

Abstract
Glioblastoma is the most frequent primary brain tumor in adults and a highly lethal malignancy with a median survival of about 15 months. The aggressive invasion of the surrounding normal brain makes complete surgical resection impossible, increases the resistance to radiation and chemotherapy, and assures tumor recurrence. Thus, there is an urgent need to develop innovative therapeutics to target the invasive tumor cells for improved treatment outcomes of this disease. Expression of TROY (TNFRSF19), a member of the tumor necrosis factor (TNF) receptor family, increases with increasing glial tumor grade and inversely correlates with patient survival. Increased expression of TROY stimulates glioblastoma cell invasion in vitro and in vivo and increases resistance to temozolomide and radiation therapy. Conversely, silencing TROY expression inhibits glioblastoma cell invasion, increases temozolomide sensitivity, and prolongs survival in an intracranial xenograft model. Here, a novel complex is identified between TROY and EGFR which is mediated predominantly by the cysteine-rich CRD3 domain of TROY. Glioblastoma tumors with elevated TROY expression have a statistically positive correlation with increased EGFR expression. TROY expression significantly increases the capacity of EGF to stimulate glioblastoma cell invasion, whereas depletion of TROY expression blocks EGF stimulation of glioblastoma cell invasion. Mechanistically, TROY expression modulates EGFR signaling by facilitating EGFR activation and delaying EGFR receptor internalization. Moreover, the association of EGFR with TROY increases TROY-induced NF-κB activation. These findings substantiate a critical role for TROY-EGFR complex in regulation of glioblastoma cell invasion.
Keywords: TROY, EGFR, glioma, migration, invasion
Introduction
Glioblastoma multiforme (GBM) is the most common primary central nervous system tumor in human adults and accounts for approximately 40% of primary brain tumors (1). The median survival of glioblastoma patients with newly diagnosed disease is 14.6 months and declines to 8 months for patients with recurrent disease. The overall five-year survival is approximately 7% which starkly underlines the limited efficacy of current treatment regimens. Among the primary hurdles for successful clinical control is the highly aggressive invasion of malignant cells into the surrounding normal brain parenchyma, which renders complete surgical resection impossible, increases resistance to radiation and cytotoxic chemotherapy, and virtually assures recurrent tumor growth. However, the mechanisms of glioma cell invasion are complex and remain largely undefined. Further therapeutic advances in the treatment of glioblastoma will ultimately require a greater understanding of the signaling pathways that drive glioma cell invasion as well as the identification and specific targeting of the critical signaling effectors.
TROY (TNFRSF19), a member of the tumor necrosis factor receptor (TNFR) superfamily that are characterized by the highly conserved cysteine-rich domains (CRD) in their extracellular regions, is widely expressed during embryonic development but its postnatal expression is tightly restricted (2–7). TROY has been implicated in several invasive cancers. For example, TROY is not expressed in normal melanocytes but is highly expressed in primary and metastatic melanoma (8). TROY has been identified as a susceptibility factor for nasopharyngeal carcinoma and metastatic lung cancer in two recent genome-wide association studies (9,10) and is overexpressed in colorectal cancer (11). We have previously reported that TROY expression increases with glial tumor grade and is inversely correlated with patient survival (12). Furthermore, TROY expression is up-regulated on invasive glioma cells relative to cells residing in the primary tumor core in GBM (13). Overexpression of TROY stimulated glioma cell migration and invasion through Pyk2-Rac1 signaling pathways (12). In contrast, silencing TROY expression inhibited glioma cell migration in vitro and prolonged survival in a glioma xenograft model (12,13). Moreover, TROY promotes glioma cell survival through nuclear factor kappa B (NF-κB) activation and an AKT survival pathway (13). However, it is much less well-understood through which downstream effectors TROY enhances glioma cell migration and invasion.
To further identify downstream effectors and/or signaling pathways responsible for TROY-induced cell migration and invasion in GBM, we performed immunoprecipitation of the TROY receptor from TROY expressing T98G glioma cells and analyzed the precipitates with MALDI-TOF and MS/MS analysis. We identified the epidermal growth factor receptor (EGFR/ErbB1) as a novel binding partner of TROY. Co-immunoprecipitation studies verified the interaction between TROY and EGFR, and this direct interaction is mediated predominantly by CRD3 domain of TROY. In addition, mRNA analysis from two different glioblastoma genomic datasets showed a positive correlation between TROY and EGFR expression. Notably, TROY expression significantly increased the capacity of EGF to stimulate glioblastoma cell invasion, whereas knockdown of TROY expression blocked EGF stimulation of glioma cell migration. TROY expression modulated EGFR signaling by facilitating EGFR activation and delaying EGFR receptor internalization. Moreover, the association of EGFR with TROY enhanced TROY-induced NF-κB activation. These results support a novel role for the TROY-EGFR complex in regulation of GBM migration and invasion and suggest that the TROY-EGFR complex represents an unappreciated therapeutic target to inhibit glioma invasion and decrease therapeutic resistance.
Materials and Methods
Antibodies and reagents
The anti-TROY (EPR3214(2)) polyclonal antibody was obtained from Abcam. Antibodies to HA (C29F4), EGFR (D38B1), phospho-EGFR (cat. no. 2234), ErbB2 (29D8), ErbB3 (D22C5) and ErbB4 (111B2) were obtained from Cell Signaling Technologies (Beverly, MA). The goat anti-AU1 antibody (cat. no. A190-124A) was obtained from Bethyl Laboratories (Montgomery, TX). The anti-β-actin (BA3R) monoclonal antibody (1:5000 dilution) was obtained from ThermoFisher Scientific. All antibodies were used at a dilution of 1:1000 unless otherwise indicated. Collagen was obtained from Advanced Biomatrix. EGF was obtained from Invitrogen.
Expression constructs
The 3X HA epitope-tagged wild-type (WT) TROY construct was constructed as previously described (12). The cDNAs for TROYΔECD, TROYΔCD, TROY-CRD1, TROY-CRD2, and TROY-CRD3, each with a C-terminal 3X HA epitope, were amplified by splice overlap extension PCR and subcloned into the pcDNA3 expression vector. The TROY variant designated TROY-TRAFm containing a mutation of the TRAF binding domain (SLQE -> SLAA) was generated using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). A bacterial plasmid (Clone: HsCD00022359) containing the coding sequence of human ErbB2 (14) was obtained from DNASU plasmid repository (http://DNASU.org). A fragment containing the coding sequencing was subcloned into pcDNA3 adding an AU1 epitope tag (DTYRYI) on the carboxyl terminus. All constructs were verified by DNA sequencing. For stable transduction of glioma cell lines, the HA epitope-tagged TROY fragment and TROYΔECD fragment were excised from pcDNA3 and separately ligated into the lentiviral transfer plasmid pCDH (System Biosciences) that contains a second transcriptional cassette for the expression of green fluorescent protein (GFP). An empty pCDH vector expressing only the GFP vector was used as a control. Recombinant lentiviruses were produced as described (15). An EGFR-GFP retroviral plasmid construct was generated as previously described (16) and was a kind gift from Dr. Steven Rosenfeld (Mayo Clinic Florida).
Generation of a NF-κB response element-driven firefly luciferase reporter stable cell line
A cDNA fragment containing five copies of a NF-κB response element (NF-κB-RE) and the firefly luciferase reporter gene luc2P was excised from the expression plasmid pGL4.32 [luc2P/NF-κB-RE/Hygro] (Promega) (vector accession number: www.ncbi.nlm.nih.gov/nuccore/EU581860) and subcloned into the pCDH lentiviral vector. Recombinant lentiviruses were produced as described (15). Q293 cells were transduced with the recombinant lentivirus and selected with puromycin to generate the reporter cell line designated Q293/NF-κB-luc.
Cell Culture
The human glioma cell lines A172, LN229, T98G, U87, breast cancer cell line SK-BR-3, ovarian cancer cell line SK-OV-3 (American Type Culture Collection), the 293FT lentiviral packing cell line (Life Technologies), and Q293 cells expressing a NF-κB driven firefly luciferase reporter gene (Q293/NF-κB-luc) were maintained in Dulbecco’s Modified Eagle Medium (DMEM) (Invitrogen) supplemented with 10% heat-inactivated FBS (Invitrogen), 1% nonessential amino acids, 2 mM glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C with 5% CO2. When indicated, cells were serum starved by replacing the culture medium with DMEM supplemented with 0.1% bovine calf serum. GBM8 and GBM39 are primary GBM lines obtained from the Mayo Clinic Brain SPORE. These lines were established directly from patient surgical samples and maintained as subcutaneous flank xenografts through serial passaging in immune deficient mice (17,18). Fresh flank tumors were resected, processed to single cell suspension by mechanical dissociation, and maintained in neurosphere media (DMEM/F12 supplemented with 2% B-27, 20 ng/ml bFGF and 20 ng/ml EGF).
Transfection
Sub-confluent cultures of cells were transfected 0.4 μg of plasmid DNA using the Effectene reagent (Qiagen, Chatsworth, CA). Four hours after the addition of transfection reagents, the media was replaced with fresh DMEM. Transfected cells were routinely harvested for analysis 24 h after transfection.
