

Abstract
Colorectal carcinomas (CRC) harbor well-defined genetic abnormalities including aberrant activation of Wnt/β-catenin and MAPK pathways, often simultaneously. While the MAPK pathway can be targeted using potent small molecule drugs, including BRAF and MEK inhibitors, β-catenin inhibition has been historically challenging. RNA interference (RNAi) approaches have advanced to the stage of clinical viability, and are especially well-suited for transcriptional modulators such as β-catenin. In this study, we report therapeutic effects of combined targeting of these pathways with pharmacological agents. Using a recently-described tumor-selective nanoparticle containing a β-catenin-targeting RNAi trigger, in combination with the FDA-approved MEK inhibitor (MEKi) trametinib, we demonstrate synergistic tumor growth inhibition in in vivo models of CRC, melanoma and hepatocellular carcinoma. At dose levels which were insufficient to significantly impact tumor growth as monotherapies, combination regimens resulted in synergistic efficacy and complete tumor growth inhibition. Importantly, dual MEKi/RNAi therapy dramatically improved survival of mice bearing CRC liver metastases. In addition, pharmacological silencing of β-catenin mRNA was effective against tumors which are inherently resistant or which acquire drug-induced resistance to trametinib. These results provide a strong rationale for clinical evaluation of this dual-targeting approach for cancers harboring Wnt/β-catenin and MAPK pathway mutations.
Introduction
Colorectal cancer (CRC) is the third most common cancer and the second leading cause of cancer deaths in the United States, despite advances in early detection and targeted therapeutics (1). Targeted therapies that have been incorporated into current standard of care regimens for CRC include anti-epidermal growth factor receptor (EGFR) monoclonal antibodies. While EGFR is expressed in approximately 85% of CRCs (2–4), anti-EGFR antibodies were found to only confer a survival benefit to patients with wild-type KRAS and BRAF (5–7). This leaves a large population of CRC patients with KRAS or BRAF mutations, at least 50%, ineligible for the anti-EGFR therapies cetuximab and panitumumab (8–10). For this and other reasons (11–15), refractory CRC remains a severe unmet medical need and the focus on dozens of current clinical trials.
One approach to treating this poor-prognosis population is to target effectors which are functionally downstream of KRAS and BRAF. The best characterized targets in this category are the MEK1 and MEK2 kinases, which regulate mitogen-activated protein kinases (MAPKs) via direct phosphorylation. Small molecule MEK inhibitors are already approved for BRAF-mutant melanoma, and are being evaluated for CRC as part of various combination regimens (16). In one recent Phase I/II trial, triple-inhibition of EGFR, BRAF and MEK in BRAF-mutant CRC with panitumumab, dabrafenib and trametinib, respectively, showed improved progression-free survival (PFS) compared to double-inhibition of EGFR and BRAF (17). However, even this complex drug regimen yielded only modest gains, underscoring many observations suggesting that such potent drug cocktails are insufficient to address refractory CRC. Additionally, clinical experience in melanoma and other tumors has shown that development of resistance to MEK inhibitors remains a key challenge, even when these agents are used in combination (18).
Strikingly, constitutive activation of Wnt/β-catenin signaling, an evolutionarily-conserved network found in nearly every tissue type with broad roles in development and homeostasis, is implicated in approximately 90% of CRCs (19). Although this activation sometimes occurs through direct gain-of-function mutation of CTNNB1, the gene that encodes β-catenin, the more common genetic lesion in this pathway is loss-of-function mutation of upstream negative regulator APC (20). The role of unchecked Wnt/β-catenin signaling in tumor initiation and tumor maintenance is suggested in many preclinical rodent models, including the recent demonstration that restoring APC function in advanced-stage CRC was sufficient to induce differentiation of the colonic epithelium back to the normal physiological state (21). In addition to the well-characterized effects of canonical Wnt pathway activation, new mechanisms by which β-catenin contributes to tumor progression are emerging including a role in evasion of the immune system(22–24). It has been proposed that crosstalk between the Wnt/β-catenin and KRAS/BRAF-driven MAPK pathways were shown to cooperate in tumor initiation and tumor progression (25). Importantly, several studies have identified the aberrant activation of Wnt signaling as a primary cause of CRCs and as a resistance mechanism in MAPK pathway-activated CRCs (26, 27). Despite this knowledge, none of the therapeutic agents specifically targeting the WNT pathway has yet been approved to-date (28).
Despite the central role of Wnt/β-catenin in CRC, pharmacological intervention of this critical pathway has proven to be challenging, and consequently there is a dearth of drug development programs that have proceeded to advanced clinical trials (29, 30). With few exceptions, the majority of prior efforts to inhibit Wnt signaling do not target β-catenin itself, but rather target specific Wnt ligands or ligand secretory pathways, Wnt receptors or transcriptional co-activators where pathway redundancies may limit efficacy (31). In addition to difficulty associated with direct targeting of β-catenin by conventional pharmaceutical modalities, there is also a concern that non-selectively blocking its function in normal tissues will interfere with essential homeostasis functions, particularly in the gastrointestinal tract (31).
RNA interference technology (RNAi) is a promising alterative to small molecules and biologics for cancer therapy, as it offers the potential to potently silence any expressed gene at the mRNA level. As a drug modality, RNAi triggers are currently progressing through advanced clinical trials, where they have demonstrated sufficient therapeutic index, potency, duration and safety to enable expansion beyond their early successes targeting genetic diseases of the liver (31). We have recently shown robust preclinical activity in Wnt-dependent tumors of diverse origin with an RNAi agent targeting CTNNB1 (32). This agent, hereby termed DCR-BCAT, is an optimized Dicer-substrate siRNA (DsiRNA) formulated in a tumor-selective nanoparticle, and demonstrates dose-dependent silencing of β-catenin after systemic administration in preclinical tumor models (32). A DCR-BCAT precursor featuring an earlier-generation lipid nanoparticle, DCR-MYC, achieved RNAi target engagement in diverse tumor types at well-tolerated doses in a Phase I clinical trial (33).
To address whether inhibition of both pathways simultaneously yields benefit, we sought to evaluate a combination of the novel β-catenin RNAi-based inhibitor, DCR-BCAT and the MEK inhibitor (MEKi) trametinib (34) in multiple preclinical CRC models. Importantly, in addition to primary tumor models, metastasis models were included as this setting represents the source of most genetic complexity (35), mortality and medical need for this disease (36–39). This combination demonstrated synergistic efficacy in three CRC models, all with Wnt and MAPK activation. DCR-BCAT was also efficacious in models which are inherently resistant to MEKi, or which acquire resistance to MEKi after several cycles of therapy. We also investigate the potential of dual therapy in other tumor types with relevant genetics, particularly melanoma and hepatocellular carcinoma (HCC). Together, these findings suggest that direct RNAi-mediated inhibition of β-catenin could be an effective strategy to overcome the current limitations of combination therapy for refractory CRC and other cancers.
Materials and Methods
Materials
All DsiRNAs were synthesized by Integrated DNA Technologies, Inc. (IDT, Coralville, IA). Primer and probe oligonucleotides used in quantitative real-time polymerase chain reaction (qRT-PCR) detection were synthesized by IDT or Life Sciences (Carlsbad, CA). Trametinib was purchased from Selleckchem (Houston, TX) with > 99% purity.
