

Abstract
A novel set of 64 analogues based on our lead compound 1 was designed and synthesized with an initial objective of understanding the structural requirements of ligands binding to a highly perplexing substrate-binding site of P-gp and their effect on modulating the ATPase function of the efflux pump. Compound 1, a stimulator of P-gp ATPase activity, was transformed to ATPase inhibitory compounds 39, 53 and 109. The ATPase inhibition by these compounds was predominantly contributed by the presence of a cyclohexyl group in lieu of the 2-aminobenzophenone moiety of 1. The 4,4-difluorocyclohexyl analogues, 53 and 109, inhibited the photolabeling by [125I]-IAAP, with IC50 values of 0.1 and 0.76 μM, respectively. Selected compounds were shown to reverse paclitaxel resistance in HEK293 cells overexpressing P-gp and were selective toward P-gp over CYP3A4. Induced-fit docking highlighted a plausible binding pattern of inhibitory compounds in the putative-binding pocket of P-gp. The current study underscores the stringent requirement by P-gp to bind to chemically similar molecules.
INTRODUCTION
Failure of chemotherapy to treat various types of cancer is often linked to the phenomenon of multidrug resistance (MDR), wherein the cancer becomes resistant to structurally and functionally diverse classes of drugs.1 Amongst the several mechanisms responsible for MDR phenotype, such as decline in drug uptake, alterations in cell cycle progression and drug metabolism, augmented DNA damage repair, and decreased apoptosis, the increase in active efflux of a drug by ATP-binding cassette (ABC) transporters appears as a major contributing factor for acquired resistance.2 Amongst the superfamily of ABC transporters which cause MDR, P-glycoprotein (P-gp/ABCB1/MDR1) has been considered the most prevalent.3
Human P-gp is a 1280 amino acid long membrane protein folded into two similar halves. Each half consist of a nucleotide-binding domain (NBD) and a transmembrane domain (TMD). Each TMD can further be divided into six transmembrane (TM) helices. P-gp, in normal tissues, functions to protect cells from xenobiotic insults. However, MDR cells develop an overexpression of P-gp, which leads to the efflux of amphipathic substrate chemotherapeutic drugs out of the cell, limiting the ability of these drugs to kill the cancer cells.4 P-gp has a broad substrate selectivity ranging from small molecules to macrocycles with diverse chemical structures and high lipophilicity.5 The substrate/drug-binding pocket, located in the TMDs, can accommodate two to three molecules simultaneously.6, 7 Moreover, the same substrate can bind to multiple regions within the hydrophobic binding cavity of P-gp.8 Powered with the energy from ATP binding/hydrolysis process at the NBDs, P-gp operates via an alternating access mechanism for the efflux of substrates. According to this model, a conformational change occurs upon binding of the drug to P-gp in an inward open state that brings the NBDs together forming an inward closed conformation. Drug translocation is induced during the closed conformation of P-gp, followed by the release of drug on the extracellular side where the efflux pump acquires an outward open conformation. Subsequently, ATP hydrolysis at the NBD facilitates P-gp to attain its original drug-binding competent inward open state conformation.9 Recent cryo-EM analyses of the catalytic cycle of P-gp supports these open and closed conformational states of P-gp.10
Extensive research has been documented in the development of MDR reversal compounds, leading to three generations of P-gp modulators over the years;11 however, these modulators have proved ineffective in clinical trials due to wide range of reasons. These failures were partly ascribed to insufficient bioavailability at tumor sites, non-specific toxicities, indiscriminate inhibition of multiple ABC transporters, including P-gp in the intestine, liver, kidney, and the blood-brain barrier as well as one of the major drug metabolizing enzymes, CYP3A4.12–14 Moreover, certain concerns have been raised regarding the relevant selection of the patient population in clinical investigation of the P-gp modulators. The criteria for patient selection should consider the causal factor for MDR, including the specifically overexpressed ABC transporter for which the modulator was developed. Additionally, single nucleotide polymorphisms (SNPs) in these efflux pumps should be determined during patient selection process.15 Accompanying conditions in patients such as inflammatory diseases may also have significant effect on the treatment outcomes.16, 17 Despite these issues, the key role played by these efflux transporters in MDR development warrants further research for ultimate clinical success.
Development of new classes of P-gp inhibitors, devoid of the limitations associated with previously disclosed compounds, is a significant need in oncologic settings for the treatment of resistant cancers. Consequently, new approaches are devised to develop MDR reversal compounds with high P-gp affinity and selectivity.18 Using one such approach, we incorporated chemical moieties that are often present in P-gp modulators to develop a lead compound, (S)-N-(2-benzoylphenyl)-2-(2-methyl-1-(3,4,5-trimethoxybenzamido)propyl)thiazole-4-carboxamide (TTT-28; 1), which features 3,4,5-trimethoxybenzoyl and 2-aminobenzophenone moieties at the N- and C-termini, respectively, of the (S)-valine-derived thiazole scaffold.19 Compound 1 increased intracellular paclitaxel concentration in SW620/Ad300 drug-resistant cell lines and demonstrated a significant reversal of resistance to paclitaxel, doxorubicin and vincristine at a concentration of 10 μM in vitro. It also had preferential selectivity toward P-gp than other tested ABC transporters. Moreover, it has shown desirable profile in vivo as evidenced from reduced tumor volume and tumor weight with no apparent side-effects in mice.20 Additionally, biochemical assays such as [125I]-IAAP and ATP hydrolysis indicated compound 1 to interact with the TMDs of P-gp at the drug-binding site with a potent stimulation of ATPase catalytic activity.19
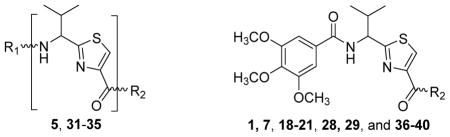
One of the approaches to determine the direct interaction of test compounds with P-gp is to measure their effect on basal ATPase activity. P-gp utilizes ATP hydrolysis as a mechanism to energize its efflux function.21, 22 The membrane vesicles prepared from cells expressing P-gp exhibit basal ATPase activity, which may have resulted from either endogenous substrates or uncoupled ATPase activity.23, 24 Compounds which are P-gp substrates and likely to be transported usually are stimulators of basal P-gp ATPase activity. Alternatively, these compounds could competitively inhibit transport of other drug substrates. Inhibitors of basal and substrate-stimulated ATPase activity tend to be incompetent for transport by P-gp, yet capable of averting the transport of other substrate drugs. Moreover, inhibitors of ATP hydrolysis by P-gp are shown to act for a longer duration of time in vitro than their stimulatory counterparts to reverse the multi-drug resistance.25–27 Compounds lacking interactions with P-gp are devoid of both stimulatory and inhibitory effects on basal ATPase activity. The affinity and/or the reversing ability of compounds, within the same class of ATPase modulators, can be better correlated with the concentrations required for half maximal stimulation (EC50) or inhibition (IC50) of the P-gp ATPase activity than their extent of stimulation or inhibition.23 Most P-gp modulators stimulate ATPase activity, except for a few clinically tested inhibitors such as tariquidar, elacridar and zosuquidar.28, 29 These clinical candidates potently inhibited ATPase activity, which is an indication of their high binding affinity toward P-gp. In addition, site-directed mutagenesis experiments have shown that the loss of ATPase inhibitory activity for these clinical candidates also resulted in loss of their ability to reverse efflux of a fluorescent substrate (NBD-cyclosporine A) by P-gp. This correlation implies the significance of ATPase inhibition for efficient reversal of efflux function.29 The absence of high resolution human P-gp crystal structure and known promiscuity of the drug-binding site i.e., the presence of multiple ligand-binding pockets, poses significant difficulties in elucidating common chemical pharmacophore/groups for efficient binding. With this backdrop, we pursued a two-pronged approach that involved: 1) transformation of an ATPase stimulator 1 into a compound with inhibitory action on ATPase activity and 2) probing the transport substrate-binding sites of P-gp through a multitude of analogue syntheses. Implementation of these approaches led to medicinal chemistry optimization program that entailed modifications at the: 1) C-terminus, 2) N-terminus and 3) central region (α-carbon and carboxamide groups) of the lead compound 1 (Figure 1).
Figure 1.

Modification of various structural elements of a lead compound 1.
