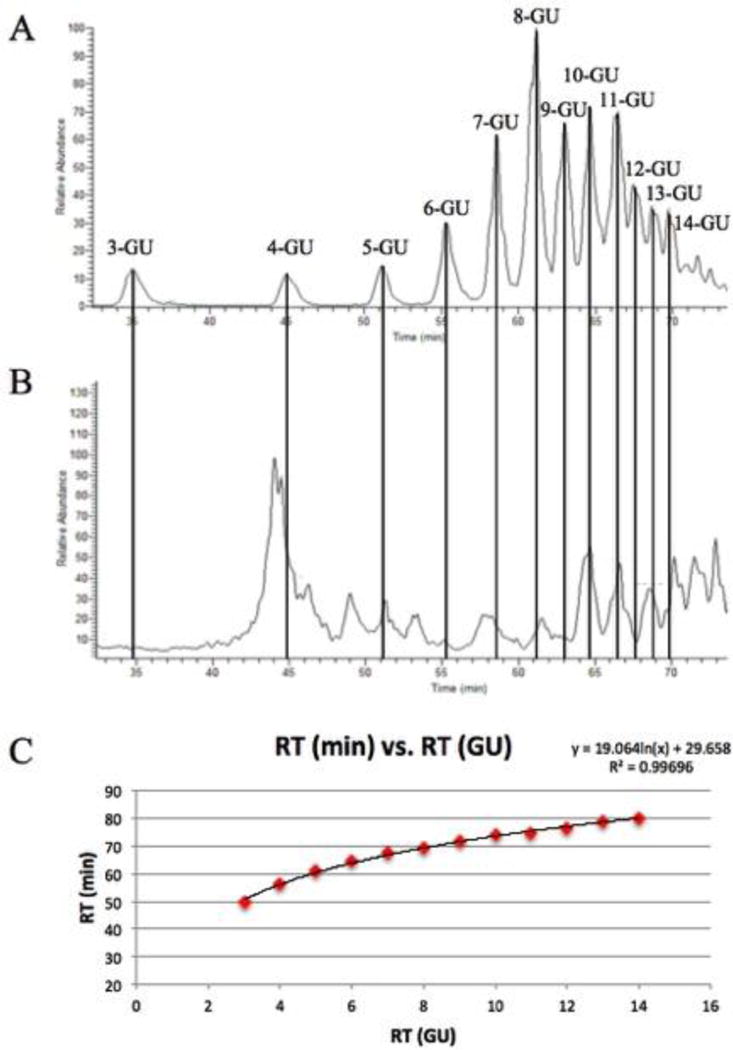
Procainamide-labeled dextran samples served as a retention time calibrant to the peptides used in the model. Monosaccharides elute in terms of increasing linkage (A) and then peptide retention times (B) were converted from minutes to glucose units (GU) using the logarithmic fit of the dextran units (C).
There have been previously made models that relied on other calibration methods, such as Gilar’s, which used the percentage of organic solvent at the time of peptide elution [2]. While this technique has shown to be suitable for retention prediction on a single LC-MS system, it may be unreliable across multiple systems due to differing dwell volumes and variations in the accuracy of the percentage reading from system to system. One of the main focuses of this model was for it to be able to be used on completely different LC-MS systems, and running a retention time calibrant on each system mitigates this problem. Instead of having to worry about the accuracy of the percentage reading or accounting for the dwell volume, all that is required for accurate calibration is a simple dextran run on the system.
2.2 Test Peptides
To test the model’s accuracy of prediction, H. pylori samples were run on the same LC-MS setup as the peptides used to create the model. From the test samples, 64 peptides fit the selection criteria and were investigated. Figure 3 shows the actual times of the test peptides plotted against the predicted times, which yielded a high correlation coefficient of 0.96444, slightly higher than the correlation coefficient of the model itself. This shows that the model is more than capable of predicting retention times for biologically relevant samples. Of the 64 test peptides, 38 of them had lower actual retention times than their predicted ones (59%), which were calculated using Equation 1. Over the course of a 90-minute long gradient the average deviation from actual to predicted times was only 0.35 GU, or 1.72 min, indicating the accuracy of prediction.
Figure 3.
