

Abstract
Ductal carcinoma in situ (DCIS) is the most common form of breast cancer, with 50,000 cases diagnosed every year in the United States. Over-treatment and under-treatment remain significant clinical challenges in patient care. Identifying key mechanisms associated with DCIS progression could uncover new biomarkers to better predict patient prognosis and improve guided treatment. Chemokines are small soluble molecules that regulate cellular homing through molecular gradients. CCL2-mediated recruitment of CCR2+ macrophages are a well-established mechanism for metastatic progression. While the CCL2/CCR2 pathway is a therapeutic target of interest, little is known about the role of CCR2 expression in breast cancer. Here, using a Mammary Intraductal injection (MIND) model to mimic DCIS formation, the role of CCR2 was explored in minimally-invasive SUM225 and highly-invasive DCIS.com breast cancer cells. CCR2 overexpression increased SUM225 breast cancer survival and invasion associated with accumulation of CCL2 expressing fibroblasts. CCR2-deficient DCIS.com breast cancer cells formed fewer invasive lesions with fewer CCL2+ fibroblasts. Co-grafting CCL2-deficient fibroblasts with DCIS.com breast cancer cells in the subrenal capsule model inhibited tumor invasion and survival associated with decreased expression of aldehyde dehydrogenase (ALDH1), a pro-invasive factor, and decreased expression of HTRA2, a pro-apoptotic serine protease. Through data mining analysis, high expression of CCR2 and ALDH1 and low HTRA2 expression were correlated with poor prognosis of breast cancer patients.
Implications: This study demonstrates that CCR2 overexpression in breast cancer drives early-stage breast cancer progression through stromal-dependent expression of CCL2 with important insight into prognosis and treatment of DCIS.
Keywords: Breast cancer, CCL2, CCR2, invasion, stroma
Introduction
Ductal carcinoma in situ (DCIS) is the most common form of pre-invasive breast cancer in the US, with over 50,000 cases diagnosed every year. Standard treatment for DCIS involves a combination of lumpectomy and radiation therapy (1,2). Yet, 10 to 35% of patients experience disease recurrence, often accompanied by invasive ductal carcinoma (IDC) (3,4), indicating that under-treatment and over-treatment remain significant concerns in patient care. Few approaches exist to evaluate prognosis of DCIS. Compared to IDC, the use of biomarkers in DCIS has not been well studied. Small or low grade lesions may still become invasive (4,5). Estrogen receptor (ER), Her2, Ki67, p16 and Cox2 are associated with disease recurrence but not with development of invasive breast cancer (6). Identifying key mechanisms associated with DCIS progression could lead to better prognostic factors and tailored treatments for patients with DCIS.
Chemokines are small soluble molecules (8kda), which form molecular gradients to mediate homing of immune cells to tissues during inflammation. Chemokines signal to seven transmembrane receptors that couple to G protein dependent and independent pathways to promote cell migration (7,8). CCL2/CCR2 chemokine signaling is a critical regulator of macrophage recruitment during wound healing and infection (9). CCL2 and CCR2 are overexpressed in multiple cancer types including: pancreatic, prostate and colon cancers and breast cancer correlating with poor patient prognosis (10,11). In breast cancer, animal models have demonstrated that CCL2 recruits CCR2+ macrophages to promote tumor growth and metastasis (11,12). The CCL2/CCR2 pathway is a current therapeutic target of interest (11), but little is known about mechanisms of this pathway in cancer beyond signaling in immune cells.
We recently found that CCR2 is overexpressed in breast cancer cells and regulates CCL2-induced cell survival and migration (13), indicating a macrophage-independent role for CCL2 in breast cancer. Using a novel Mammary Intraductal injection (MIND) model of DCIS, we demonstrate that CCR2 overexpression in DCIS lesions enhances invasive progression associated with accumulation of CCL2-expressing fibroblasts. Using the subrenal capsule model, we demonstrate that fibroblasts derived from DCIS promote breast cancer survival and invasion through CCL2 dependent mechanisms. Furthermore, increased CCL2/CCR2 signaling in DCIS is associated with increased expression of ALDH1, a pro-invasive factor, and decreased expression of HTRA2, a pro-apoptotic serine protease, factors associated with poor prognosis of breast cancer patients (14,15). These studies identify a key mechanism of DCIS progression involving CCL2/CCR2 signaling between fibroblasts and breast epithelial cells, with important clinical implications.
Materials and Methods
Cell culture
Human fibroblasts were isolated from reduction mammoplasty or DCIS tissues obtained from the Biospecimen Core Facility at the University of Kansas Medical Center (KUMC), and immortalized by expression of human teleromerase reverse transcriptase as described (16). Fibroblasts were authenticated by expression of: Platelet Derived Growth Factor Receptor-α (PDGFR-α), Fibroblast Specific Protein 1 (Fsp1) and α-smooth muscle actin (α-sma) and absence of pan-cytokeratin. DCIS.com cells originated from Dr. Fred Miller’s laboratory (17). These cell lines were cultured in Dulbecco’s modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS) (Atlas Biological cat no. FR-0500-A), 2 mM L-glutamine (Cellgro cat no. 25-005-CI), 100 I.U/ml penicillin and 100 μg/ml streptomycin (Cellgro cat. no. 10-080). SUM225 cells originated from Dr. Steven Ethier’s laboratory (18), and were cultured in Ham’s F12 media containing 10% FBS, 5 μg/ml insulin, 1 μg/ml cortisone and antibiotics. Cells were passaged no longer than 6 months, and tested for mycoplasma after thawing using the MycoAlert™ Plus Kit (Lonza cat no. LT07-701).
Lentiviral transduction
For CCR2 overexpression, full length CCR2 cDNA was obtained from University of Missouri-Rolla cDNA Resource Center (clone id no. CCR200000), and subcloned into pHAGE–CMV-MCS-IRES-zsgreen lentiviral plasmid (PLASMID, Harvard University) using NHEI and XbaI restriction sites. pHAGE empty vector was used as a vehicle control. CCR2 and non-silencing control shRNAs in pGFP-c-shlenti lentivirus vectors were purchased from Origene (cat no. TL321181). The CCR2 targeting sequence was: 5′-TATTGTCATTCTCCTGAACACCTTCCAGG3′. CCL2 and non-silencing control shRNAs in pGFP-c-shlenti lentivirus vectors were obtained from Origene (cat no. TL316716). The CCL2 targeting sequence (Origene) was: 5′-ACTTCACCAATAGGAAGATCTCAGTGCAG-3′. CCL2 and non-silencing control shRNAs in GIPZ shRNA lenvirus vectors were obtained from Dharmacon (cat no. V2LHS31298). The CCL2 targeting sequence was: 5′-TAAGTTAGCTGCAGATTCT-3′.
To generate lentivirus, 3.33 μg of PMD2G (Addgene cat no.12260), 6.66 μg PDPAX2 (Addgene cat no.12259) and 10 μg target vectors were co-transfected in HEK-293T cells using Lipofectamine 2000 (ThermoFisher cat no. 11668027). Medium was removed 48 hours later and used to transduce cells, which were sorted for Green Fluorescent Protein (GFP) expression by fluorescence activated cell sorting (FACS).
Gene deletion by Clustered regularly interspaced short palindromic repeats (CRISPR)
The CCR2 guide RNA was cloned into the pSpCAs9(BB)-2A-GFP(PX458) vector (Addgene cat no.48318) using BsmBI enzyme. The CCR2 guide RNA sequence was: 5′-TTCACAGGGCTGTATCACATCGG-3′, which targeted the exon encoding the extracellular loop between the 2nd and 3rd transmembrane domains of human CCR2. The vector was transfected into DCIS.com breast cancer cells using jetPei® transfection reagent (Polyplus cat no. 101-01), with N/P 7.5. 48 hours later, GFP positive cells were FACS sorted, cultured as single cell clones in 96 well plates, and expanded into 6 well plates. Genomic DNA of individual colonies was screened by PCR to detect mutant colonies. The detection primer pair spanning the CCR2 targeting site was: 5′- ACATGCTGGTCGTCCTCATC, 3′-AAACCAGCCGAGACTTCCTG. The PCR product of the wildtype gene was 901bp, and contained one DdeI enzyme digestion site to yield 231 and 670bp fragments. Exon excision introduced an additional DdeI restriction site resulting in fragment sizes of 181, 231 and 468 bp upon DdeI digestion.
Enzyme-linked immunosorbent assay (ELISA)
40,000 cells/well were seeded in 24 well plates in DMEM/10% FBS for 24 hours, washed in PBS and incubated in serum free DMEM for 24 hours in 500 μl/well. Conditioned media were assayed for human CCL2 by ELISA (Peprotech cat no.900-M31). Reactions were catalyzed using tetramethylbenzidine substrate (cat no. 34028, Pierce). Absorbance was read at OD450 nM using a BioTek Microplate Reader.
