

Abstract
The ETS family transcription factor ETV4 is aberrantly expressed in a variety of human tumors and plays an important role in carcinogenesis through upregulation of relevant target gene expression. Here, it is demonstrated that ETV4 is overexpressed in pancreatic cancer (PC) tissues as compared to the normal pancreas, and is associated with enhanced growth and rapid cell cycle progression of PC cells. ETV4 expression was silenced through stable expression of a specific short-interfering hairpin RNA (shRNA) in two PC cell lines (ASPC1 and Colo357), while it was ectopically expressed in BXPC3 cells. Silencing of ETV4 in ASPC1 and Colo357 cells reduced the growth by 55.3 % and 38.9 %, respectively; while forced expression of ETV4 in BXPC3 cells increased the growth by 46.8 % in comparison to respective control cells. Furthermore, ETV4-induced cell growth was facilitated by rapid transition of cells from G1- to S-phase of the cell cycle. Mechanistic studies revealed that ETV4 directly regulates the expression of cyclin D1 (CCND1), a protein crucial for cell cycle progression from G1- to S-phase. These effects on the growth and cell cycle were reversed by the forced expression of cyclin D1 in ETV4-silenced PC cells. Altogether, these data provide the first experimental evidence for a functional role of ETV4 in pancreatic cancer growth and cell cycle progression.
Implications
The functional and mechanistic data presented here regarding ETV4 in pancreatic cancer growth and cell cycle progression suggest that ETV4 could serve as a potential biomarker and novel target for pancreatic cancer therapy.
Keywords: ETV4, Pancreatic Cancer, Cyclin D1, Cell Cycle
Introduction
Pancreatic cancer (PC) is the third leading cause of cancer-related death in the United States with ∼8 % 5-year survival post-diagnosis (1). According to an estimate by American Cancer Society, ∼53,670 patients are expected to be diagnosed with PC this year and about 43,090 people will succumb to this disease (1). Furthermore, it is expected to become the second leading cause of cancer-related death by the year 2030 or even earlier, if no breakthrough is made in approaches to its clinical management (2). Clearly, there is an urgent need to make efforts at several fronts to tackle this extremely lethal malignancy effectively. In that regard, identification of aberrantly expressed and functionally involved genes in PC pathogenesis is highly significant from the perspective of the development of novel approaches for early detection and therapy.
ETV4, also known as PEA3 and E1AF, is an ETS-oncogene family transcription factor (3). It is broadly expressed in tissues during embryogenesis; however, its expression is low or highly restricted to specific organs in most adult tissues (4). During early development, ETV4 promotes morphogenesis of epithelial organs like kidney, lungs and mammary gland (5-7). Moreover, ETV4 transcription factor also plays a role in cellular growth, proliferation, migration and apoptosis (3). An aberrant expression of ETV4 is also reported in various cancer tissues including, breast, gastric, prostate, colon, and ovarian cancers at both mRNA and protein levels (8-12). The overexpression of ETV4 is associated with advanced and more aggressive forms of the tumor with worse prognosis (9,12). Moreover, a role of ETV4 has also been established in anchorage-independent growth (13), enhanced motility and invasiveness (14), epithelial to mesenchymal transition (10) and tumorigenesis (10,15).
In the present study, we examined the expression and functional significance of ETV4 in pancreatic cancer. A high expression of ETV4 was reported in pancreatic tumor tissues as compared to the normal pancreas. Furthermore, high ETV4 expression was associated with increased growth and rapid cell cycle progression in a Cyclin D1-dependent manner. ETV4 regulated cyclinD1 expression by directly binding to its gene promoter, and the effects of ETV4 silencing in PC cells were partly abrogated by the rescue of Cyclin D1 expression. Taken together, these findings suggest an important role of ETV4 in PC pathogenesis.
Materials and Methods
Cell lines and pancreatic cancer tissue specimens
The pancreatic cancer cell lines, Panc10.05, Panc02.03, Panc03.27, Capan1, SW1990, HPAF, MiaPaCa, CFPAC, ASPC1, Colo357, Panc1 and BXPC3, were procured from ATCC (Manassas, VA) and maintained under normal culturing conditions as described previously (16). All the cells were tested and determined to be free from mycoplasma contamination every alternate month. Frozen pancreatic tissue samples (normal and malignant) were obtained through cooperative human tissue network (CHTN) at the University of Alabama at Birmingham (UAB) under an Institutional Review Board (IRB) approved protocol.
Microarray data analysis
Oncomine cancer microarray database (17) (http://www.oncomine.org) was used to study the gene expression of ETV4 in human pancreatic cancer and normal pancreas tissue samples. A gene was considered as overexpressed when its mean value in tumor samples was significantly higher than mean value in the normal tissue counterpart using a Student's t-test (P<0.05).
Antibodies and plasmids
Antibody against Cyclin D1 (rabbit polyclonal) was purchased from Cell Signaling Technology (Beverly, MA). Anti -ETV4 (rabbit polyclonal) antibody and respective anti-rabbit horseradish peroxidase (HRP)-conjugated secondary antibodies were procured from Santa Cruz Biotechnology (Santa Cruz, CA). The β-actin (mouse monoclonal) antibody was purchased from Sigma-Aldrich (St. Louis MO). ETV4 knockdown short hairpin RNA (shRNA) expression constructs (pGFP-V-RS-shETV4) along with scrambled control (pGFP-V-RS-NTScr) and ETV4 overexpression construct (pCMV6-ETV4) and scramble control (pCMV6-Neo) were purchased from Origene (Rockville, MD). Cyclin D1 overexpression construct (pCMV6-CCND1) and control vector (pCMV6) were also purchased from Origene.
Constructs, transfections and treatments
For ETV4 silencing, Colo357 and ASPC1 cell lines were transfected with pGFP-V-RS-shETV4, and for ectopic overexpression of ETV4, BXPC3 cell lines were transfected with pCMV6-ETV4, along with their respective control plasmids, using X-tremeGENE HP DNA Transfection reagent (Roche, Indianapolis, IN) as per the manufacturer's instructions. Stable pooled population of transfected cells were selected in media containing Puromycin (2 μg/ml; for shRNA) or G148 (200 μg/ml; for overexpression), expanded and examined for stable ETV4 silencing or overexpression. The construct -962 human cyclin D1 promoter pGL3Basic (Addgene plasmid #32727) was from Frank McCormick laboratory and was procured through Addgene.
Luciferase Reporter Assays
Cells were cultured at a density of 1×106 cells/well in 6-well plate and transfected with -962 human cyclin D1 promoter pGL3Basic and mutant variation of -962 human cyclin D1 promoter pGL3Basic reporter constructs. After 24 h of transfection, the cells were harvested in passive lysis buffer and luciferase activity was measured using the Dual Luciferase Assay System (Promega Madison, WI).
Site-directed mutagenesis
Site-directed mutagenesis was performed using QuickChange Multi site-directed mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer's instructions. The cyclin D1 luciferase reporter vector was used as template and the predicted target site A sequence 5′- GGATGGCT-3′ was mutated to 5′-CGTTGCCA-3′ by PCR using the following primers: FP: GCAGGCGGGCGCCTCAGGGATGCCATTTGGGCTCTGC; RP: GCAGAGCCCAAATGGCATCCCTGAGGCGCCCGCCTGC. The mutagenized plasmids were isolated using the Qiagen Miniprep Kit (Qiagen) and DNA sequencing (Eurofins, Louisville, KY) confirmed mutations of the region containing the mutation.
Gene expression analysis
Total RNA was extracted using TRIzol reagent (Invitrogen) and reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA) as described in detail earlier (18). The list of primer pairs is presented in table S1.