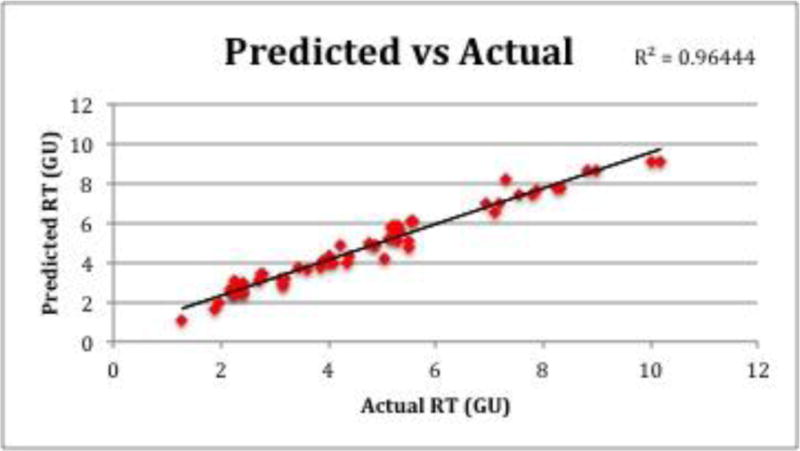
Predicted vs. actual times of H. pylori test peptides
A 4000 Q-Trap with a Nexera UFLC system was used to test the accuracy of prediction of the model on a completely different LC-MS system. BSA and carbonic anhydrase were run on this system that had a different column size, flow rate, gradient, column temperature, and length of analysis. Peptides identified on both LC-MS systems only differed by an average of 2.29 minutes (0.52 GU) and were within 3.73% of each other, indicating that despite the LC-MS system and numerous gradient conditions being different from the setup used to create the model, predicted retention times were still very close to actual retention times. This allows researchers to use the model for prediction even if there are contrasting gradient conditions.
2.3 Peptide Retention Prediction Purpose and Correlation with Database Searching
The purpose for peptide retention prediction is threefold. First, it can provide a quicker data analysis as peptides can be identified from their m/z ratio as well as their retention time, eliminating the need for database searching, which can be time-consuming. This is similar to accurate mass and time (AMT) tagging technology. Second, retention prediction is able to filter out false positives and lead to more confident identifications by comparing actual retention times to theoretical retention times. When MS2 is insufficient for identifying peptides with the same m/z ratio, retention time prediction offers an additional identification layer. Finally, it can help in isomeric identification. In a recent study, our lab was able to fully separate the n-Asp and isoAsp versions of the peptide GFYPSDIAVEWESNGQPENNYK, which are indistinguishable in the mass spectrometer. The derived retention coefficients for these two modifications were different, allowing the prediction model to distinguish between the two peptides [19]. This shows that when MS2 data is inadequate for separately identifying different species, retention time prediction can help.
In database searching, peptides are scored based on the “match” between experimental data and their database sequence. The higher the score of the peptide match, the less likely it is a random match. To test if our model was similar in this aspect, namely that peptides with lower deviations from actual to predicted retention times would have a lower probability of being a random match, 100 peptides from H. Pylori proteins were ran on the same LTQ setup as the peptides used to create the model, and their deviations were compared to their Mascot scores. Figure 4 shows this comparison, as peptides were grouped based on their Mascot score and plotted against their average deviations. The resulting data shows an agreement with Mascot score and deviation from predicted to actual retention times, as the peptides with lower deviations have higher Mascot scores and vice versa. It also shows that peptides with lower Mascot scores and higher deviations between actual and predicted times have much larger standard deviations. This indicates that peptides with actual retention times that are very close to theoretical ones are less likely to be false positives.
Figure 4.

Deviation of actual retention times and theoretical retention times plotted against Mascot score
2.4 The Effect of Amino Acid Location
Site-specific trends were investigated in the dataset of 297 peptides, specifically at the N-terminus due to the use of trypsin on most of the samples. It was found that 44 out of 70 (63%) peptides with hydrophobic residues at their N-terminus eluted earlier than predicted and optimized coefficients were created for this, as shown in Table 2. Using an iterative process that maximized the correlation coefficient, it was found that a 10% decrease in the value of the original hydrophobic amino acids (phenylalanine, isoleucine, leucine, tryptophan, and tyrosine) resulted in optimized coefficients that had a R-squared value of 0.95552, which is a 0.00952 increase in the original R-squared value. With these optimized coefficients, the average deviation between actual and predicted retention times dropped from 0.255 GU to 0.246 GU, and the sum of deviation went from −5.133 GU to −0.259 GU, increasing the fit of the model. With this alteration, 37 out of the 70 (53%) peptides eluted earlier than predicted, evening the distribution of predicted retention times greater and smaller than actual retention times. These coefficients are only to be used for the first hydrophobic amino acids at the N-terminus of a peptide and no others. Hydrophilic amino acids at the N-terminus were also investigated, but although there was a slight trend (40 of 73, 55%) of peptides with actual retention times larger than their predicted ones, the optimization of the coefficients would be negligible and would not help increase the correlation coefficient.