Immunoblotting and immunoprecipitation
Immunoblotting of cell lysates and protein determination were performed as described (19). Briefly, cells were lysed on ice in the presence of protease and phosphatase inhibitors, lysates were clarified by centrifugation, and protein concentrations were determined using bicinchoninic acid (BCA) assay (Pierce). Where indicated, the isolation of cell membrane proteins was carried out using the Mem-PER Plus Membrane Protein Extraction Kit (Thermo Scientific) according to the manufacturer’s protocol. For immunoblotting, equal amounts of cell lysates were resolved by SDS-PAGE, transferred to nitrocellulose, and incubated with the appropriate primary antibody. Protein detections were conducted using IRDye-conjugated secondary antibodies with the Odyssey Infrared Imaging System (LI-COR Biosciences). Antibody signals on immunoblots were quantitated using the LI-COR Image Studio 5.0 software.
For immunoprecipitation experiments, cells were lysed 24 h after transfection with RIPA buffer (50 mM Tris, pH 8.0, 135 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate and 5% glycerol) containing protease and phosphatase inhibitors. Eight hundred micrograms of cell lysates was precleared with protein G-agarose beads (Millipore) for 1 h at 4 °C. Precleared lysates were incubated with the appropriate antibodies with a dilution of 1:100 overnight at 4°C followed by incubation for 1 h with protein G-Agarose. The immune complexes were washed five times with ice-cold RIPA buffer, eluted with 2X SDS sample buffer and boiled in the presence of 2-mercaptoethanol (Sigma), and resolved by SDS-PAGE. Immunoblotting of resolved immunoprecipitates was performed as described (19).
Gene expression analysis of TROY and EGFR correlation
TROY and EGFR gene expression were mined in the publicly available REMBRANDT dataset (NCBI Gene Expression Omnibus dataset GSE4290 containing 195 clinically annotated brain tumor specimens) and The Cancer Genome Atlas (TCGA) database. Expression values were filtered, and principal component analysis to investigate the relationship between samples was performed as described previously (12). Spearman correlation test was applied to determine the correlation between mRNA expression of TROY and EGFR.
GBM tissue microarray and immunohistochemistry
The preparation of the GBM tissue microarray and immunohistochemistry protocols used to examine TROY and EGFR expression in glioblastoma tumor samples has been described previously (20). A standard histological scoring system of 0, negative; 1, weak; 2, moderate; and 3, strong was used to grade the staining by individuals blinded to the sample identity. Kendall’s tau correlation test was used to determine the correlation between protein expression of TROY and EGFR.
Luciferase reporter assays
Q293/NF-κB-luc cells were plated in complete DMEM media in 6-well plates. The cells were transfected in triplicate with either the pcDNA3 vector alone, or 3X HA epitope-tagged TROY, TROY-TRAFm, or the TROY∆ECD constructs (0.4 μg/well) using Effectene as described above. Cells were 0.1% serum starved for 16 hours and then lysed in Reporter Lysis Buffer (Promega). Luciferase assays were performed using the luciferase reporter assay system (Promega) according to the manufacturer’s instructions.
Matrigel invasion assays
Glioma cells were seeded in 100-mm diameter dishes and incubated overnight at 37 °C. Subsequently, the culture media was replaced with DMEM, 0.1% FCS for additional 16 hr at 37 °C. Cells were harvested, resuspended in growth factor reduced Matrigel (BD) (1.0 × 105 cells/50 μL), added in triplicate to collagen-coated transwell chambers, and allowed to invade through Matrigel towards 20 nM EGF. After incubation for 24 hr at 37 °C, non-invaded cells were scrapped off the upper side of the membrane and cells invaded to the other side of the membrane were fixed with 4% paraformaldehyde (PFA) and stained with ProLong® Gold Antifade reagent with DAPI (Invitrogen). Nuclei of invaded cells were counted in five high power fields (HPF) with a 20X objective.
Flow cytometric analysis of EGFR cell surface expression
T98G cells, T98G cells stably expressing TROY and T98G cells stably expressing TROYΔECD were serum-starved overnight (16 hours), followed by 20 nM EGF stimulation for varying time periods at 37 °C and then harvested with 0.02% EDTA in Ca2+ and Mg2+-free PBS, and washed three times with staining buffer (PBS containing 0.5% BSA). Cells were fixed with 4% paraformaldehyde in PBS for 10 min, washed, and incubated for 1 h with a PE-conjugated EGFR monoclonal antibody (BD Pharmingen; cat. no. 555997) or a PE-conjugated mouse IgG2b isotype control antibody (BD Pharmingen; cat. no. 555743). Samples were run on the BD Biosciences FACSCelesta System (BD Biosciences) and the data were analyzed using BD FACSDiva software (BD Biosciences). EGFR surface expression at each time point was normalized to surface EGFR expression in serum starved cells in the absence of EGF.
Statistical analysis
Statistical analyses were conducted by the two-sample t test using GraphPad Prism 7.0 (GraphPad, Inc.). Tests for correlations using Spearman’s correlation coefficient and Kendall’s tau correlation coefficient were calculated using the cor.test function in the R statistical package. A p value less than 0.05 was considered significant.
Results
1. Identification of EGFR and EGFRvIII as binding partners for TROY
To investigate the mechanism through which TROY expression stimulates GBM cell invasion, we performed immunoprecipitation of the TROY receptor from T98G glioma cells over-expressing TROY and analyzed the precipitates with MALDI-TOF and MS/MS analysis to identify proteins that interact with TROY and potentially mediate TROY signaling (12). EGFR was among the proteins identified by mass spectrometry in the TROY immunoprecipitates. To validate the interaction between TROY and EGFR, we immunoprecipitated endogenous EGFR from T98G lysates with anti-EGFR antibody and observed co-immunoprecipitation of TROY (Fig. 1A). Moreover, overexpression of the related TNFRSF family member Fn14 (TNFRSF12A) did not lead to co-immunoprecipitation with EGFR (Supplementary figure 1). To further confirm the association of EGFR with TROY observed in the cultured glioma cells, we utilized patient derived GBM xenografts possessing either EGFR amplification or expression of the EGFR variant EGFRvIII (17,21). Primary GBM xenograft GBM8, which expresses EGFR, and GBM39, which expresses EGFRvIII, were lysed, immunoprecipitated with anti-EGFR antibody, and the precipitates were immunoblotted with an anti-TROY antibody. We observed that endogenous TROY co-immunoprecipitated with EGFR and also co-immunoprecipitated with EGFRvIII (Fig. 1B).
Figure 1.
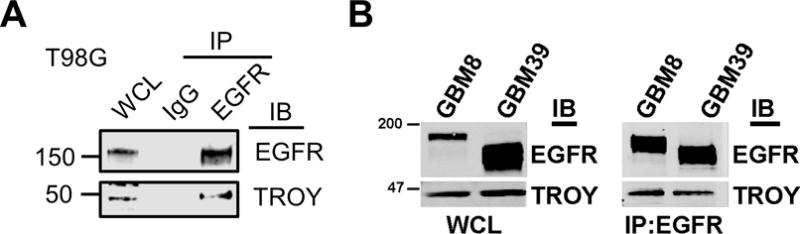
TROY associates with EGFR and EGFRvIII. (A) Cell extracts from T98G cells were immunoprecipitated with an anti-EGFR antibody and the immunoprecipitates were immunoblotted with either anti-EGFR or anti-TROY antibodies. WCL, whole cell lysate. The positions of molecular standards are shown on the left (in KDa). (B) Primary GBM xenograft line GBM8 expressing EGFR or GBM39 expressing the EGFR variant EGFRvIII were lysed, immunoprecipitated with anti-EGFR antibody, and the precipitates were immunoblotted with the indicated antibodies. WCL, whole cell lysate. The positions of molecular standards are shown on the left (in KDa).
We previously reported that TROY mRNA expression is low in non-neoplastic brain but increases with glial tumor grade and is inversely correlated with patient survival (12). Analysis of patient glioblastoma tumor samples indicated increased TROY protein expression relative to expression in non-neoplastic brain and significantly elevated TROY expression in tumor biopsy samples relative to non-neoplastic brain in situ (13). We examined the correlation between expression levels of TROY and EGFR among glial tumor specimens in the TCGA dataset and REMBRANDT dataset (Henry Ford data set, National Center for Biotechnology Information Gene Expression Omnibus data set GSE4290). Of note, correlation analysis demonstrated a significant positive correlation between the mRNA expression of EGFR and TROY in both GBM datasets (Spearman correlation coefficient = 0.4255 or 0.5133, p < 0.0001, respectively; Fig. 2A), further supporting an association between TROY and EGFR in advanced glioma. The results were further validated at the protein level by IHC analysis of a tissue microarray (TMA) of 44 GBM specimens (20). IHC staining indicated that 70% of samples had elevated TROY expression and 50% of samples had increased EGFR expression (IHC score 2-3; Fig. 2B). Those tumors with elevated TROY expression had a statistically positive correlation with increased EGFR expression (Fig. 2C, Kendall’s tau correlation coefficient =0.63, p < 0.0001). Together, this data demonstrates that the association of TROY and EGFR observed in cultured cells in vitro was consistent with that in patient GBM tumors.