Western blot analysis
Proteins isolated from PC cells and frozen tissues were estimated using DC Protein Assay Kit (Bio-Rad, Hercules, CA), resolved by SDS-PAGE and subjected to immunoblot analysis using specific antibodies against various proteins (19). Primary antibodies were detected with respective anti-mouse or anti-rabbit horseradish peroxidase-conjugated secondary antibodies (1:2000; Santa Cruz Biotechnology, Santa Cruz, CA). The signal of secondary antibodies was detected using ECL plus Western Blotting detection kit (Thermo Scientific, Logan, UT) and LAS-3000 image analyzer (Fuji Photo Film Co., Tokyo, Japan).
Growth kinetics assay
Cells (1×104/well) were seeded in 6-well plates (in triplicate) and allowed to grow for 8 days. The growth rate was determined every day by counting the number of cells on a hemocytometer. Cell population doubling time (dt) was calculated during exponential growth phase (120–168 h) using the following formula: dt = 0.693 t/ln (Nt/N0), where t is time (in h), Nt is the cell number at time t, and N0 is the cell number at initial time (20). The percent growth inhibition on the 8th day was calculated using the formula: 100-[(NshETV4 or Nneo/NNTScr) × 100], where NshETV4, Nneo and NNTScr are the number of cells on the 8th day (20).
Colony formation assay
For anchorage dependent colony formation, cells (1×103/well) were seeded in 6-well plates for colony formation. After two weeks, colonies were fixed with methanol, stained with crystal violet, photographed and counted using Image analysis software (Gene Tools, Syngene, Frederick, MD) as described in detail earlier (21).
Cell cycle analysis
Cell cycle analysis was executed on synchronized cells by culturing them in serum-free media for 72 h, and then incubated in regular media for 24 h. Cells were washed, trypsinized and fixed with 70 % ethanol overnight at 4 °C. Next day cells were washed with cold PBS, stained with Propidium iodide using PI/RNase staining buffer for 1 h at 37 °C and analyzed by flow-cytometry (Becton-Dickinson, San Jose, CA).
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed using a ChIP-IT enzymatic kit as previously described (22). Briefly, cells were fixed with paraformaldehyde (37 %) for DNA-protein cross-linking, lysed and sonicated to obtain small chromatin fragments and subjected to immunoprecipitation using anti-ETV4 or normal rabbit IgG (as control) antibodies. Subsequently, immunoprecipitates were subjected to reverse cross-linking and DNA was isolated. ChIPed DNA was analyzed by PCR using specific primers: cyclin D1 promoter site (-343), CCND1-Chip-F-5′-ACCGGACTACAGGGGCAAC-3′, CCND1-Chip-R-5′-AAACGCCGGGAGCAGCGA-3′; cyclin D1 promoter site (-794), CCND1-Chip-F-5-CTTGGGCATTTGCAACGACG-3′, CCND1-Chip-R-5′-CACACAACCCCTGTGCAAGT-3′; cyclin D1 promoter site (-941), CCND1-Chip-F-5-GAAGAGTCTCCAGGCTAGAAG-3′, CCND1-Chip-R-5′-GCAGCAGCCCAAGATGGTG-3′; cyclin D1 promoter site (-1038), CCND1-Chip-F-5-CAGAGGTGTGTTTCTCCCG-3′ and CCND1-Chip-R-5′-CTTCCTACCTTGACCAGTCG-3′. Input DNA (without immunoprecipitation) and normal IgG-precipitated DNA were used as positive and negative controls, respectively.
Statistical Analysis
All the experiments were performed at least three times, independently and all data are expressed as “mean ± SD”. Wherever appropriate, the data were also subjected to unpaired two tailed Student's t-test. p< 0.05 was considered statistically significant.
Results
ETV4 is aberrantly expressed in pancreatic cancer clinical specimens and cell line models
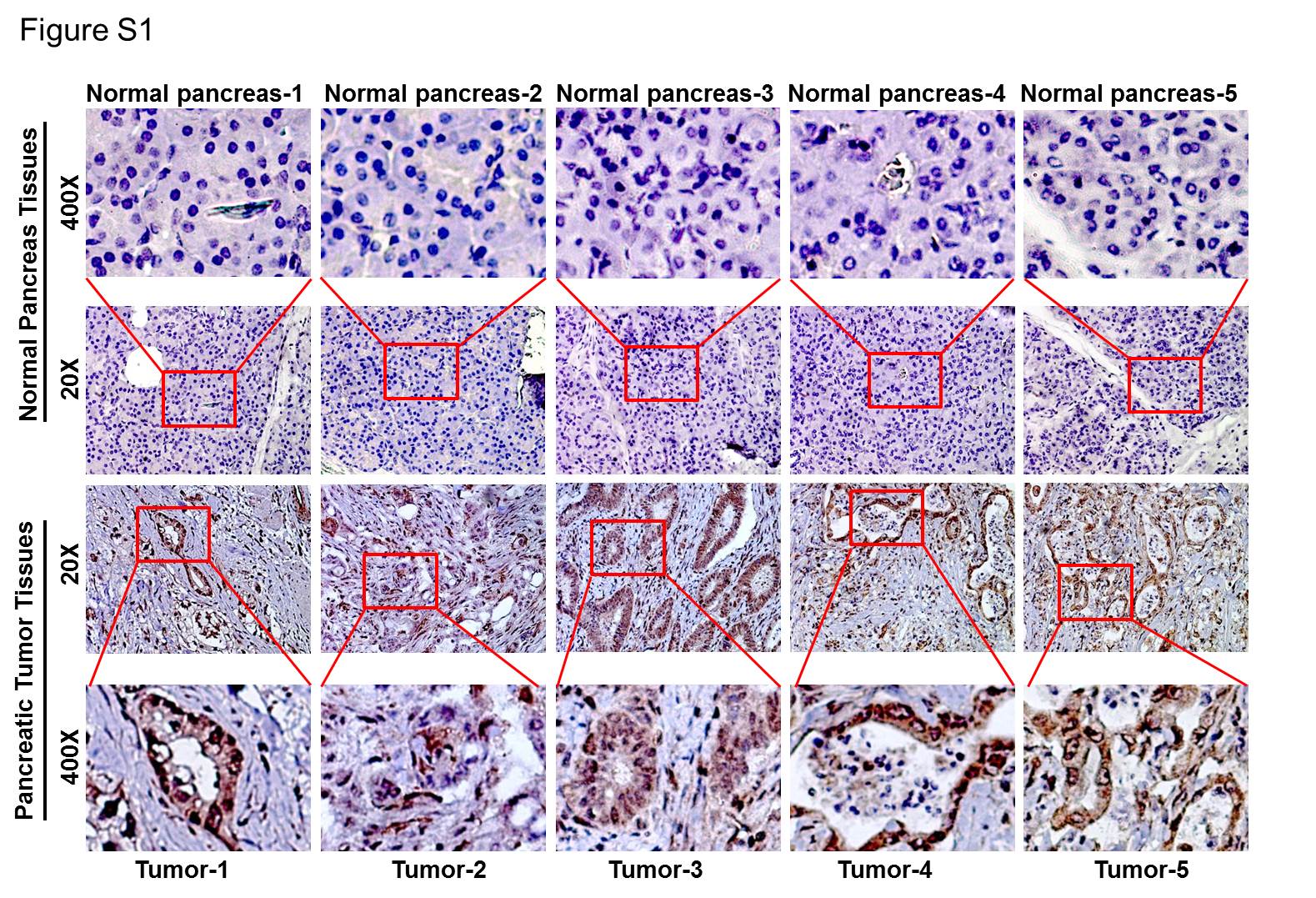
We searched the ONCOMINE databases for the expression of ETV4 in PC tissues. In three microarray expression studies (23-25), ETV4 mRNA expression level was higher in PC tissues than in normal pancreas tissues (Figure 1A). To investigate the role of ETV4 in pancreatic cancer pathogenesis, we first tested the expression of ETV4 in human PC tissue specimens (n=20) along with normal (n=7) pancreatic tissue samples using quantitative real-time RT-PCR and western blot analysis. The expression of ETV4 at mRNA level in PC tissue specimens was significantly higher (p< 0.01) than in normal samples (Figure 1B). In addition, we observed an aberrantly high expression of ETV4 in all PC tissues, whereas no or minimal expression was observed in normal pancreatic tissue specimens (Figure 1C). To observe the staining pattern of ETV4, we performed immunohistochemistry (IHC) on a small set of human pancreatic tumor tissues and normal pancreas (n=5, each). We detected an intense nuclear staining of ETV4 with some diffuse staining in the cytoplasm in all PC tissues, while no or minimal staining was observed in normal tissue samples (Figure S1). We also examined the expression of ETV4 in a panel of PC cell lines of varying tumorigenic and metastatic potential (26) and detected high expression of ETV4 in majority of the cell lines. BXPC3, a poorly tumorigenic cell line (27), showed minimal expression of ETV4 (Figure 1D). These findings imply that ETV4 is overexpressed in PC and might play a role in PC pathogenesis.