MIND model
Non-Obese Diabetic Severe Combined Immunodeficient interleukin receptor-γ2 null female mice (NOD-SCID) 8–10 weeks of age were purchased from Jackson Laboratories. MIND injections were performed as described (19). Briefly, 4000 cells/μl breast epithelial cells were prepared in 50 μl phosphate buffered saline (PBS) containing 0.1% trypan blue. A Y incision was made on the abdomen of mice anesthetized with ketamine/xylazine [100mg/Kg(K)+10mg/kg(X)] to expose the 4–5 and 9–10 inguinal glands. The inguinal nipples were snipped. A 30-gauge Hamilton syringe with a blunt-ended ½ inch needle was used to deliver 5 μl (20,000) cells/nipple. Skin flaps were closed with wound clips. Mice were palpated for lesions twice weekly. SUM225 injected mice were sacrificed 7 weeks post-injection. DCIS.com injected mice were sacrificed 4 weeks post-injection.
Subrenal graft
Transplantation into subrenal capsules of NOD–SCID female mice (6–8 weeks old) was performed as described (20). Briefly, 250,000 fibroblasts were re-suspended with 100,000 DCIS.com cells in 50 μl rat tail collagen I (BD Pharmingen), and cultured in DMEM/10% FBS for 24 hours. Mice were anesthetized by ketamine/xylazine, a 1–1.5 cm midline incision was made in the back 3 cm from the base of the tail, and the lateral or contralateral kidney was exposed. A small incision was made in the capsule layer using forceps and small spring loaded scissors. The graft was inserted using a glass pipette. The body wall was closed with gut absorbable sutures and the skin was closed with wound clips. Mice were monitored twice weekly and sacrificed 3 weeks post-transplantation.
DAB immunostaining
Tissues were fixed in 10 % neutral formalin buffer and embedded in wax as described (21). For DAB immunostaining, 5 micron sections were dewaxed and heated in 10 mM sodium citrate buffer pH 6.0 for 2 minutes. Endogenous peroxidases were quenched in PBS/60 % methanol/3% H2O2, blocked in PBS/3% FBS, and incubated with primary antibodies (1:100) overnight at 4°C: collagen IV (Novus Biologicals NB120-6586SS), cleaved caspase-3 Asp175 (Cell Signaling Technology cat no. 9579), Von Willebrand Factor 8 (VWF8) (Millipore cat no. Ab7356), PDGFR-α (Cell Signaling Technology cat no. 5241), KI67 (Santa Cruz Biotechnology cat no. 1307), HTRA2 (Cell Signaling Technology cat no. 2176), CCL2 (Santa Cruz Biotechnology, cat .no cat no. 1304) or F4/80 (Abcam cat no. ab6640). Fsp1 antibodies (Abcam cat no. 27427) were diluted 1:3. Slides were incubated for 2 hours at 1:1000 with: anti-rabbit-biotinylated (Vector Laboratories cat no. BA-5000), anti-goat biotinylated (Vector Laboratories cat no. BA-5000), or anti-rat-biotinylated (cat no.BA-9401, Vector Laboratories). For laminin staining, slides were treated with 20 μg/ml Proteinase K for 1 hour at 37°C prior to incubation with 1:100 pan-specific antibodies (Novus Biologicals cat no. NB300-144AF700). Slides were incubated with streptavidin peroxidase (Vector Laboratories cat no. PK-4000), developed with 3,3′-diaminobenzidine (DAB) substrate (Dako cat no. K346711), counterstained with Mayers’ hematoxylin and mounted with Cytoseal. Proliferating Cell Nuclear Antigen (PCNA) (cat no. sc25280, Santa Cruz Biotechnology) and ALDH1A1 (RnD Systems cat no. MAB5869) proteins were detected using the Mouse on Mouse (MOM) kit (Vector Laboratories cat no. BMK-2202).
Immunofluorescence
For CK/α-sma co-staining, slides were heated in 10 mM sodium citrate buffer pH 6.0 for 2 minutes. Slides were incubated with antibodies 1:100 overnight at 4°C to: α-sma, (Spring Biosciences cat no. SP171) and CK5 (ThermoFisher cat no. MA5-12596) or CK19 (ThermoFisher cat no. MS198). Slides were incubated for 2 hours at 1:200 with anti-rabbit-IgG-AlexaFluor®568 (ThermoFisher cat no. A10042) and anti-mouse IgG-AlexaFluor®488 (ThermoFisher cat no. A-11001). For pan-cytokeratin/phalloidin co-staining, slides were heated in 10 mM sodium citrate pH 6.8 for 5 minutes. Slides were incubated with 1:100 AlexaFluor®488-phalloidin (ThermoFisher cat no. A12379) and anti-pan-cytokeratin (Santa Cruz Biotechnology cat no. 8018) overnight at 4°C, and incubated with secondary anti-mouse-AlexaFluor®647 (ThermoFisher cat no. 31571) using the MOM kit. Sections were counterstained with 4′,6-Diamidino-2-Phenylindole,Dihydrochloride (DAPI) and mounted with PBS/glycerol.
Image quantification
Five random fields/section were captured at 10× magnification using the FL-Auto EVOS System (Invitrogen). DAB staining was quantified as described (10). Briefly, images were imported into Adobe Photoshop, DAB staining was selected using the Magic Wand tool, copied and saved a separate file. Images were opened in Image J (NIH), and converted to grey scale. Background pixels were removed by threshold adjustment. Images were subject to particle analysis. Positive DAB values were normalized to total area values, expressed as arbitrary units. To quantify stromal staining, epithelial tissues were cropped out in Adobe Photoshop. DAB staining was selected in stroma, copied to a new window and saved as a separate file. Images were opened in Image J and quantified. Stromal DAB values were normalized to total stromal values.
Scoring of tumor invasion
Tissues were sectioned at 3 depths approximately 50 microns apart. 2–3 serial sections per depth were stained. Images were captured at 4× and 10× magnification and scored in a blinded fashion: 1 (non-invasive), 2 (lowly invasive) or 3 (highly invasive). For CK/α-sma CO-IF: 1 indicated no invasion, with intact α-sma+ myoepithelium and confinement of epithelial cells within the duct. 2 indicated 50% or less disappearance of the α-sma surrounding the duct and/or appearance of 3 or fewer cells invading through the duct. 3 indicated more than 50% α-sma disappearance, with appearance of more than 3 cells invaded through the duct and making contact with the periductal stroma. For collagen IV and laminin immunostaining: 1 indicated well defined expression in the basement membrane, 2 indicated additional low level expression in lesion. 3 indicated higher expression in epithelium, with poor definition between epithelium and stroma. For phalloidin/pan-cytokeratin staining: 1 indicated a well-defined border between tumor and kidney, with a few tumor cells invaded into kidney tissue. 2 indicated some tumor cell invasion, characterized by viable tumor cells present in kidney tissue; the border between kidney and tumor tissue was less defined. 3 indicated high invasion characterized by extensive number of tumor cells in kidney tissue; tumor was embedded in kidney, and the border between kidney tissue and tumor were undefined.
Flow cytometry
Flow cytometry staining for CCR2 expression was conducted as described (13). Briefly, adherent cells were detached from plastic by Accutase® (ThermoFisher cat no. A1110501), washed in PBS and incubated with anti-CCR2-PE for 1 hour on ice. Samples were washed in PBS 3 times and analyzed on a LSRII Flow cytometer, normalized to unstained controls.
Fibroblast proliferation assay
Fibroblasts were seeded 30,000/well in 24 well plates overnight. DCIS.com cells (500,000) were seeded in 10 cm dishes, and incubated with 5 ml serum free DMEM for 24 hours. Fibroblasts were treated with 500 μl of DMEM or tumor conditioned medium for 24 or 48 hours. Fibroblasts were detached through trypsinization, quenched in DMEM/10% FBS and pelleted by microcentifugation. Fibroblasts were re-suspended in 50 μl PBS and counted by hemocytometer.
Statistical Analysis
Cell culture experiments were repeated a minimum of three times. Data are expressed as mean± standard error of the mean (SEM). Statistical analysis was determined using Two-Tailed T-test or ANOVA with Bonferonni’s post-hoc comparisons for normal distributions and Kruskall-Wallis Test with Dunn’s post-hoc comparison for non-Gaussian distributions. Statistical analysis was performed using Graphpad Software. Significance was determined by p<0.05. *p<0.05, **p<0.01, ***p<0.0001, n.s=not significant or p>0.05.
Ethics approval and consent to participate
All animal experiments were performed at KUMC according to guidelines from the Association for Assessment and Accreditation of Laboratory Animal Care. Experiments were approved by the Institutional Animal Care and Use Committee. Patient samples were collected under approval by Institutional Review Board (IRB) at KUMC. All samples were de-identified by the Biospecimen Core, an IRB approved facility, prior to distribution.
Results
CCR2 overexpression in SUM225 cells enhances DCIS progression
In DCIS, cancer cells grow, but remain within the boundaries of ducts and lobules, which are lined by α-sma+ myoepithelial cells and basement membrane, structural barriers between the stroma and duct. Progression from DCIS to IDC is characterized by disappearance of the myoepithelium and appearance of invading ductal carcinoma cells into the surrounding stroma (22). To clarify the role of epithelial CCR2 expression in DCIS progression, we utilized MIND models established through injection of SUM225 and DCIS.com breast cancer cells. SUM225 breast cancer cells are lowly-invasive, of a luminal/Her2+ subtype (23). DCIS.com breast cancer cells, a basal-like subtype, are more highly-invasive (19). By flow cytometry, CCR2 expression was significantly lower in SUM225 cells compared to DCIS.com breast cancer cells (Figure 1A). We first examined the effects of CCR2 overexpression on progression of SUM225 lesions. By lentivirus transduction, two different SUM225 cell lines were generated to overexpress CCR2 (CCR2-L and CCR2-H), and compared with SUM225 cells expressing pHAGE vehicle control (Figure 1A). These cells were MIND injected into NOD-SCID mice, and examined 7 weeks post-injection, when lesions were palpable. CCR2-overexpressing xenografts showed no significant changes in mammary tissue mass compared to pHAGE control (Figure 1B).