Lipid nanoparticle (LNP) and Trametinib formulations
DCR-BCAT and Placebo LNPs were prepared as previously described (32). Trametinib was dissolved in DMSO to make 5 mg/ml solution. This solution was further diluted to the appropriate concentration for p.o. dosing with the solvent mixture containing 10% Ethanol, 10% Kolliphor EL and 80% water. Vehicle used in the studies was 10% DMSO in the solvent mixture.
Cell lines
Human (CRC) cell lines LS411N, Ls174t and SW403, hepatocellular carcinoma cell line Hep3B and human melanoma cell line A2058 were obtained from ATCC (Manassas, VA). CRC cell lines were grown in RPMI medium supplemented with 10% fetal bovine serum (FBS). Hep3B and A2058 cells were grown in DMEM medium supplemented with 10% FBS. Mice were obtained from Harlan Laboratories (Indianapolis, IN). All cell lines used in this study were authenticated originally by ATCC using short tandem repeat analysis. Cells were expanded and frozen at low passage within one month after the receipt of the original stocks, and used within 6 passages after thawing. All cell lines were subjected to mycoplasma testing (IMPACT 1 profile by IDEXX BioResearch) before being released for use in animal studies.
Cell line-derived xenograft models
6–8 week old Hsd:Athymic Nude-Foxn1nu mice (hereby referred to as nude mice) were injected subcutaneously with LS411N (5x106 cells), SW403 (5x106 cells), Ls174t (5x106 cells), Hep3B (5x106 cells + matrigel) under the right shoulder. Tumor volume was measured twice a week to monitor tumor growth/suppression. Dosing was initiated when the tumors reached 200 mm3. For tumor growth inhibition studies, animals were randomized and assigned to one of four cohorts and subjected to dosing cycles as described in Results (n=6/cohort, plus 3 additional animals for pharmacokinetic/pharmacodynamics [PK/PD] analysis after the first cycle). CRC liver metastasis models were generated by surgically implanting 1–2x106 cells in the spleen of nude mice after midline abdominal incision. After surgery, the abdominal incision was closed with 5-0 to 6-0 absorbable, nonbraided suture and the skin was closed with a single wound clip. Mice were anesthetized with isoflurane before initiating the surgery and during surgery. Buprenorphine was given preoperatively and post-operatively at 0.1 mg/kg subcutaneously for pain relief. All intravenous dosing was performed via lateral tail vein at a total volume of 10 ml/kg. Trametinib was given orally at 10 ml/kg. Mice were held in a pathogen-free environment and all procedures involving animals were performed according to protocols approved by Dicerna Pharmaceuticals’ Institutional Animal Care and Use Committee (Dicerna-IACUC).
Quantitative RT-PCR and fluorescence in-situ hybridization (FISH)
Animal tissues were preserved by either snap-freezing or RNA-later fixative (Life Technologies, Carlsbad, CA) and were homogenized using a bead mill (TissueLyzer, Qiagen, Germantown, MD). After total RNA isolation, representative RNA samples were subjected to QC and determination of the RNA Integrity Number (RIN) score by Agilent 2100 Bioanalyzer. 100 ng of total RNA was used to make complementary DNA (cDNA) using high capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA). The cDNA was then diluted 4x for qRT-PCR using TaqMan Fast Advanced Master Mix (Applied Biosystems) and gene-specific primer-probe sets. Primer/probe sets were obtained from Life Technologies and included CTNNB1 (Hs00170025_m1) PP1A (4326316E), MYC (Hs00153408_m1) AXIN2 (Hs00610344_m1). In some experiments, SuperScript III quantitative one-step RT-PCR kit (Applied Biosystems, #11732-088) was used as well. FISH (Supplementary figure S1) for CTNNB1 mRNA was performed exactly as described (32).
Immunohistochemistry
β-catenin immunohistochemistry was performed as described, using a β-catenin antibody (Cell Signaling, Danvers, MA, 8480) at a concentration of 1:500 and Signal Stain DAB substrate kit (Cell Signaling, #8059). Ki67 immunochemistry was performed using an anti-Ki67 antibody (Abcam, Cambridge, MA, #ab16667) at a concentration of 1:100 and DAB under standard conditions. Image intensity quantitation was performed using Nikon Elements Software.
Western blot analysis
Tumor tissues (30–50 mg/mouse) were resuspended in lysis buffer containing protease and phosphatase inhibitors, and homogenized using a bead mill (Qiagen TissueLyser (Germantown, MD). The lysates were then clarified by centrifugation at 14,000 x g for 15 min at 4°C. Protein concentrations of the lysates were determined using the bicinchoninic acid protein assay. Equal amounts of total proteins were loaded on SDS-PAGE, transferred onto nitrocellulose membrane and probed with primary antibodies overnight at 4°C. The primary antibodies against ERK, pERK and β-catenin were obtained from Cell Signaling Technology. Blots were visualized using the Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE).
Statistics
For the synergy analysis, initial assessments of group comparisons were made using ANOVA testing. Prior to ANOVA, normality was determined using the Shapiro-Wilk Test. Combination cohorts showing evidence of a difference between them and their single-agent counterparts were then further tested for synergy. Combination cohorts determined to not have synergy were tested for additivity using Tukey’s test for additivity. All models for additivity included a statement for drug interaction. Further testing for additivity and synergy was performed using the Loewe method of determining a combination index (40). Evidence of synergy was determined if the combination index was found to be less than 1.
Results
DCR-BCAT monotherapy improves the survival of mice bearing CRC liver metastases
β-catenin as a target for CRC and other tumors is strongly supported by both human genetic and functional preclinical data. DCR-BCAT, based on the formulation previously referred to as EnCore-R/CTNNB1 DsiRNA (32) is an intravenously-administered RNAi drug product. DCR-BCAT causes specific, dose-dependent silencing of CTNNB1 mRNA in preclinical murine and human tumors in vivo (32). In xenografted human CRC tumors, robust mRNA silencing occurs throughout the tumor parenchyma, indicating efficient tumor penetration of the lipid nanoparticle ((32) and Supplementary figure S1). Importantly, DCR-BCAT is well-tolerated in wild-type mice at doses as high as 10 mg/kg/week, with no elevation of liver transaminases, cumulative tissue exposure upon repeat dosing, or accelerated blood clearance that would suggest an anti-drug antibody response (Supplemental Figure S2). Since the target site shares complete identity between the human and murine genomes, mechanism-based and nonspecific causes of toxicity can both be considered. This tolerability profile enables a sufficient therapeutic window in preclinical models to evaluate efficacy as a monotherapy and in combination with other targeted therapeutics.