Figure 2.
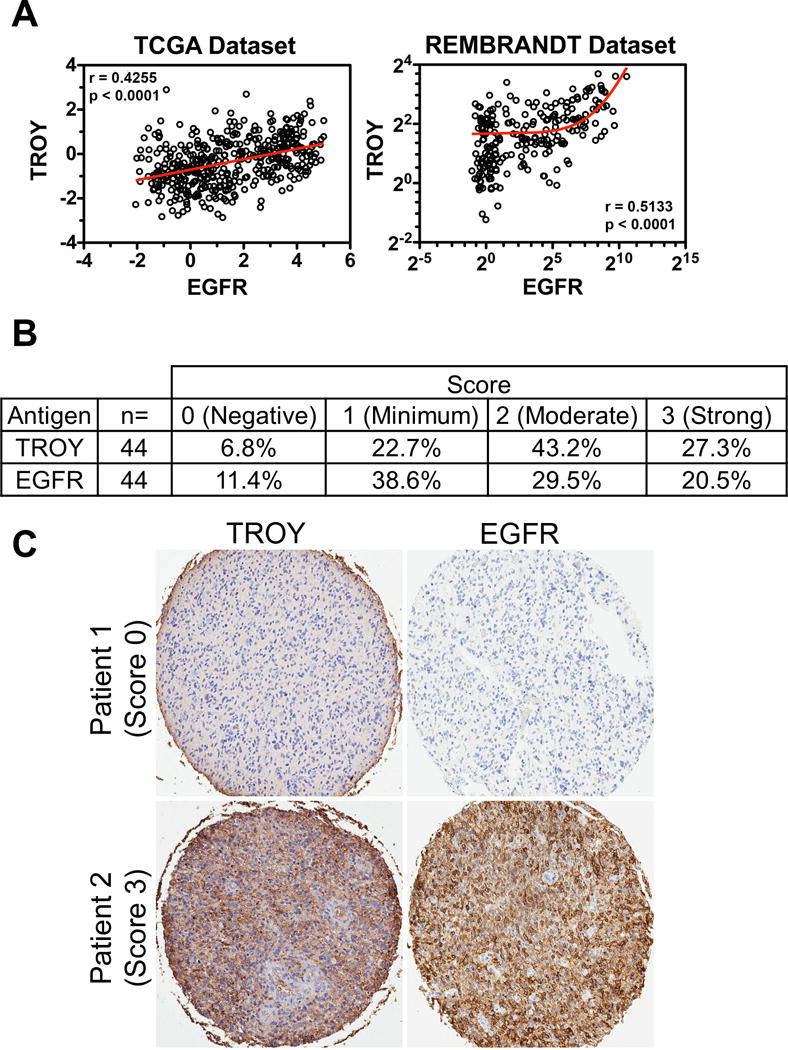
TROY expression positively correlates with EGFR in GBM. (A) TROY mRNA expression in GBM correlates with EGFR mRNA expression determined by Spearman’s correlation. The mRNA expression level of TROY and EGFR was examined in the RNA seq database from TCGA (data depicted as relative Z-score value) and the REMBRANDT GBM microarray dataset (data depicted as log2 ratio). (B) A total of 44 samples on a tissue microarray were scored for TROY and EGFR expression and the percentage distribution of staining intensity of TROY and EGFR was assessed. (C) TROY and EGFR staining on representative samples from two patients with GBM (X5 objective, Aperio GL Scanner). The correlation between the two stains was analyzed using Kendall’s tau correlation test (Kendall Tau = 0.63; p < 0.0001).
2. TROY interacts with ErbB2 and ErbB3
ErbB family members are capable of forming both homodimer and heterodimer complexes with other ErbB family members and complex composition can impact signaling output. For example, ErbB2, which lacks an identified high affinity ligand, is the preferred binding partner for other ErbB family members and ErbB2 containing heterodimers are particularly strong activators of downstream pathways due to reduced receptor internalization and rapid receptor recycling (22,23). Increased expression of ErbB2 has been described in a significant percentage of primary GBM and was associated with poor prognosis (24,25). ErbB3 is a component of a signature gene set of the proneural subtype of GBM (26).
To determine the specificity of the interaction of TROY with EGFR, we utilized GBM cell lines T98G, U87, LN-229, A172, breast cancer cell line SK-BR-3, and ovarian cancer cell line SK-OV-3. The expression pattern of members of the ErbB family in these cell lines was detected by immunoblotting. All cell lines express EGFR protein. ErbB2 is overexpressed in SK-BR-3 and SK-OV-3 cells, whereas ErbB3 is overexpressed in LN-229. ErbB4 protein is not expressed in the four GBM cell lines but is detectable in SK-OV-3 cells (Fig. 3A).
Figure 3.
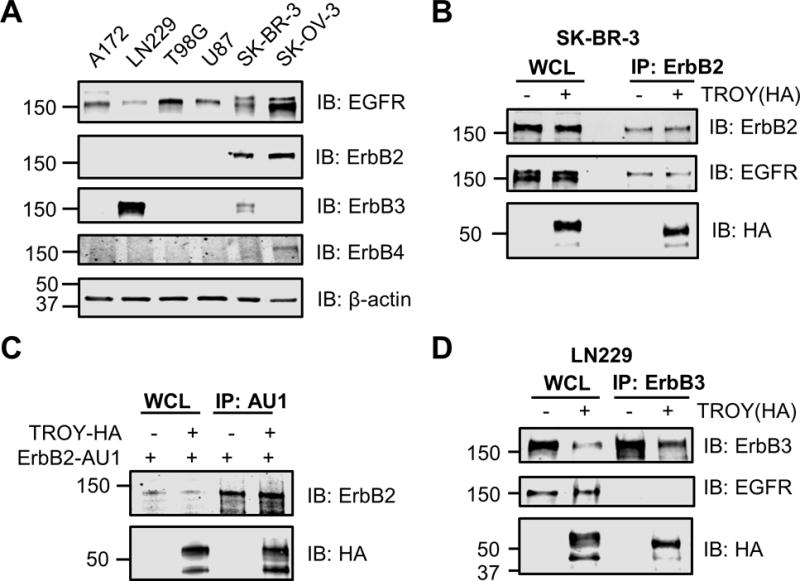
TROY co-immunoprecipitates with ErbB3 and with ErbB2. (A) The expression pattern profile of erbB family members in T98G, U87, LN-229, A172, SK-BR-3 and SK-OV-3 cells. Cell lysates were immunoblotted with the indicated antibodies. (B) TROY co-immunoprecipitates with ErbB2 in SK-BR-3 cells. SK-BR-3 cells were transfected with or without HA tagged TROY plasmid. Twenty-four hours after transfection, cells were lysed and immunoprecipitated with anti-ErbB2 antibody. Immunoprecipitates or whole cell lysate (WCL) were immunoblotted (IB) with the indicated antibodies. The positions of molecular standards are shown on the left (in KDa). (C) TROY co-immunoprecipitates with ErbB2 in Q293 cells. Q293 cells were co-transfected with AU1 tagged ErbB2 plasmid plus/minus HA tagged TROY plasmid. Twenty-four hours after transfection, cells were lysed and immunoprecipitated with anti-AU1 antibody. Immunoprecipitates or whole cell lysate (WCL) were immunoblotted (IB) with the indicated antibodies. The positions of molecular standards are shown on the left (in KDa). (D) TROY co-immunoprecipitates with ErbB3 in LN229 cells. LN229 cells were transfected with either empty expression vector or HA-tagged TROY plasmid. Twenty-four hours after transfection, cells were lysed and immunoprecipitated with anti-ErbB3 antibody. Immunoprecipitates or whole cell lysate (WCL) were immunoblotted (IB) with the indicated antibodies. The positions of molecular standards are shown on the left (in KDa).
To explore whether TROY could interact with ErbB2, SK-BR-3 cells were transfected or untransfected with HA tagged TROY. Immunoprecipitation of the transfected or untransfected cell lysates with an anti-HER2/ErbB2 antibody followed by immunoblotting of the precipitates with the anti-HA antibody showed that TROY was present in the ErbB2 immunoprecipitates (Fig. 3B). EGFR was also present in the ErbB2 immunoprecipitates indicating the presence of ErbB2/EGFR heterodimers. This prevented determination of whether TROY interacts directly with ErbB2 or indirectly through its interaction with EGFR indicating a potential multimeric complex of TROY-EGFR-ErbB2. To examine whether TROY can interact directly with ErbB2 we performed co-immunoprecipitation experiments in Q293 cells which lack detectable EGFR expression. Immunoprecipitation of ErbB2 from 293 cells co-transfected with ErbB2 and TROY demonstrated that TROY was present in the immunoprecipitates (Fig. 3C) supporting its capacity to associate directly with ErbB2. Similarly, LN-229 cells were transfected or untransfected with HA tagged TROY to determine whether TROY could interact with ErbB3. Immunoprecipitation of the transfected or untransfected cell lysates with an anti-ErbB3 antibody followed by immunoblotting of the precipitates with the anti-HA antibody showed that TROY co-immunoprecipitated with ErbB3 (Fig. 3D). Notably, EGFR was not found in the immunoprecipitates, indicating that TROY directly interacts with ErbB3 (Fig. 3D). Together, these data demonstrate that in addition to EGFR, TROY associates with ErbB2 and with ErbB3, providing insights into the potential signal diversification mediated by the complex.