Figure 1. CCR2 overexpression in SUM225 breast cancer cells enhances invasive progression.
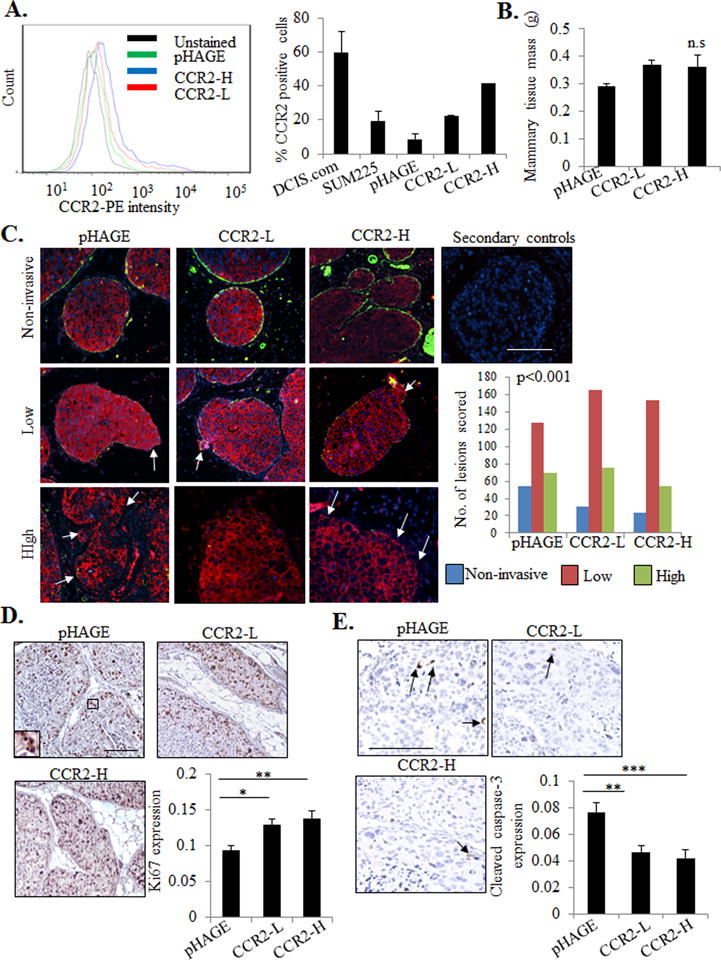
A. Flow cytometry analysis for CCR2 expression in parental DCIS.com or SUM225 parental cells or SUM225 cells expressing vehicle pHAGE control or CCR2 (CCR2-L, CCR2-H). Histogram analysis shown on left. Graph shows percentages of positive cells. B. Tissue mass of SUM225 MIND injected mammary glands C. SUM225 lesions were co-stained for CK-19 (red) and α-sma (green), and scored for the number of invasive lesions n=252 lesions for control pHAGE, 272 lesions for CCR2-L, and 231 for CCR2-H group. Representative images are shown with secondary antibody control panel of anti-rabbit-Alexa-fluor488/anti-mouse-Alexa-fluor-568/DAPI overlay. Arrows indicate invasive foci. D-E. Image J quantification of immunostaining for Ki67 (D) or cleaved caspase-3 (E) in SUM225 lesions (arbitrary units). Arrows point to examples of positive staining. Statistical analysis was performed using One way ANOVA with Bonferonni post-hoc comparison (B, D, E) or Chi square test (C). Statistical significance was determined by p<0.05. *p<0.05. ns= not significant. Mean±SEM values are shown. Scale bar=200 microns.
Extent of epithelial invasion in the mammary gland or breast tissue has been determined by evaluating myoepithelial integrity through α-sma expression, and examining for presence of carcinoma cells contacting the surrounding stroma (19,24–26). To evaluate the effects of CCR2 overexpression on ductal invasion, we co-stained for α-sma to define ductal myoepithelium, and for human specific CK19 to define SUM225 cells. Lesions were scored for invasiveness. Non-invasive lesions had intact α-sma+ myoepithelium, lowly-invasive lesions showed reduced α-sma expression, lining the breast duct, and a few invasive cancer cells. Highly-invasive lesions showed minimal α-sma expression and multiple invasive cancer cells. In the pHAGE controls, 21 % were non-invasive, 51% were lowly-invasive and 28% were highly-invasive. Of CCR2-L MIND lesions, 12% were non-invasive, 57% were lowly-invasive and 31% were highly-invasive. Of CCR2-H MIND lesions, 8% were non-invasive, 66% were lowly-invasive and 26% were highly-invasive (Figure 1C). The decrease in lowly-invasive lesions and increase in lowly-invasive lesions indicate a shift towards invasion.
To further characterize invasion through basement membrane, mammary tissues were stained for laminin and collagen IV, which are basement membrane proteins associated with invasiveness in breast cancer (27,28). In non-invasive lesions, laminin and collagen IV were expressed in the basement membrane and surrounding stroma. In lowly-invasive lesions, laminin and collagen IV were also detected in the epithelium, correlating with a few invading epithelial cells. In highly-invasive lesions, laminin and collagen IV expression in lesions resulted in poorly defined borders between stroma and epithelium. Consistent with α-sma/CK19 CO-IF, laminin and collagen IV staining revealed that CCR2 overexpression in SUM225 cells decreased the number of lowly-invasive lesions and increased the number of highly-invasive lesions (Supplemental Figure S1A-B). CCR2 overexpression was also associated with increased cell proliferation and decreased apoptosis as indicated by Ki67 and cleaved caspase-3 staining (Figure 1D–E). These data indicate that CCR2 overexpression in SUM225 cells enhances the progression of MIND xenografts.
Knockdown or knockout of CCR2 in DCIS.com cells inhibits DCIS progression
We next examined the effects of CCR2 deficiency on progression of DCIS.com lesions. Of the 4 shRNA sequences tested, 1 induced significant knockdown of CCR2 expression in DCIS.com cells (Figure 2A). MIND injection of DCIS.com cells in NOD-SCID mice resulted in palpable mammary lesions at 4 weeks. CCR2 knockdown (CCR2-KD) decreased mammary tissue tumor growth compared to control shRNA expressing xenografts (Figure 2B). To examine for changes in ductal invasion, sections were CO-IF stained for α-sma and human specific CK5 to identify DCIS.com cells, and scored. While the percentage of lowly-invasive lesions (75%) was higher in the CCR2-KD group compared to control (68%), the percentage of highly-invasive lesions dropped from 20% in the control shRNA group to 6% in the CCR2-KD group. The percentage of non-invasive lesions increased from 12% in controls to 19% in the CCR2-KD group. These data indicated a shift from high to less invasive lesions with CCR2-KD (Figure 2C). This trend was also observed in analysis of collagen IV and laminin expression in DCIS.com MIND lesions (Supplemental Figure S2A-B). CCR2 KD was also associated with decreased tumor cell proliferation and increased apoptosis (Figure 2D-E).
Figure 2. shRNA mediated CCR2 knockdown in DCIS.com breast cancer cells inhibits invasive progression.
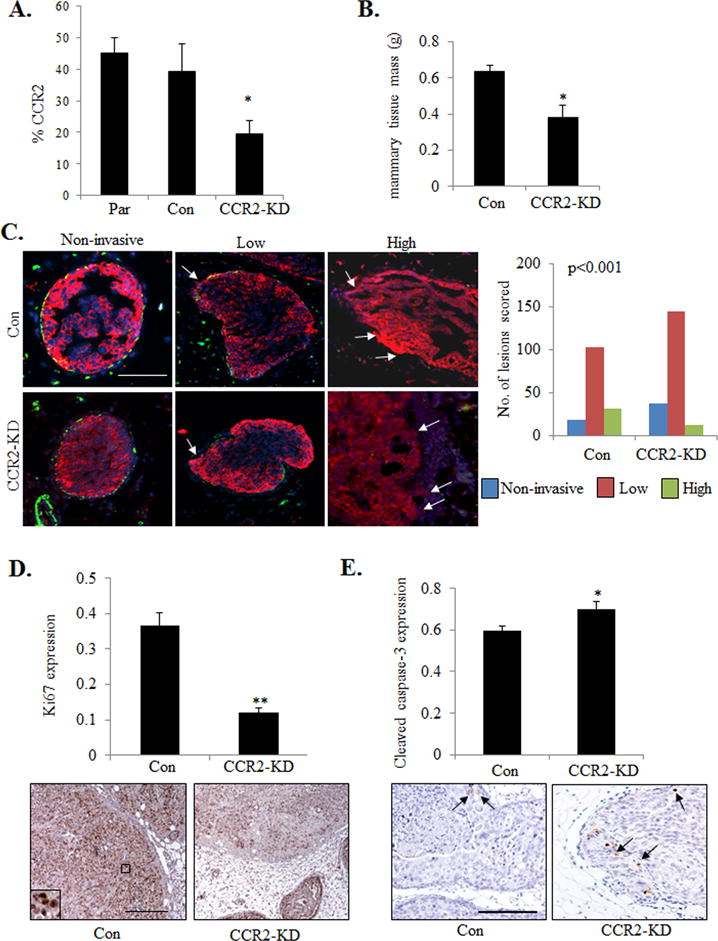
A. Flow cytometry analysis for CCR2 expression in Parental (Par) or DCIS.com cells expressing control (Con) or CCR2 shRNA (CCR2-KD). B. Tissue mass of DCIS.com MIND injected mammary glands C. DCIS.com MIND lesions were co-stained for CK5 (red) and α-sma (green) and counterstained with DAPI (blue). Representative images are shown with arrows pointing to invading tumor cells. Lesions were scored for invasiveness. n=152 lesions for control shRNA group, 193 lesions for CCR2-KD group. n=8 mice/group. D-E. Image J quantification of Ki67 (D) or cleaved caspase-3 (E) immunostaining in DCIS.com lesions. Arbitrary units are shown. Arrows point to examples of positive staining. Statistical analysis was performed using One way ANOVA with Bonferonni post-hoc comparison (B, D,E) or Chi square test (C). Statistical significance was determined by p<0.05. *p<0.05, **p<0.01. Scale bar=200 microns.