To determine the effect of DCR-BCAT monotherapy in aggressive models of CRC liver metastasis, Ls174t and LS411N cells were surgically implanted into spleen of nude mice as described previously (32). These Wnt-activated human CRC cell lines colonize the liver and form multiple metastatic lesions (Figure 1A), resulting in progressive disease and mortality within approximately one to three months (32). Systemic administration of DCR-BCAT results in significant reduction (50%) of CTNNB1 mRNA in these liver metastases one day after a single dose (Figure 1B). Ls174t tumors progress more rapidly than LS411N tumors, enabling a comparison drug response between models with different growth kinetics. For both models, tumor-bearing mice were randomly cohorted into 3 groups (n=8) and treated with PBS, DCR-Placebo or DCR-BCAT at 3 mg/kg/dose intravenously for a short regimen lasting 17 days (Figure 1C–1D). DCR-Placebo contains identical nucleic acid content and chemistry in a scrambled sequence, formulated in the same lipid nanoparticle composition as DCR-BCAT. Animals were monitored daily for lethality or health deterioration to the point of moribund status, at which point they were euthanized according to IACUC protocol. In both the Ls174t model (Figure 1C) and the LS411N model (Figure 1D), DCR-BCAT treatment conferred a significant survival advantage over either DCR-Placebo or the PBS vehicle. For example, median survival of LS411N tumor-bearing mice increased by >11 weeks relative to placebo, for extended for >5 months after the last dose was administered (Figure 1D). These data demonstrate that silencing of CTNNB1 mRNA in an acute regimen slows the progression of both primary and metastatic lesions.
Figure 1. DCR-BCAT monotherapy improves the survival of mice bearing CRC liver metastases.

A, Development of liver metastases 2–6 weeks after orthotopic implantation of Ls174t human CRC cells into the spleen. Animals were euthanized at the indicated times after implantation and representative liver images are shown. B, Knockdown of CTNNB1 mRNA in liver metastases of Ls174t tumors. 3 weeks after implantation, a single 10 mg/kg dose i.v. dose of DCT-BCAT or PBS was administered and animals were euthanized 24h post-dose. CTNNB1 mRNA was measured by qPCR (n=5/group). Each data point represents an individual animal. C–D, Ls174t (C) or LS411N (D) cells were implanted into the spleen as described in Materials and Methods. Animals were randomized and dosed intravenously with PBS, DCR-Placebo or DCR-BCAT (n=8/cohort, 3 mg/kg/dose). Dosing days are shown by the black arrows. A representative image of a liver from a PBS animal at the time of takedown is shown, indicating the typical pattern of metastatic lesions. Animals were monitored for health daily, Kaplan-Meier plots show the day each subject was found either dead or moribund. The median survival for each cohort is displayed in the figure legend. P values for statistical significance were generated using the Gehan-Breslow-Wilcoxon test in GraphPad Prism software.
DCR-BCAT efficacy in the CRC liver metastasis model improves in combination with a MEK inhibitor
Mutations that affect the Wnt/β-catenin and/or MAPK pathways are the most common genetic lesions in CRC, and are identified in the overwhelming majority of patient biopsies collected from primary or metastatic tumors. Ls174t tumors harbor activating mutations in both the CTNNB1 and KRAS genes (41), and therefore are expected to be sensitive to inhibition of the Wnt/β-catenin and MAPK pathways, respectively. We sought to explore if inhibition of both pathways simultaneously is advantageous in the preclinical metastatic CRC setting. Trametinib, a MEK inhibitor, was chosen as the MAPK pathway modulator based on its specificity profile and extensive preclinical and clinical data availability (34). First, trametinib monotherapy was tested in this model and exhibited dose-dependent efficacy (Figure 2A). Based on this single-agent trametinib data and the single-agent DCR-BCAT data (Figure 1C), we selected a dose level of 2 mg/kg/dose of each agent for combination therapy in the Ls174t metastasis model. We also used our experience in the single-agent settings to tune the kinetics of disease progression and treatment. Namely, we used a high cell implantation number (2 x 106) and waited three weeks after implantation before initiation of therapy. These experimental conditions serve to accelerate tumor progression and ensure that significant metastatic disease is present in the liver at the time of treatment, as shown in Figure 1A. Strikingly, the DCR-BCAT and trametinib combination provided a dramatic survival benefit compared to the single agents tested at the same dose level, improving the median survival time by 30 days vs. each single agent. The median survival was 44–45 days for monotherapy, and 74 days for dual therapy (Figure 2B). Several animals in the combination cohort survived for longer than 100 days, even though the final dose was administered on Day 37. Importantly, the nanoparticle-formulated placebo DsiRNA (DCR-Placebo) offered no benefit over MEKi monotherapy alone, demonstrating that the lipid excipients or off-target activity did not contribute to the observed efficacy. These data offer proof-of-concept that direct inhibition of β-catenin is a viable strategy to sensitize CRC to MEK inhibition.
Figure 2. Combined inhibition of CTNNB1 and MEK significantly improves the survival of mice bearing Ls174t liver metastases.

A, Effect of trametinib monotherapy on survival of mice bearing Ls174t liver metastases. Drug was administered p.o. at two dose levels, 3x/week as indicated by the arrows. B, Effect of combination therapy on survival of mice bearing Ls174t liver metastases. Animals were dosed i.v. with DsiRNA formulations (2 mg/kg/dose), and p.o. with trametinib (2 mg/kg/dose) on the same days, 3x weekly as indicated by the arrows (n=8/cohort). Median survival values are indicated on the figure legends. Note that, in the monotherapy experiment, the cell implantation number was lower (1 x 106 cells) than in the combination experiment and in Fig 1A (2 x 106 cells for both).
Low-dose administration of DCR-BCAT yields synergistic efficacy with trametinib in different genetic subtypes of Wnt/MAPK-activated CRC
The subcutaneous tumor xenograft setting enables rapid evaluation of therapeutic regimens for tumor growth inhibition. To determine if the survival benefit in the metastatic setting can translate into tumor growth inhibition (TGI) when implanted subcutaneously, we performed an efficacy study in mice harboring subcutaneous Ls174t tumors (Figure 3A). The dose levels for this study were chosen based on the single-dose TGI curves for DCR-BCAT (32) and trametinib (Supplementary figure S3). At a trametinib dose level of 0.3 mg/kg in the monotherapy setting, we observed sub-maximal TGI and also sub-maximal effects on MYC mRNA, a recently-reported indicator of MEK inhibitor activity due to the stabilizing effect of ERK phosphorylation on MYC activity (Supplementary figure S3) (42–44). While treating the subcutaneous Ls174t tumors with either agent alone yielded approximately 60% TGI after one dosing cycle, >90% TGI was observed in the combination-treated cohort (p<0.001, Loewe’s combination index <1). As an important control, we also readily observe MEK inhibition by phospho-ERK1/2 western blot in both the trametinib monotherapy and combination settings, but not with DCR-BCAT alone (Figure 3A, bottom panel). These data not only corroborate the findings from the survival study (Figure 2), but also exemplify the ability of the optimized lipid nanoparticle formulation to deliver the RNAi trigger to extrahepatic tumor sites.
Figure 3. DCR-BCAT in combination with trametinib leads to synergistic efficacy in human CRC xenografts of varying genetic backgrounds.

A–C, Ls174t (A), LS411N (B) and SW403 (C) cells were implanted subcutaneously into the flank of nude mice. Animals were dosed with one of 6 different monotherapy or combination regimens as indicated (n=6/cohort) on the days shown by the arrows. For the combination regimens, subjects received both the DsiRNA (i.v.) first, immediately followed by trametinib (p.o.). Tumor volumes were measured frequently by caliper. Mean values are plotted, error bars represent the SEM. The percentages on the plots reflect the degree of tumor growth inhibition (TGI) at the time of the final measurement. P values and combination indices for synergy were determined as described in Materials and Methods. A, lower panel: parallel cohorts were necropsied 8 h after the last dose of the first cycle (Day 10). Tumor homogenates were subjected to western blots for phospho-ERK1/2 and total-ERK1/2. D, Immunohistochemistry for Ki67 for SW403 tumors. Ki67 staining was performed using FFPE tumor sections collected at the terminal time point (n=3), representative 10x images are shown. Scale bar = 600 μm. E, Quantitation of Ki67 mean intensity; each data point represents an individual animal.