3. Structural basis of the interaction between TROY and EGFR
To investigate the structural basis of the interaction between TROY and EGFR, we initially generated two constructs, TROY ΔECD, a TROY variant lacking the extracellular domain and TROY ΔCD, a TROY variant lacking its cytoplasmic domain (Fig. 4A). These variants were separately co-transfected with EGFR into Q293 cells. The transfected cells were lysed 24 h later and the lysates were then subjected to immunoprecipitation followed by immunoblot analysis. Immunoprecipitation of the transfected cell lysates with an anti-EGFR antibody followed by immunoblotting of the precipitates with an anti-HA antibody showed that HA-tagged TROY wild-type and TROY ΔCD were detected in the EGFR precipitates, whereas TROY ΔECD was not detected, indicating that the extracellular domain of TROY plays an important role in its association with EGFR (Fig. 4B).
Figure 4.
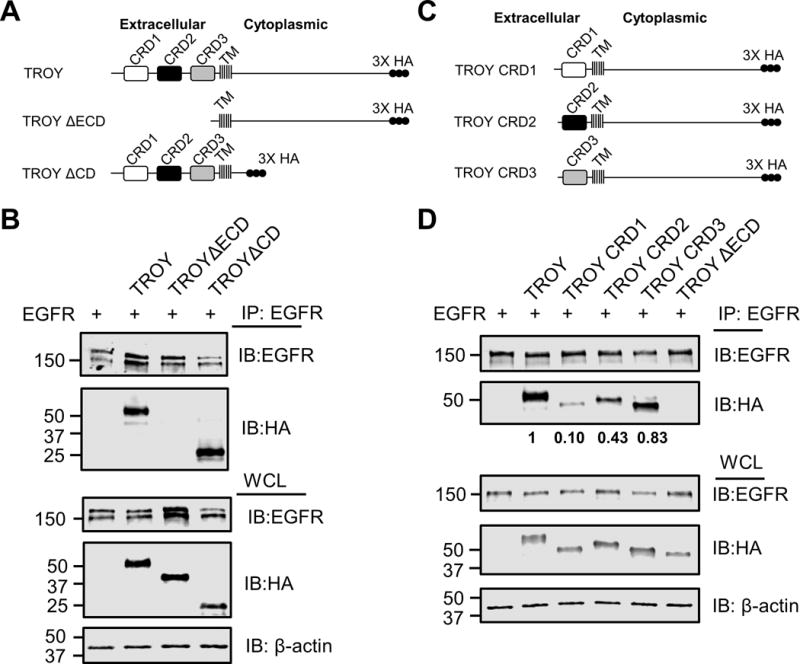
TROY interacts with EGFR through its extracellular domain. (A) Schematic representation of the expression constructs encoding TROY WT, TROY ΔECD, or TROY ΔCD protein. The extracellular domain, transmembrane (TM) domain, and cytoplasmic domain are indicated and the 3X HA epitope tag is shown. (B) Q293 cells were co-transfected with EGFR and the indicated TROY constructs and 24 hr later cells were lysed and immunoprecipitated with anti-EGFR antibody. Immunoprecipitates or whole cell lysate (WCL) were immunoblotted (IB) with the indicated antibodies. The positions of molecular standards are shown on the left (in KDa). (C) Schematic representation of the expression constructs encoding TROY CRD1, or TROY CRD2 or TROY CRD3 protein. The transmembrane (TM) domain and cytoplasmic domain are indicated and the 3X HA epitope tag is shown. (D) Q293 cells were co-transfected with EGFR and the indicated TROY constructs and 24 hr later cells were lysed and immunoprecipitated with anti-EGFR antibody. Immunoprecipitates or whole cell lysate (WCL) were immunoblotted (IB) with the indicated antibodies. The positions of molecular standards are shown on the left (in KDa). The relative densitometry values of TROY variants in the anti-EGFR immunoprecipitates are shown.
A hallmark of members of the TNFR family of receptors is the presence of a variable number of relatively short, conserved cysteine rich domains (CRD) in the extracellular domain that are involved both in ligand binding and receptor oligomerization. The extracellular domain of TROY contains two highly conserved cysteine-rich TNFR-like domains (CRD1, CRD2) and one incomplete TNFR-like domain (CRD3) (2). To identify whether these extracellular domains are responsible for the interaction between TROY and EGFR, we generated a set of HA-tagged TROY variants containing only the CRD1 domain (TROY CRD1), only the CRD2 domain (TROY CRD2), or only the CRD3 domain (TROY CRD3) (Fig. 4C). These variants were separately co-transfected with EGFR into Q293 cells. Immunoblotting of the whole cell lysates of the transfected cells with an anti-HA antibody indicated that each of the variants was expressed in an equivalent manner to wild type TROY. Immunoprecipitation of the transfected cell lysates with an anti-EGFR antibody followed by immunoblotting of the precipitates with the anti-HA antibody indicated that each of the TROY variants were each detected in the EGFR immunoprecipitates, however there was a significant difference in the amount of each of the variants present in the immunoprecipitates (Fig. 4D). Notably, the amount of TROY CRD3 in the anti-EGFR immunoprecipitates was nearly that observed for the full-length wild type TROY. In contrast, the amount of TROY CRD2 present in the anti-EGFR immunoprecipitate was significantly reduced compared to that of wild type TROY or TROY CRD3 while only minimal amounts of TROY CRD1 were present in the EGFR immunoprecipitates. Quantitation of the amounts of TROY CRD1, TROY CRD2, and TROY CRD3 in the anti-EGFR immunoprecipitates were 0.10, 0.43, and 0.83, respectively, normalized to the amount of wild type TROY in the anti-EGFR immunoprecipitates. Together, these data indicate that the interaction between EGFR and TROY is mediated the extracellular domain of TROY particularly the CRD3 domain.
4. TROY is essential for EGF-induced GBM invasion
It is well appreciated that EGFR signaling is associated with GBM tumorigenesis and growth (27,28). Previously, we demonstrated that increased expression of TROY stimulated GBM cell migration in vitro and cell invasion ex vivo in an organotypic brain slice invasion assay (12). The identification of TROY as a novel EGFR binding partner suggests the possibility that GBM cells with increased TROY expression may have an added survival benefit due to facilitated activation of classical EGFR oncogenic signaling pathways along with the activation of TROY signaling stimulating migration/invasion. Therefore, we examined the role of the TROY-EGFR signaling complex in glioma cell migration/invasion. Control T98G cells, T98G cells overexpressing TROY, T98G cells overexpressing a shRNA targeting TROY, and T98G cells overexpressing TROYΔECD were serum starved, then treated with or without EGF and analyzed using a Matrigel invasion assay. In control T98G cells, which express endogenous EGFR, addition of EGF resulted in a small but significant stimulation of cell invasion relative to serum-starved cells. Increased expression of TROY significantly stimulated the invasion of serum-starved T98G cells, which was further significantly increased by EGF stimulation (Fig. 5A). Conversely, knockdown of TROY expression completely blocked EGF-mediated stimulation of invasion (Fig. 5A). Unlike increased expression of wild type TROY, increased expression of the variant TROYΔECD that does not associate with EGFR had no effect on T98G cell invasion either in the absence or presence of EGF. Immunoblotting analysis showed the expression of HA-tagged TROY and HA-tagged TROYΔECD and knockdown of TROY in the indicated cell lines (Fig. 5B and 5C). These results suggest that TROY expression significantly increases the capacity of EGF to stimulate glioblastoma cell invasion while knockdown of TROY expression by shRNA or loss of the association between TROY and EGFR by removal of extracellular domain of TROY blocks EGF stimulation of glioma cell invasion, suggesting that the TROY-EGFR complex may represent an unappreciated therapeutic target to inhibit glioma invasion.
Figure 5.
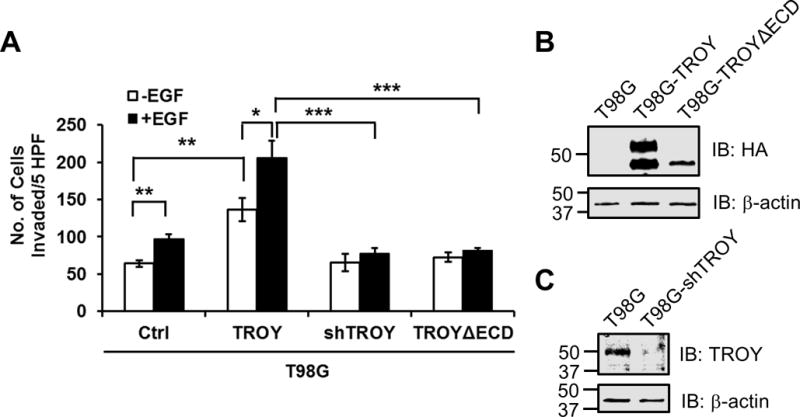
TROY is essential for EGF-induced GBM invasion. (A) Parental T98G (ctrl), T98G cells overexpressing TROY (TROY WT), T98G cells expressing a shRNA targeting TROY (TROY shRNA), and T98G cells overexpressing TROYΔECD were serum starved, placed into the top chamber of a Transwell chamber coated with Matrigel, and left untreated or stimulated with 20 nM EGF for 24 hours. The numbers of cells migrated into the bottom chamber were quantitated. *, p < 0.05. (B) Cell lysates of T98G, T98G cells overexpressing TROY (T98G-TROY) and T98G cells overexpressing TROYΔECD (T98G-TROYΔECD) cells were immunoblotted with either an anti-HA antibody or an anti-α-actin antibody. The positions of molecular standards are shown on the left (in KDa). (C) Cell lysates of T98G and T98G cells expressing a shRNA targeting TROY (T98G-shTROY) were immunoblotted with either an anti-TROY antibody or an anti-α-actin antibody. The positions of molecular standards are shown on the left (in KDa).