Figure 1. Expression of ETV4 in pancreatic cancer.
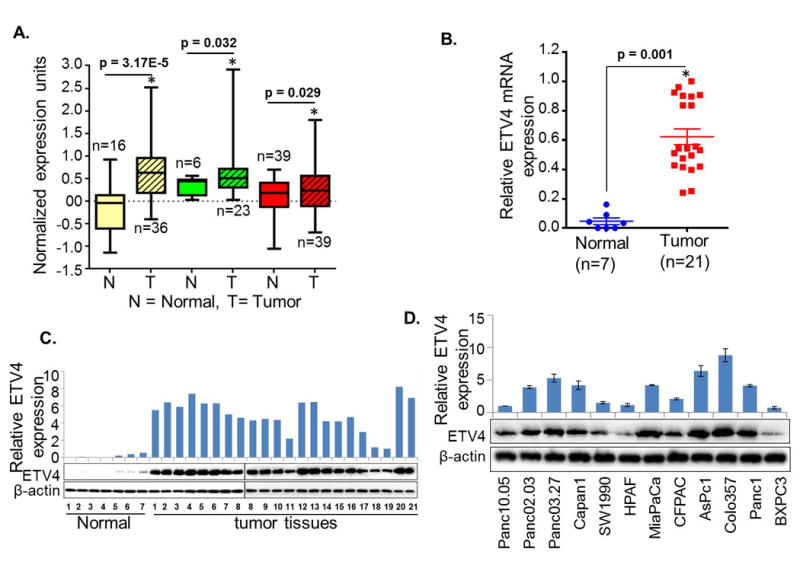
(A) Oncomine database analysis for the expression of ETV4. Analysis of ETV4 expression in pancreatic cancer (n=21) and normal pancreas (n=7) tissue samples (B) at mRNA level by quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR) (p<0.5) and (C) at the protein level by immunoblot analysis (n=1). GAPDH and β-actin were used as internal controls. (D) ETV4 expression in pancreatic cancer cell lines. Bars represent mean ± S.D. n=3; *, p< 0.05.
ETV4 expression is associated with the growth of PC cells
To explore the functional role of ETV4 in PC, we silenced ETV4 in two high ETV4-expressing tumorigenic cell lines, Colo357 and ASPC1, while overexpressed it in a poorly tumorigenic and low ETV4-expressing cell line, BXPC3. We used four shRNA constructs to knockdown ETV4 expression, of which two (shETV4 #1 and shETV4 #3) were able to cause significant ETV4 silencing in both cell lines (Figure 2A). Therefore, we performed subsequent functional studies with their resulting transfectants and/or their pooled population (Colo 357-shETV4 and ASPC1-shETV4). Moreover, in a converse approach, we forcefully overexpressed ETV4 in poorly tumorigenic BxPC3 cell line by stable transfection (Figure 2A). Subsequently, we studied the growth kinetics of high (Colo357-Scr, ASPC1-Scr, and BxPC3-ETV4) and low (Colo357-shETV4, ASPC1-shETV4, and BxPC3-Neo) ETV4-expressing PC cells. Our findings reveal that silencing of ETV4 led to a decrease in the growth of Colo357 and ASPC1 cells, while its ectopic expression promoted the growth of BXPC3 cells. After 8 days, the cell growth reduced by 38.9 % in Colo357-shETV4 and 55.3 % in ASPC1-shETV4 cells as compared to their respective control cells, while we recorded 46.8 % enhancement in the growth of ETV4 overexpressing cells (BXPC3-ETV4) as compared to BXPC3-Neo cells (Figure 2B). These changes in growth were due to altered population doubling times (dt), which increased from 21.4 h to 32.1 h and 29.7 h to 40.1 h in ETV4-silenced Colo357-shETV4 and ASPC1-shETV4 cells, respectively, as compared to their control sublines. In contrast, population doubling time of BXPC3 cells was decreased from 36.1 h to 27.2 h upon ectopic expression of ETV4 (Figure 2B). To monitor the effect of ETV4 on growth in the long term, we performed the colony-forming assay. Overexpression of ETV4 enhanced the clonogenic ability by 3.3 fold in BXPC3-ETV4 cells while ETV4-silenced (Colo357-shETV4 and ASPC1-shETV4) cells showed diminished clonogenic ability (3.4 and 2.4 folds, respectively) compared to their respective control cells (Figure 2C). These findings thus suggest that ETV4 plays an important role in the growth of PC cells.
Figure 2. ETV4 enhances pancreatic cancer cell growth and clonogenic ability.
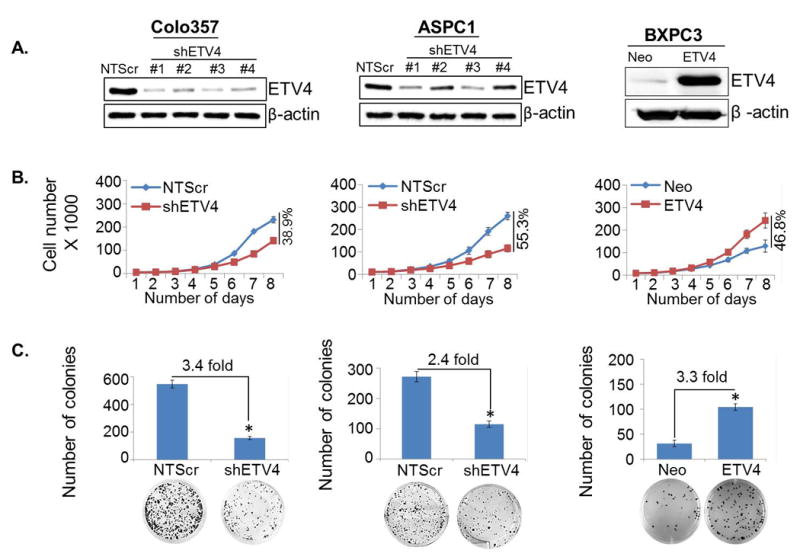
(A) Stable ETV4-silenced (Colo357-shETV4 and ASPC1-shETV4) and overexpressed (BXPC3-ETV4) cell lines with their scrambled control (Colo357-NTScr and ASPC1-NTScr and BXPC3-Neo) cells were generated, and ETV4 expression was examined by immunoblot analyses. (B) For growth kinetics, cells (1×104) were seeded in 6-well plates and growth was monitored by counting the cell number up to 8 days. (C) For anchorage-dependent colony formation assay, cells were seeded at low density (1×103cells/well) in regular media. After 2 weeks, colonies were stained with crystal violet and visualized and photographed using imaging system. Bars represent mean ± S.D. n=3; *, p< 0.05.