Table 2.
Optimized coefficients for the first hydrophobic amino acid at the N-terminus
| Amino Acid | Coefficient |
|---|---|
| Phenylalanine (F) | −1.063 |
| Isoleucine (I) | −0.676 |
| Leucine (L) | −0.879 |
| Tryptophan (W) | −1.252 |
| Tyrosine (Y) | −0.473 |
| R-Squared Value | 0.94620 |
Peptides with hydrophobic residues one position over from the N-terminus were also investigated for trends, and it was found that 11 of 15 (73%) of peptides that fit this description had actual retention times that were shorter than their predicted ones. Using the same iterative process, it was found that a 5% decrease in the value of the original hydrophobic coefficients resulted in optimized coefficients that had an elevated R-squared value of 0.95563, which is a 0.00963 increase in the original R-squared value. These optimized coefficients are found in Table 3 and are only for the hydrophobic residue of a peptide that is one position over from the N-terminus, while the first residue at the N-terminus is also a hydrophobic residue. In addition to an increased R-squared value, the average deviation dropped from 0.199 GU to 0.193 GU and the sum of deviation went from −1.593 GU to −0.254 GU using the optimized coefficients, with a more even distribution of predicted retention times that were greater and smaller than actual retention times (8 out of 15 (53%) peptides had actual retention times shorter than predicted ones). Hydrophilic residues in this position were also investigated, but it was found again that even though there was a significant trend (10 of 15, 67%), the optimization of the coefficients would again be negligible and would not help the correlation of the model.
Table 3.
Optimized coefficients for the second hydrophobic amino acid at the N-terminus
| Amino Acid | Coefficient |
|---|---|
| Phenylalanine (F) | −1.015 |
| Isoleucine (I) | −0.646 |
| Leucine (L) | −0.839 |
| Tryptophan (W) | −1.195 |
| Tyrosine (Y) | −0.451 |
| R-Squared Value | 0.94600 |
A potential reason that the hydrophobic residues are having a greater impact than the hydrophilic residues at the N-terminus is due to the fact that the N-terminus is already charged and hydrophilic, allowing hydrophobic residues to change the interactions with the stationary phase to a greater extent than the hydrophilic residues. There have been some models that have incorporated optimized coefficients that are based on the distance from the termini, but excluding the coefficients derived from hydrophobic residues one spot over from the N-terminus, there were no other identified trends that suggested that doing the same would improve the fit of the model [4,5,17].
2.5 The Effect of Peptide Length
Although amino acid composition contributes the most to peptide retention, other models have shown that length has an effect as well [4,20–22]. Mant, et al. showed that the retention of peptides that have 15 or more residues in their sequence deviated more than expected and cannot be overlooked [21,22]. Table 4 shows peptides from standard proteins that were not used in this model due to their length, and the average deviations (1.06 GU, or 4.80 min.) are 3–4 times higher than peptides with shorter sequences that were used in the model. A potential reason for this could be due to longer peptides more easily forming second order structures and interacting with the stationary phase in a way that cannot be predicted accurately. This consideration was applied to the creation of this model, as the cutoff for peptide size was 15 amino acids in length.
Table 4.