To validate the effects of CCR2-KD on DCIS.com progression, the CCR2 gene was knocked out by CRISPR. Two wildtype clones and 1 homozygous knockout clone (CCR2-KO) were identified from 70 clones (Supplemental Figure S3A). By flow cytometry, wildtype clones showed similar CCR2 expression levels to parental cells, while CCR2-KO cells showed a significant reduction in CCR2 expression (Supplemental Figure S3B). MIND injection of CCR2-KO cells resulted in a significant reduction in mammary tissue mass and fewer invasive lesions (Supplemental Figure 3C-D), compared to WT xenografts. These data indicate that CCR2 KD or KO inhibits the progression from DCIS to IDC in DCIS.com MIND xenografts.
Increased angiogenesis, fibrosis and macrophage recruitment are associated with invasive breast cancer (22). To determine how epithelial CCR2 expression affected the surrounding mammary stroma, immunostaining was performed to analyze expression of biomarkers for macrophages (F4/80) and endothelial cells (VWF8). To account for fibroblast heterogeneity, we immunostained for two different markers: Fsp1 and PDGFR-α (29,30). DAB expression of stromal biomarkers was quantified by pixel density analysis and normalized to total stromal area, using an Image J protocol described previously (10). There were no significant changes in VWF8 or F4/80 expression with CCR2 overexpression or knockdown (Supplemental Figure S4A-B). Fsp1 and PDGR-α were expressed in fibroblastic stroma and in epithelial cells, consistent with studies showing mesenchymal marker expression in breast cancer cells (10,31). CCR2 overexpressing SUM225 xenografts showed increased stromal expression of Fsp1 and PDGFR-α (Figure 3A-B), associated with stromal CCL2 expression (Figure 3C). Conversely, CCR2 deficient DCIS.com MIND xenografts showed a significant decrease in fibroblastic cells and decreased CCL2 expression in the stroma (Figure 4A-B). These data indicate that CCR2 overexpression or knockdown is associated with changes in CCL2 expressing fibroblasts in the DCIS stroma.
Figure 3. CCR2 overexpression in SUM225 MIND xenografts increases the levels of CCL2 expressing fibroblasts.
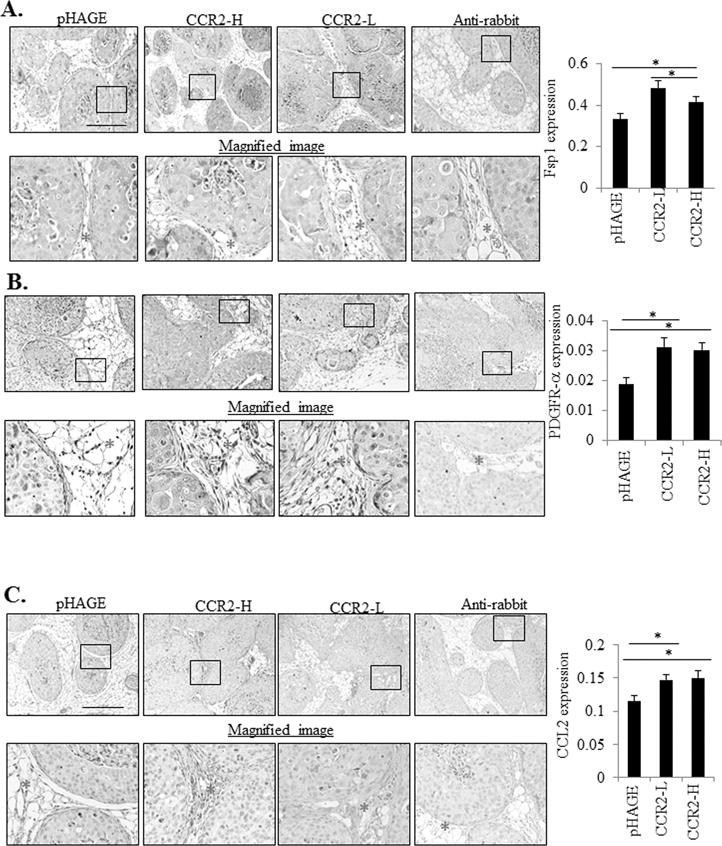
Sum 225 MIND lesions were immunostained for A. Fibroblast Specific Protein 1 (Fsp1), B. Progesterone Growth Factor Receptor-α (PDGFR-α). or C. CCL2 expression. Representative images are shown with magnified image underneath. The stroma is marked with an asterisk in the magnified image. Expression in the stroma was quantified by Image J, in arbitrary units. Statistical analysis was performed using One way ANOVA with Bonferonni post-hoc comparison. Statistical significance was determined by p<0.05. *p<0.05, ***p<0.001. Mean±SEM values are shown. Scale bar=400 microns.
Figure 4. CCR2 shRNA knockdown in DCIS.com MIND xenografts reduces the levels of CCL2 expressing fibroblasts.
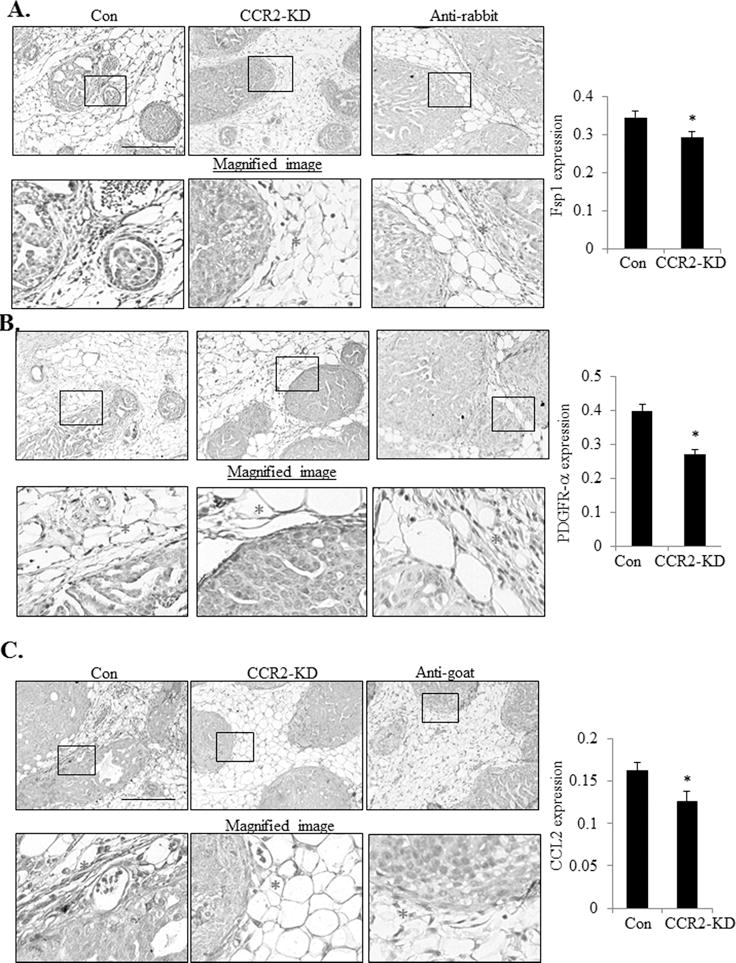
DCIS.com MIND lesions were immunostained for A. Fibroblast Specific Protein 1 (Fsp1), B. Progesterone Growth Factor Receptor-α (PDGFR-α) or C. CCL2 expression. Representative images are shown with magnified image underneath. The stroma is marked with an asterisk in the magnified image. Expression in the stroma was quantified by Image J, in arbitrary units. Statistical analysis was performed using Two-Tailed T-test (B). Statistical significance was determined by p<0.05. *p<0.05. Mean±SEM values are shown. Scale bar= 400 microns.