Published data on P-gp modulators has firmly established how the substrate-binding region of P-gp distinguishes various structural classes of compounds on the basis of varied affinity.5, 30 However, the present study reveals that moderate structural changes, including strictly classical and non-classical isosteric modifications in a particular series of compounds, can impact P-gp function to produce no effect, and induce stimulation or inhibition of ATPase activity. Therefore, structure-activity relationship (SAR) investigations pertaining to our previously published lead stimulator (1) of ATPase function of P-gp led to the comprehensive synthesis of 64 target compounds with varying impact on ATPase activity. This report highlights the rigid requirement of P-gp relevant to binding to chemical analogues belonging to the same chemical series. Moreover, this study also provides an insight for surmounting the P-gp efflux problem associated with the substrate behavior of many drugs and clinical development candidates by conducting subtle structural modifications in the lead candidate structures. According to February 2012 FDA guidance for industry, all investigational drugs should be tested in vitro to determine whether they are substrates of ABC transporters. Consequently, our SAR study may provide tools for converting a P-gp substrate structure into a compound with no affinity toward P-gp. Further induced-fit docking of representative ATPase inhibitory target compounds was conducted within the drug/substrate-binding pocket of homology modeled structure of human P-gp to analyze the observed SAR trends.
RESULTS AND DISCUSSION
Chemistry
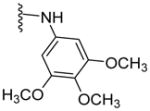
Altogether, a set of 64 (S)-amino acid-derived thiazole target compounds were synthesized to assess their interactions with P-gp using ATPase activity assay. The SAR study was instigated based on our lead compound 1, which is a (S)-valine thiazole-derived P-gp ATPase stimulator. Compounds 231 and 619 required for the synthesis of modifications of 1 were synthesized as per our previous reports. Scheme 1 shows the synthesis of target compounds 5 and 7. The swapped analogue 5 was obtained over three steps: i) coupling of the acid 2 with the 3,4,5-trimethoxyaniline using HOBt, HCTU and DIEA; ii) Boc deprotection of the coupled product 3 using TFA and iii) subsequent coupling of generated free amine 4 with the 2-benzoylbenzoic acid. Compound 7 was synthesized by coupling the acid 6 with the 3-aminobenzophenone in the presence of HOBt, HCTU and DIEA.
Scheme 1. Synthesis of Target Compounds 5 and 7a.

aReagents and conditions: (a) Appropriate aniline, HCTU, HOBt, DIEA, DMA, 0°C to rt, overnight; (b) TFA, CH2Cl2, 0°C to rt, 12 h; (c) 2-Benzoyl benzoic acid, HCTU, HOBt, DIEA, DMA, 0°C to rt, overnight.
Scheme 2 represents the synthesis of key intermediates required for coupling reactions to obtain various N- and C-terminal substitutions of (S)-valine-derived thiazole scaffold. Anthranilic acid was converted to an amide 8 by first activating the carboxyl group with the thionyl chloride and subsequent treatment with aniline.32 2-Nitroaniline in pyridine was stirred with the benzoyl chloride and subsequent palladium-catalyzed hydrogenation of the nitro group gave the reverse amide 9.33 In the past, we observed low yields for coupling reaction with the 2-aminobenzophenone, which may have resulted from the electron withdrawing nature of the benzoyl group.20 To solve this problem, we reduced the carbonyl group to produce corresponding benzhydrol intermediates, 10 and 11, with desired nucleophilicity of the 2-amino group to facilitate amide bond formation during the coupling reactions, using previously reported procedure.20 Next, 3-aminobenzoic acid was protected by reaction with Boc-anhydride in the presence of triethylamine to furnish 12.34 The amino substituted benzoic acids were converted to corresponding azido substituted benzoic acids (13–15) using diazotization reaction conditions followed by sodium azide treatment.35 The 1-benzylpiperidin-4-amine, 17, was prepared over two steps: i) benzylation of the piperidine ring nitrogen of a tert-butyl piperidin-4-yl-carbamate to provide 16 and ii) Boc deprotection.36
Scheme 2. Synthesis of Key Intermediates Required for the Coupling Reactions to Obtain Various N- and C-terminal Substitutions on (S)-Valine Thiazole Amino Acida.

aReagents and conditions: (a) (i) SOCl2, Et2O, reflux, 2 h; (ii) Aniline, pyridine, Et2O, reflux, 3 h; (b) (i) Benzoyl chloride, pyridine, THF, rt. Overnight; (ii) H2, Pd/C, MeOH, rt, overnight; (c) NaBH4, EtOH, reflux, 2 h; (d) Di-tert-butyl dicarbonate, Et3N, dioxane, H2O, rt, 24 h; (e) (i) HCl, H2O, 0°C; (ii) NaNO2, H2O, rt, 15 min; (iii) NaN3, H2O, rt; (f) Benzyl chloride, Et3N, THF, reflux, overnight; (g) TFA, CH2Cl2, 0°C to rt, 4 h.
The synthesis of isosteric analogues of benzophenone is depicted in Scheme 3. The trimethoxybenzoyl-containing acid 6 was reacted with several synthesized as well as commercially available amines in the presence of HOBt, HCTU and DIEA to obtain final test compounds 18–21.
Scheme 3. Synthesis of Target Compounds 18–21a.

aReagents and conditions: (a) Appropriate amine, HCTU, HOBt, DIEA, DMA, 0°C to rt, overnight.
The benzophenone analogues with N- and C-termini substitutions were synthesized according to Scheme 4. Acid 2 was reacted with amino benzhydrol derivatives 10 and 11 to provide coupled products 22 and 23, respectively, which were then subjected to Dess-Martin periodinane oxidation to produce 24 and 25 and subsequent Boc removal provided 26 and 27. Amine 27 was reacted with 3,4,5-trimethoxybenzoyl chloride furnishing the 4′-bromobenzophenone derivative 28. The 4′-bromo was then converted to 4′-azido derivative 29 using sodium azide in the presence of proline and copper iodide.37 Compound 31 was prepared by coupling 26 with acid intermediate 12 and subsequent Boc group removal. The N-terminal azide compounds 32–34 and 35 were synthesized starting from amines 26 and 27, respectively, and reacting with appropriate azide intermediates 13–15.
Scheme 4. Synthesis of Target Compounds 28–29 and 31–35a.

aReagents and conditions: (a) Appropriate amine, HCTU, HOBt, DIEA, DMA, 0°C to rt, overnight; (b) Dess-Martin periodinane, THF, rt, 30 min; (c) TFA, CH2Cl2, 0°C to rt, 4–12 h; (d) 3,4,5-Trimethoxybenzoyl chloride, DIEA, THF, 0°C to rt, overnight; (e) NaOH, NaN3, (S)-proline, CuI, H2O, EtOH, reflux, 24 h; (f) Appropriate acid, HCTU, HOBt, DIEA, DMA, 0°C to rt, overnight.
Scheme 5 illustrate the synthesis of alicyclic analogues 36–40. The acid 6 was coupled with appropriate amines in the presence of HOBt, HCTU and DIEA coupling reagents.
Scheme 5. Synthesis of Target Compounds 36–40a.

aReagents and conditions: (a) Appropriate amine, HCTU, HOBt, DIEA, DMA, 0°C to rt, overnight.
The cyclohexyl and related analogues were synthesized according to Scheme 6. First, commercially available Boc-protected amines were coupled with the acid 6 to obtain compounds 41, 45, 47, and 49 followed by Boc deprotection to yield free amine derivatives 42, 46, 48, and 50. Compounds 43, 44, and 51–67 were prepared by reacting the acid 6 with various synthesized and commercially available amines.
Scheme 6. Synthesis of Target Compounds 41–44, 46, 48, and 50–67a.

aReagents and conditions: (a) Appropriate amine, HCTU, HOBt, DIEA, CH2Cl2, 0°C to rt, overnight; (b) TFA, CH2Cl2, 0°C to rt, 12 h.
Variedly substituted N-terminus analogues were prepared according to Scheme 7. The acid 2 was coupled with the 4,4-difluorocyclohexylamine and then Boc-deprotected to the amine 77. This amine was then coupled with commercially available carboxylic acids or acid chlorides providing compounds 69–76 and 78–80.
Scheme 7. Synthesis of Target Compounds 69–76 and 78–80 with Various Substitutions on the N-terminus of (S)-Valine Thiazole Amino Acid while Retaining the 4,4-Difluorocyclohexyl Substitution on the C-terminusa.

aReagents and conditions: (a) 4,4-Difluorocyclohexylamine, HCTU, HOBt, DIEA, CH2Cl2, 0°C to rt, overnight; (b) TFA, CH2Cl2, 0°C to rt, 12 h; (c) Appropriate acid, HCTU, HOBt, DIEA, CH2Cl2, 0°C to rt, overnight or appropriate acid chloride, DIEA, THF, 0°C to rt, overnight.