Approximately 50% of human CRCs harbor genetic lesions that cause dysregulation of both Wnt/β-catenin and MAPK signaling. We had previously shown that only Wnt-activated tumors are sensitive to DCR-BCAT monotherapy (32). The mutations that affect Wnt/β-catenin signaling are most commonly found in the CTNNB1 and APC genes, while the mutations that affect MAPK signaling are most commonly found in KRAS and BRAF. To investigate whether these specific genetic backgrounds affects the response to dual-therapy, we evaluated DCR-BCAT and trametinib in two additional models (Figure 3B–C). While Ls174t has mutations in CTNNB1 and KRAS, LS411N carries APC/BRAF mutations and SW403 carries APC/KRAS mutations. To determine if we can achieve even greater mathematical synergy than in the Ls174t experiment (Figure 3A), we decreased the dose of DCR-BCAT to levels that were not expected to show efficacy in the monotherapy setting (0.1–0.3 mg/kg/dose; corresponding to approximately one-tenth of the doses required for single-agent efficacy). In both LS411N and SW403 tumors, near-complete tumor stasis was achieved under conditions where single-agent efficacy was negligible. Synergy was demonstrated using Loewe’s drug combination index (Figures 3B–C) (40). Furthermore, immunohistochemical staining of the SW403 tumor sections for Ki67 demonstrates significantly reduced cell proliferation in the combination cohort, therefore identifying inhibition of cell proliferation as a primary mechanism (Figure 3D–E). These data offer preliminary mechanistic insight into dual-therapy, and suggest the possibility of achieving a high therapeutic window and tolerability profile with exceeding low doses of the CTNNB1-targeting RNAi trigger.
DCR-BCAT treatment overcomes acquired and pre-existing resistance to trametinib in CRC and melanoma models
Acquired resistance to trametinib and other MEK inhibitors is well-documented, owing to compensatory secondary genetic lesions in receptor tyrosine kinases, RAS/RAF family members, MEK1/2 itself, or activation of ERK-independent pathways (45). This ultimately limits the clinical potential of this promising therapeutic class (46). We sought to determine if we could model resistance to trametinib in a preclinical CRC model, and if the RNAi combination therapy would be efficacious in this setting. Figure 4A shows that xenografted SW403 tumors are initially sensitive to trametinib, but fail to respond robustly after progressive dosing cycles. Indeed, the third dosing cycle shows little or no TGI. However, adding DCR-BCAT (3 mg/kg/dose) to the regimen yielded rapid tumor regression after the tumors have acquired the MEKi resistance (Figure 4A). The tumors fully recover within two weeks of dosing, but an additional cycle of combination therapy is again effective at inducing tumor regression. These data demonstrate that DCR-BCAT activity is not affected by the acquired resistance mechanisms that affect MEKi. Additionally, these data demonstrate the potential for this combination to treat established large established tumors.
Figure 4. DCR-BCAT treatment overcomes acquired and pre-existing resistance to trametinib.

A, Nude mice bearing SW403 xenograft tumors were dosed with trametinib at 3 mg/kg/dose for consecutive cycles as indicated by the arrows (n=5). For cycles 4 and 5, DCR-BCAT (3 mg/kg/dose) was added to the regimen. The mean tumor volumes are displayed, error bars represent the SEM. B, In additional cohorts, animals were necropsied at the indicated days and tumor RNA was prepared. qPCR was performed for CTNNB1, AXIN2, and MYC mRNAs (left panel) or BMF mRNA (right panel). Data were normalized to the PPIA housekeeping gene and tumors from time-matched PBS-treated animals. C, Nude mice bearing A2058 xenografts were dosed with one of 6 different monotherapy or combination regimens as indicated on the days shown by the arrows (n=5–6/cohort). Mean values are plotted, error bars represent the SEM. The percentages on the plots reflect the degree of tumor growth inhibition (TGI) at the time of the final measurement. D, Immunohistochemistry for Ki67 for A2058 tumors. Ki67 staining was performed using FFPE tumor sections collected at the terminal time point (n=3). Representative 10x images are shown. E, Quantitation of Ki67 mean intensity; each data point represents an individual animal
To further investigate the mechanism by which DCR-BCAT overcomes trametinib resistance, we generated tumor samples at different time points corresponding to the experiment in Figure 4A. Tumor RNA was prepared from the time of pretreatment (Day 15), trametinib sensitivity (Day 26), trametinib resistance (Days 36–42), and reversal of resistance with DCR-BCAT (Day 45). Interesting, onset of resistance was associated with increases in CTNNB1, AXIN2, and MYC mRNAs, all of which were subsequently decreased upon addition of DCR-BCAT to re-sensitize the tumors (Figure 4B). These data suggest that the tumor employs modulation of Wnt/β-catenin signaling as a strategy to overcome MAPK pathway inhibition, consistent with recent data generated using a BRAF inhibitor in melanoma (47) Finally, another mechanism that has been reported to enable acquired resistance to MAPK pathway inhibitors is maintenance of the eIF4F translation initiation complex, in part through transcriptional suppression of pro-apoptotic Bcl-2 family member BMF (48) To that end, we have also observed a large increase in BMF mRNA during reversal of resistance, indicating that at least two distinct mechanisms appear to be involved in sensitization to the drug combination (Figure 4B, right panel).
In addition to acquired resistance, some tumors are inherently resistant to MEK inhibition, even if they harbor BRAF or KRAS mutations. The BRAF/PTEN mutant human melanoma cell line A2058 is insensitive to BRAF and MEK inhibition due to a stabilized elF4F translation initiation complex (48). Unlike the acquired resistance mechanisms, A2058 innate resistance involves a point of convergence of several oncogenic pathways by dysregulating translation of signal-responsive mRNAs (48). Using a dose level of trametinib sufficient to yield complete stasis in the CRC models (Supplementary figure S3), 3 mg/kg/dose, only 36% TGI was achieved in subcutaneously-xenografted A2058 tumors. DCR-BCAT monotherapy (3 mg/kg/dose) also yielded a partial response, while the combination led to complete TGI (105%, Figure 4B). This result is confirmed by Ki67 immunohistochemistry for cell proliferation, which correlates well with the TGI data (Figure 4D–E). In the combination cohort, complete TGI is associated with a 70% decrease in Ki67 staining intensity, compared to a ~50% decrease in the RNAi monotherapy cohort (n=3–5/group). Therefore, RNAi therapy is not affected by multiple clinically-relevant resistance mechanisms, increasing its probability of success in combination with MEKi.