ETV4 promotes G1/S phase transition by modulating cell-cycle associated genes
Having observed a role of ETV4 in the growth of pancreatic cancer cells, we next examined the effect of ETV4 expression on cell cycle progression. For this, we first synchronized cells by culturing them in serum-free media for 72 h and then cultured in regular serum containing medium for 24 h. Cells were processed for staining with Propidium iodide using the PI/RNase Kit and analyzed by flow cytometry. The data from flow cytometry analysis indicate that ETV4-silencing causes a G1 phase cell cycle arrest in PC cells, while a greater fraction of ETV4-overexpressing cells is detected in S-phase (Figure 3A). In low ETV4 expressing ((Colo357-shETV4, ASPC1-shETV4 and BXPC3-Neo) cell lines, enhanced fraction of cells in G1-phase (50.7 %, 56.1 and 63.5 % respectively) were observed as compared with high ETV4-expressing (Colo357-NTScr (38.1 %), ASPC1- NTScr (30.5 %) and BXPC3-ETV4 (63.5 %) cells. Further, drastic reduction in the fraction of cells in S-phase in cells exhibiting low expression of ETV4(Colo357-shETV4 (25.7 %), ASPC1-shETV4 (15.7 %) and BXPC3-Neo (24.5 %) was seen as compared to Colo357-NTScr (46.3 %), ASPC1- NTScr (42.1 %) and BXPC3-ETV4 (47.5 %) cells (Figure 3A). These data indicate a role of ETV4 in cell cycle progression by facilitating G1 to S phase transition. To decipher the unbiased mechanistic basis of ETV4-induced cell cycle progression, we next examined the expression profile of 48 genes implicated in cell cycle regulation in high and low ETV4-expressing PC cells by quantitative RT-PCR. We observed that 6 genes (CCND1, CCNE1, CUL1, CDK4, CDK6 and SUMO1) primarily involved in G1 phase and G1/S phase transition (Table 1) of cancer cells were significantly altered (fold ≥ 2; p<0.001) and cyclin D1 was the most downregulated gene exhibiting ∼18-fold decreased expression in ETV4-silenced Colo357 PC cells (Figure 3B). The expression of Cyclin D1 was further confirmed at mRNA and protein level in all the three PC cell lines engineered for low and high expression of ETV4 (Colo357-NTScr/-shETV4, ASPC1-NTScr/-shETV4 and BXPC3-Neo/-ETV4). As expected, we detected reduced expression of cyclin D1 in low ETV4-expressing PC cells, while its high expression was observed in ETV4-overexpressing BXPC3 cells (Figure 3C). Taken together, these findings suggest that the ETV4 facilitates cell cycle progression of PC cells by modulating the expression of genes associated with the G1-S transition phase of cell cycle.
Figure 3. ETV4-silencing arrests cell cycle in G1/S phase by regulating the expression of genes involved in regulation and progression of cell cycle.
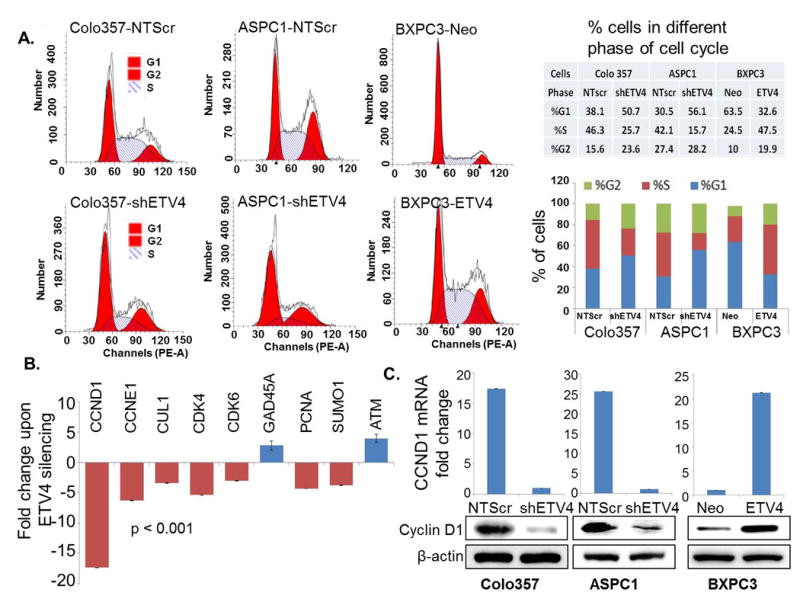
(A) ETV4-silenced and -overexpressing cells were synchronized by culturing them in serum-free media for 48h and then incubated in regular medium for 24h. Subsequently, cells were fixed in methanol and stained with Propidium iodide. The distribution of cells in different phases of cell cycle was analyzed by flow cytometry. (B) Expression profile of selected cell cycle associated genes by quantitative RT-PCR upon ETV4 silencing in Colo357 cells and (C) expression of Cyclin D1 at mRNA and protein level in low and high ETV4-expressing Colo357, ASPC1 and BXPC3 cells. GAPDH and β-actin were used as loading controls.
Table 1. List of selective cell cycle regulatory genes with symbol, functions, and folds change in high and low ETV4-expressing PC cells.
| Gene | Function | Fold change | p Value* |
|---|---|---|---|
| CCND1 | CCND1 interacts with CDK4 and CDK6 and control cell cycle at the G1/S transition | -17.3643 | 0.000172144 |
| CCNE1 | CCNE1 interacts with CDK2 and control cell cycle at the G1/S transition | -6.28749 | 0.000477381 |
| CUL1 | CUL1 regulate cell cycle at the G1/S transition by ubiquitinylation of cyclins D1 and E | -3.42371 | 0.001956218 |
| CDK4 | Interacts with cyclin D1 | -5.36261 | 1.44007E-05 |
| CDK6 | Interacts with cyclin D1 | -3.02619 | 0.001454547 |
| GADD45A | GADD45A inhibit entry of cells into S phase | 2.81315 | 0.051256666 |
| PCNA | PCNA controls DNA replication | -4.28021 | 0.000373706 |
| SUMO1 | SUMO1 stablize CDK6 by SUMOylation in G1 phase and control cell cycle through G1/S transition | -3.77065 | 0.001100455 |
| ATM | ATM activates checkpoint and phosphorylate p53 | 3.980067 | 0.015487116 |
p < 0.001 was considered significant.
Cyclin D1 is a direct transcriptional target of ETV4
Having observed the transcriptional regulation of Cyclin D1 by ETV4, and Cyclin D1 being an essential cell cycle regulatory protein required for rapid progression of cell cycle from G1 to S phase of the cycle (28,29), we next examined if cyclin D1 is under direct transcriptional control of ETV4. For this, we first performed an in silico analysis of ∼1 kb DNA region 5′ upstream of coding DNA sequence (CDS) of CCND1 (GenBank™ accession number Z29078) using available online analytical applications (TF-Bind, JASPAR and ALGGEN-PROMO). Our analysis revealed four putative ETV4 binding sites within the promoter region of CCND1 (Figure 4A). To confirm the direct binding of ETV4 to the CCND1 promoter, we performed ChIP analysis using ETV4-specific antibody. Normal IgG was used as negative control. The ChIPed DNA was subjected to the PCR amplification using primer sets covering putative ETV4-binding sites (Figure 4A). The data demonstrated that ETV4 most strongly bound to the predicted promoter region -343 to -336 that lies in the closest proximity to the transcriptional initiation site of cyclin D1, while it's binding to other sites was less significant (Figure 4B). To determine whether the predicted target site (-343 to -336) for ETV4 binding is a functional target site we mutated the ETV4 binding sequence 5′- GGATGGCT-3′ to 5′- CGTTGCCA -3′ (Figure 4C) using site-directed mutagenesis kit and mutation was confirmed by DNA sequencing of the region containing the mutation. The PC cells, Colo357 and ASPC1, were transfected with mutant or wild type Cyclin D1, and a control promoter-reporter constructs. Estimation of relative luciferase activity demonstrated 70.1 % and 72.4 % reduction in mutant construct-transfected Colo357 and ASPC1 cells, respectively (Figure 4C and D) suggesting that the mutated sequence in the CCND1 promoter represents the functional target site of ETV4. In accordance to these findings, we also detected reduced PCR amplification signal for cyclin D1 in ETV4-silenced cells. The relative ETV4 binding to CCND1 promoter at -343 to -336 site is reduced by 12.1-fold in Colo357-shETV4 and 12.7-fold in ASPC1-shETV4 cells, supporting the specificity of ETV4-dependent chromatin pull-down (Figure 4E). Together, these data suggest that ETV4 positively regulates cyclin D1 at the transcriptional level in PC cells.