Retention of peptides with 15 or more amino acids
| Peptide | Length | Deviation (min) | Deviation (GU) |
|---|---|---|---|
| RPCFSALTPDETYVPK | 16 | 6.01 | 1.88 |
| LFTFHADICTLPDTEK | 16 | 8.90 | 2.35 |
| NTDGSTDYGILQINSR | 16 | 0.58 | 0.16 |
| EDLIWELLNQAQEHFGK | 17 | 0.37 | 0.09 |
| GITWGEETLMEYLENPK | 17 | 5.96 | 0.98 |
| VYACEVTHQGLSSPVTK | 17 | 24.24 | 4.33 |
| TTPPVLDSDGSFFLYSK | 17 | 6.06 | 0.95 |
| GITWGEETLMEYLENPKK | 18 | 7.54 | 1.79 |
| TVAAPSVFIFPPSDEQLK | 18 | 3.33 | 0.56 |
| RTVAAPSVFIFPPSDEQLK | 19 | 1.20 | 0.29 |
| AAPSVTLFPPSSEELQANK | 19 | 0.16 | 0.04 |
| ANPTVTLFPPSSEELQANK | 19 | 0.91 | 0.24 |
| EVQLVQSGGGLVQPGGSLR | 19 | 5.45 | 1.28 |
| DLILQGDATTGTDGNLELTR | 20 | 3.84 | 1.10 |
| VDNALQSGNSQESVTEQDSK | 20 | 0.36 | 0.16 |
| GLVLIAFSQYLQQCPFDEHVK | 21 | 7.96 | 1.64 |
| GFYPSDIAVEWESNGQPENNYK | 22 | 0.89 | 0.27 |
| SPDSHPADGIAFFISNIDSSIPSGSTGR | 28 | 2.61 | 0.89 |
Applying an elevated column temperature could disrupt a peptide’s secondary structure so that it interacts in a more predictable manner with the stationary phase. Long peptides from human IgGs, BSA, transferrin, concanavalin A, lysozyme, and cytochrome C were run at column temperatures of 25°C and 60°C, and the data are shown in Table 5. It is clear from this data that the higher column temperature decreases the deviation from predicted times. However, some peptides run at 25°C were closer to the predicted times, suggesting that not all of the longer peptides may have had second order structure. Regardless, applying the column temperature decreases the deviation from 1.06 GU to 0.60 GU, allowing for better prediction for peptides over 15 amino acids in length.
Table 5.
Retention of long peptides with and without a column oven
| Peptide | Length | Predicted RT (GU) | RT (Without Oven) | RT (With 60° Oven) |
|---|---|---|---|---|
| RPCFSALTPDETYVPK | 16 | 6.39 | 8.28 | 6.63 |
| LFTFHADICTLPDTEK | 16 | 5.05 | 7.40 | 4.56 |
| NTDGSTDYGILQINSR | 16 | 6.27 | 6.43 | 6.35 |
| EDLIWELLNQAQEHFGK | 17 | 5.47 | 5.39 | 5.05 |
| GITWGEETLMEYLENPK | 17 | 4.33 | 3.35 | 4.04 |
| VYACEVTHQGLSSPVTK | 17 | 6.69 | 2.37 | 4.07 |
| TTPPVLDSDGSFFLYSK | 17 | 3.19 | 4.14 | 3.18 |
| GITWGEETLMEYLENPKK | 18 | 6.45 | 4.66 | 6.52 |
| TVAAPSVFIFPPSDEQLK | 18 | 3.65 | 4.22 | 3.92 |
| RTVAAPSVFIFPPSDEQLK | 19 | 5.49 | 5.78 | 4.92 |
| AAPSVTLFPPSSEELQANK | 19 | 5.77 | 5.81 | 5.18 |
| ANPTVTLFPPSSEELQANK | 19 | 6.24 | 6.00 | 5.87 |
| EVQLVQSGGGLVQPGGSLR | 19 | 4.84 | 6.11 | 4.60 |
| DLILQGDATTGTDGNLELTR | 20 | 6.27 | 7.38 | 5.73 |
| VDNALQSGNSQESVTEQDSK | 20 | 10.70 | 10.54 | 10.04 |
| GLVLIAFSQYLQQCPFDEHVK | 21 | 4.01 | 5.65 | 4.69 |
| GFYPSDIAVEWESNGQPENNYK | 22 | 6.82 | 7.08 | 6.13 |
| SPDSHPADGIAFFISNIDSSIPSGSTGR | 28 | 7.65 | 8.54 | 5.62 |
It is also evident from the dataset that long peptides with actual retention times closer to predicted retention times at 25°C had a smaller average deviation (0.904 GU) than long peptides with closer retention times at 60°C (1.291 GU). This indicates that applying the elevated column temperature produces a more significant change in interaction with the stationary phase, and further supports our reasoning that many of these long peptides have second order structure that unravels at higher temperatures. There were only 3 cases out of 18 where a peptide had a longer retention time at 65°C in comparison to 25°C, and in all cases they were closer to the predicted times. Applying a higher temperature to a column will decrease the retention times to peptides without higher order structure, but in these cases there is significant evidence that their structure and/or interaction with the stationary phase changed due to the increase in retention times.