DCR-BCAT-containing regimens are efficacious in a MYC-dependent hepatocellular carcinoma model
The MYC oncogene is amplified or overexpressed in the majority of human tumors, and is particularly well-characterized as a critical driver of hepatocellular carcinoma (HCC) (49). Wnt/β-catenin is one of several signaling networks that regulate its expression and activity (50), and therefore RNAi-mediated silencing of CTNNB1 is highly effective in HCC preclinical models (51, 52). Interestingly, MEK inhibition has been shown to destabilize MYC protein by regulating its ERK-dependent phosphorylation (53). We reasoned that indirectly targeting MYC transcription and MYC stability simultaneously using DCR-BCAT and trametinib, respectively, may yield significant benefit over monotherapy. Indeed, this is the case in subcutaneously-xenografted MYC-dependent Hep3B human HCC tumors, where 90% TGI was achieved with the combination (Figure 5A). To determine the relationship between MYC suppression and efficacy, MYC mRNA was measured after one dosing cycle. While DCR-BCAT and trametinib (2 mg/kg/dose of either agent) suppressed expression by 37% and 59%, respectively, the combination treatment yielded an 85% decrease in MYC compared to vehicle-treated tumors (Figure 5B). Finally, we sought to determine if the potentiation of RNAi-mediated efficacy by trametinib could be reproduced by targeting MYC directly with an RNAi trigger. MYC-targeting DsiRNA (33) was formulated EnCore-R, the same LNP composition used for DCR-BCAT. Strikingly, synergistic efficacy was observed with the MYC/CTNNB1 RNAi combination in Hep3B tumors, even though the dose of each RNAi trigger was reduced by half (from 2 mg/kg/dose to 1 mg/kg/dose) compared to the single-agent cohorts (Figure 5C). These data suggest strong potential for RNAi trigger combinations in diverse human tumors.
Figure 5. DCR-BCAT combination therapy in Hep3B HCC tumors.
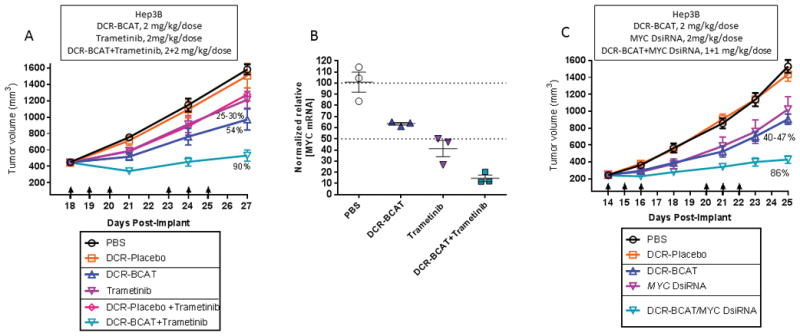
A, Nude mice bearing subcutaneous Hep3B tumors were treated as in Figure 3. 2 mg/kg/dose was used for all agents (n=6). B, qPCR measurements of MYC mRNA in Hep3B tumors collected 24h after one cycle of qdx3 dosing at 2 mg/kg/dose (n=3); each data point represents an individual animal. C, Nude mice bearing subcutaneous Hep3B tumors were treated with one of two formulated DsiRNAs, targeting CTNNB1 (DCR-BCAT), or MYC at 2 mg/kg/dose. The combination was administered at 1 mg/kg/dose of each DsiRNA. Tumor volumes were measured frequently by caliper. Mean values are plotted, error bars represent the SEM.
Discussion
RNAi is an emerging drug modality which has progressed to advanced clinical trials. Specific target engagement in patients has been demonstrated for oligonucleotide therapeutics in cardiovascular diseases (54), viral infection (55), rare genetic diseases (56) and cancer (57). For the latter, safe and efficient tumor-selective drug delivery technology remains a key limitation. LNP-based approaches have been explored to overcome the harsh tumor microenvironment, poor pharmacokinetics, and cell-trafficking properties of nucleic acid-containing drugs. Previous RNAi candidates tested in the clinic for hepatocellular carcinoma contained derivatives of LNP formulations which were initially clinically developed for normal liver applications, not for oncology. EnCore LNP technology (32, 52) was optimized for tumor selectivity, enabling messenger RNA knockdown of well-validated oncogenic targets. β-catenin is an example of a cancer target with an abundance of human genetic evidence, but which has been difficult to drug using conventional small molecules or biologics. The translatability of preclinical efficacy results to human cancers has yet to be fully investigated for RNAi.
Numerous preclinical CRC models are sensitive to Wnt pathway blockers of varying quality and specificity (32, 58, 59). However, several lines of evidence point to the potential benefits of blocking MAPK and Wnt signaling simultaneously. In transgenic mice, activating KRAS in a mutant APC background increases nuclear localization of β-catenin and accelerates CRC tumorigenesis (60). Several reports demonstrated synergistic activity in vitro after targeting both pathways using combinations of small-molecule research grade compounds or marketed drugs (41, 61), or combinations of shRNAs and small molecules (27). In vivo proof-of-concept was achieved using CRC subcutaneous xenograft models carrying inducible short hairpin RNAs (shRNAs) targeting CTNNB1 and KRAS (62). To our knowledge, this concept has never been translated into a pharmacological approach using a specific β-catenin inhibitor prior to the current report, and has not previously been evaluated in the genetically-distinct and clinically important setting of CRC metastasis.
In addition to primary and metastatic CRC, we also explored the combination approach in models of melanoma and HCC. The diversity of preclinical settings for which dual-therapy yields benefit suggests the potential for broad applicability in Wnt/β-catenin and MAPK driven tumors, as well as MYC-dysregulated tumors (Figures 3, 4 and 5). While BRAF inhibitors are frequently employed in melanoma therapy, there is some controversy regarding their potential in combination with Wnt pathway modulators. Inducible CTNNB1 shRNA expression increases sensitivity to BRAFi (47) and inhibits metastasis in melanoma (63), but surprisingly, positive Wnt/β-catenin signaling was actually required for BRAFi-induced apoptosis (64) or associated with decreased proliferation (65) in specific contexts. These observations suggest that there may be some exceptions to the benefits of dual therapy. Indeed, one recent report strongly suggests that PTEN status in BRAF-activated melanoma affects whether Wnt signaling promotes or suppresses metastasis (66). = Additional rigorous preclinical and clinical experimentation will be necessary to determine which genetic backgrounds are most sensitive to dual therapy in melanoma.
The precise molecular interactions which enable the effects of this dual therapy approach are still emerging. Our data suggests a role for transcriptional upregulation of Wnt effector genes by MEKi, as well as regulation of the eIF4a transcription initiation complex (Figure 4B). However, we cannot exclude additional mechanisms. Cross-talk between the Wnt/β-catenin and MAPK networks appears to be extremely context-dependent. For example, β-catenin has been reported to mediate resistance to BRAFi in a manner dependent on a physical interaction with transcription factor Stat3 (47). In another report, a key negative regulator of β-catenin activity, glycogen synthase kinase 3 (GSK3), has been implicated in stabilizing Ras family members and therefore offering a point of convergence between the two networks (67). Other potential intermediates include dual-specificity phosphatases (DUSPs), which negatively regulate the Ras-Raf-MAPK axis and have β-catenin-responsive promoter elements (68). Finally, ERK can directly phosphorylate and regulate LRP6, a co-activator of the Wnt receptor Frizzled (69). Taken together, multiple bi-directional intermediates facilitate MAPK/Wnt cross-talk and could offer mechanistic insight into the effects of this and other drug combinations.