5. TROY expression facilitates EGFR activation and delays its internalization
TROY interacts with EGFR and its expression can regulate EGF stimulated invasion, suggesting that TROY can modulate EGFR signaling. One possible explanation for TROY potentiation of EGFR signaling is that EGFR interaction with TROY results in a conformation that facilitates EGFR activation. To test whether TROY induces activation of EGFR with or without EGF treatment, we first examined the phosphorylation state of endogenous EGFR associated with TROY in T98G cells. Parental T98G or T98G cells overexpressing TROY were serum starved, left untreated or stimulated with EGF and plasma membrane proteins were extracted and immunoprecipitated with anti-EGFR antibody and followed by immunoblotting of the precipitates with the anti-phosphorylated-EGFR (Y1068) antibody. The phosphorylation of EGFR was significantly enhanced upon EGF stimulation in the precipitates of T98G cells expressing TROY, suggesting that TROY expression can promote EGFR activation (Fig. 6A).
Figure 6.
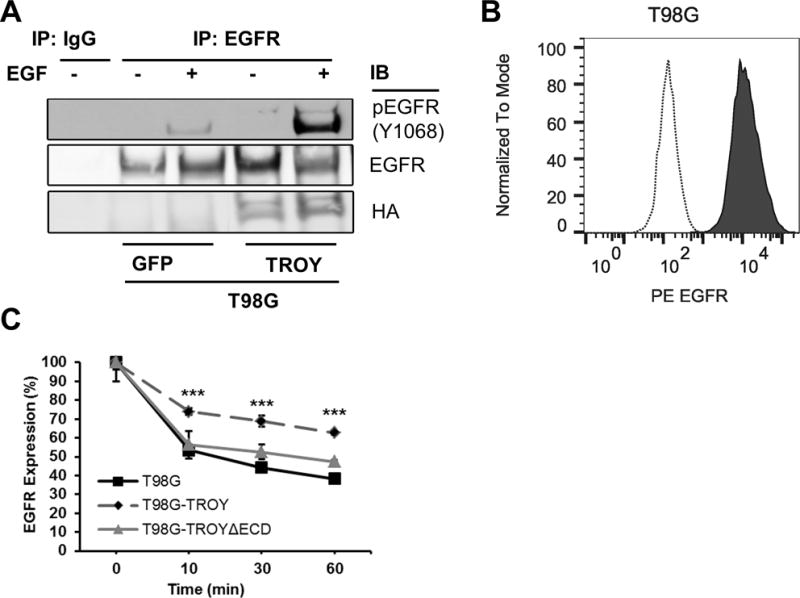
Enhanced EGFR phosphorylation and delayed EGFR internalization in TROY overexpressing cells. (A) T98G cells overexpressing GFP (control cells) or T98G cells overexpressing TROY were serum starved, left untreated or stimulated with 20 nM EGF for 10 mins. The plasma membrane proteins were extracted and immunoprecipitated with anti-EGFR antibody, and the precipitates were immunoblotted with the indicated antibodies. (B) EGFR surface expression was analyzed by flow cytometry. Depicted are histograms of EGFR surface expression on fixed T98G cells with a PE-labeled anti-EGFR antibody (solid line) or an isotype control antibody (dotted line). (C) Surface EGFR expression. Serum starved T98G cells or T98G cells overexpressing TROY or T98G cells overexpressing TROYΔECD were incubated with 20 nM EGF at 37 °C. At 10, 30 and 60 minutes after EGF addition, surface expression of EGFR was analyzed using mouse anti-EGFR PE conjugated antibody by flow cytometry. Data is normalized to surface expression of untreated cells. ***, p < 0.001.
Activation of EGFR by EGF leads to its internalization and downregulation of EGFR signaling (29). A potential explanation for TROY potentiation of EGFR signaling is that association of EGFR with TROY may alter the internalization of EGFR induced by EGF leading to prolonged EGFR signaling similar to the ligand independent, low level constitutive activation of the EGFRvIII variant which exhibits defective receptor internalization and prolonged oncogenic signaling (30,31). Conversely, antibody-induced internalization and degradation of surface expressed EGFR caused inhibition of tumor growth (32). To investigate this possibility, we examined the effect of TROY expression on the maintenance of EGFR surface expression using flow cytometry with an anti-EGFR antibody (Fig. 6B). T98G glioma cells or T98G glioma cells expressing TROY variants were left untreated or stimulated with 20 nM EGF for 10, 30 and 60 minutes and analyzed by flow cytometry. The amount of EGFR expressed on the cell surface was significantly higher in T98G cells overexpressing TROY at all time points relative to control T98G cells without increased TROY expression (Fig. 6C). In contrast, the amount of EGFR expressed on the cell surface of T98G cells overexpressing the TROYΔECD variant, which does not associate with EGFR (Fig. 4B), was similar to control T98G cells indicating that the internalization of EGFR was delayed by its association with TROY. Together, these data suggest that TROY can modulate EGFR signaling by facilitating EGFR phosphorylation and delaying EGFR internalization.
6. Association of EGFR with TROY enhances TROY induced NF-κB activation
Previously, we demonstrated that TROY expression activates the NF-κB pathway in glioblastoma cell lines (13). To explore the effect of TROY variants on NF-κB activation, Q293 cells expressing a NF-κB-luciferase reporter (Q293/NF-κB-luc) were transiently transfected with vector or expression plasmids encoding either TROY, TROY ΔECD, or TROY-TRAFm, a TROY mutant that contains a mutation of the TRAF binding domain at the C-terminus of the TROY cytoplasmic domain (SLQE -> SLAA) (Fig. 7A). Twenty-four hours after transfection, cells were serum starved (0.1% FBS) for 16 h and processed for immunoblot analysis and luciferase reporter assay. Results of the NF-κB reporter assay demonstrated that transient overexpression of TROY resulted in induction of the NF-κB pathway relative to cells transfected with vector alone (Fig. 7B). Immunoblotting analysis showed equivalent expression levels of TROY, TROYΔECD and TROY-TRAFm in the transfected cells (Fig. 7C). As anticipated, the reporter assay demonstrated that activation of the NF-κB pathway requires recruitment of TRAF proteins since mutation of the TRAF binding domain of TROY blocked the induction of NF-κB reporter. Increased expression of the TROYΔECD variant, which retains the TROY cytoplasmic domain, also resulted in significant activation of the NF-κB pathway.
Figure 7.
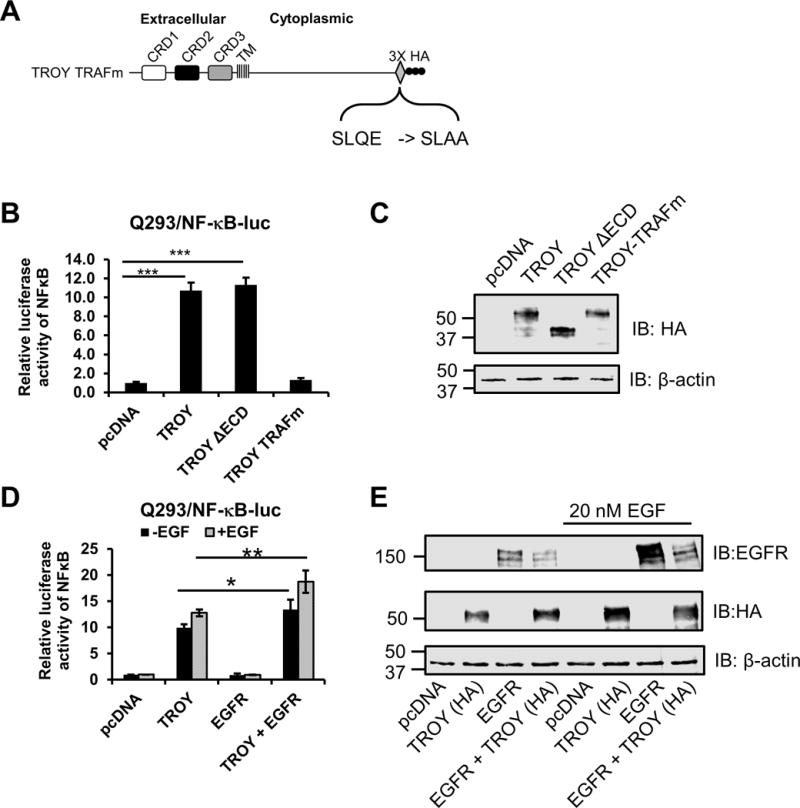
The association of EGFR with TROY enhances TROY-induced NF-κB activation. (A) Schematic representation of the expression construct encoding TROY TRAFm protein. The extracellular domain, transmembrane (TM) domain, cytoplasmic domain, and mutations of TRAF binding sites are indicated and the 3X HA epitope tag is shown. (B) Q293/NF-κB-luc cells were transfected with the indicated plasmids. Twenty-four hours after transfection, cells were serum starved (0.1% FBS) for 16 h and lysed and NF-κB-luc reporter expression was measured using luciferase reporter assay kit. Luciferase activity was normalized to the vector-transfected cells. The values shown are mean ± SD from three independent experiments. ***, p < 0.001. (C) The lysates for the luciferase assay (B) were immunoblotted with the indicated antibodies. (D) Q293/NFκB-luc cells were transfected with the indicated plasmids. Twenty-four hours after transfection, cells were serum starved (0.1% FBS) for 16 h and then left untreated or treated with 20 nM EGF for 90 mins. Cells were lysed and NF-κB-luc reporter expression was measured using luciferase reporter assay kit. Luciferase activity was normalized to the vector-transfected cells. The values shown are mean ± SD from three independent experiments. *, p < 0.05; **, p < 0.01. (E) The lysates for the luciferase assay (D) were immunoblotted with the indicated antibodies.