CONCLUSION
A peptide retention model based on amino acid composition was created using a HALO® penta-HILIC column with gradient elution. This model was shown to have a high correlation coefficient (0.946), on par with previously reported RP and HILIC models. It also includes optimized coefficients for hydrophobic residues at the N-terminus and hydrophobic residues one residue over from the N-terminus. The use of dextran as a retention time calibrant was essential for making this model capable of being used on any LC-MS system and adopters of this model can easily create an excel table using the derived coefficients that can be customized to fit into their workflow.
We are currently deriving coefficients for peptides with post-translational modifications that can be separated from unmodified peptides using the HILIC column. Many of these modifications cannot be separated by RP chromatography and they include oxidation, deamidation, and O-linked glycosylation, among others [23]. Our results have shown that HILIC is suitable for separating the modified peptides from their unmodified counterparts due to the hydrophilicity of the modifications. Coefficients for the oxidation of methionine and deamidation of asparagine, as well as the O-glcNAcylation, O-galNAcylation, and O-fucosylation of serine and threonine residues have been derived [19,23]. We hope to further develop our glycopeptide retention prediction model by combining this model with a glycan retention prediction model that is currently being developed in our laboratory, and we also hope to create an easy-to-use public tool that can predict the retention of unmodified peptides and peptides with hydrophilic modifications using the HILIC column.
Supplementary Material
HIGHLIGHTS.
A novel HILIC peptide retention prediction model based on amino acid composition was created.
The model exhibited a high correlation (0.946) between predicted and actual retention times.
Peptides over 15 amino acids in length were shown to deviate from predicted times more than shorter peptides
Length and position were shown to impact peptide retention, and site-specific corrections were created for hydrophobic residues at the first two positions at the N-terminus.
Acknowledgments
We would like to thank Rudradatt Persaud for his help with the early visualization of the project. We would also like to thank Dr. Mary Elizabeth Thompson and Dr. T. Colin Campbell for their help with the data sorting and regression analysis. Finally, we would like to thank Clayton Seigel for his help with the data processing. This work was supported in part by National Institute of General Medical Sciences [GM93747 to BEB]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST DISCLOSURE
The authors declare no competing financial interest.
References
- 1.Yoshida T. Prediction of peptide retention time in normal-phase chromatography. J Chromatogr A. 1998;811:61–67. doi: 10.1016/s0021-9673(99)00284-8. [DOI] [PubMed] [Google Scholar]
- 2.Gilar M, Jaworski A. Retention behavior of peptides in hydrophilic-interaction chromatography. J Chromatogr A. 2011;1218:8890–8896. doi: 10.1016/j.chroma.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 3.Wang Y, Lehmann R, Lu X, Zhao X, Zu G. Novel, fully automatic hydrophilic interaction/reversed-phase column-switching high-performance liquid chromatographic system for the complementary analysis of polar and apolar compounds in complex samples. J Chromatogr A. 2008;1204:28–34. doi: 10.1016/j.chroma.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 4.Krokhin OV, Craig R, Spicer V, Ens W, Standing KG, Beavis RC, Wilkins JA. An improved model for prediction of retention times of tryptic peptides in ion pair reversed-phase HPLC. Mol Cell Proteomics. 2004;3.9:908–919. doi: 10.1074/mcp.M400031-MCP200. [DOI] [PubMed] [Google Scholar]
- 5.Tripet B, Cepeniene D, Kovacs JM, Mant CT, Krokhin OV, Hodges RS. Requirements for prediction of peptide retention time in reversed-phase high-performance liquid chromatography: hydrophilicity/hydrophobicity of side-chains at the N- and C- termini of peptides are dramatically affected by the end-groups and location. J Chromatogr A. 2007;1141:212–225. doi: 10.1016/j.chroma.2006.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Hare MJ, Nice EC. Hydrophobic high-performance liquid chromatography of hormonal polypeptides and proteins on alkylsilane-bonded silica. J Chromatogr. 1979;171:209–226. doi: 10.1016/s0021-9673(01)95300-2. [DOI] [PubMed] [Google Scholar]