Figure 4. ETV4 transcriptionally upregulates cyclin D1 via direct binding to its promoter region.
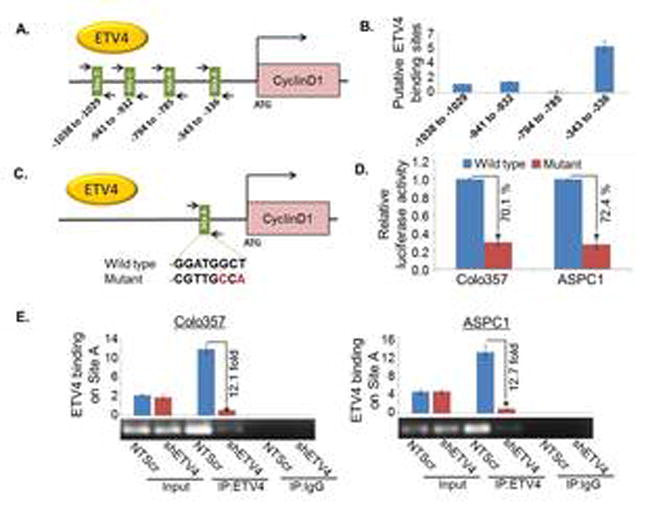
(A) Schematic diagram of human cyclin D1-promoter showing putative ETV4 binding sites. Arrows indicate the position and orientation of forward and reverse primers. The number below the bars represents the position of putative binding sites. (B) The direct binding of ETV4 to Cyclin D1 promoter was shown using ChIP assay. PCR was performed using specific primers as indicated. (C) Site A (-343 to -336) sequence 5′- GGATGGCT-3′ was mutated to 5′- CGTTGCCA -3′ using site-directed mutagenesis kit. (D) The wild type and mutated cyclin D1 promoter construct was transfected into PC cells and luciferase assay was performed 24h after transfection using the dual Luciferase Reporter Assay kit to determine the luciferase activity. (E) PCR amplification signal in low and high ETV4 expressing Colo357 and ASPC1 cells, suggesting the specificity of ETV4-dependent chromatin pull-down. Input DNA (without immunoprecipitation) and normal IgG-precipitated DNA were used as positive and negative controls, respectively. Bars represent mean ± S.D, n=3; *, (p<0.05).
ETV4 regulates cell cycle progression and overall growth, at least in part, through upregulation of cyclin D1
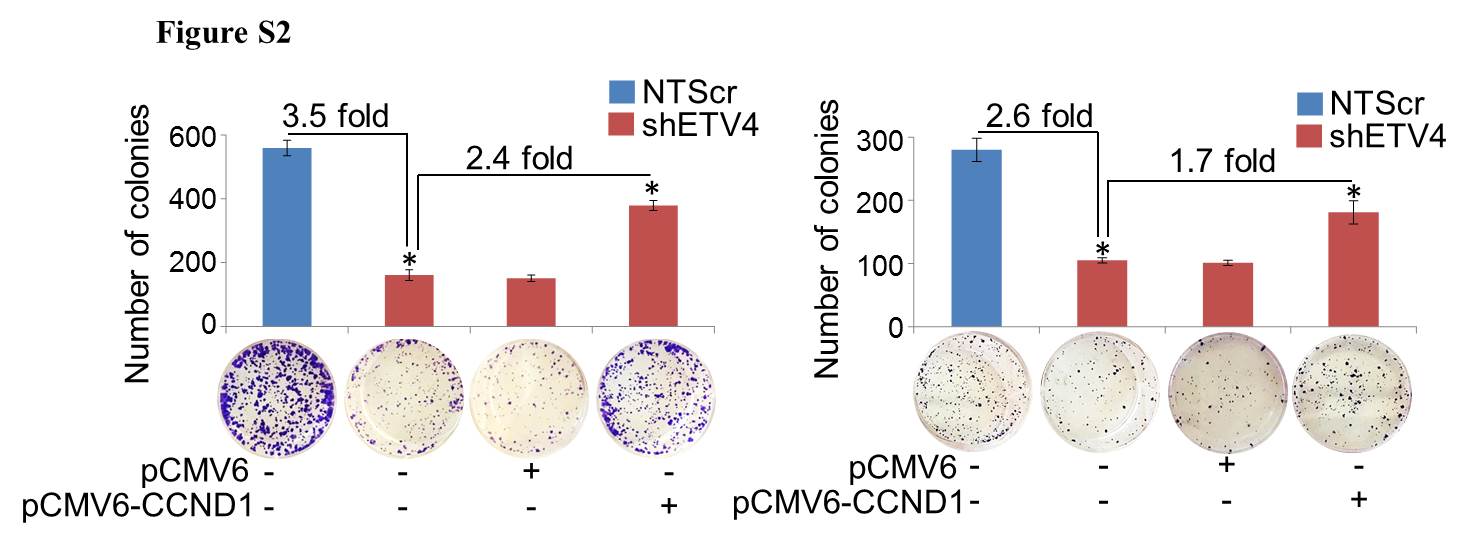
To determine the involvement of cyclin D1 in ETV4-induced effect on the PC cells, low ETV4 expressing (Colo357-shETV4 and ASPC1-shETV4) cells were transiently transfected with CCND1 overexpression construct and the effect on cell cycle and growth was examined. First, we confirmed the overexpression of cyclin D1 by immunoblot analysis in ETV4-silenced (Colo357 and APSC1) cells transiently transfected with CCND1 construct (pCMV6-CCND1) (Figure 5A). The findings were further confirmed by reading the transcriptional activity of cyclin D1 promoter by luciferase reporter assay (Figure 5B). Next, we tested whether overexpression of cyclin D1 in ETV4-silenced PC cells could reverse the growth inhibitory effect of ETV4-silencing in PC cells. Our data revealed that overexpression of cyclin D1 significantly abrogated the growth inhibitory effect of ETV4 silencing in both Colo357 and APSC1 cells (Figure 5C). Furthermore, overexpression of cyclin D1 in ETV4-silenced cells released the cells from G1/S phase arrest (Figure 5D) and partially rescued the long-term inhibitory effect on growth (Figure S2) of PC cells. These data suggest that ETV4 promotes PC cell growth and cell cycle progression, at least in part, through upregulation of cyclin D1.
Figure 5. ETV4 regulates pancreatic cancer cell growth through cyclin D1.