The concept of targeting β-catenin to overcome MEKi resistance is also consistent with Bardelli’s “trunk and branch” model of the cancer evolutionary tree (70). This model states that the tumor remains addicted to the ‘trunk’, or primary mutations (for example, APC or CTNNB1 in colorectal cancer), even as secondary genetic lesions (‘branches’) arise. Therapies that target the branch mutations (e.g. KRAS, BRAF, P13K) can produce dramatic responses at first, but then the resistance develops because of the selection of pre-existing clones with trunk mutations (71). The model supports multiple parallel mechanisms of resistance and of dual-agent synergy. Trunk mutations are often undruggable by conventional modalities, but may be among the most critical points of intervention for many tumor types.
Potent combinations of novel targeted therapeutics are undoubtedly going to be incorporated into standard-of-care treatment for numerous cancer types in the coming years, and will lead to improved outcomes. However, a key clinical limitation of these approaches is the presence of overlapping drug-induced toxicities. This is particularly apparent when combining multiple kinase inhibitors, such as BRAF and MEK inhibitors,(72), MEK and MTOR inhibitors and MEK and EGFR inhibitors (73). We believe that combining conventional kinase inhibitors with innovative drug modalities, including RNAi, has unexplored potential to improve outcomes while minimizing class-related adverse effects.
Supplementary Material
Acknowledgments
Grant Support
This work was funded in part by the Center for Strategic Scientific Initiatives, NCI (IR43CA186410-01A1 and IR43CA186410-02) to Bob D. Brown and colleagues at Dicerna Pharmaceuticals, Inc.
Abbreviations
- CRC
colorectal cancer
- DMSO
dimethyl sulfoxide
- DsiRNA
Dicer substrate small interfering RNA
- FISH
fluorescence in situ hybridization
- HCC
hepatocellular carcinoma
- LNP
lipid nanoparticle
- MAPK
Mitogen activated protein kinase
- MEK
MAPK/ERK kinase
- MEKi
MEK inhibitor
- qPCR
quantitative polymerase chain reaction
- RNAi
RNA interference
- TGI
tumor growth inhibition
Footnotes
Disclosure of Potential Conflicts of Interest:
All authors are full-time employees and shareholders of Dicerna Pharmaceuticals.
References
- 1.Cunningham D, Atkin W, Lenz HJ, Lynch HT, Minsky B, Nordlinger B, et al. Colorectal cancer. Lancet. 2010;375:1030–47. doi: 10.1016/S0140-6736(10)60353-4. [DOI] [PubMed] [Google Scholar]
- 2.Jean GW, Shah SR. Epidermal growth factor receptor monoclonal antibodies for the treatment of metastatic colorectal cancer. Pharmacotherapy. 2008;28:742–54. doi: 10.1592/phco.28.6.742. [DOI] [PubMed] [Google Scholar]
- 3.Loong HH, Ma BB, Chan AT. Update in antiepidermal growth factor receptor therapy in the management of metastatic colorectal cancer. J Oncol. 2009;2009:967920. doi: 10.1155/2009/967920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Venook AP. Epidermal growth factor receptor-targeted treatment for advanced colorectal carcinoma. Cancer. 2005;103:2435–46. doi: 10.1002/cncr.21123. [DOI] [PubMed] [Google Scholar]
- 5.Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–65. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 6.Hsu KH, Tseng JS, Wang CL, Yang TY, Tseng CH, Chen HY, et al. Higher frequency but random distribution of EGFR mutation subtypes in familial lung cancer patients. Oncotarget. 2016;7:53299–308. doi: 10.18632/oncotarget.10715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Knickelbein K, Zhang L. Mutant KRAS as a critical determinant of the therapeutic response of colorectal cancer. Genes Dis. 2015;2:4–12. doi: 10.1016/j.gendis.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Douillard JY, Rong A, Sidhu R. RAS mutations in colorectal cancer. N Engl J Med. 2013;369:2159–60. doi: 10.1056/NEJMc1312697. [DOI] [PubMed] [Google Scholar]
- 9.Wilson PM, Labonte MJ, Lenz HJ. Molecular markers in the treatment of metastatic colorectal cancer. Cancer J. 2010;16:262–72. doi: 10.1097/PPO.0b013e3181e07738. [DOI] [PubMed] [Google Scholar]
- 10.Berlin J. Beyond exon 2--the developing story of RAS mutations in colorectal cancer. N Engl J Med. 2013;369:1059–60. doi: 10.1056/NEJMe1307992. [DOI] [PubMed] [Google Scholar]
- 11.Allegra CJ, Rumble RB, Hamilton SR, Mangu PB, Roach N, Hantel A, et al. Extended RAS Gene Mutation Testing in Metastatic Colorectal Carcinoma to Predict Response to Anti-Epidermal Growth Factor Receptor Monoclonal Antibody Therapy: American Society of Clinical Oncology Provisional Clinical Opinion Update 2015. J Clin Oncol. 2016;34:179–85. doi: 10.1200/JCO.2015.63.9674. [DOI] [PubMed] [Google Scholar]
- 12.Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, Zanon C, Moroni M, Veronese S, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res. 2007;67:2643–8. doi: 10.1158/0008-5472.CAN-06-4158. [DOI] [PubMed] [Google Scholar]
- 13.Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–3. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 14.Misale S, Arena S, Lamba S, Siravegna G, Lallo A, Hobor S, et al. Blockade of EGFR and MEK intercepts heterogeneous mechanisms of acquired resistance to anti-EGFR therapies in colorectal cancer. Sci Transl Med. 2014;6:224ra26. doi: 10.1126/scitranslmed.3007947. [DOI] [PubMed] [Google Scholar]
- 15.Jimeno A, Messersmith WA, Hirsch FR, Franklin WA, Eckhardt SG. KRAS mutations and susceptibility to cetuximab and panitumumab in colorectal cancer. Cancer J. 2009;15:110–3. doi: 10.1097/PPO.0b013e31819e3202. [DOI] [PubMed] [Google Scholar]
- 16.Zhao Y, Adjei AA. The clinical development of MEK inhibitors. Nat Rev Clin Oncol. 2014;11:385–400. doi: 10.1038/nrclinonc.2014.83. [DOI] [PubMed] [Google Scholar]
- 17.Atreya CE, Cutsem EV, Bendell JC, Andre T, Schellens JHM, Gordon MS, et al. Updated efficacy of the MEK inhibitor trametinib (T), BRAF inhibitor dabrafenib (D), and anti-EGFR antibody panitumumab (P) in patients (pts) with BRAF V600E mutated (BRAFm) metastatic colorectal cancer (mCRC) Journal of Clinical Oncology. 2015;33:103. [Google Scholar]
- 18.Welsh SJ, Rizos H, Scolyer RA, Long GV. Resistance to combination BRAF and MEK inhibition in metastatic melanoma: Where to next? Eur J Cancer. 2016;62:76–85. doi: 10.1016/j.ejca.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 19.Giles RH, van Es JH, Clevers H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim Biophys Acta. 2003;1653:1–24. doi: 10.1016/s0304-419x(03)00005-2. [DOI] [PubMed] [Google Scholar]