CCL2 from DCIS fibroblasts is important for CCR2 mediated breast cancer survival and invasion
To further characterize the expression of CCL2 in DCIS stroma, fibroblasts were isolated from patient samples of normal breast or DCIS tissues and analyzed for CCL2 expression. By ELISA, 2 out of 3 DCIS fibroblast lines (1213-249, 80H) expressed higher levels of CCL2 compared to normal fibroblasts (hNAF2525, hNAF8727) and DCIS.com cells (Figure 5A). While previous studies have established an important role for carcinoma-associated fibroblasts from invasive breast cancers (32,33), the role of fibroblasts derived from DCIS tissues remain poorly understood. To determine the functional role of CCL2 derived from DCIS fibroblasts in CCR2 mediated breast cancer progression, we utilized the subrenal capsule model. The advantage of this model is that is devoid of fibroblasts, enabling us to determine the relative contribution of co-grafted fibroblasts without interference from host stroma. Mammary carcinoma cells grafted in the subrenal capsule form tumors similarly to orthotopic injection (34,35). 1213-249 DCIS derived fibroblasts, which showed the highest level of CCL2, were co-grafted with DCIS.com breast cancer cells in the renal capsule of NOD-SCID mice, and analyzed for changes in tumor growth and invasion. Fibroblasts co-grafted with parental DCIS.com cells showed increased tumor mass compared to DCIS.com cells grafted alone (Figure 5B). CCR2 deficient DCIS.com cells co-grafted with 1213-249 fibroblasts showed a significant 20% decrease in tumor mass compared to fibroblasts co-grafted with control DCIS.com cells (Figure 5B). To examine for changes in tumor invasion into normal kidney tissue, we performed CO-IF staining for pan-cytokeratin and phalloidin to distinguish tumor cells from kidney tissues. In the subrenal capsule model, pan-cytokeratin antibodies stained DCIS.com tumors more clearly than CK5 antibodies used in the MIND model. Pan-cytokeratin antibodies recognized CK:4,5,6,8,10,13 and 18, and preferentially stained breast cancer cells over kidney tissues, which expressed fewer of the cytokeratins (36). Using this approach, tumor invasion was characterized by a lack of defined border between tumor and kidney tissues, and scattering of tumor cells throughout the kidney, as observed in control lesions (Figure 5C). CCR2 deficient cells co-grafted with fibroblasts showed a reduction in tumor invasion, characterized by more cohesive tumors and a clearer delineation between tumor and kidney tissues (Figure 5C). Decreased invasiveness was associated with decreased tumor cell proliferation and increased apoptosis as indicated by PCNA and cleaved caspase-3 immunostaining (Figures 5D-E). CCR2-KD DCIS.com cells grafted alone did not show significant differences in tumor growth or invasion compared to control shRNA cells grafted alone (Supplemental Figure S5A-C). These studies indicate that CCR2 knockdown in DCIS.com breast cancer cells inhibits fibroblast-mediated tumor growth and invasion.
Figure 5. CCR2 knockdown in DCIS.com cells inhibits fibroblast mediated cancer progression.
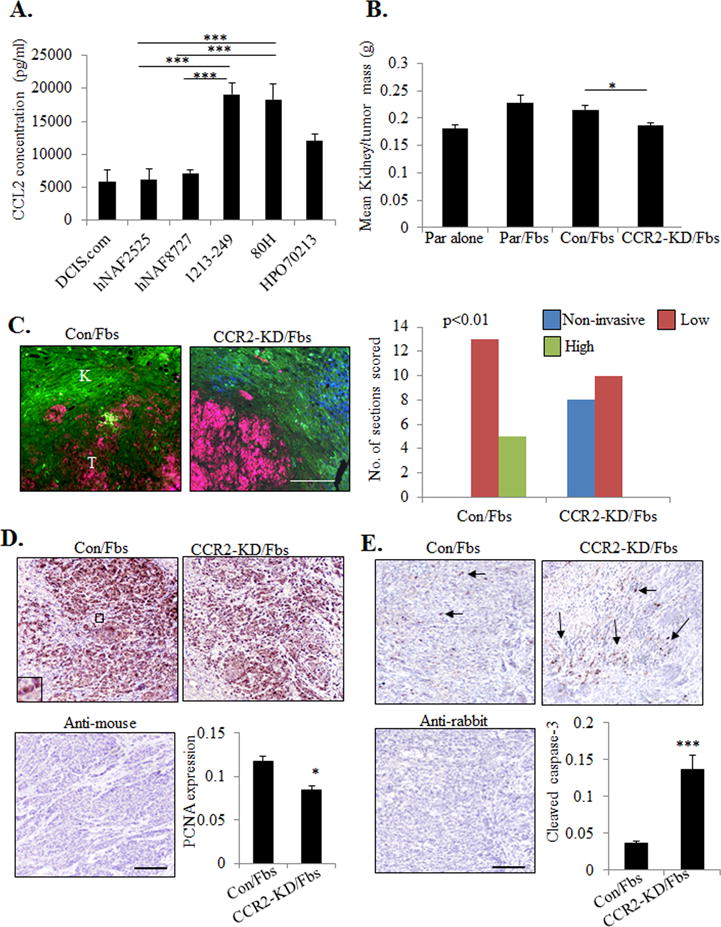
A. CCL2 ELISA of conditioned medium from fibroblasts derived from normal breast (hNAF2525, hNAF8727) or DCIS tissues (1213-249, 80H, HPO70213), in comparison with DCIS.com breast cancer cells. B-E. 1213-249 fibroblasts (Fbs) were co-grafted with parental (Par) DCIS.com cells or DCIS.com cells expressing control (Con) or CCR2 shRNAs (CCR2-KD) in the subrenal capsule of NOD-SCID mice for 21 days. Kidney tissues were measured for tumor mass (B), scored for tumor invasion into normal kidney by pan-cytokeratin (red) and phalloidin (green) staining (C), tumor cell proliferation by PCNA immunostaining (D), tumor cell apoptosis by cleaved caspase-3 immunostaining (E). Scale bar=400 microns. Arrows point to examples of positive staining. Expression in tissues was quantified by Image J. n=7 mice per group. Statistical analysis was performed using One way ANOVA with Bonferonni post-hoc comparison (B) or Two tailed T-test (D,E). Statistical significance was determined by p<0.05. *p<0.05, ***p<0.001. Mean±SEM are shown.
To determine the relevance of CCL2 expression in DCIS fibroblasts, fibroblasts were immortalized by hTERT expression to enable stable shRNA expression. Two CCL2 deficient fibroblast lines were generated from two different shRNA systems. A 47% decrease in CCL2 expression was observed using the GFP-c-shLenti Origene system (CCL2-pLenti). A 30% decrease in CCL2 expression using the GIPZ Dharmacon system (CCL2/GIPZ) (Figure 6A). CCL2 deficient or control fibroblasts were co-grafted with DCIS.com breast cancer cells in the subrenal capsule and analyzed for changes in tumor progression. CCL2 deficient fibroblasts co-grafted with DCIS.com cells resulted in smaller tumors and reduced tumor invasion, associated with decreased tumor cell proliferation and increased apoptosis (Figures 6B-C). These studies indicate that CCL2 derived from DCIS fibroblasts enhances progression of DCIS.com breast tumors.
Figure 6. CCL2 derived from DCIS fibroblasts is important for progression of DCIS.com breast cancer cells.
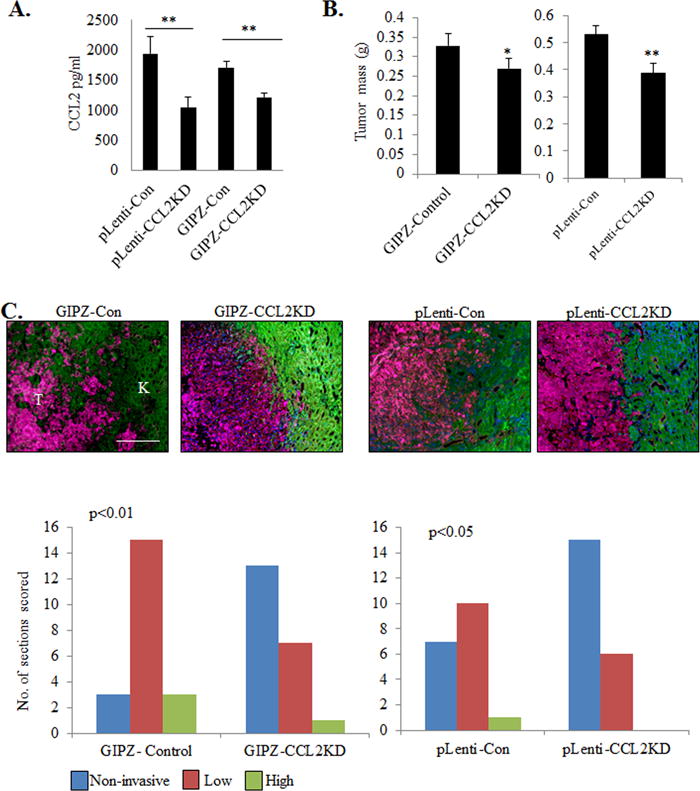
A. CCL2 ELISA of 1213-249 fibroblasts expressing control shRNA (Con) or CCL2 shRNAs from pLenti or GIPZ lentivirus systems. B-C. Control or CCL2 deficient 1213-249 fibroblasts were co-grafted with DCIS.com breast cancer cells in the subrenal capsule and analyzed for changes in tumor growth (B), and scored for tumor invasion into normal kidney tissue by pan-cytokeratin (red) and phalloidin (green) staining (C). N=7 mice/group. Scale bar=400 microns. K=kidney, T=tumor. Statistical analysis was performed using One way ANOVAwith Bonferonni post-hoc comparison. Statistical significance was determined by p<0.05. **p<0.01. Mean±SEM are shown.