The thioamide derivatives were synthesized by reacting 39 or 53 independently, with Lawesson’s reagent as shown in Scheme 8. Both di- and mono-thioamide derivatives were obtained and characterized by 1H-NMR and LCMS analyses. Compounds 82 and 84 were designated as thiazole-4-thioamides based on the ‘NH’ chemical shift of the thioamide. Comparison of 1H-NMR spectrums of 53 and 77 allowed us to assign cyclohexyl amide peak as a doublet at ~8 ppm, and hence, the trimethoxybenzoyl amide ‘NH’ can be assigned as doublet at ~9 ppm (Supporting Information Figure S1). For mono-thioamide 84, a doublet at 8.9 ppm suggests preservation of trimethoxybenzoyl amide oxygen and the absence of a doublet at ~8 ppm with appearance of a doublet at 10 ppm indicated occurrence of a thioamide conversion near the 4,4-difluorocyclohexyl ring. Such chemical shift trend is also applicable to compounds 39 and 82. Moreover, 1H-NMR spectrums of 81 and 83 showed chemical shifts of both the doublets at around ~9.8 and ~10.6 ppm.
Scheme 8. Synthesis of the Thioamide Analogues 81–82 and 83–84 Starting from carboxamides 39 and 53, Respectivelya.

aReagents and conditions: (a) Lawesson’s reagent, THF, rt, 8 h.
Valine residue in 53 was replaced with several amino acids as shown in Scheme 9. The thiazole rings were constructed starting from the amino protected (S)-amino acids by first converting these to respective amides 85–89, further to thioamides 90–9438 and subsequently, to bis-protected thiazoles 95–99.39 The thiazoles 95–99 were synthesized employing a procedure where calcium carbonate was used to neutralize in situ generated hydrobromic acid.39 This procedure was opted against cumbersome three step method used previously for (S)-valine-derived thiazole construction to improve yields, simplify purification process and achieve rapid reaction output. However, the calcium carbonate method led to partial racemization at the chiral center for thiazoles 96–99. This was inferred based on the smaller optical rotation values for compounds 96 and 97 as compared to those reported by Bredenkamp and colleagues.40 The racemization at the α-carbon may have occurred via acid catalyzed imine-enamine transition states during the final aromatization step.41–43 Deprotection using TFA for thiazoles 95–98 or 5% piperidine in DMF44 for Fmoc-proline derivative 99 yielded respective amino derivatives. Further commercially available ethyl 2-amino-thiazole-4-carboxylate and the synthesized amines were each reacted with 3,4,5-trimethoxybenzoyl chloride to furnish 100 and 102–106. Respective ethyl esters were then hydrolyzed and coupled with the 4,4-difluorocyclohexylamine using amide coupling reagents (HCTU, HOBt and DIEA) to provide target compounds 101 and 107–111.
Scheme 9. Synthesis of the a-Carbon Modified Target Compounds 101 and 107–111a.

aReagents and conditions: (a) (i) Isobutyl chloroformate, N-methyl morpholine, THF, −20°C, 45 min-2 h; (ii) 30% NH4OH in excess, −20°C to rt, 30 min-2 h; (b) Lawesson’s reagent, THF, rt, 8 h; (c) Ethyl bromopyruvate, CaCO3, EtOH, −20°C to rt, 8 h; (d) TFA, CH2Cl2, 0°C to rt, 12 h or 5% piperidine in DMF, rt, 2 h; (e) 3,4,5-Trimethoxybenzoyl chloride, DIEA, THF, 0°C to rt, overnight; (f) (i) NaOH, THF:Methanol:H2O (10:2:3), 0°C to rt, overnight; (ii) 4,4-Difluorocyclohexylamine, HCTU, HOBt, DIEA, CH2Cl2, 0°C to rt, overnight.
Structure-ATPase Activity Relationship
A novel set of 64 target compounds was synthesized and tested in an ATPase assay (at different concentrations: 0.05 μM, 0.5 μM, and 2.5 μM) using native membranes (High Five insect cells) that express human P-gp to determine their affinity toward P-gp. The biological readouts for these compounds are reported as percentage (stimulation/inhibition) of P-gp basal ATPase activity. In the following SAR discussion, percentage of P-gp’s ATPase activity was reported as stimulation of the inherent/basal activity unless otherwise specified. The ATPase activity can be interpreted as: 1) compounds stimulating basal ATPase activity are considered as substrates; 2) no effect on basal ATPase activity means potentially no interaction with P-gp and 3) inhibition of basal ATPase activity indicates inhibition of P-gp catalytic activity and subsequent inhibition of its efflux function.
To analyze the SAR trends, we categorized target compounds into three clusters: 1) modifications at the C-terminus and 2) the N-terminus of the (S)-valine-derived thiazole scaffold, and 3) the α-carbon of the valine amino acid, including bioisosteric replacement of the C- and N-terminus carboxamide with the thiocarboxamide group. A majority of the synthesized compounds showed stimulation of ATPase activity, which suggests these compounds to be potential P-gp substrates. The comparisons between ATPase stimulatory compounds were described with respect to binding affinity. This description is based on the percentage stimulation at the lowest identical concentration tested for the analogues being discussed i.e. higher the stimulation (at the same concentration), higher the binding affinity. Some of the target compounds inhibited ATPase activity, which indicated improved affinity toward P-gp and inhibition of its efflux function. Certain analogues exhibited neither stimulation nor inhibition of ATPase activity suggesting the loss of their affinity toward P-gp. In this work, we used high-affinity P-gp inhibitor, zosuquidar, as a positive control.
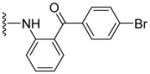
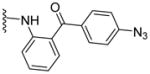
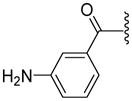
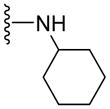
Based on our previous molecular modeling studies,19 we sought to improve the binding efficiency of our lead compound 1 through a complementary fit in the proposed P-gp substrate/drug-binding pocket. Consequently, our initial efforts were mainly focused on performing structural modifications on the existing N- and C-terminal substituents of compound 1 and subsequently, to analyze their activity based on modulation of the basal ATPase activity of P-gp (Table 1). Firstly, compound 5 was synthesized with the same 3,4,5-trimethoxyphenyl and benzophenone groups, as in lead compound 1, except their positions are switched from the N- to the C-terminus and vice-versa. This switch led to the loss of affinity to P-gp as evidenced from no activity at 0.05 μM compared to that of compound 1, which had high stimulation (54% at 0.05 μM, 83% at 0.5 μM and 88% at 2.5 μM) of basal ATPase activity. However, compound 5 displayed a moderate (46%) stimulation of basal ATPase activity at 0.5 μM concentration, which indicated its substrate nature toward P-gp. Based on this result, the next round of SAR efforts retained the positions of the aryl moieties similar to those seen in compound 1. Prior to varying the aryl substitutions, we decided to find an optimal positional isomer of the benzophenone attachment to the C-terminus. Since the 2- and 4-aminobenzophenone modifications at the C-terminus were previously assessed,19 we synthesized the only remaining 3-aminobenzophenone regioisomer 7, which showed a weak stimulation of ATPase activity (13% at 0.05 μM). This data suggests the key contribution of an optimally positioned C-terminal 2-benzoyl moiety for affinity toward P-gp. Therefore, we retained an ortho-substituted arrangement for the benzophenone moiety in the next array of structural modifications on the C-terminus. Subsequently, we explored the influence of classical and non-classical isosteric replacements of the carbonyl group present in C-terminal 2-aminobenzophenone moiety. The replacement of the carbonyl bridge of the benzophenone moiety with an amide (18, 21% at 0.05 μM), reverse amide (19, NA at 0.05 μM), ether (20, NA at 0.05 μM) and methylene (21, 15% at 0.05 μM) resulted in loss of affinity toward P-gp compared to 1. The stimulation at concentration of 0.05 μM for these compounds indicated substrate type interactions, albeit with low binding affinity as compared to compound 1. Thereafter, a set of seven compounds was prepared representing modifications at the aryl moieties of both N- and C-termini of compound 1. These compounds (28, 149% at 0.5 μM; 29, 148% at 0.5 μM; 31, 308% at 0.5 μM; 32, 142% at 0.5 μM; 33, 122% at 0.5 μM; 34, 135% at 0.5 μM and 35, 99% at 0.5 μM) showed high stimulation of ATPase activity. Collectively, this data indicated the importance of the carbonyl group, in addition to the sharp angular shape produced by an ortho-benzophenone moiety, for potent ATPase stimulation. The azido analogues, 29 and 32–35 were primarily synthesized as potential photoprobes to map the residues in the binding pocket at the TMDs of P-gp. To our disappointment, we could not obtain any covalently bound P-gp adduct for compounds 1 (contains benzophenone as a photoprobe), and 29 and 32–35 (contains azide as a photoprobe). This may have resulted from a significant reduction in the affinity of these compounds for P-gp and/or degradation of the compounds upon exposure to UV light, which is required for photo crosslinking. All the compounds synthesized up to this point were either regioisomers, isosteres or modifications of the N- and C-terminal aryl substituents and showed ATPase stimulatory effect. Therefore, we may consider these analogues as potential P-gp substrates and, hence, by a mechanism similar to the resistant cancer drugs, might eventually undergo expulsion by the efflux pump. Based on the above data, we hypothesize that the respective aryl moieties at both N- and C-termini are involved in interactions within the hydrophobic substrate-binding site of P-gp, which causes adaptation of the P-gp conformation that results in stimulation of basal ATPase activity (vide infra). This observation prompted us to explore non-aromatic groups with a goal to obtain P-gp ATPase inhibitors. Toward this goal, first we decided to determine the impact of replacing the C-terminal benzophenone moiety with various alicyclic ring systems. Compounds 36–40 with saturated aliphatic cycles of increasing steric bulk and lipophilicity were synthesized. The ATPase assay showed either no activity or low stimulation of basal ATPase activity for four of the five compounds (36, NA at 0.5 μM; 37, 25% at 0.5 μM and NA at 2.5 μM; 38, NA at 0.5 μM; and 40, NA at 0.5 μM), an indication of a complete loss of affinity toward P-gp. Encouragingly, compound 39, a C-terminal cyclohexyl analogue, showed 20% inhibition of basal ATPase activity at 0.05 μM concentration. Thus, the first P-gp ATPase inhibitor of the series was realized. It is interesting to find that an increase in the three-dimensionality with reduction in steric bulk in compound 39 as compared to lead 1 resulted in increased affinity toward P-gp with an inhibition of the catalytic process. P-gp ATPase inhibitory activity, although weak, of compound 39 with insertion of a cyclohexyl ring in lieu of the 2-aminobenzophenone moiety in compound 1 prompted us to investigate additional saturated ring systems at the C-terminus.