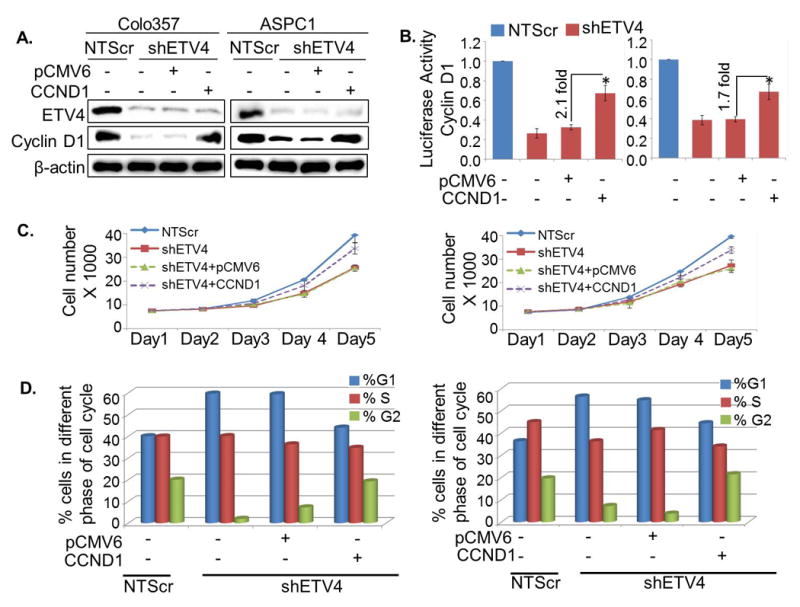
(A) Cyclin D1 cDNA construct (pCMV6-CCND1) was transfected in ETV4-silenced Colo357 and ASPC1 cells for its overexpression and confirmed by immunoblot analyses. (B) After 48 h of transfection, cells further transfected with Cyclin D1 promoter reporter construct and transcriptional activity of Cyclin D1 was examined. Data are presented as normalized relative luciferase activity (mean ± SD; n = 3, *p < 0.05). (C-D) Overexpression of cyclin D1 in ETV4-silenced cells rescued the inhibitory effect of ETV4-silencing on (C) growth and (D) cell cycle progression of PC cells.
Discussion
The present study revealed, for the first time, the oncogenic involvement of ETV4 in pancreatic cancer. Specifically, using malignant and normal pancreatic tissues and established cancer cell lines, we were able to show the aberrant expression of ETV4 in pancreatic cancer. This data is also supported by analysis of ONCOMINE datasets from three independent studies (23-25). Oncomine datasets analyses; however, also showed one contradictory report citing a downregulation of ETV4 at the mRNA level in PC and pancreatitis tissues as compared to the normal pancreas (30). This could either be sample-specific or tumor-subset-specific as the study used a very small sample size (n=5). In a small pilot study, we also performed immuno-staining for ETV4 to examine its in situ expression and subcellular localization in normal and malignant pancreatic tissues (n=5, each). None of the normal tissues exhibited ETV4 staining (except some faint background staining), while varying levels of ETV4 were detected in malignant tissues of mixed grades suggesting its tumor-specific expression. However, clear determination of its specificity as well as overall incidence and extent of dysregulated expression will require a study that examines its expression in larger set of normal, adjacent normal and cancerous tissues of pancreas. It would also be interesting to investigate if ETV4 expression exhibits any correlation with increasing tumor-grades. Regardless, there are numerous studies in the literature that report an overexpression of ETV4 in a variety of other tumor types as well, including breast, lung, colon and prostate etc., which clearly suggests it's important roles in cancer pathogenesis (8-12,31).
Pancreatic cancer is characterized by several genetic aberrations (32). Of which, genetic mutations in K-Ras (33,34), inactivating mutation or deletion of TP53 (35,36), homozygous deletion of SMAD4 (37,38), INK4A (39), PTEN (40) are frequently detected, and their roles in etiology and progression of pancreatic cancer have also been reported (32-40). However, precise mechanisms through which gene aberrations promote pancreatic cancer initiation and progression remain to be clearly understood. It is believed that the accumulation of these genetic changes leads to altered expression of several tumor-specific genes through activation of oncogenic signaling pathways. Importantly, a regulatory connection of K-Ras signaling with ETV4 expression has also been reported (41). Furthermore, ETV4-mediated regulation of PI3-kinase and Ras signaling has also been reported in a mouse model of advanced prostate cancer (42). A role of ETV4 as a downstream effector of MET signaling pathway has also been reported, whereby it is involved in the regulation of migration and invasiveness (14). In another report, a positive correlation of ETV4 with HER2/Neu overexpression, tumor grade and higher recurrence in human breast cancer patients is also demonstrated (43) suggesting a pathologic involvement of ETV4 in multiple cancer types.
Our data suggested an oncogenic function of ETV4 in PC by demonstrating its role in growth and cell cycle progression of PC cells. Similarly, earlier studies have also documented a pathological role of ETV4 overexpression in variety of other cancers. Pellecchia et al (2012) reported that inhibition of ETV4 retarded the proliferation of prostate cancer cells (10). Moreover, ETV4 is shown to promote growth, proliferation and cell cycle progression in breast cancer cells as well (15). Uncontrolled cell proliferation is one of the hallmarks of cancer. This is achieved by the tumor cells typically through abnormal expression and functions of genes involved in cell cycle regulation. In this regard, we also observed the cell cycle regulatory role of ETV4 in pancreatic cancer, whereby it promoted cell-cycle progression by facilitating transition of cells from G1 to S phase. Furthermore, we also observed changes in expression of genes associated with G1 or G1/S phase transition.
Cyclin D1 is a critical cell cycle regulator, which is rapidly synthesized in G1 phase and degraded as the cells enter into the S phase suggesting its important role in G1-S progression (28,29). Under normal scenario, expression of cyclin D1 is tightly regulated; however, high expression of Cyclin D1 is frequently reported in cancer cells leading to dysregulation of cell cycle and overall growth of the cells (44). Several lines of evidence support that aberrant overexpression of Cyclin D1, either through genetic (gene amplification) or non-genetic (epigenetic, transcriptional) mechanisms, is a common event in a variety of cancers including pancreatic cancer (45-47). Cyclin D1 is overexpressed in ∼ 42-82 % human pancreatic cancers. However, its gene amplification is reported only in 25 % cases (48) suggesting that its overexpression is likely regulated through other mechanisms in PCs. One such mechanism may be the enhanced transcription of Cyclin D1 in PC cells. Our findings are quite intriguing as we have identified a novel mechanism of Cyclin D1 regulation in PC by ETV4. We observed that ETV4 binds to the promoter region of Cyclin D1 and upregulates the transcription of Cyclin D1. Our findings are in agreement with the previous study which demonstrated that ETV4 transcriptionally activates Cyclin D3, a member of the Cyclin D family, in breast cancer cells (15). We also observed that Cyclin D1 was required, at least in part, for ETV4-induced pancreatic cancer cell growth and cell cycle progression.
Having observed the pathological significance of ETV4 in PC in terms of cell cycle regulation, it would be of interest to conduct additional studies in animal model systems to investigate its role in pancreatic tumorigenicity and metastasis. In a previous related study, ETV4 has been shown to be pathologically involved in tumorigenesis and metastasis of prostate cancer (42). In other reports, overexpression of ETV4 was also shown to drive metastasis in lung, mammary and colorectal carcinomas (49-51). Prognostic significance of ETV4 has also been reported in some solid malignancies (52-54). Considering that ETV4 is able to influence PC progression, it can also potentially exhibit an association with patient's prognosis, and be used as a biomarker for disease progression and therapeutic planning. Moreover, from the mechanistic standpoint, more comprehensive studies, such as ChIP-seq and RNA-seq needs to be performed in PC model systems to define its global as well as promoter-specific impact on gene regulation. Resulting data may not only identify novel ETV4-regulated gene targets, but also yield important mechanistic insights into pancreatic tumor biology to support future ETV4-focussed basic and translational studies.