CCL2/CCR2 mediated invasion is associated with increased ALDH1A1 and decreased HTRA2 expression
Lastly, we analyzed the relationship between expression of downstream CCL2/CCR2 signaling proteins and increased breast cancer survival and invasion. Through candidate screening of factors related to breast cancer survival and invasion, we found that CCL2 treatment of DCIS.com breast cancer cells over time increased expression of ALDH1A1, a stem cell and pro-invasive factor (14), and reduced expression of HTRA2, a pro-apoptotic mitochondrial serine protease (15) (Supplemental Figure S6A-B). CCR2 knockdown in DCIS.com breast cancer cells decreased expression of ALDH1A1 and increased HTRA2 in MIND xenografts by immunohistochemistry staining (Figure 7A-B). Conversely, CCR2 overexpression in Sum255 MIND xenografts enhanced ALDH1A1 expression and decreased HTRA2 expression (Supplemental Figure S7A-B), indicating that epithelial CCR2 can regulate ALDH1A1 and HTRA2 expression. Furthermore, CCL2 knockdown in fibroblasts increased HTRA2 expression and decreased ALDH1 expression in DCIS.com cells in the subrenal capsule model (Figures 7C-D), indicating that paracrine CCL2 signaling from the fibroblastic stroma was important for regulating ALDH1A1 and HTRA2 expression. Through KM Plotter analysis (37), increased CCR2 and ALDH1A1 and decreased HTRA2 expression were significantly associated with decreased metastasis free survival of breast cancer patients (Figure 7E). These data demonstrate a clinical relevance for CCL2/CCR2 signaling proteins in breast cancer.
Figure 7. CCL2/CCR2 mediated DCIS progression is associated with increased ALDH1 and decreased HTRA2 expression.
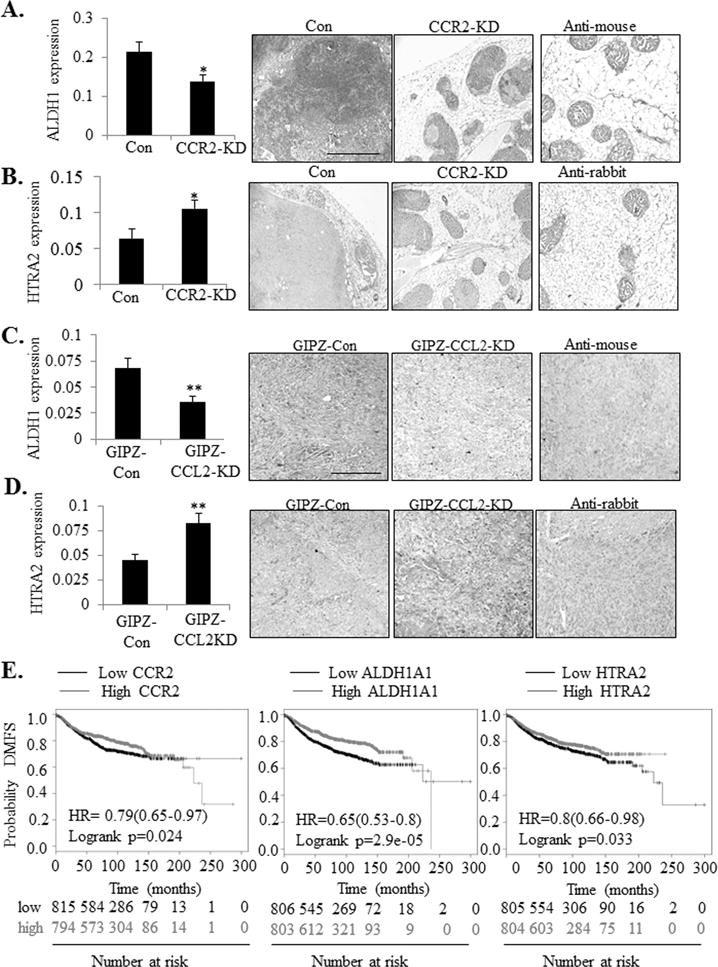
A-D. ALDH1 and HTRA2 expression was examined by immunostaining of tumor tissues in the DCIS.com MIND Model (A-B) and subrenal capsule model (C-D). Expression in tissues was quantified by Image J. K=kidney tissue. T= tumor. Scale bar= 400 microns. E. RNA Expression of CCR2 (Affyid 207794_at), ALDH1A1 (Affyid 212224_at) and HTRA2 (Afftyid 2030809_s_at) were analyzed for associations with Distance Metastasis Free Survival (DMFS) through KM plotter. Statistical analysis was performed using Two Tailed-T-test (A-B) or Log-rank Test (C). HR= Hazard Ratio. Statistical significance was determined by p<0.05. *p<0.05, **p<0.01. Mean±SEM are shown.
Discussion
The role of fibroblasts in DCIS progression is poorly understood. Fibroblasts derived from invasive breast ductal carcinomas promote tumor growth, invasion, metastasis and chemoresistance (32,33). One study showed that fibroblasts from normal, IDC or arthritic tissues enhanced progression of MCF10A cell lines in a subcutaneous injection model through Transforming Growth Factor-β and Hedgehog dependent mechanisms (38). For the first time, we show that fibroblasts derived from DCIS patient samples accelerate progression from DCIS to IDC through CCR2 dependent mechanisms. Moreover, CCL2/CCR2 mediated breast cancer progression is associated with increased expression of clinical relevant pro-invasive factors (ALDH1A1) and decreased expression of pro-apoptotic factors (HTRA2).
Here, we noted some complementary and conflicting phenotypes through CCR2 overexpression and knockdown. CCR2 overexpression in SUM225 cells enhanced formation of invasive lesions and increased the presence of CCL2+ fibroblasts, associated with increased ALDH1 and decreased HTRA2. CCR2 knockdown and knockout in DCIS.com cells inhibited invasive progression and decreased the presence of CCL2+ fibroblasts, associated with decreased ALDH1 and increased HTRA2 expression. However, whereas CCR2 knockdown significantly affected mammary tumor mass, CCR2 overexpression did not. While CCR2 overexpression increased tumor cell proliferation and survival of SUM225 lesions, these levels were still lower than the cell proliferation and survival detected in DCIS.com MIND xenografts. CCR2 expression levels in overexpressing cells did not reach the levels detected in DCIS.com breast cancer cells. Therefore it is possible that the increase in cell proliferation and survival in CCR2 overexpressing cells was not sufficient to affect overall mammary tissue mass. The levels of CCR2 expression in DCIS.com cells are consistent with previous studies showing that CCR2 expression levels are higher in basal-like breast cancer cells compared to luminal breast cancer cells (13). Because SUM225 cells are luminal/Her2+, additional oncogenic pathways may be important to DCIS progression of this subtype. Regardless of subtype, by analyzing the effects of CCR2 overexpression in SUM225 cells with CCR2 knockdown in DCIS.com cells, we demonstrate a critical role for epithelial CCR2 receptor expression in DCIS progression.
Despite a 2 fold increase in the number of CCR2+ cells in the CCR2-H SUM225 cell line, CCR2-H cells did not show increased invasion, proliferation or survival compared to CCR2-L cells. It is possible a threshold of receptor expression modulates cellular activity. Such a threshold has been detected in T cells whereby 8000 T cell receptors/cell are needed for a commitment to proliferate (39,40). A threshold also exists for EGFR levels in regulating Cbl and Grb2 dependent signaling in epithelial cells (41). In our studies, CCR2-L cells may have reached a threshold for CCR2 expression in determining cellular invasion. While more cells expressed CCR2 in the CCR2-H cell line, the level of expression may not have been sufficient to commit these cells to invade. Histogram analysis revealed that while more cells overexpressed CCR2 in the CCR2-H cell line, expression levels did not vary highly between CCR2-L and CCR2-H cells. In addition to a receptor threshold, heterogeneity in expression of intracellular signaling components in breast cancer (42) may also explain why CCR2-H cells did not result in further DCIS progression. As we are unable to control which SUM225 cells express CCR2, it is possible some CCR2 overexpressing cells did not exhibit the necessary downstream signaling components to induce invasion. As CCR2 overexpression in SUM225 cells did not reach the levels of invasion detected in DCIS.com cells, it is likely that other oncogenic factors would be required to further enhance carcinoma invasion. Several oncogenic signaling pathways including Notch, EGF, and HGF signaling are associated with DCIS progression (43,44). It would be of interest to further understand how CCL2/CCR2 coordinates DCIS progression with other oncogenic factors.