Table 1.
Effect of Positional Isomers, Isosteres, and Alicyclic Analogues of Compound 1 on the ATPase Activity of Human P-glycoprotein
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Compd | R1 | R2 | ATPase activitya % Stimulation or b Inhibition |
AlogPc | ||
|
| ||||||
| 0.05 μM | 0.5 μM | 2.5 μM | ||||
| 1d | - |
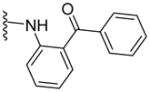
|
54 | 83 | 88 | 4.98 |
| 5 |
|
|
NAe | 46 | NDf | 4.98 |
| 7 | - |
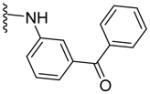
|
13 | 143 | ND | 4.98 |
| 18 | - |
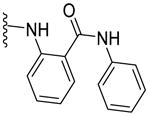
|
21 | 142 | ND | 4.36 |
| 19 | - |
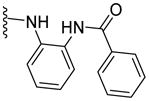
|
NA | 52 | ND | 4.36 |
| 20 | - |
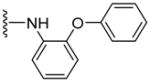
|
NA | 88 | ND | 5.14 |
| 21 | - |
|
15 | 112 | ND | 5.55 |
| 28 | - |
|
309 | 149 | ND | 5.73 |
| 29 | - |
|
ND | 147.8±24.4 | 169.4±44.5 | 3.81 |
| 31 |
|
|
376 | 308 | ND | 4.29 |
| 32 |
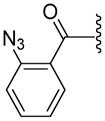
|
|
ND | 142.0±8.3 | 182.0±29.2 | 3.86 |
| 33 |
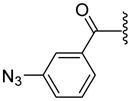
|
|
ND | 121.7±15.8 | 194.5±24.5 | 3.86 |
| 34 |
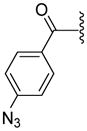
|
|
ND | 134.9±10.3 | 164.2±40.5 | 3.86 |
| 35 |
|
|
ND | 99.4±3.5 | 84.7±8.6 | 4.61 |
| 36 | - |
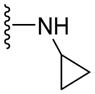
|
NA | NA | ND | 2.49 |
| 37 | - |
|
ND | 25.3±13.2 | NA | 2.95 |
| 38 | - |
|
ND | NA | NA | 3.41 |
| 39 | - |
|
20b | 18b | ND | 3.86 |
| 40 | - |
|
NA | NA | ND | 4.15 |
ATPase activity measured using High Five insect cells membranes expressing P-gp. Mean ± SD values shown are obtained from three independent experiments performed in duplicates. For some compounds, average values from two independent experiments in triplicates are given. Basal ATPase activity is considered as ‘0’. The values in the ATPase activity column indicates stimulation or
inhibition of basal ATPase activity. Zosuquidar was used as a positive control, which showed inhibition of ATPase activity as 40% at 0.05 μM and 58% at 0.5 μM.
Calculated using calculate property functionality in Maestro v10.1 (Schrödinger, LLC, New York, NY, 2015).
Compound reported in our previous published work.19
ATPase values below or equal to 10% are indicated as
NA and
ND means Not Determined.
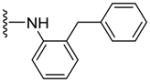
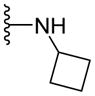
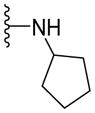
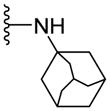
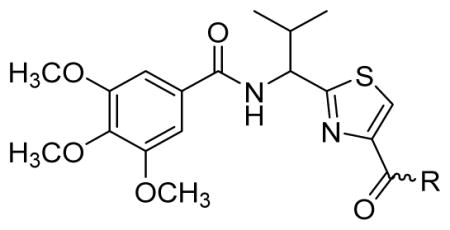
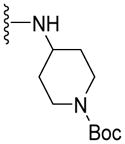
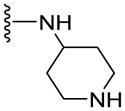
Since smaller (cyclopropyl, cyclobutyl, and cyclopentyl) or larger (adamantyl) ring size than the cyclohexyl ring proved detrimental, we focused our attention on inserting six membered rings at the C-terminus of the (S)-valine-derived thiazole scaffold. In our previous study, we found that hydrogen bonding interactions in the drug-interacting site of P-gp played a critical role in offering high affinity to clinical candidates.29 With this notion, we synthesized hydrogen bond donor- and/or acceptor group-containing cyclohexyl analogues to maximize interaction probability in the drug/substrate-binding pocket of P-gp (Table 2). A tert-butyl carbamate-protected piperidine-4-yl analogue, 41, exhibited a stimulatory ATPase activity as evidenced from 48% stimulation at 2.5 μM. Isosteric replacement of the cyclohexyl ring with the piperidine ring (42, 15% at 2.5 μM) and pyran ring (43, NA at 2.5 μM) showed complete loss of their ability to inhibit ATPase activity. Additional set of compounds, wherein we replaced the cyclohexyl amine moiety of 39 with various saturated six-membered ring systems such as 4-piperidone (44, NA at 2.5 μM), piperidin-4-methylamine (46, NA at 2.5 μM), 4-aminopiperidine (48, NA at 2.5 μM), 4-aminocyclohexyl methylamine (50, NA at 2.5 μM) and 4-aminothiopyran-1,1-dioxide (51, NA at 2.5 μM), had no inhibitory effect on the ATPase activity. This data has prompted us to revisit the cyclohexyl ring pattern as in compound 39 with the intent of bringing in substitutions such as fluoro, methyl and keto at the 4-position of the cyclohexyl ring. These efforts led to synthesis of cyclohexan-4-one analogue 52 (NA at 2.5 μM), 4,4-difluorocyclohexyl derivative 53 (34% inhibition at 2.5 μM) and 4,4-dimethylcyclohexyl analogue 54 (34% at 2.5 μM). This piece of SAR resulted in identification of 53 with ATPase inhibitory potential that was higher than that of the first ATPase inhibitory compound, 39, of the series. It is worth mentioning that P-gp can discriminate between the 4,4-difluorocyclohexyl (53) versus 4,4-dimethylcyclohexyl (54) isosteric analogues for binding preference and inhibition versus stimulation of the basal ATPase activity. The bioactivities displayed by the alicyclic ring analogues imply that the cyclohexane portion of the molecule may fit favorably in the substrate-binding pocket of P-gp with non-polar residues around it. To probe the steric requirements of the substrate-binding pocket of P-gp in a cyclohexyl interacting region, we prepared a small set of compounds with varying steric bulk such as dicyclohexyl amine (55, 134% at 2.5 μM), piperidinyl piperidine (56, 47% at 2.5 μM), N-benzyl piperidine-4-amine (57, 154% at 2.5 μM), pyrimidinyl piperidine-4-amine (58, 83% at 2.5 μM), and two enantiomers of tetralane amines (59, 123% at 2.5 μM and 60, 31% at 2.5 μM). The difference in the activities of diastereoisomers, 59 and 60, suggested chiral discrimination by the C-terminus fragment binding region of P-gp. Moderate to high stimulation of ATPase activity by these analogues suggested no tolerance for bulky substituents around the cyclohexyl binding site with respect to inhibition of ATPase activity, which reinforces the existence of a sterically forbidden region around the C-terminal cyclohexyl binding pocket (vide infra).