To investigate the effect of the association of EGFR with TROY on TROY-induced NF-κB activation, Q293/NF-κB-luc reporter cells were transiently transfected with empty vector, expression plasmids encoding either TROY or EGFR-GFP, or co-transfected with a combination of TROY and EGFR-GFP. Twenty-four hours after transfection, cells were serum starved (0.1% FBS) for 16 h, left untreated or stimulated with EGF for 90 mins. Cell lysates were then processed for immunoblot analysis and luciferase reporter assay. The luciferase reporter assay demonstrated that EGFR expression alone did not induce NF-κB activation in serum starved Q293/NF-κB-luc cells relative to control Q293/NF-κB-luc cell transfected with vector alone and did not significantly change following addition of EGF. However, co-expression of EGFR with TROY significantly enhanced NF-κB activation above that of TROY expression alone both in the serum starved cells and following addition of EGF (Fig. 7D). Immunoblotting analysis showed the expression levels of EGFR and TROY in the transfected cells as controls for transfection (Fig. 7E). These results suggest that the association of EGFR with TROY enhanced the capacity of TROY to promote NF-κB activation.
Discussion
Our previous studies have provided several compelling lines of evidence that support an important role for TROY expression and signaling in GBM cell invasion and therapeutic resistance (12,13). In the current study, we further investigated the underlying mechanism of how TROY regulates glioblastoma migration and invasion. The major findings of this study are as follows: (a) TROY forms a novel complex with EGFR, which is mediated by the extracellular domain of TROY, (b) GBM tumors with elevated TROY expression had a statistically positive correlation with increased EGFR expression, as evidence by IHC analysis of GBM specimens, (c) TROY expression significantly enhanced EGF-stimulated GBM cell invasion, while knockdown of TROY expression inhibited EGF stimulation of glioma cell invasion, (d) TROY expression enhanced EGFR phosphorylation and retarded EGFR internalization, and (e) the association of EGFR with TROY enhanced TROY-induced NF-κB activation. Our study is the first to report the identification of a TROY-EGFR complex, which may function as a novel signalsome in GBM cells. Together, these results support a critical role for the TROY-EGFR complex in regulation of glioma invasion and suggest that targeting the TROY-EGFR complex and its signaling pathways represent a potential approach to inhibit glioma invasion and decrease therapeutic resistance.
It is well appreciated that EGFR signaling is associated with GBM tumorigenesis and growth (27,28). Amplification and overexpression of EGFR is observed in a significant fraction of glioblastoma tumors making it a potential therapeutic target. While small molecule tyrosine kinase inhibitors have demonstrated efficacy in several cancers with increased EGFR activity (33,34), they have been relatively ineffective in GBM (35–37). The reasons for the poor performance are not clearly defined and possibilities include poor blood-brain barrier permeability, tumoral heterogeneity, cell-type specific compensatory pathways for EGFR signaling, the association of EGFR in multi-protein signaling complexes, and the expression of distinct EGFR variants with differential conformational requirements for EGFR activity (28,36,38,39). Alternative compensatory signaling pathways for inhibition of EGFR signaling are under investigation. Guo et al. recently demonstrated that EGFR inhibition triggers a rapid adaptive response via a TNF-JNK-AXL-ERK signaling in GBM (40) and that inhibition of the TNF-JNK-AXL-ERK signaling renders EGFR-expressing glioma cells sensitive to erlotinib. Wykosky et al. found that the uPA-uPAR-ERK1/2 signaling axis mediates inhibition of the pro-apoptotic protein Bim during EGFR inhibition in GBM (41). Inhibition of MEK or a BH3 mimetic to replace Bim function confers gefitinib sensitivity on tyrosine kinase inhibitors-resistant GBMs. These alternative compensatory pathways may bypass or evade inhibition of EGFR signaling, thereby enabling combination therapies to simultaneously attack multiple molecular targets for GBM invasion. Therefore, combination therapy is likely required to achieve the most therapeutic benefit.
In this study, we have identified a novel TROY-EGFR signaling complex in GBM cells that increases cell invasion. Identification of the discrete region(s) of TROY that mediates its interaction with EGFR would suggest potential targets for inhibitor development. Because EGFR co-immunoprecipitates with the TROY∆CD variant, we conclude that the cytoplasmic domain is not critical for complex formation and focused on the extracellular region. The extracellular region of TROY includes two highly conversed cysteine-rich TNFR-like domains (CRD1, CRD2) and one incomplete TNFR-like domain (CRD3) (2). TNFR motifs share a structure fold similar to EGFR repeats and could potentially mediate interaction in a manner similar to EGFR dimerization (42). Co-immunoprecipitation assays indicated that the association of EGFR with TROY was complex as it was observed to associate with TROY CRD1, TROY CRD2, and TROY CRD3 however, the interaction was strongest with CRD3 suggesting that the association of TROY with EGFR could be potentially destabilized by inhibitors targeting the TROY CRD3 domain. Inhibitors targeting TNFR CRDs have been previously reported. It has been shown that selective inhibition of mouse TNFR1 via targeting CRD1 of the TNFR1 by a monovalent domain antibody DMS5540 inhibits the progression of collagen-induced arthritis in vivo (43). In addition, it has also been reported that selective inhibition of TNFR1 via targeting the CRD1 and CRD2 domains by the humanized antibody ATROSAB inhibits NF-κB-induced IL-6 and IL-8 production in vitro (44). Since TROY was observed to co-immunoprecipitate with EGFRvIII in primary GBM xenografts, it would appear that amino acids 6-273 of EGFR or EGF ligand binding capacity are not critical for complex formation and suggests the complex formation may be linked to EGFR conformation. Further investigations are needed to determine which domain(s) of EGFR are responsible for the interaction of EGFR and TROY. Compensatory expression and activation of ErbB2 and ErbB3 in response to EGFR inhibition has recently been reported to mediate resistance of GBM cancer stem-like cells (45). In addition to interaction with EGFR, TROY can interact with ErbB2 and with ErbB3, suggesting that TROY may possibly be involved in compensatory activation of ErbB2 and ErbB3 following EGFR inhibition.
Consistent with the temporal dichotomy between the invasive and the proliferative phenotypes, increased TROY expression did not increase glioma cell proliferation but significantly increased the resistance to ionizing radiation and to temozolomide-induced apoptosis (13). Resistance was dependent on TROY induced activation of AKT and NF-κB (13), which are strongly implicated in resistance to apoptosis (46–48). Conversely, knockdown of TROY expression decreased Akt and NF-κB activation and increased glioma cell sensitivity to temozolomide relative to control cells. Results from the current study showed that while increased TROY expression stimulated NF-κB activation, the association of EGFR with TROY further enhanced TROY induced NF-κB activation. One possibility is that EGFR may act to facilitate the oligomerization of TROY that has been implicated in TROY-mediated NF-κB activation (13). Alternatively, the TROY-EGFR complex may increase NF-κB activation by fostering increased signaling diversity. That is, the complex may recruit additional signaling molecules that may utilize alternative pathways to increase NF-κB activation. Defining and comparing the set of genes activated by NF-κB in response to TROY signaling as well as the set of genes activated by NF-κB in response to signaling from the TROY-EGFR complex is an area under current investigation. Together, the results suggest that glioma cells expressing both TROY and EGFR could have more survival benefits than those glioma cells expressing either TROY or EGFR alone. Thus, TROY expression in the presence of EGFR stimulated glioma cell invasion relative to control cells supporting the concept that the association of TROY and EGFR recruits additional intracellular components and facilitates activation of additional pathways related to cell motility which in turn may be further enhanced by ligand binding to EGFR. Thus, the knockdown of TROY expression could block EGF stimulation of glioma cell invasion by preventing the recruitment of specific critical signaling effectors to this novel complex. TROY expression promotes EGFR signaling by facilitating EGFR phosphorylation and delaying its internalization. Together, these results suggest that GBM cells with increased TROY expression may have added survival benefit due to facilitated activation of classical EGFR oncogenic signaling pathways and the activation of TROY signaling pathways stimulating migration and invasion. Our previous study showed that silencing TROY expression by shRNA prolongs survival in a glioma xenograft model (13). Furthermore, our recent study demonstrated that the repurposed small molecule propentofylline inhibits glioblastoma cell invasion and survival by modulating TROY expression and downstream signaling pathways (49). Therefore, combinatorial inhibition of TROY and EGFR may provide an additional survival benefit to patients with GBM with increased TROY and EGFR expression.