We also observed that CCR2 overexpression and knockdown affected the levels of fibroblasts in DCIS stroma. We expected that CCR2 signaling in breast cancers modulated fibroblast growth through expression of soluble growth factors such as PDGF and WNT5A, positive regulators of fibroblast proliferation (45,46). However, cultured DCIS fibroblasts treated with conditioned medium from CCR2 deficient or control DCIS.com control cells showed no significant changes in cell growth (Supplemental Figure 8). Furthermore, there were no changes in blood vessel density or macrophage recruitment, indicating that epithelial CCR2 would not regulate fibroblast accumulation indirectly through these stromal cell types. It is possible that epithelial CCR2 acts on other stromal components to indirectly modulate fibroblast growth, including adipocytes or granulocytes. Another possibility may involve the extracellular matrix. Hyaluronan and fibronectin increase fibroblast cell growth, while collagen suppresses fibroblast growth through mechano-signal transduction mechanisms (47,48). These factors would be present in mammary tissues, but not in conditioned medium. Studies are currently underway to understand how CCL2/CCR2 signaling breast cancer cells modulate the surrounding breast tumor microenvironment.
We show that increased ALDH1 and decreased HTRA2 expression are associated with CCL2/CCR2 mediated DCIS progression. Previous studies have implicated ALDH1 expression in cancer stem cell renewal, invasion and drug resistance (49). Emerging studies indicate an important role for HTRA2 in positively regulating mitochondrial dependent apoptosis (50). The increased expression of HTRA2 in CCR2 deficient lesions is consistent with the increased expression of cleaved caspase-3, as an indicator of apoptosis. CCL2/CCR2 signaling in breast cancer cells may promote DCIS progression by enhancing ALDH1+ tumor initiating cells, or activating invasive pathways through ALDH1 activity in breast cancer cells. CCL2/CCR2 signaling may facilitate survival of DCIS lesions through suppression of HTRA2 mediated apoptosis pathways.
In summary, these studies identify a novel role for CCL2/CCR2 signaling in cancer progression, identify potentially new prognostic factors for DCIS, and potentially new molecular targets for the prevention of invasive breast cancer.
Supplementary Material
Acknowledgments
Funding: This study was supported by the Susan G. Komen Foundation (CCR13261859) to Cheng and National Cancer Institute (R01CA172764) to Behbod and Cheng.
Financial support: This study was supported by the Susan G. Komen Foundation (CCR13261859) to N Cheng and National Cancer Institute (R01CA172764) To F. Behbod and N. Cheng.
Abbreviations List
- ALDH1
Aldehyde Dehydrogenase 1
- ANOVA
Analysis of Variance
- CCL2
C-C Chemokine Ligand 2
- CCR2
C-C Chemokine Receptor 2
- CRISPR
Clustered regularly interspaced short palindromic repeats
- CK
Cytokeratin
- CO-IF
co-immunofluorescence
- DAB
diaminobenzidine
- DAPI
4′,6-Diamidino-2-Phenylindole Dihydrochloride
- DMEM
Dulbecco’s modified Eagle medium
- DCIS
ductal carcinoma in Situ
- ELISA
Enzyme-linked immunosorbent assay
- FBS
fetal bovine serum
- Fsp1
Fibroblast Specific Protein 1
- GFP
Green Fluorescent Protein
- HTRA2
High Temperature Requirement Protein A2
- IDC
invasive ductal carcinoma
- IRB
Institutional Review Board
- KD
knockdown
- KUMC
University of Kansas Medical Center
- MIND
Mammary Intra-ductal Injection
- MOM
Mouse on Mouse, Non-Obese Diabetic Severe Combined Immunodeficient interleukin receptor-γ2
- NOD-SCID, PDGFR-α
Platelet Derived Growth Factor Receptor-α
- PCNA
Proliferating Cell Nuclear Antigen
- α-sma
α-smooth muscle actin
- VWF8
Von Willebrand Factor 8
Footnotes
Conflict of interest disclosure: The authors have no conflicts of interest to disclose.
Competing Interests: The authors declare no competing interests.
References
- 1.Pang JM, Gorringe KL, Fox SB. Ductal carcinoma in situ - update on risk assessment and management. Histopathology. 2016;68(1):96–109. doi: 10.1111/his.12796. [DOI] [PubMed] [Google Scholar]
- 2.Park TS, Hwang ES. Current Trends in the Management of Ductal Carcinoma In Situ. Oncology (Williston Park) 2016;30(9) [PubMed] [Google Scholar]
- 3.Virnig BA, Tuttle TM, Shamliyan T, Kane RL. Ductal carcinoma in situ of the breast: a systematic review of incidence, treatment, and outcomes. J Natl Cancer Inst. 2010;102(3):170–8. doi: 10.1093/jnci/djp482. [DOI] [PubMed] [Google Scholar]
- 4.Khan S, Epstein M, Lagios MD, Silverstein MJ. Are We Overtreating Ductal Carcinoma in Situ (DCIS)? Ann Surg Oncol. 2016 doi: 10.1245/s10434-016-5501-z. [DOI] [PubMed] [Google Scholar]
- 5.Goldstein SN. Controversies in pathology in early-stage breast cancer. Seminars in radiation oncology. 2011;21(1):20–5. doi: 10.1016/j.semradonc.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 6.Kerlikowske K, Molinaro AM, Gauthier ML, Berman HK, Waldman F, Bennington J, et al. Biomarker expression and risk of subsequent tumors after initial ductal carcinoma in situ diagnosis. J Natl Cancer Inst. 2010;102(9):627–37. doi: 10.1093/jnci/djq101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palomino DC, Marti LC. Chemokines and immunity. Einstein (Sao Paulo) 2015;13(3):469–73. doi: 10.1590/S1679-45082015RB3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balkwill FR. The chemokine system and cancer. The Journal of pathology. 2012;226(2):148–57. doi: 10.1002/path.3029. [DOI] [PubMed] [Google Scholar]
- 9.Ansari AW, Kamarulzaman A, Schmidt RE. Multifaceted Impact of Host C-C Chemokine CCL2 in the Immuno-Pathogenesis of HIV-1/M. tuberculosis Co-Infection. Front Immunol. 2013;4:312. doi: 10.3389/fimmu.2013.00312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yao M, Yu E, Staggs V, Fan F, Cheng N. Elevated expression of chemokine C-C ligand 2 in stroma is associated with recurrent basal-like breast cancers. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2016 doi: 10.1038/modpathol.2016.78. [DOI] [PubMed] [Google Scholar]
- 11.Lim SY, Yuzhalin AE, Gordon-Weeks AN, Muschel RJ. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget. 2016;7(19):28697–710. doi: 10.18632/oncotarget.7376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borsig L, Wolf MJ, Roblek M, Lorentzen A, Heikenwalder M. Inflammatory chemokines and metastasis–tracing the accessory. Oncogene. 2014;33(25):3217–24. doi: 10.1038/onc.2013.272. [DOI] [PubMed] [Google Scholar]
- 13.Fang WB, Jokar I, Zou A, Lambert D, Dendukuri P, Cheng N. CCL2/CCR2 chemokine signaling coordinates survival and motility of breast cancer cells through Smad3 protein- and p42/44 mitogen-activated protein kinase (MAPK)-dependent mechanisms. J Biol Chem. 2012;287(43):36593–608. doi: 10.1074/jbc.M112.365999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ginestier C, H M, Charafe-Jauffet E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer C, Liu S, Schott A, Hayes D, Birmbaum D, Wicha MS, Dontu G. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1(5):555–67. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jeyaraju DV, Cisbani G, De Brito OM, Koonin EV, Pellegrini L. Hax1 lacks BH modules and is peripherally associated to heavy membranes: implications for Omi/HtrA2 and PARL activity in the regulation of mitochondrial stress and apoptosis. Cell Death Differ. 2009;16(12):1622–9. doi: 10.1038/cdd.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Hare MJ, Bond J, Clarke C, Takeuchi Y, Atherton AJ, Berry C, et al. Conditional immortalization of freshly isolated human mammary fibroblasts and endothelial cells. Proc Natl Acad Sci U S A. 2001;98(2):646–51. doi: 10.1073/pnas.98.2.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller FR, Santner SJ, Tait L, Dawson PJ. MCF10DCIS.com xenograft model of human comedo ductal carcinoma in situ. J Natl Cancer Inst. 2000;92(14):1185–6. doi: 10.1093/jnci/92.14.1185a. [DOI] [PubMed] [Google Scholar]
- 18.Ethier SP, Ammerman CA, Dziubinski ML. Isolation and Culture of Human Breast Cancer Cells from Primary Tumors and Mestases. In: Margot M, Ip BBA, editors. Methods in Mammary Gland Biology and Breast Cancer Research. New York: Kluwer Academic/Plenum Publishers; 2000. [Google Scholar]