Table 2.
Effect of the Cyclohexyl Analogues on ATP Hydrolysis by P-gp
| ||||
|---|---|---|---|---|
|
| ||||
| Compd | R | ATPase activitya % Stimulation or b Inhibition |
AlogPc | |
|
| ||||
| 0.5 μM | 2.5 μM | |||
| 39 |
|
18b | NDf | 3.86 |
| 41 |
|
52.7±11.2 | 48.0±13.9 | 3.47 |
| 42 |
|
46.3±4.9 | 14.5±5.3 | 1.58 |
| 43 |
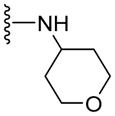
|
26.9±18.9 | NAe | 1.84 |
| 44 |
|
NA | NA | 1.83 |
| 46 |
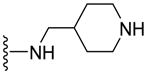
|
23.0±3.6 | NA | 2.16 |
| 48 |
|
45.4±13.2 | NA | 1.35 |
| 50 |
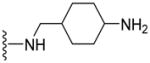
|
NA | NA | 2.67 |
| 51 |
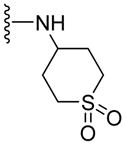
|
NA | NA | 1.57 |
| 52 |
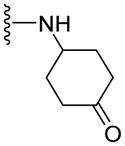
|
13.1±0.3 | NA | 2.30 |
| 53 |
|
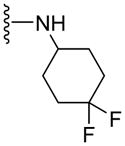
30.7±4.9b | 33.9±2.9b | 3.11 |
| 54 |
|
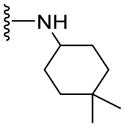
NA | 34.3±3.6 | 4.32 |
| 55 |
|
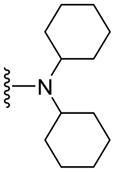
109 | 134 | 5.93 |
| 56 |
|
37.2±1.6 | 47.2±20.7 | 3.24 |
| 57 |
|
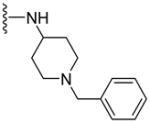
169.1±16.3 | 153.5±21.2 | 3.70 |
| 58 |
|
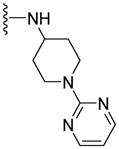
17.1±6.6 | 83.0±18.2 | 2.53 |
| 59 |
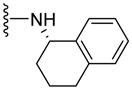
|
108.5±17.3 | 122.8±24.3 | 4.60 |
| 60 |
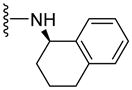
|
33.9±4.3 | 31.3±19.1 | 4.60 |
same as Table 1 footnotes.
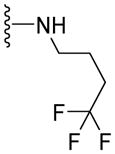
The 4,4-difluorocyclohexyl being established as the optimal moiety at the C-terminus for ATPase inhibitory effect, we proceeded to probe optimal binding conformation of the fluoro containing rings within the P-gp drug-binding pocket. Working toward this hypothesis, several analogues containing the fluoro groups on various ring systems were linked to the C-terminus of the (S)-valine-derived thiazole scaffold as shown in Table 3. Compounds (61, 56% at 2.5 μM and 62, 76% at 2.5 μM) with the piperidine rings, had moderate to high stimulation of ATPase activity. This clearly explains the importance of the conformational disposition of the cyclohexyl moiety in 53 for inhibitory action on ATPase activity. Two analogues, representing a conformationally flexible extension at the cyclohexane ring, were synthesized. Compound 63 with the methylene insertion between the 4,4-difluorocyclohexyl ring and an amide ‘NH’ inhibited ATPase activity by 27% at 0.5 μM but stimulated by 21% at 2.5 μM. Conversely, the 4-trifluoromethyl cyclohexyl analogue (64, 22% inhibition at 2.5 μM) weakly inhibited ATPase activity. The inhibition efficiency of the compound decreases when the 4,4-difluorocyclohexyl or the fluoro groups are extended by one linker atom. To determine whether ATPase inhibition can be improved by retaining the fluoro groups while eliminating saturated ring structure, we synthesized two analogues containing fluorinated aromatic rings (65, 244% at 2.5 μM and 66, 176% at 2.5 μM) and a flexible non-cyclic 4,4,4-trifluorobutyl analogue (67, 77% at 2.5 μM). These analogues have the same number of bonds between the fluorine and the amide group, as in compound 53, but with different orientations. All of these compounds stimulated basal ATPase activity. The vector for the fluorine atoms in various fluorinated analogs is clearly distinct from that present in 53, which demonstrates the contribution of an optimal axial and equatorial arrangement of the fluorines in 53 for P-gp affinity. Compounds 61, 62, 65, 66, and 67 substantiates the importance of a cyclohexyl ring whereas compounds 63 and 64 suggest the requirement for precise positioning of the fluorine atoms in the substrate-binding site of P-gp for inhibition of basal ATPase activity. It is evident from the above SAR that any deviation from the 4,4-difluorocyclohexyl group results in a loss of ATPase inhibitory activity. Therefore, in the next round of SAR, we retained the 4,4-difluorocyclohexyl ring structure on the C-terminus and conducted structural modifications at the N-terminus.
Table 3.
Modulation of ATPase Activity of P-gp by Difluorocyclohexyl Analogues
| ||||
|---|---|---|---|---|
|
| ||||
| Compd | R | ATPase activitya % Stimulation or b Inhibition |
AlogPc | |
|
| ||||
| 0.5 μM | 2.5 μM | |||
| 53 |
|
30.7±4.9b | 33.9±2.9b | 3.11 |
| 61 |
|
40.9±3.2 | 55.9±16.9 | 3.03 |
| 62 |
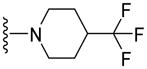
|
13.3±2.2 | 75.6±2.5 | 3.73 |
| 63 |
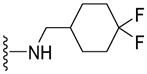
|
27.0±3.5b | 20.7±10.3 | 3.44 |
| 64 |
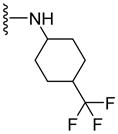
|
NAe | 21.7±9.0b | 4.47 |
| 65 |
|
59.5±3.5 | 244.4±3.4 | 3.79 |
| 66 |
|
57.0±0.4 | 176.3±6.3 | 4.20 |
| 67 |
|
17.1±0.4 | 77.3±1.1 | 3.59 |
same as Table 1 footnotes.
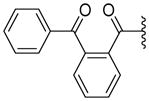
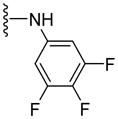
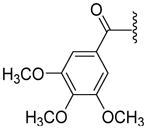
Toward this goal, we have synthesized 12 compounds as shown in Table 4. At the outset, various unsaturated and saturated ring systems were installed in lieu of the trimethoxyphenyl moiety. The aromatic heterocycles such as 4-thiazolyl (69, 50% at 2.5 μM) and 3-quinolinyl (71, 84% at 2.5 μM) produced a switch in the ATPase activity from the inhibition to a stimulation. The 5-pyrimidinyl analogue, 70 (NA at 2.5 μM), failed to interact with P-gp. Amongst the saturated ring analogues, pyran 72 (NA at 2.5 μM) proved inactive and the 4,4-difluorocyclohexyl analogue, 73 (16% inhibition at 2.5 μM), showed a weak inhibition of ATPase activity. These analogues substantiated the importance of a 3,4,5-trimethoxybenzoyl moiety for its affinity enhancing interactions at the substrate-binding pocket of P-gp (vide infra). This result prompted us to probe further SAR around the 3,4,5-trimethoxybenzoyl moiety by synthesizing analogues that feature sequential removal of the methoxy groups in compound 53 to obtain analogues 74 (12% at 2.5 μM), 75 (36% at 2.5 μM), 76 (11% at 2.5 μM) and complete elimination of 3,4,5-trimethoxybenzoyl moiety as in 77 (NA at 2.5 μM). These compounds produced no appreciable effect on the ATPase activity. It can be concluded that a 3,4,5-trimethoxybenzoyl moiety at the N-terminus along with the 4,4,-difluorocyclohexyl substituent at the C-terminus are indispensable for ATPase inhibition. However, it remains to be determined whether methoxy groups can be replaced with other hydrogen bond acceptor groups. In pursuit of this investigation, we synthesized a 3,4,5-trifluorobenzoyl-containing compound 78 (23% inhibition at 2.5 μM), which expectedly retained the ATPase inhibitory effect; however, not to the extent produced by 53. Hence, it is anticipated that the methoxy oxygen atoms may be involved in hydrogen bonding interactions within the drug-binding pocket of P-gp (vide infra). To probe the influence of an altered positioning of the hydrogen bond forming methoxy groups, we decided to separate the 3,4,5-trimethoxyphenyl ring and the carboxamide group by a flexible methylene linker to produce compound 79 (70% at 2.5 μM) and by a trans-favored rigid carbonyl linker (sterically restricted vicinal dicarbonyl functionality) to obtain 80 (176% at 2.5 μM); both had shown stimulation of the ATPase activity. It is anticipated that these extended analogues may have undergone steric clash within the substrate-binding pocket of P-gp, and thus, led to loss of the inhibitory interactions. Based on this data, we believe that the N-terminus aromatic ring in compound 53 provide requisite hydrophobicity for recognition by P-gp as well as proper orientation of the hydrogen bond forming methoxy groups for ATPase inhibitory effect. As a result, hereafter, we retained the 3,4,5-trimethoxybenzoyl and the 4,4-difluorocyclohexyl moieties at the N- and C-termini of the (S)-valine-derived thiazole scaffold, respectively.