In summary, our study established the role of ETV4, for the first time, in PC growth and cell cycle progression through transcriptional activation of Cyclin D1. These results give new insights into the role of ETV4 in pancreatic cancer and suggests that it could serve as a novel biomarker for pancreatic cancer stratification at the molecular level and exploited as a target for pancreatic cancer therapy.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
Grant Support: This work was supported by funding from NIH/NCI [1R03CA169829 (to SS) and CA175772 (to APS)] and USAMCI.
Footnotes
Conflict of Interest: “The authors declare no potential conflicts of interest.”
Authors' Contributions: Conception and design: SS, APS, NT, SKD, SKS
Development of methodology: SS, APS, NT, SKS, SKD, SA, AA
Acquisition of data (provided animals acquired and managed patients, provided facilities, etc.): SS, APS, NT, SKD, SKS, BW, JE
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): SS, APS, NT, SKS, SKD, SA, BW.
Writing, review, and/or revision of the manuscript: SS, APS, NT, SKS, SKD, SA.
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): NT, SKS, SKD, SA, AA
Study supervision: SS, APS
All authors read and approved the final manuscript.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–21. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 3.Oh S, Shin S, Janknecht R. ETV1, 4 and 5: an oncogenic subfamily of ETS transcription factors. Biochim Biophys Acta. 2012;1826:1–12. doi: 10.1016/j.bbcan.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maroulakou IG, Bowe DB. Expression and function of Ets transcription factors in mammalian development: a regulatory network. Oncogene. 2000;19:6432–42. doi: 10.1038/sj.onc.1204039. [DOI] [PubMed] [Google Scholar]
- 5.Sonnenberg E, Meyer D, Weidner KM, Birchmeier C. Scatter factor/hepatocyte growth factor and its receptor, the c-met tyrosine kinase, can mediate a signal exchange between mesenchyme and epithelia during mouse development. J Cell Biol. 1993;123:223–35. doi: 10.1083/jcb.123.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chotteau-Lelievre A, Dolle P, Peronne V, Coutte L, de LY, Desbiens X. Expression patterns of the Ets transcription factors from the PEA3 group during early stages of mouse development. Mech Dev. 2001;108:191–5. doi: 10.1016/s0925-4773(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 7.Andermarcher E, Surani MA, Gherardi E. Co-expression of the HGF/SF and c-met genes during early mouse embryogenesis precedes reciprocal expression in adjacent tissues during organogenesis. Dev Genet. 1996;18:254–66. doi: 10.1002/(SICI)1520-6408(1996)18:3<254::AID-DVG6>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 8.Yuan ZY, Dai T, Wang SS, Peng RJ, Li XH, Qin T, et al. Overexpression of ETV4 protein in triple-negative breast cancer is associated with a higher risk of distant metastasis. Onco Targets Ther. 2014;7:1733–42. doi: 10.2147/OTT.S66692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qi M, Liu Z, Shen C, Wang L, Zeng J, Wang C, et al. Overexpression of ETV4 is associated with poor prognosis in prostate cancer: involvement of uPA/uPAR and MMPs. Tumour Biol. 2015;36:3565–72. doi: 10.1007/s13277-014-2993-7. [DOI] [PubMed] [Google Scholar]
- 10.Pellecchia A, Pescucci C, De LE, Luceri C, Passaro N, Sica M, et al. Overexpression of ETV4 is oncogenic in prostate cells through promotion of both cell proliferation and epithelial to mesenchymal transition. Oncogenesis. 2012;1:e20. doi: 10.1038/oncsis.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nishikawa A, Iwasaki M, Akutagawa N, Manase K, Yamashita S, Endo T, et al. Expression of various matrix proteases and Ets family transcriptional factors in ovarian cancer cell lines: correlation to invasive potential. Gynecol Oncol. 2000;79:256–63. doi: 10.1006/gyno.2000.5944. [DOI] [PubMed] [Google Scholar]
- 12.Keld R, Guo B, Downey P, Cummins R, Gulmann C, Ang YS, et al. PEA3/ETV4-related transcription factors coupled with active ERK signalling are associated with poor prognosis in gastric adenocarcinoma. Br J Cancer. 2011;105:124–30. doi: 10.1038/bjc.2011.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hollenhorst PC, Paul L, Ferris MW, Graves BJ. The ETS gene ETV4 is required for anchorage-independent growth and a cell proliferation gene expression program in PC3 prostate cells. Genes Cancer. 2011;1:1044–52. doi: 10.1177/1947601910395578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kherrouche Z, Monte D, Werkmeister E, Stoven L, de LY, Cortot AB, et al. PEA3 transcription factors are downstream effectors of Met signaling involved in migration and invasiveness of Met-addicted tumor cells. Mol Oncol. 2015;9:1852–67. doi: 10.1016/j.molonc.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang J, Wei Y, Liu D, Zhou J, Shen J, Chen X, et al. E1AF promotes breast cancer cell cycle progression via upregulation of Cyclin D3 transcription. Biochem Biophys Res Commun. 2007;358:53–8. doi: 10.1016/j.bbrc.2007.04.043. [DOI] [PubMed] [Google Scholar]
- 16.Tyagi N, Bhardwaj A, Singh AP, McClellan S, Carter JE, Singh S. p-21 activated kinase 4 promotes proliferation and survival of pancreatic cancer cells through AKT- and ERK-dependent activation of NF-kappaB pathway. Oncotarget. 2014;5:8778–89. doi: 10.18632/oncotarget.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tyagi N, Marimuthu S, Bhardwaj A, Deshmukh SK, Srivastava SK, Singh AP, et al. p-21 activated kinase 4 (PAK4) maintains stem cell-like phenotypes in pancreatic cancer cells through activation of STAT3 signaling. Cancer Lett. 2016;370:260–7. doi: 10.1016/j.canlet.2015.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deshmukh SK, Srivastava SK, Bhardwaj A, Singh AP, Tyagi N, Marimuthu S, et al. Resistin and interleukin-6 exhibit racially-disparate expression in breast cancer patients, display molecular association and promote growth and aggressiveness of tumor cells through STAT3 activation. Oncotarget. 2015;6:11231–41. doi: 10.18632/oncotarget.3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tyagi N, Bhardwaj A, Srivastava SK, Arora S, Marimuthu S, Deshmukh SK, et al. Development and Characterization of a Novel in vitro Progression Model for UVB-Induced Skin Carcinogenesis. Sci Rep. 2015;5:13894. doi: 10.1038/srep13894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deshmukh SK, Srivastava SK, Zubair H, Bhardwaj A, Tyagi N, Al-Ghadhban A, et al. Resistin potentiates chemoresistance and stemness of breast cancer cells: Implications for racially disparate therapeutic outcomes. Cancer Lett. 2017;396:21–9. doi: 10.1016/j.canlet.2017.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhardwaj A, Srivastava SK, Singh S, Tyagi N, Arora S, Carter JE, et al. MYB Promotes Desmoplasia in Pancreatic Cancer through Direct Transcriptional Up-regulation and Cooperative Action of Sonic Hedgehog and Adrenomedullin. J Biol Chem. 2016;291:16263–70. doi: 10.1074/jbc.M116.732651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Badea L, Herlea V, Dima SO, Dumitrascu T, Popescu I. Combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia. Hepatogastroenterology. 2008;55:2016–27. [PubMed] [Google Scholar]