- 19.Behbod F, Kittrell FS, LaMarca H, Edwards D, Kerbawy S, Heestand JC, et al. An intraductal human-in-mouse transplantation model mimics the subtypes of ductal carcinoma in situ. Breast Cancer Res. 2009;11(5):R66. doi: 10.1186/bcr2358. doi bcr2358 [pii] 10.1186/bcr2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng N, Lambert DL. Mammary transplantation of stromal cells and carcinoma cells in C57BL/6J mice. Journal of visualized experiments : JoVE. 2011;(54) doi: 10.3791/2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng N, Bhowmick NA, Chytil A, Gorksa AE, Brown KA, Muraoka R, et al. Loss of TGF-beta type II receptor in fibroblasts promotes mammary carcinoma growth and invasion through upregulation of TGF-alpha-, MSP- and HGF-mediated signaling networks. Oncogene. 2005;24(32):5053–68. doi: 10.1038/sj.onc.1208685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mardekian SK, Bombonati A, Palazzo JP. Ductal carcinoma in situ of the breast: the importance of morphologic and molecular interactions. Hum Pathol. 2016;49:114–23. doi: 10.1016/j.humpath.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 23.Sambade MJ, Kimple RJ, Camp JT, Peters E, Livasy CA, Sartor CI, et al. Lapatinib in combination with radiation diminishes tumor regrowth in HER2+ and basal-like/EGFR+ breast tumor xenografts. Int J Radiat Oncol Biol Phys. 2010;77(2):575–81. doi: 10.1016/j.ijrobp.2009.12.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Valdez KE, Fan F, Smith W, Allred DC, Medina D, Behbod F. Human primary ductal carcinoma in situ (DCIS) subtype-specific pathology is preserved in a mouse intraductal (MIND) xenograft model. The Journal of pathology. 2011;225(4):565–73. doi: 10.1002/path.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Damiani S, Ludvikova M, Tomasic G, Bianchi S, Gown AM, Eusebi V. Myoepithelial cells and basal lamina in poorly differentiated in situ duct carcinoma of the breast. An immunocytochemical study. Virchows Arch. 1999;434(3):227–34. doi: 10.1007/s004280050332. [DOI] [PubMed] [Google Scholar]
- 26.Lazard D, Sastre X, Frid MG, Glukhova MA, Thiery JP, Koteliansky VE. Expression of smooth muscle-specific proteins in myoepithelium and stromal myofibroblasts of normal and malignant human breast tissue. Proc Natl Acad Sci U S A. 1993;90(3):999–1003. doi: 10.1073/pnas.90.3.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patarroyo M, Tryggvason K, Virtanen I. Laminin isoforms in tumor invasion, angiogenesis and metastasis. Semin Cancer Biol. 2002;12(3):197–207. doi: 10.1016/S1044-579X(02)00023-8. [DOI] [PubMed] [Google Scholar]
- 28.Ioachim E, Charchanti A, Briasoulis E, Karavasilis V, Tsanou H, Arvanitis DL, et al. Immunohistochemical expression of extracellular matrix components tenascin, fibronectin, collagen type IV and laminin in breast cancer: their prognostic value and role in tumour invasion and progression. Eur J Cancer. 2002;38(18):2362–70. doi: 10.1016/s0959-8049(02)00210-1. [DOI] [PubMed] [Google Scholar]
- 29.Park SY, Kim HM, Koo JS. Differential expression of cancer-associated fibroblast-related proteins according to molecular subtype and stromal histology in breast cancer. Breast Cancer Res Treat. 2015;149(3):727–41. doi: 10.1007/s10549-015-3291-9. [DOI] [PubMed] [Google Scholar]
- 30.Sugimoto H, Mundel TM, Kieran MW, Kalluri R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol Ther. 2006;5(12):1640–6. doi: 10.4161/cbt.5.12.3354. [DOI] [PubMed] [Google Scholar]
- 31.Carvalho I, Milanezi F, Martins A, Reis RM, Schmitt F. Overexpression of platelet-derived growth factor receptor alpha in breast cancer is associated with tumour progression. Breast Cancer Res. 2005;7(5):R788–95. doi: 10.1186/bcr1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bussard KM, Mutkus L, Stumpf K, Gomez-Manzano C, Marini FC. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016;18(1):84. doi: 10.1186/s13058-016-0740-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fang WB, Yao M, Cheng N. Priming cancer cells for drug resistance: role of the fibroblast niche. Front Biol (Beijing) 2014;9(2):114–26. doi: 10.1007/s11515-014-1300-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang C, Yan Z, Arango ME, Painter CL, Anderes K. Advancing bioluminescence imaging technology for the evaluation of anticancer agents in the MDA-MB-435-HAL-Luc mammary fat pad and subrenal capsule tumor models. Clin Cancer Res. 2009;15(1):238–46. doi: 10.1158/1078-0432.CCR-08-0897. [DOI] [PubMed] [Google Scholar]
- 35.Cheng N, Chytil A, Shyr Y, Joly A, Moses HL. Enhanced hepatocyte growth factor signaling by type II transforming growth factor-beta receptor knockout fibroblasts promotes mammary tumorigenesis. Cancer Res. 2007;67(10):4869–77. doi: 10.1158/0008-5472.CAN-06-3381. [DOI] [PubMed] [Google Scholar]
- 36.Livasy CA, Karaca G, Nanda R, Tretiakova MS, Olopade OI, Moore DT, et al. Phenotypic evaluation of the basal-like subtype of invasive breast carcinoma. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2006;19(2):264–71. doi: 10.1038/modpathol.3800528. [DOI] [PubMed] [Google Scholar]
- 37.Szasz AM, Lanczky A, Nagy A, Forster S, Hark K, Green JE, et al. Cross-validation of survival associated biomarkers in gastric cancer using transcriptomic data of 1,065 patients. Oncotarget. 2016 doi: 10.18632/oncotarget.10337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu M, Yao J, Carroll DK, Weremowicz S, Chen H, Carrasco D, et al. Regulation of in situ to invasive breast carcinoma transition. Cancer Cell. 2008;13(5):394–406. doi: 10.1016/j.ccr.2008.03.007. doi S1535-6108(08)00091-3 [pii] 10.1016/j.ccr.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kimachi K, Croft M, Grey HM. The minimal number of antigen-major histocompatibility complex class II complexes required for activation of naive and primed T cells. European journal of immunology. 1997;27(12):3310–7. doi: 10.1002/eji.1830271230. [DOI] [PubMed] [Google Scholar]
- 40.Viola A, Lanzavecchia A. T cell activation determined by T cell receptor number and tunable thresholds. Science. 1996;273(5271):104–6. doi: 10.1126/science.273.5271.104. [DOI] [PubMed] [Google Scholar]
- 41.Capuani F, Conte A, Argenzio E, Marchetti L, Priami C, Polo S, et al. Quantitative analysis reveals how EGFR activation and downregulation are coupled in normal but not in cancer cells. Nature communications. 2015;6:7999. doi: 10.1038/ncomms8999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Norton N, Advani PP, Serie DJ, Geiger XJ, Necela BM, Axenfeld BC, et al. Assessment of Tumor Heterogeneity, as Evidenced by Gene Expression Profiles, Pathway Activation, and Gene Copy Number, in Patients with Multifocal Invasive Lobular Breast Tumors. PLoS One. 2016;11(4):e0153411. doi: 10.1371/journal.pone.0153411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jedeszko C, Victor BC, Podgorski I, Sloane BF. Fibroblast hepatocyte growth factor promotes invasion of human mammary ductal carcinoma in situ. Cancer Res. 2009;69(23):9148–55. doi: 10.1158/0008-5472.CAN-09-1043. doi 0008-5472.CAN-09-1043 [pii] 10.1158/0008-5472.CAN-09-1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farnie G, Clarke RB, Spence K, Pinnock N, Brennan K, Anderson NG, et al. Novel cell culture technique for primary ductal carcinoma in situ: role of Notch and epidermal growth factor receptor signaling pathways. J Natl Cancer Inst. 2007;99(8):616–27. doi: 10.1093/jnci/djk133. [DOI] [PubMed] [Google Scholar]
- 45.Grotendorst GR, Duncan MR. Individual domains of connective tissue growth factor regulate fibroblast proliferation and myofibroblast differentiation. FASEB J. 2005;19(7):729–38. doi: 10.1096/fj.04-3217com. [DOI] [PubMed] [Google Scholar]
- 46.Vuga LJ, Ben-Yehudah A, Kovkarova-Naumovski E, Oriss T, Gibson KF, Feghali-Bostwick C, et al. WNT5A is a regulator of fibroblast proliferation and resistance to apoptosis. Am J Respir Cell Mol Biol. 2009;41(5):583–9. doi: 10.1165/rcmb.2008-0201OC. doi 2008-0201OC [pii] 10.1165/rcmb.2008-0201OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meran S, Thomas DW, Stephens P, Enoch S, Martin J, Steadman R, et al. Hyaluronan facilitates transforming growth factor-beta1-mediated fibroblast proliferation. J Biol Chem. 2008;283(10):6530–45. doi: 10.1074/jbc.M704819200. [DOI] [PubMed] [Google Scholar]
- 48.Mariggio MA, Cassano A, Vinella A, Vincenti A, Fumarulo R, Lo Muzio L, et al. Enhancement of fibroblast proliferation, collagen biosynthesis and production of growth factors as a result of combining sodium hyaluronate and aminoacids. International journal of immunopathology and pharmacology. 2009;22(2):485–92. doi: 10.1177/039463200902200225. [DOI] [PubMed] [Google Scholar]
- 49.Rodriguez-Torres M, Allan AL. Aldehyde dehydrogenase as a marker and functional mediator of metastasis in solid tumors. Clin Exp Metastasis. 2016;33(1):97–113. doi: 10.1007/s10585-015-9755-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kilbride SM, Prehn JH. Central roles of apoptotic proteins in mitochondrial function. Oncogene. 2013;32(22):2703–11. doi: 10.1038/onc.2012.348. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.