Table 4.
Effect of Amino Terminus Modification of Compound 53 on the ATPase Activity of P-gp
| ||||
|---|---|---|---|---|
|
| ||||
| Compd | R | ATPase activitya % Stimulation or b Inhibition |
AlogPc | |
|
| ||||
| 0.5 μM | 2.5 μM | |||
| 53 |
|
30.7±4.9b | 33.9±2.9b | 3.11 |
| 69 |
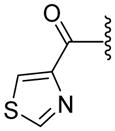
|
NDf | 49.7±27.18 | 1.86 |
| 70 |
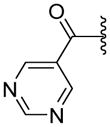
|
ND | NAe | 1.38 |
| 71 |
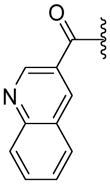
|
ND | 83.9±27.0 | 3.34 |
| 72 |
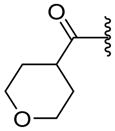
|
ND | NA | 1.85 |
| 73 |
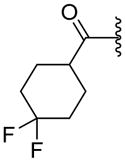
|
NA | 16.4±13.7b | 2.87 |
| 74 |
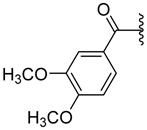
|
ND | 11.9±8.03 | 3.13 |
| 75 |
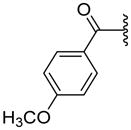
|
ND | 35.8±14.2 | 3.14 |
| 76 |
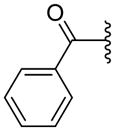
|
ND | 10.5±11.12 | 3.16 |
| 77 |
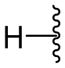
|
NA | NA | 1.47 |
| 78 |
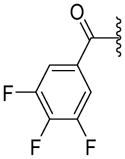
|
16.6±1.3b | 23.3±0.1b | 3.77 |
| 79 |
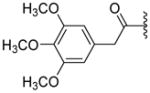
|
ND | 70.2±19.7 | 3.14 |
| 80 |
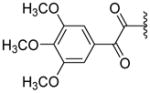
|
ND | 177.5±26.7 | 3.00 |
same as Table 1 footnotes.
Subsequent investigations sought to identify the impact of structural modifications at the central region of compound 53, including the carboxamide to thiocarboxamide isosteric replacements (Table 5). In anticipation of obtaining additional ATPase inhibitory compounds, we replaced the carboxamide group at both N- and C-terminus with isosteric thiocarboxamide. This structural change would also provide analogues that are stable to hydrolytic cleavage. Similarly, replacement of hydrophobic valine side chain with analogous proteinogenic amino acids is anticipated to produce additional inhibitors of ATPase activity. Toward these objectives, we synthesized isosteric thioamide analogues 81–84, which were anticipated to behave similar to the carboxamide analogues 39 and 53. Unexpectedly, the dithioamide analogues (81, 92% at 2.5 μM and 83, 64% at 2.5 μM) stimulated basal ATPase activity whereas monothioamide 82 (NA at 0.5 μM) lost the affinity. We postulate that change of oxygen to sulfur atom may have resulted in loss or weakening of hydrogen bonding ability of the compounds (81–83) indicating their contribution in the interactions in the substrate-binding pocket of P-gp. Nevertheless, 84 (26% inhibition at 2.5 μM) with one thioamide retained the inhibitory potency comparable to that of carboxamide 53 presumably due to favorable 4,4-difluoro substitution. Surprising activity profile of the thioamide compounds demonstrates how P-gp discriminates between molecular changes as small as amide to thioamide.
Table 5.
Effect of Bioisosteres and Amino Acid Analogues of compound 53 on P-gp ATPase Activity
| Compd | Structure | ATPase activitya % Stimulation or b Inhibition |
AlogPc | |
|---|---|---|---|---|
|
| ||||
| 0.5 μM | 2.5 μM | |||
| 53 |
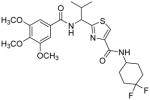
|
30.7±4.9b | 33.9±2.9b | 3.11 |
| 81 |
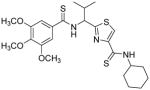
|
65 | 92 | 6.05 |
| 82 |
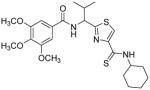
|
NAe | 27 | 4.96 |
| 83 |
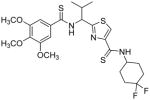
|
25 | 64 | 5.30 |
| 84 |
|
37.4±5.5b | 25.9±2.1b | 4.20 |
| 101 |
|
53.9±0.6 | 60.2±2.7 | 2.12 |
| 107 |
|
NA | 13 | 1.78 |
| 108 |
|
16 | 68 | 3.50 |
| 109 |
|
39.6±4.7b | 42.5±6.3b | 3.82 |
| 110 |
|
22.4±4.9b | 23.0±6.4b | 3.55 |
| 111 |
|
NA | 21 | 2.58 |
same as Table 1 footnotes.
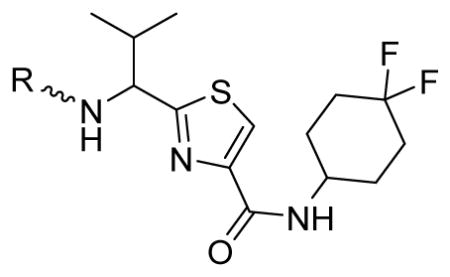
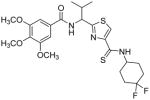
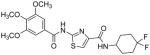
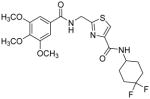
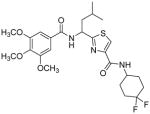
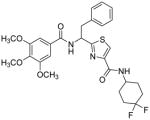
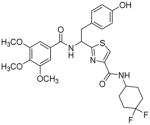
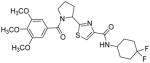
Subsequently, we determined the role of the α-carbon and its side chain toward ATPase activity. Compound 101, lacking the α-carbon, stimulated basal ATPase activity by 60% at 2.5 μM and glycine analogue, 107 (13% at 2.5 μM), devoid of the isopropyl side chain, had a weak interaction with P-gp. Moreover, replacement of the isopropyl with an isobutyl group as in leucine analogue, 108 (68% at 2.5 μM), led to a switch from inhibition to the stimulation of ATPase activity. We presume that compound 101 may have lost some of the ATPase inhibitory interactions due to the truncation by one bond length and a lack of side chain. Further the valine side chain may be responsible for proper orientation of the molecule in the drug/substrate-binding pocket of P-gp as evidenced from the lack of affinity of 107 toward P-gp. To further probe the isopropyl interacting region topology of P-gp, we replaced the isopropyl side chain with the benzyl and weakly acidic 4-hydroxybenzyl side chains yielding ATPase inhibitory compounds 109 (TTT-150; 43% inhibition at 2.5 μM) and 110 (23% inhibition at 2.5 μM). The preference for the aromatic amino acid residues, as in 109 and 110, may have resulted from favorable pi-pi interactions in the isopropyl binding region of P-gp for ATPase inhibitory activity (vide infra). Analogue 110 showed slightly less ATPase inhibition as compared to 109, an observation hinted toward unfavorable contribution of the weakly acidic and polar 4-hydroxyl group. To explore the impact of replacing flexible valine by a rigid amino acid such as proline, we prepared (S)-proline-derived thiazole analogue, 111 (21% at 2.5 μM), which lost the affinity toward P-gp. It may be noted that these α-carbon modified analogues (108–111) were partially racemized. In general, the side chain fragment of these compounds play an important role in interactions at the drug-binding region of P-gp.