The association of TROY with EGFR and EGFR family members adds to a growing list or proteins outside of the EGFR family that are involved in the formation of alternative heterodimers with EGFR family members (reviewed in (50)). Notably, a number of distantly related receptor tyrosine kinases including MET, IGF-1R, and AXL were each observed to interact with EGFR, ErbB2, and ErbB3. Although the structural basis of these interactions remains to be determined, these alternative receptor interactions have significant potential to increase the intracellular interactome relative to ErbB family heterodimers with resulting effects on cellular localization and diversification of signaling pathways through transactivation. Indeed, in triple negative breast cancer cells, the interaction of AXL with EGFR diversified EGFR signaling by activating additional pathways other than those activated by EGF-EGFR alone and was required for EGF stimulated cell motility (51). These novel receptor pairings have significant implications for ligand blocking therapies and the therapeutic efficacy of single agent tyrosine kinase inhibitors (52,53).
In summary, the current data substantiate an important role for the TROY-EGFR complex in the regulation of GBM migration and invasion. The results of the current study show that TROY interacts with EGFR, expression of TROY potentiates EGF stimulated GBM cell invasion in vitro and can modulate EGFR signaling, and EGFR enhances TROY-induced NF-κB activation. Taken together, defining the mechanistic basis of the formation of the TROY-EGFR signalsome and understanding its functional implications in GBM may have a potential to identify new targets in signaling pathways to limit GBM growth, inhibit invasion, and improve clinical outcome.
Supplementary Material
Implications.
TROY-EGFR signaling complex emerges as a potential therapeutic target to inhibit glioblastoma cell invasion.
Acknowledgments
The authors thank Dr. Steven Rosenfeld (Mayo Clinic Florida) for the gift of the EGFR-GFP retroviral plasmid.
Funding
This works was supported in part by NIH grant R01 NS086853 (J.C. Loftus and N.L. Tran).
Abbreviations
- GBM
Glioblastoma multiforme
- TNFR
tumor necrosis factor receptor
- CRD
cysteine-rich domains
- NF-κB
the nuclear factor kappa B
- EGFR
the epidermal growth factor receptor
Footnotes
Conflict of interest
The authors declare no potential conflict of interest.
References
- 1.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Hu S, Tamada K, Ni J, Vincenz C, Chen L. Characterization of TNFRSF19, a novel member of the tumor necrosis factor receptor superfamily. Genomics. 1999;62(1):103–7. doi: 10.1006/geno.1999.5979. [DOI] [PubMed] [Google Scholar]
- 3.Park JB, Yiu G, Kaneko S, Wang J, Chang J, He XL, et al. A TNF receptor family member, TROY, is a coreceptor with Nogo receptor in mediating the inhibitory activity of myelin inhibitors. Neuron. 2005;45(3):345–51. doi: 10.1016/j.neuron.2004.12.040. [DOI] [PubMed] [Google Scholar]
- 4.Pispa J, Mikkola ML, Mustonen T, Thesleff I. Ectodysplasin, Edar and TNFRSF19 are expressed in complementary and overlapping patterns during mouse embryogenesis. Gene Expr Patterns. 2003;3(5):675–9. doi: 10.1016/s1567-133x(03)00092-9. [DOI] [PubMed] [Google Scholar]
- 5.Hisaoka T, Morikawa Y, Kitamura T, Senba E. Expression of a member of tumor necrosis factor receptor superfamily, TROY, in the developing mouse brain. Brain Res Dev Brain Res. 2003;143(1):105–9. doi: 10.1016/s0165-3806(03)00101-9. [DOI] [PubMed] [Google Scholar]
- 6.Hisaoka T, Morikawa Y, Kitamura T, Senba E. Expression of a member of tumor necrosis factor receptor superfamily, TROY, in the developing olfactory system. Glia. 2004;45(4):313–24. doi: 10.1002/glia.10323. [DOI] [PubMed] [Google Scholar]
- 7.Shao Z, Browning JL, Lee X, Scott ML, Shulga-Morskaya S, Allaire N, et al. TAJ/TROY, an orphan TNF receptor family member, binds Nogo-66 receptor 1 and regulates axonal regeneration. Neuron. 2005;45(3):353–9. doi: 10.1016/j.neuron.2004.12.050. [DOI] [PubMed] [Google Scholar]
- 8.Spanjaard RA, Whren KM, Graves C, Bhawan J. Tumor necrosis factor receptor superfamily member TROY is a novel melanoma biomarker and potential therapeutic target. Int J Cancer. 2007;120(6):1304–10. doi: 10.1002/ijc.22367. [DOI] [PubMed] [Google Scholar]
- 9.Bei JX, Li Y, Jia WH, Feng BJ, Zhou G, Chen LZ, et al. A genome-wide association study of nasopharyngeal carcinoma identifies three new susceptibility loci. Nat Genet. 2010;42(7):599–603. doi: 10.1038/ng.601. [DOI] [PubMed] [Google Scholar]
- 10.Hu Z, Wu C, Shi Y, Guo H, Zhao X, Yin Z, et al. A genome-wide association study identifies two new lung cancer susceptibility loci at 13q12.12 and 22q12.2 in Han Chinese. Nat Genet. 2011;43(8):792–6. doi: 10.1038/ng.875. [DOI] [PubMed] [Google Scholar]
- 11.Schon S, Flierman I, Ofner A, Stahringer A, Holdt LM, Kolligs FT, et al. beta-catenin regulates NF-kappaB activity via TNFRSF19 in colorectal cancer cells. Int J Cancer. 2014;135(8):1800–11. doi: 10.1002/ijc.28839. [DOI] [PubMed] [Google Scholar]
- 12.Paulino VM, Yang Z, Kloss J, Ennis MJ, Armstrong BA, Loftus JC, et al. TROY (TNFRSF19) is overexpressed in advanced glial tumors and promotes glioblastoma cell invasion via Pyk2-Rac1 signaling. Mol Cancer Res. 2010;8(11):1558–67. doi: 10.1158/1541-7786.MCR-10-0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loftus JC, Dhruv H, Tuncali S, Kloss J, Yang Z, Schumacher CA, et al. TROY (TNFRSF19) promotes glioblastoma survival signaling and therapeutic resistance. Mol Cancer Res. 2013;11(8):865–74. doi: 10.1158/1541-7786.MCR-13-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park J, Hu Y, Murthy TV, Vannberg F, Shen B, Rolfs A, et al. Building a human kinase gene repository: bioinformatics, molecular cloning, and functional validation. Proc Natl Acad Sci U S A. 2005;102(23):8114–9. doi: 10.1073/pnas.0503141102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loftus JC, Yang Z, Tran NL, Kloss J, Viso C, Berens ME, et al. The Pyk2 FERM domain as a target to inhibit glioma migration. Mol Cancer Ther. 2009;8(6):1505–14. doi: 10.1158/1535-7163.MCT-08-1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun Y, Goderie SK, Temple S. Asymmetric distribution of EGFR receptor during mitosis generates diverse CNS progenitor cells. Neuron. 2005;45(6):873–86. doi: 10.1016/j.neuron.2005.01.045. [DOI] [PubMed] [Google Scholar]
- 17.Sarkaria JN, Yang L, Grogan PT, Kitange GJ, Carlson BL, Schroeder MA, et al. Identification of molecular characteristics correlated with glioblastoma sensitivity to EGFR kinase inhibition through use of an intracranial xenograft test panel. Mol Cancer Ther. 2007;6(3):1167–74. doi: 10.1158/1535-7163.MCT-06-0691. [DOI] [PubMed] [Google Scholar]
- 18.Sarkaria JN, Carlson BL, Schroeder MA, Grogan P, Brown PD, Giannini C, et al. Use of an orthotopic xenograft model for assessing the effect of epidermal growth factor receptor amplification on glioblastoma radiation response. Clin Cancer Res. 2006;12(7 Pt 1):2264–71. doi: 10.1158/1078-0432.CCR-05-2510. [DOI] [PubMed] [Google Scholar]
- 19.Tran NL, McDonough WS, Savitch BA, Sawyer TF, Winkles JA, Berens ME. The tumor necrosis factor-like weak inducer of apoptosis (TWEAK)-fibroblast growth factor-inducible 14 (Fn14) signaling system regulates glioma cell survival via NFkappaB pathway activation and BCL-XL/BCL-W expression. J Biol Chem. 2005;280(5):3483–92. doi: 10.1074/jbc.M409906200. [DOI] [PubMed] [Google Scholar]
- 20.Fortin SP, Ennis MJ, Savitch BA, Carpentieri D, McDonough WS, Winkles JA, et al. Tumor necrosis factor-like weak inducer of apoptosis stimulation of glioma cell survival is dependent on Akt2 function. Mol Cancer Res. 2009;7(11):1871–81. doi: 10.1158/1541-7786.MCR-09-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giannini C, Sarkaria JN, Saito A, Uhm JH, Galanis E, Carlson BL, et al. Patient tumor EGFR and PDGFRA gene amplifications retained in an invasive intracranial xenograft model of glioblastoma multiforme. Neuro Oncol. 2005;7(2):164–76. doi: 10.1215/S1152851704000821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baulida J, Kraus MH, Alimandi M, Di Fiore PP, Carpenter G. All ErbB receptors other than the epidermal growth factor receptor are endocytosis impaired. J Biol Chem. 1996;271(9):5251–7. doi: 10.1074/jbc.271.9.5251. [DOI] [PubMed] [Google Scholar]