- 20.Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- 21.Dow LE, O’Rourke KP, Simon J, Tschaharganeh DF, van Es JH, Clevers H, et al. Apc Restoration Promotes Cellular Differentiation and Reestablishes Crypt Homeostasis in Colorectal Cancer. Cell. 2015;161:1539–52. doi: 10.1016/j.cell.2015.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–5. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 23.Spranger S, Gajewski TF. A new paradigm for tumor immune escape: beta-catenin-driven immune exclusion. J Immunother Cancer. 2015;3:43. doi: 10.1186/s40425-015-0089-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spranger S, Gajewski TF. Tumor-intrinsic oncogene pathways mediating immune avoidance. Oncoimmunology. 2016;5:e1086862. doi: 10.1080/2162402X.2015.1086862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guardavaccaro D, Clevers H. Wnt/beta-catenin and MAPK signaling: allies and enemies in different battlefields. Sci Signal. 2012;5:pe15. doi: 10.1126/scisignal.2002921. [DOI] [PubMed] [Google Scholar]
- 26.Tentler JJ, Nallapareddy S, Tan AC, Spreafico A, Pitts TM, Morelli MP, et al. Identification of predictive markers of response to the MEK1/2 inhibitor selumetinib (AZD6244) in K-ras-mutated colorectal cancer. Mol Cancer Ther. 2010;9:3351–62. doi: 10.1158/1535-7163.MCT-10-0376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spreafico A, Tentler JJ, Pitts TM, Tan AC, Gregory MA, Arcaroli JJ, et al. Rational combination of a MEK inhibitor, selumetinib, and the Wnt/calcium pathway modulator, cyclosporin A, in preclinical models of colorectal cancer. Clin Cancer Res. 2013;19:4149–62. doi: 10.1158/1078-0432.CCR-12-3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kahn M. Can we safely target the WNT pathway? Nat Rev Drug Discov. 2014;13:513–32. doi: 10.1038/nrd4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sawa M, Masuda M, Yamada T. Targeting the Wnt signaling pathway in colorectal cancer. Expert Opin Ther Targets. 2016;20:419–29. doi: 10.1517/14728222.2016.1098619. [DOI] [PubMed] [Google Scholar]
- 30.Lu B, Green BA, Farr JM, Lopes FC, Van Raay TJ. Wnt Drug Discovery: Weaving Through the Screens, Patents and Clinical Trials. Cancers (Basel) 2016:8. doi: 10.3390/cancers8090082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blagodatski A, Poteryaev D, Katanaev VL. Targeting the Wnt pathways for therapies. Mol Cell Ther. 2014;2:28. doi: 10.1186/2052-8426-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ganesh S, Koser ML, Cyr WA, Chopda GR, Tao J, Shui X, et al. Direct Pharmacological Inhibition of beta-Catenin by RNA Interference in Tumors of Diverse Origin. Mol Cancer Ther. 2016;15:2143–54. doi: 10.1158/1535-7163.MCT-16-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tolcher AW, Papadopoulos KP, Patnaik A, Rasco DW, Martinez D, Wood DL, et al. Safety and activity of DCR-MYC, a first-in-class Dicer-substrate small interfering RNA (DsiRNA) targeting MYC, in a phase I study in patients with advanced solid tumors. Journal of Clinical Oncology. 2015;33:11006. [Google Scholar]
- 34.Lugowska I, Kosela-Paterczyk H, Kozak K, Rutkowski P. Trametinib: a MEK inhibitor for management of metastatic melanoma. Onco Targets Ther. 2015;8:2251–9. doi: 10.2147/OTT.S72951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vakiani E, Janakiraman M, Shen R, Sinha R, Zeng Z, Shia J, et al. Comparative genomic analysis of primary versus metastatic colorectal carcinomas. J Clin Oncol. 2012;30:2956–62. doi: 10.1200/JCO.2011.38.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nordlinger B, Van Cutsem E, Rougier P, Kohne CH, Ychou M, Sobrero A, et al. Does chemotherapy prior to liver resection increase the potential for cure in patients with metastatic colorectal cancer? A report from the European Colorectal Metastases Treatment Group. Eur J Cancer. 2007;43:2037–45. doi: 10.1016/j.ejca.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 37.Altendorf-Hofmann A, Scheele J. A critical review of the major indicators of prognosis after resection of hepatic metastases from colorectal carcinoma. Surg Oncol Clin N Am. 2003;12:165–92. xi. doi: 10.1016/s1055-3207(02)00091-1. [DOI] [PubMed] [Google Scholar]
- 38.Luu C, Arrington AK, Schoellhammer HF, Singh G, Kim J. Targeted therapies in colorectal cancer: surgical considerations. J Gastrointest Oncol. 2013;4:328–36. doi: 10.3978/j.issn.2078-6891.2013.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Cutsem E, Cervantes A, Nordlinger B, Arnold D, Group EGW. Metastatic colorectal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2014;25(Suppl 3):iii1–9. doi: 10.1093/annonc/mdu260. [DOI] [PubMed] [Google Scholar]
- 40.Loewe S. The problem of synergism and antagonism of combined drugs. Arzneimittelforschung. 1953;3:285–90. [PubMed] [Google Scholar]
- 41.Mologni L, Brussolo S, Ceccon M, Gambacorti-Passerini C. Synergistic effects of combined Wnt/KRAS inhibition in colorectal cancer cells. PLoS One. 2012;7:e51449. doi: 10.1371/journal.pone.0051449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hayes TK, Neel NF, Hu C, Gautam P, Chenard M, Long B, et al. Long-Term ERK Inhibition in KRAS-Mutant Pancreatic Cancer Is Associated with MYC Degradation and Senescence-like Growth Suppression. Cancer Cell. 2016;29:75–89. doi: 10.1016/j.ccell.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ryu SH, Heo SH, Park EY, Choi KC, Ryu JW, Lee SH, et al. Selumetinib Inhibits Melanoma Metastasis to Mouse Liver via Suppression of EMT-targeted Genes. Anticancer Res. 2017;37:607–14. doi: 10.21873/anticanres.11354. [DOI] [PubMed] [Google Scholar]
- 44.Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell. 2012;149:307–21. doi: 10.1016/j.cell.2012.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov. 2014;13:928–42. doi: 10.1038/nrd4281. [DOI] [PubMed] [Google Scholar]
- 46.Sullivan RJ, Flaherty KT. Resistance to BRAF-targeted therapy in melanoma. Eur J Cancer. 2013;49:1297–304. doi: 10.1016/j.ejca.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 47.Sinnberg T, Makino E, Krueger MA, Velic A, Macek B, Rothbauer U, et al. A Nexus Consisting of Beta-Catenin and Stat3 Attenuates BRAF Inhibitor Efficacy and Mediates Acquired Resistance to Vemurafenib. EBioMedicine. 2016;8:132–49. doi: 10.1016/j.ebiom.2016.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boussemart L, Malka-Mahieu H, Girault I, Allard D, Hemmingsson O, Tomasic G, et al. eIF4F is a nexus of resistance to anti-BRAF and anti-MEK cancer therapies. Nature. 2014;513:105–9. doi: 10.1038/nature13572. [DOI] [PubMed] [Google Scholar]
- 49.Lin CP, Liu CR, Lee CN, Chan TS, Liu HE. Targeting c-Myc as a novel approach for hepatocellular carcinoma. World J Hepatol. 2010;2:16–20. doi: 10.4254/wjh.v2.i1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barker N, Morin PJ, Clevers H. The Yin-Yang of TCF/beta-catenin signaling. Adv Cancer Res. 2000;77:1–24. doi: 10.1016/s0065-230x(08)60783-6. [DOI] [PubMed] [Google Scholar]