Herein, we summarize the key SAR trends. Despite large, flexible and tolerant substrate/drug-binding region of P-gp, the SAR data on the (S)-amino acid-derived thiazole compounds in this report suggests a unique ability of P-gp to distinguish amongst essentially considered as isosteric structures in medicinal chemistry. Alicyclic analogues 39, 42, and 43 showed preference for the cyclohexyl ring over isosteric piperidine and pyran rings. Only the difluoro substitution at the 4-position of the cyclohexyl ring (53), amongst different hydrogen bonding groups (42, 51 and 52), improves inhibitory efficiency. Moreover, structural changes with either removal or addition of one atom resulted in a switch from ATPase inhibition to stimulation as depicted by analogues 53 versus 61, 79, and 108. Unanticipated effects of the thioamides (81, 82 and 83) demonstrated subtle ability of P-gp to recognize specific groups. Moreover, glycine analogue 107 indicates the significance of hydrophobic contacts in the substrate-binding site of P-gp. Additionally, positioning of the trimethoxybenzoyl moiety, commonly present in several P-gp modulators, is critical at the N-terminus and is involved in key hydrogen bonding interactions (vide infra). Introduction of an aryl ring at the C-terminus of the thiazole scaffold resulted in stimulation of ATPase activity and only all carbon six membered saturated ring resulted in inhibitory activity.
Furthermore, a variety of molecular descriptors, reviewed by Didziapetris et. al.,45 were calculated based on 2D and 3D structures of the compounds (Table S1). Albeit weak correlation, atom based logP (AlogP/octanol-water partition coefficient) analysis indicates that compounds with AlogP ≥3 exhibits higher affinity (moderate stimulation or inhibition of ATPase) with few exceptions (38, 40, 42, 48, 58, 73, 74, 76, 82, and 101) paralleling similar reports by others and our group.19, 46–49 All compounds except 77 have molecular weight >400 Da. Majority of the target compounds are well within the acceptable range of Lipinski’s Rule-of-Five. The values for the calculated descriptors spans to a similar range for ATPase stimulatory, inhibitory and no affinity compounds. No apparent discrimination was found for these properties against the biological behavior of the compounds. It is intriguing that such similar compounds render different effects on interaction with P-gp. Finally, we examined, in silico, our target compounds against a reference set to identify any substructure commonly found in Pan Assay Interference Compounds (PAINS).50 None of the target compounds, with the exception of five azide-containing compounds 29, and 32–35, were identified as potential PAINS in our analysis. These azide-containing compounds were primarily synthesized for photoaffinity labeling experiments.
[125I]-IAAP Photolabeling Assay
To determine whether the ATPase inhibitors are interacting with P-gp at its substrate-binding site, we conducted binding competition assay of compounds 53 and 109 against [125I]-IAAP. The UV-light-induced covalent labeling of [125I]-IAAP to P-gp was inhibited by both compounds 53 and 109 in a concentration-dependent fashion (Figure 2). Compound 53 inhibited a maximum of 90% labeling at >5 μM concentrations (Figure 2A). The [125I]-IAAP labeling IC50 for compound 53 was 0.1 μM, which is improved compared to lead compound 1 (0.72 μM).19 However, the IC50 for compound 109 was 0.76 μM, which is close to that of compound 1 (Figure 2B). These results and ATPase activity data show that compound 53 inhibited basal ATPase activity by binding to TMDs of human P-gp with higher affinity than compound 1.
Figure 2.

Photoaffinity labeling competition assay with compounds 53 (A) and 109 (B) inhibiting the incorporation of [125I]-IAAP to human P-gp. [125I]-IAAP incorporation into P-gp band in the absence of compounds (only DMSO) was taken as 100%. The data, in the graphs, were fitted for (A) R2 = 0.94 and (B) R2 = 0.92 with a one-phase decay equation using GraphPad Prism 7 with points representing the average of three independent experiments and error bars denote SD.
MDR Reversal Assay
MDR reversal assay was carried out using representative analogues (1, 31, 39, 53, 60, and 109) to determine the translation from biochemical assay results (ATPase and [125I]-IAAP) to clinical implication. Compounds were selected based on their effect on ATPase activity: potent stimulators (1 and 31), inhibitors (39, 53 and 109), and moderate stimulator (60). We determined the effect on cytotoxicity to the parental cell line (HEK 293-pcDNA 3.1) and ABCB1 (P-gp) transfected cell line (HEK-ABCB1) upon treatment of each compound in combination with paclitaxel. Paclitaxel was used as the ABCB1 substrate chemotherapy drug. Verapamil (10 μM) and zosuquidar (0.25 μM) were used as positive reversal compounds against the HEK-ABCB1 cell line (Table 6). The non-toxic test concentration of 10 μM for reversal compounds was selected because the preliminary cytotoxicity data showed at least 80% cell survival in both parental and resistant cell lines when treated alone (data not shown).
Table 6.
Reversal Effect of the Representative Compounds on the Cytotoxicity of Paclitaxel to Parental HEK 293-pcDNA 3.1 and P-gp transfected HEK-ABCB1 Cell Lines
| Treatment | Conc. | HEK 293-pcDNA 3.1 | HEK-ABCB1 | ||
|---|---|---|---|---|---|
|
| |||||
| IC50 ± SD (μM)a | FRb | IC50 ± SD (μM)a | FRb | ||
| Paclitaxel | 0.071 ± 0.015 | 1 | 2.054 ± 0.087 | 28.9 | |
| + 1 | 10 μM | 0.072 ± 0.021 | 1 | 0.046 ± 0.017 | 0.6 |
| + 31 | 10 μM | 0.037 ± 0.013 | 0.5 | 1.272 ± 0.190 | 17.9 |
| + 39 | 10 μM | 0.068 ± 0.014 | 1 | 0.042 ± 0.020 | 0.6 |
| + 53 | 10 μM | 0.041 ± 0.020 | 0.6 | 0.042 ± 0.016 | 0.6 |
| + 60 | 10 μM | 0.099 ± 0.031 | 1.4 | 0.207 ± 0.097 | 2.9 |
| + 109 | 10 μM | 0.037 ± 0.019 | 0.5 | 0.118 ± 0.015 | 1.7 |
| + Verapamil | 10 μM | 0.066 ± 0.012 | 0.9 | 0.052 ± 0.018 | 0.7 |
| + Zosuquidar | 0.25 μM | 0.013 ± 0.008 | 0.2 | 0.143 ± 0.010 | 2.0 |
Half maximal inhibitory concentration (IC50) values calculated for paclitaxel concentration, with shown treatment, required for 50% inhibition of cell survival. Standard Deviation (±SD) values calculated from four independent experiments each performed in triplicates.
Fold Resistance (FR) values calculated by dividing the IC50 value of paclitaxel in the presence of indicated compound, with shown treatment, in the ABCB1 expressing HEK-ABCB1 cells by the IC50 value in the presence of paclitaxel alone in HEK293 cells with control plasmid.
The cytotoxicity results demonstrate that the ABCB1-expressing cells exhibit 28.9-fold resistance (FR) to paclitaxel, with an IC50 of 2.054 μM, as compared to the parental HEK 293-pcDNA 3.1 cells, which showed an IC50 of 0.071 μM when treated with paclitaxel alone. Pre-treatment with test compounds significantly reduced the IC50 value of ABCB1 substrate, paclitaxel, in the HEK-ABCB1 resistant cells. While the effect of the reversal compounds in combination with paclitaxel showed a negligible toxic effect in the parental HEK 293-pcDNA 3.1 cells, they proved to diminish the resistance significantly in the resistant, P-gp overexpressing cells. The relative resistance of the HEK-ABCB1 cells reduced from 28.9-fold to 0.6, 0.6, 0.6, 2.9 and 1.7-fold in the presence of compounds 1, 39, 53, 60, and 109 (10 μM), respectively. These compounds performed comparably to our positive controls at their respective concentrations: verapamil, a first-generation P-gp inhibitor and known ATPase stimulator (10 μM); and zosuquidar, a third generation potent P-gp ATPase inhibitor (0.25 μM). The lowered test concentration of zosuquidar was necessary due to the potency of the compound and because it was cytotoxic at 5 μM concentration. Competitive inhibition of the P-gp can be reversible or irreversible throughout the 72 h incubation period. We envision the ATPase stimulators may reverse MDR through competition with the substrate paclitaxel while the ATPase inhibitors prevent the paclitaxel efflux by non-competitive inhibition. Compound 31 has a weak reversal effect at 10 μM (fold-resistance of 17.8) suggesting it might be effluxed out at a rapid rate. Overall, compounds 1, 39, 53, 60, and 109 were found to overcome P-gp-mediated resistance to paclitaxel.