- 24.Buchholz M, Braun M, Heidenblut A, Kestler HA, Kloppel G, Schmiegel W, et al. Transcriptome analysis of microdissected pancreatic intraepithelial neoplastic lesions. Oncogene. 2005;24:6626–36. doi: 10.1038/sj.onc.1208804. [DOI] [PubMed] [Google Scholar]
- 25.Pei H, Li L, Fridley BL, Jenkins GD, Kalari KR, Lingle W, et al. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell. 2009;16:259–66. doi: 10.1016/j.ccr.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deer EL, Gonzalez-Hernandez J, Coursen JD, Shea JE, Ngatia J, Scaife CL, et al. Phenotype and genotype of pancreatic cancer cell lines. Pancreas. 2010;39:425–35. doi: 10.1097/MPA.0b013e3181c15963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Delesque N, Buscail L, Esteve JP, Saint-Laurent N, Muller C, Weckbecker G, et al. sst2 somatostatin receptor expression reverses tumorigenicity of human pancreatic cancer cells. Cancer Res. 1997;57:956–62. [PubMed] [Google Scholar]
- 28.Alao JP. The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention. Mol Cancer. 2007;6(24):24. doi: 10.1186/1476-4598-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bertoli C, Skotheim JM, de Bruin RA. Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol. 2013;14:518–28. doi: 10.1038/nrm3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Logsdon CD, Simeone DM, Binkley C, Arumugam T, Greenson JK, Giordano TJ, et al. Molecular profiling of pancreatic adenocarcinoma and chronic pancreatitis identifies multiple genes differentially regulated in pancreatic cancer. Cancer Res. 2003;63:2649–57. [PubMed] [Google Scholar]
- 31.Moss AC, Lawlor G, Murray D, Tighe D, Madden SF, Mulligan AM, et al. ETV4 and Myeov knockdown impairs colon cancer cell line proliferation and invasion. Biochem Biophys Res Commun. 2006;345:216–21. doi: 10.1016/j.bbrc.2006.04.094. [DOI] [PubMed] [Google Scholar]
- 32.Khan MA, Azim S, Zubair H, Bhardwaj A, Patel GK, Khushman M, et al. Molecular Drivers of Pancreatic Cancer Pathogenesis: Looking Inward to Move Forward. Int J Mol Sci. 2017;18:E779. doi: 10.3390/ijms18040779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.di Magliano MP, Logsdon CD. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology. 2013;144:1220–9. doi: 10.1053/j.gastro.2013.01.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guerra C, Barbacid M. Genetically engineered mouse models of pancreatic adenocarcinoma. Mol Oncol. 2013;7:232–47. doi: 10.1016/j.molonc.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Redston MS, Caldas C, Seymour AB, Hruban RH, da CL, Yeo CJ, et al. p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res. 1994;54:3025–33. [PubMed] [Google Scholar]
- 36.Morton JP, Timpson P, Karim SA, Ridgway RA, Athineos D, Doyle B, et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci U S A. 2010;107:246–51. doi: 10.1073/pnas.0908428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilentz RE, Iacobuzio-Donahue CA, Argani P, McCarthy DM, Parsons JL, Yeo CJ, et al. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res. 2000;60:2002–6. [PubMed] [Google Scholar]
- 38.Iacobuzio-Donahue CA, Fu B, Yachida S, Luo M, Abe H, Henderson CM, et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J Clin Oncol. 2009;27:1806–13. doi: 10.1200/JCO.2008.17.7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldstein AM, Fraser MC, Struewing JP, Hussussian CJ, Ranade K, Zametkin DP, et al. Increased risk of pancreatic cancer in melanoma-prone kindreds with p16INK4 mutations. N Engl J Med. 1995;333:970–4. doi: 10.1056/NEJM199510123331504. [DOI] [PubMed] [Google Scholar]
- 40.Wartenberg M, Centeno I, Haemmig S, Vassella E, Zlobec I, Galvan JA, et al. PTEN alterations of the stromal cells characterise an aggressive subpopulation of pancreatic cancer with enhanced metastatic potential. Eur J Cancer. 2016;65:80–90. doi: 10.1016/j.ejca.2016.06.013. [DOI] [PubMed] [Google Scholar]
- 41.Plotnik JP, Budka JA, Ferris MW, Hollenhorst PC. ETS1 is a genome-wide effector of RAS/ERK signaling in epithelial cells. Nucleic Acids Res. 2014;42:11928–40. doi: 10.1093/nar/gku929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aytes A, Mitrofanova A, Kinkade CW, Lefebvre C, Lei M, Phelan V, et al. ETV4 promotes metastasis in response to activation of PI3-kinase and Ras signaling in a mouse model of advanced prostate cancer. Proc Natl Acad Sci U S A. 2013;110:E3506–E3515. doi: 10.1073/pnas.1303558110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Myers E, Hill AD, Kelly G, McDermott EW, O'Higgins NJ, Young LS. A positive role for PEA3 in HER2-mediated breast tumour progression. Br J Cancer. 2006;95:1404–9. doi: 10.1038/sj.bjc.6603427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bates S, Peters G. Cyclin D1 as a cellular proto-oncogene. Semin Cancer Biol. 1995;6:73–82. doi: 10.1006/scbi.1995.0010. [DOI] [PubMed] [Google Scholar]
- 45.Gillett C, Fantl V, Smith R, Fisher C, Bartek J, Dickson C, et al. Amplification and overexpression of cyclin D1 in breast cancer detected by immunohistochemical staining. Cancer Res. 1994;54:1812–7. [PubMed] [Google Scholar]
- 46.Inomata M, Uchino S, Tanimura H, Shiraishi N, Adachi Y, Kitano S. Amplification and overexpression of cyclin D1 in aggressive human esophageal cancer. Oncol Rep. 1998;5:171–6. doi: 10.3892/or.5.1.171. [DOI] [PubMed] [Google Scholar]
- 47.Zhang YJ, Jiang W, Chen CJ, Lee CS, Kahn SM, Santella RM, et al. Amplification and overexpression of cyclin D1 in human hepatocellular carcinoma. Biochem Biophys Res Commun. 1993;196:1010–6. doi: 10.1006/bbrc.1993.2350. [DOI] [PubMed] [Google Scholar]
- 48.Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011;11:558–72. doi: 10.1038/nrc3090. [DOI] [PubMed] [Google Scholar]
- 49.Trimble MS, Xin JH, Guy CT, Muller WJ, Hassell JA. PEA3 is overexpressed in mouse metastatic mammary adenocarcinomas. Oncogene. 1993;8:3037–42. [PubMed] [Google Scholar]
- 50.Mesci A, Taeb S, Huang X, Jairath R, Sivaloganathan D, Liu SK. Pea3 expression promotes the invasive and metastatic potential of colorectal carcinoma. World J Gastroenterol. 2014;20:17376–87. doi: 10.3748/wjg.v20.i46.17376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Okimoto RA, Breitenbuecher F, Olivas VR, Wu W, Gini B, Hofree M, et al. Inactivation of Capicua drives cancer metastasis. Nat Genet. 2017;49:87–96. doi: 10.1038/ng.3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qi M, Liu Z, Shen C, Wang L, Zeng J, Wang C, et al. Overexpression of ETV4 is associated with poor prognosis in prostate cancer: involvement of uPA/uPAR and MMPs. Tumour Biol. 2015;36:3565–72. doi: 10.1007/s13277-014-2993-7. [DOI] [PubMed] [Google Scholar]
- 53.Myers E, Hill AD, Kelly G, McDermott EW, O'Higgins NJ, Young LS. A positive role for PEA3 in HER2-mediated breast tumour progression. Br J Cancer. 2006;95:1404–9. doi: 10.1038/sj.bjc.6603427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Benz CC, O'Hagan RC, Richter B, Scott GK, Chang CH, Xiong X, et al. HER2/Neu and the Ets transcription activator PEA3 are coordinately upregulated in human breast cancer. Oncogene. 1997;15:1513–25. doi: 10.1038/sj.onc.1201331. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.