- 51.Tao J, Zhang R, Singh S, Poddar M, Xu E, Oertel M, et al. Targeting beta-catenin in hepatocellular cancers induced by coexpression of mutant beta-catenin and K-Ras in mice. Hepatology. 2017;65:1581–99. doi: 10.1002/hep.28975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dudek H, Wong DH, Arvan R, Shah A, Wortham K, Ying B, et al. Knockdown of beta-catenin with dicer-substrate siRNAs reduces liver tumor burden in vivo. Mol Ther. 2014;22:92–101. doi: 10.1038/mt.2013.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marampon F, Ciccarelli C, Zani BM. Down-regulation of c-Myc following MEK/ERK inhibition halts the expression of malignant phenotype in rhabdomyosarcoma and in non muscle-derived human tumors. Mol Cancer. 2006;5:31. doi: 10.1186/1476-4598-5-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang X, Lee SR, Choi YS, Alexander VJ, Digenio A, Yang Q, et al. Reduction in lipoprotein-associated apoC-III levels following volanesorsen therapy: phase 2 randomized trial results. J Lipid Res. 2016;57:706–13. doi: 10.1194/jlr.M066399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schluep T, Lickliter J, Hamilton J, Lewis DL, Lai CL, Lau JY, et al. Safety, Tolerability, and Pharmacokinetics of ARC-520 Injection, an RNA Interference-Based Therapeutic for the Treatment of Chronic Hepatitis B Virus Infection, in Healthy Volunteers. Clin Pharmacol Drug Dev. 2016 doi: 10.1002/cpdd.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zimmermann TS, Karsten V, Chan A, Chiesa J, Boyce M, Bettencourt BR, et al. Clinical Proof of Concept for a Novel Hepatocyte-Targeting GalNAc-siRNA Conjugate. Mol Ther. 2017;25:71–8. doi: 10.1016/j.ymthe.2016.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tabernero J, Shapiro GI, LoRusso PM, Cervantes A, Schwartz GK, Weiss GJ, et al. First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov. 2013;3:406–17. doi: 10.1158/2159-8290.CD-12-0429. [DOI] [PubMed] [Google Scholar]
- 58.Scholer-Dahirel A, Schlabach MR, Loo A, Bagdasarian L, Meyer R, Guo R, et al. Maintenance of adenomatous polyposis coli (APC)-mutant colorectal cancer is dependent on Wnt/beta-catenin signaling. Proc Natl Acad Sci U S A. 2011;108:17135–40. doi: 10.1073/pnas.1104182108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Masuda M, Uno Y, Ohbayashi N, Ohata H, Mimata A, Kukimoto-Niino M, et al. TNIK inhibition abrogates colorectal cancer stemness. Nat Commun. 2016;7:12586. doi: 10.1038/ncomms12586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Janssen KP, Alberici P, Fsihi H, Gaspar C, Breukel C, Franken P, et al. APC and oncogenic KRAS are synergistic in enhancing Wnt signaling in intestinal tumor formation and progression. Gastroenterology. 2006;131:1096–109. doi: 10.1053/j.gastro.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 61.Uitdehaag JC, de Roos JA, van Doornmalen AM, Prinsen MB, Spijkers-Hagelstein JA, de Vetter JR, et al. Selective Targeting of CTNBB1-, KRAS- or MYC-Driven Cell Growth by Combinations of Existing Drugs. PLoS One. 2015;10:e0125021. doi: 10.1371/journal.pone.0125021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mologni L, Dekhil H, Ceccon M, Purgante S, Lan C, Cleris L, et al. Colorectal tumors are effectively eradicated by combined inhibition of {beta}-catenin, KRAS, and the oncogenic transcription factor ITF2. Cancer Res. 2010;70:7253–63. doi: 10.1158/0008-5472.CAN-10-1108. [DOI] [PubMed] [Google Scholar]
- 63.Damsky WE, Curley DP, Santhanakrishnan M, Rosenbaum LE, Platt JT, Gould Rothberg BE, et al. beta-catenin signaling controls metastasis in Braf-activated Pten-deficient melanomas. Cancer Cell. 2011;20:741–54. doi: 10.1016/j.ccr.2011.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Biechele TL, Kulikauskas RM, Toroni RA, Lucero OM, Swift RD, James RG, et al. Wnt/beta-catenin signaling and AXIN1 regulate apoptosis triggered by inhibition of the mutant kinase BRAFV600E in human melanoma. Sci Signal. 2012;5:ra3. doi: 10.1126/scisignal.2002274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chien AJ, Moore EC, Lonsdorf AS, Kulikauskas RM, Rothberg BG, Berger AJ, et al. Activated Wnt/beta-catenin signaling in melanoma is associated with decreased proliferation in patient tumors and a murine melanoma model. Proc Natl Acad Sci U S A. 2009;106:1193–8. doi: 10.1073/pnas.0811902106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brown K, Yang P, Salvador D, Kulikauskas R, Ruohola-Baker H, Robitaille AM, et al. WNT/beta-catenin signaling regulates mitochondrial activity to alter the oncogenic potential of melanoma in a PTEN-dependent manner. Oncogene. 2017;36:3119–36. doi: 10.1038/onc.2016.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jeong WJ, Yoon J, Park JC, Lee SH, Lee SH, Kaduwal S, et al. Ras stabilization through aberrant activation of Wnt/beta-catenin signaling promotes intestinal tumorigenesis. Sci Signal. 2012;5:ra30. doi: 10.1126/scisignal.2002242. [DOI] [PubMed] [Google Scholar]
- 68.Zeller E, Mock K, Horn M, Colnot S, Schwarz M, Braeuning A. Dual-specificity phosphatases are targets of the Wnt/beta-catenin pathway and candidate mediators of beta-catenin/Ras signaling interactions. Biol Chem. 2012;393:1183–91. doi: 10.1515/hsz-2012-0130. [DOI] [PubMed] [Google Scholar]
- 69.Lemieux E, Cagnol S, Beaudry K, Carrier J, Rivard N. Oncogenic KRAS signalling promotes the Wnt/beta-catenin pathway through LRP6 in colorectal cancer. Oncogene. 2015;34:4914–27. doi: 10.1038/onc.2014.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Willyard C. Cancer therapy: an evolved approach. Nature. 2016;532:166–8. doi: 10.1038/532166a. [DOI] [PubMed] [Google Scholar]
- 71.Hata AN, Niederst MJ, Archibald HL, Gomez-Caraballo M, Siddiqui FM, Mulvey HE, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med. 2016;22:262–9. doi: 10.1038/nm.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu M, Yang X, Liu J, Zhao B, Cai W, Li Y, et al. Efficacy and safety of BRAF inhibition alone versus combined BRAF and MEK inhibition in melanoma: a meta-analysis of randomized controlled trials. Oncotarget. 2017 doi: 10.18632/oncotarget.15632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Infante JR, Cohen RB, Kim KB, Burris HA, 3rd, Curt G, Emeribe U, et al. A phase I dose-escalation study of Selumetinib in combination with Erlotinib or Temsirolimus in patients with advanced solid tumors. Invest New Drugs. 2017 doi: 10.1007/s10637-017-0459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.