- 23.Lenferink AE, Pinkas-Kramarski R, van de Poll ML, van Vugt MJ, Klapper LN, Tzahar E, et al. Differential endocytic routing of homo- and hetero-dimeric ErbB tyrosine kinases confers signaling superiority to receptor heterodimers. EMBO J. 1998;17(12):3385–97. doi: 10.1093/emboj/17.12.3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mineo JF, Bordron A, Baroncini M, Maurage CA, Ramirez C, Siminski RM, et al. Low HER2-expressing glioblastomas are more often secondary to anaplastic transformation of low-grade glioma. J Neurooncol. 2007;85(3):281–7. doi: 10.1007/s11060-007-9424-1. [DOI] [PubMed] [Google Scholar]
- 25.Schlegel J, Stumm G, Brandle K, Merdes A, Mechtersheimer G, Hynes NE, et al. Amplification and differential expression of members of the erbB-gene family in human glioblastoma. J Neurooncol. 1994;22(3):201–7. doi: 10.1007/BF01052920. [DOI] [PubMed] [Google Scholar]
- 26.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dunn GP, Rinne ML, Wykosky J, Genovese G, Quayle SN, Dunn IF, et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 2012;26(8):756–84. doi: 10.1101/gad.187922.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taylor TE, Furnari FB, Cavenee WK. Targeting EGFR for treatment of glioblastoma: molecular basis to overcome resistance. Curr Cancer Drug Targets. 2012;12(3):197–209. doi: 10.2174/156800912799277557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mosesson Y, Mills GB, Yarden Y. Derailed endocytosis: an emerging feature of cancer. Nat Rev Cancer. 2008;8(11):835–50. doi: 10.1038/nrc2521. [DOI] [PubMed] [Google Scholar]
- 30.Huang HS, Nagane M, Klingbeil CK, Lin H, Nishikawa R, Ji XD, et al. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J Biol Chem. 1997;272(5):2927–35. doi: 10.1074/jbc.272.5.2927. [DOI] [PubMed] [Google Scholar]
- 31.Schmidt MH, Furnari FB, Cavenee WK, Bogler O. Epidermal growth factor receptor signaling intensity determines intracellular protein interactions, ubiquitination, and internalization. Proc Natl Acad Sci U S A. 2003;100(11):6505–10. doi: 10.1073/pnas.1031790100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ben-Kasus T, Schechter B, Lavi S, Yarden Y, Sela M. Persistent elimination of ErbB-2/HER2-overexpressing tumors using combinations of monoclonal antibodies: relevance of receptor endocytosis. Proc Natl Acad Sci U S A. 2009;106(9):3294–9. doi: 10.1073/pnas.0812059106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. 2008;358(11):1160–74. doi: 10.1056/NEJMra0707704. [DOI] [PubMed] [Google Scholar]
- 34.Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. 2010;10(11):760–74. doi: 10.1038/nrc2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hegi ME, Diserens AC, Bady P, Kamoshima Y, Kouwenhoven MC, Delorenzi M, et al. Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib–a phase II trial. Mol Cancer Ther. 2011;10(6):1102–12. doi: 10.1158/1535-7163.MCT-11-0048. [DOI] [PubMed] [Google Scholar]
- 36.Hegi ME, Rajakannu P, Weller M. Epidermal growth factor receptor: a re-emerging target in glioblastoma. Curr Opin Neurol. 2012;25(6):774–9. doi: 10.1097/WCO.0b013e328359b0bc. [DOI] [PubMed] [Google Scholar]
- 37.Yung WK, Vredenburgh JJ, Cloughesy TF, Nghiemphu P, Klencke B, Gilbert MR, et al. Safety and efficacy of erlotinib in first-relapse glioblastoma: a phase II open-label study. Neuro Oncol. 2010;12(10):1061–70. doi: 10.1093/neuonc/noq072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vivanco I, Robins HI, Rohle D, Campos C, Grommes C, Nghiemphu PL, et al. Differential sensitivity of glioma- versus lung cancer-specific EGFR mutations to EGFR kinase inhibitors. Cancer Discov. 2012;2(5):458–71. doi: 10.1158/2159-8290.CD-11-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Furnari FB, Cloughesy TF, Cavenee WK, Mischel PS. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat Rev Cancer. 2015;15(5):302–10. doi: 10.1038/nrc3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guo G, Gong K, Ali S, Ali N, Shallwani S, Hatanpaa KJ, et al. A TNF-JNK-Axl-ERK signaling axis mediates primary resistance to EGFR inhibition in glioblastoma. Nat Neurosci. 2017 doi: 10.1038/nn.4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wykosky J, Hu J, Gomez GG, Taylor T, Villa GR, Pizzo D, et al. A urokinase receptor-Bim signaling axis emerges during EGFR inhibitor resistance in mutant EGFR glioblastoma. Cancer Res. 2015;75(2):394–404. doi: 10.1158/0008-5472.CAN-14-2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ward CW, Hoyne PA, Flegg RH. Insulin and epidermal growth factor receptors contain the cysteine repeat motif found in the tumor necrosis factor receptor. Proteins. 1995;22(2):141–53. doi: 10.1002/prot.340220207. [DOI] [PubMed] [Google Scholar]
- 43.McCann FE, Perocheau DP, Ruspi G, Blazek K, Davies ML, Feldmann M, et al. Selective tumor necrosis factor receptor I blockade is antiinflammatory and reveals immunoregulatory role of tumor necrosis factor receptor II in collagen-induced arthritis. Arthritis Rheumatol. 2014;66(10):2728–38. doi: 10.1002/art.38755. [DOI] [PubMed] [Google Scholar]
- 44.Zettlitz KA, Lorenz V, Landauer K, Munkel S, Herrmann A, Scheurich P, et al. ATROSAB, a humanized antagonistic anti-tumor necrosis factor receptor one-specific antibody. MAbs. 2010;2(6):639–47. doi: 10.4161/mabs.2.6.13583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clark PA, Iida M, Treisman DM, Kalluri H, Ezhilan S, Zorniak M, et al. Activation of multiple ERBB family receptors mediates glioblastoma cancer stem-like cell resistance to EGFR-targeted inhibition. Neoplasia. 2012;14(5):420–8. doi: 10.1596/neo.12432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Belda-Iniesta C, de Castro Carpeno J, Casado Saenz E, Cejas Guerrero P, Perona R, Gonzalez Baron M. Molecular biology of malignant gliomas. Clin Transl Oncol. 2006;8(9):635–41. doi: 10.1007/s12094-006-0033-9. [DOI] [PubMed] [Google Scholar]
- 47.Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21(21):2683–710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 48.Hayden MS, Ghosh S. NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26(3):203–34. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dhruv HD, Roos A, Tomboc PJ, Tuncali S, Chavez A, Mathews I, et al. Propentofylline inhibits glioblastoma cell invasion and survival by targeting the TROY signaling pathway. J Neurooncol. 2016;126(3):397–404. doi: 10.1007/s11060-015-1981-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kennedy SP, Hastings JF, Han JZ, Croucher DR. The Under-Appreciated Promiscuity of the Epidermal Growth Factor Receptor Family. Front Cell Dev Biol. 2016;4:88. doi: 10.3389/fcell.2016.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meyer AS, Miller MA, Gertler FB, Lauffenburger DA. The receptor AXL diversifies EGFR signaling and limits the response to EGFR-targeted inhibitors in triple-negative breast cancer cells. Sci Signal. 2013;6(287):ra66. doi: 10.1126/scisignal.2004155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mueller KL, Yang ZQ, Haddad R, Ethier SP, Boerner JL. EGFR/Met association regulates EGFR TKI resistance in breast cancer. J Mol Signal. 2010;5:8. doi: 10.1186/1750-2187-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tanizaki J, Okamoto I, Sakai K, Nakagawa K. Differential roles of trans-phosphorylated EGFR, HER2, HER3, and RET as heterodimerisation partners of MET in lung cancer with MET amplification. Br J Cancer. 2011;105(6):807–13. doi: 10.1038/bjc.2011.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.