- 7.Meek JL. Prediction of peptide retention times in high-pressure liquid chromatography on the basis of amino acid composition. Proc Natl Acad Sci USA. 1980;77:1632–1636. doi: 10.1073/pnas.77.3.1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Su SJ, Grego B, Niven B, Hearn MTW. Analysis of group retention contributions for peptides separated by reverse phase high performance liquid chromatography. J Liq Chromatogr. 1981;4:1745–1764. [Google Scholar]
- 9.Wilson KJ, Honegger A, Stotzel RP, Hughes GJ. The behavior of peptides on reversed-phase supports during high-pressure liquid chromatography. Biochem J. 1981;199:31–41. doi: 10.1042/bj1990031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sasagawa T, Okuyama T, Teller DC. Prediction of peptide retention times in reversed-phase high performance liquid chromatography during linear gradient elution. J Chromatogr. 1982;240:329–340. [Google Scholar]
- 11.Browne CA, Bennet HPJ, Solomon S. The isolation of peptides by high-performance liquid chromatography using predicted elution positions. Anal Biochem. 1982;124:201–208. doi: 10.1016/0003-2697(82)90238-x. [DOI] [PubMed] [Google Scholar]
- 12.Guo D, Mant CT, Taneja AK, Parker JMR, Hodges RS. Prediction of peptide retention times in reversed-phase high-performance liquid chromatography. J Chromatogr. 1986;359:499–517. doi: 10.1016/s0021-9673(01)94598-4. [DOI] [PubMed] [Google Scholar]
- 13.Parker JMR, Guo D, Hodges RS. New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data: correlation of predicted surface residues with antigenicity and X-ray-derived accessible sites. Biochem. 1986;25:5425–5432. doi: 10.1021/bi00367a013. [DOI] [PubMed] [Google Scholar]
- 14.Sakamoto Y, Kawakami N, Sasagawa T. Prediction of peptide retention times. J Chromatogr. 1988;442:69–79. doi: 10.1016/s0021-9673(00)94457-1. [DOI] [PubMed] [Google Scholar]
- 15.Palmblad M, Ramstrom M, Markides KE, Hakansson P, Bergquist J. Prediction of chromatographic retention and protein identification in liquid chromatography/mass spectrometry. Anal Chem. 2002;74:5826–5830. doi: 10.1021/ac0256890. [DOI] [PubMed] [Google Scholar]
- 16.Tyteca E, Periat A, Rudaz S, Desmet G, Guillarme D. Retention modeling and method development in hydrophilic interaction chromatography. J Chromatogr A. 2014;1337:116–127. doi: 10.1016/j.chroma.2014.02.032. [DOI] [PubMed] [Google Scholar]
- 17.Le Maux S, Nongonierma AB, FitzGerald RJ. Improved short peptide identification using HILIC-MS/MS: retention time prediction model based on the impact of amino acid position in the peptide sequence. Food Chem. 2015;173:847–854. doi: 10.1016/j.foodchem.2014.10.104. [DOI] [PubMed] [Google Scholar]
- 18.Yoshida T. Calculation of peptide retention coefficients in normal-phase liquid chromatography. J Chromatogr A. 1998;808:105–112. doi: 10.1016/s0021-9673(98)00092-2. [DOI] [PubMed] [Google Scholar]
- 19.Badgett MJ, Boyes BE, Orlando R. The separation and quantitation of peptides with and without oxidation of methionine and deamidation of asparagine using hydrophilic interaction liquid chromatography with mass spectrometry (HILIC-MS) JASMS. 2017;28:818–826. doi: 10.1007/s13361-016-1565-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meek JL, Rossetti ZL. Factors affecting retention and resolution of peptides in high-performance liquid chromatography. J Chromatogr. 1981;211:15–28. [Google Scholar]
- 21.Mant CT, Burke TWL, Black JA, Hodges RS. Effect of peptide chain length on peptide retention behavior in reversed-phase chromatography. J Chromatogr. 1988;458:193–205. doi: 10.1016/s0021-9673(00)90564-8. [DOI] [PubMed] [Google Scholar]
- 22.Mant CT, Zhou NE, Hodges RS. Correlation of protein retention times in reversed-phase chromatography with polypeptide chain length and hydrophobicity. J Chromatogr. 1989;476:363–375. doi: 10.1016/s0021-9673(01)93882-8. [DOI] [PubMed] [Google Scholar]
- 23.Badgett MJ, Boyes BE, Orlando R. Predicting the retention behavior of specific O-linked glycopeptides. J Biomol Tech. 2017;3:122–126. doi: 10.7171/jbt.17-2803-003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.