One of the drawbacks, with previous generations of P-gp modulators, including our lead compound 1 (CYP3A4 IC50 = 8.2 μM),20 was concomitant inhibition of CYP3A4. Therefore, we conducted in vitro CYP3A4 inhibition assay with representative compounds as per our prior report (Table 7).20 P-gp ATPase inhibitors (39, 53, and 109) did not inhibit CYP3A4 up to 25 μM whereas compound 31, which is an ATPase stimulator and benzophenone-containing analogue has CYP3A4 inhibition of 44% at 25 μM and 83% at 50 μM. Hence, replacement of the C-terminus benzophenone moiety of compound 1 with a cyclohexyl group offered selectivity toward P-gp over CYP3A4.
Table 7.
In Vitro CYP3A4 Enzyme Inhibition Assay with Selected Compounds
Molecular Modeling
Based on the SAR on P-gp ATPase activity of the synthesized analogues, we hypothesize that inhibitory compounds occupy a sterically demanding cavity in the drug-binding site and are involved in hydrogen bonding interactions with specific residues. To rationalize this hypothesis, we carried out induced-fit docking (a method that allow both protein side chains and ligand flexibility) of compounds 53 and 109 (Figure 3) within the transmembrane domain of homology modeled structure of human P-gp derived from the crystal structure of mouse P-gp (PDB ID: 4Q9H).51 We chose mouse P-gp because it is the only available mammalian crystal structure with high resolution and has 87% identical sequence to that of human P-gp. Figures 3A and 3B illustrates docking pose for compound 53. The docking data showed hydrogen bonding interaction of the 4-methoxy oxygen atom with the side chain of Gln990 (H3CO----H2N-Gln990). The amide group, connecting the trimethoxyphenyl and the Cα of the valine, entered into a hydrogen bonding interaction with the phenolic hydroxyl of Tyr307 (C=O----HO-Tyr307). The 4,4-difluorocyclohexyl moiety occupies a space that is closely surrounded by residues Phe335, Phe336, Leu339, Ile340, and Phe343. Thus, any modification of the 4,4-difluorocyclohexyl moiety at the C-terminus will result in steric hindrance and a loss of inhibitory activity. The isopropyl group of a valine residue of the compound may be involved in hydrophobic interactions with the residues, Phe728 and Ala729, in the transmembrane region of P-gp.
Figure 3. Induced-fit docking model of inhibitors within transmembrane domains of homology modeled human P-gp.

(A) Docking pose of compound 53. Amino acid residues are illustrated as thin tubes with the color representations as carbon in faded orange; hydrogen in white; nitrogen in blue; oxygen in red and sulfur in yellow. The protein is represented in faded red-orange colored ribbon form. The ball and stick model of inhibitor with the identical color scheme as above except carbon atoms represented in green and fluorine atoms in faded-green is shown. Black dashes represent protein-ligand intermolecular hydrogen bonds. (B) 2-D representation of docking pose of compound 53. The colored drops (cyan, polar and green, hydrophobic) indicate amino acid residues within 5 Å of the ligand and arrows (magenta) show hydrogen bonds. (C) Docking pose of (S)-isomer of compound 109. Representation is same as in A and aromatic interaction is shown as light blue dash. (D) 2-D representation of docking pose of (S)-isomer of compound 109. Representation is same as in B and aromatic interaction is shown by a green line. (E) A surface representation of the docked pose is shown. The ball and stick model represents (S)-isomer of inhibitor 109 and the surrounding is drug binding site surface in residue-type color scheme. (F) Superposition of docked structures of compound 53 and cis- isomer of (S)-proline analogue 111. Only residues which are interacting with compound 111 are shown. Carbon atoms are represented using faded blue color for 111 and other atoms follow the same color representation as indicated above. (G) Superposition of docked structures of compound 53 and trans-(S)-proline analogue 111. The representation is same as in F.
Binding model of (S)-isomer of 109 within the substrate-binding region of P-gp indicates the formation of three hydrogen bonds (4-H3CO----H2N-Gln838; 3-H3CO----H2N-Gln990 and the N-terminus-NH----OH-Tyr307) as shown in Figures 3C and 3D. Moreover, the side chain benzyl is stabilized by an edge-to-face (T-shape) interaction with the side chain of Tyr310. The 4,4-difluorocyclohexyl moiety is surrounded by residues Phe335, Phe336, Leu339, Ile340, and Phe343 similar to that observed for compound 53. The surface representation of bound (S)-isomer of 109 also shows its binding at the sterically demanding binding site especially at the cyclohexyl region (Figure 3E).
Since, both cis- and trans-forms for (S)-proline are reported to exist in small peptides, we performed ligand alignment using both isomers.52 Superposition of docked conformations of compound 53 and conformationally restricted cis- and trans-proline isomers (111) indicated a substantial movement of the proline analogue as compared to the valine analogue 53 (Figure 3F and 3G). The trimethoxybenzoyl portion of the molecule shifts away from the position that was discerned from the valine-derived analogue 53. Therefore, it can be concluded that the conformationally restricted proline-derived analogue, 111, presented interactions in the manner that were detrimental to its binding affinity toward P-gp. This may have resulted from the loss of key hydrogen bonding interaction with the side chains of Tyr307 and Asn842 and/or Gln990 in case of proline analogue.
It may be noted that docking experiments cannot necessarily identify the correct binding pattern of the ligand and particularly in this case due to a large and flexible binding pocket of P-gp. However, predicted binding models may facilitate future optimization efforts. The interaction pattern based on the induced-fit docking poses as well as the SAR data together suggests that the trimethoxybenzoyl group is surrounded by polar side chains of Gln725, Asn842, Gln838, and Gln990, which may facilitate proper orientation of the molecule in this region. Since the side chain benzyl group of 109 is involved in T-shape aromatic-aromatic interaction, it could be replaced with heteroaromatic ring-containing unnatural amino acids to strengthen an edge-to-face interaction with the side chain of Tyr310. Furthermore, the two amide nitrogen atoms can be linked together to form macrocyclic analogues by ring closing metathesis. Such macrocyclic analogues would bind within the substrate-binding region of P-gp in an entropically favorable manner compared to their non-cyclic counterparts. This will form the basis for our future SAR studies.
CONCLUSIONS
A series of substituted (S)-amino acid-derived thiazole analogues were synthesized to probe the substrate-binding site within the TMDs of P-gp and their effect on basal ATPase activity was determined. Starting from the lead compound 1, a potent stimulator of ATPase activity, few inhibitors (39, 53, 78, 84, 109 and 110) of ATPase activity were identified. The inhibitory activity toward ATPase was largely contributed by the cyclohexane ring. Moreover, replacement of the cyclohexane moiety with piperidine (61 and 62), aromatic (65 and 66) or open chain alkyl (67) groups resulted in a switch from inhibition to stimulation of ATPase activity. The trimethoxy group at the N-terminus was critical for preserving inhibitory function and is involved in hydrogen bonding interactions as evidenced from 74, 76 and 78. Compounds 53, 61, 79, and 108 show that variation by one bond length results in significant shift in affinity toward P-gp. Moreover, photolabeling competition data with [125I]-IAAP and compounds 53 and 109 provided evidence for interaction of these compounds with the TMDs of P-gp. MDR reversal assay data showed that ATPase stimulatory compound 31 as well as ATPase inhibitors 39, 53 and 109 act as MDR reversal compounds, presumably with different mechanisms. To our delight, these ATPase inhibitors did not inhibit CYP3A4 unlike starting lead compound 1 and previous generations of P-gp modulators. The substrate-binding pocket of P-gp is thought to be very flexible in nature and recognizes various size and type of chemical structures. The effect of the synthesized series, in this report, reinforces this fact; however, it is intriguing to discover that an extension or truncation by only one methylene group switches the inhibition to the stimulation of ATPase activity.
EXPERIMENTAL
Chemical Synthesis
Materials and Instrumentation
Chemicals were purchased from Aldrich Chemical Co. (Milwaukee, WI), AK scientific (Union City, CA), A2Z chemicals (Irvine, CA), Oakwood Products (West Columbia, SC), TCI America (Portland, OR) and Alfa Aesar (Ward Hill, MA), and were used as received. All chemicals were confirmed for uniformity by thin layer chromatography (TLC) with silica gel as the adsorbent layer (250 microns) on aluminum backed plates (Agela Technologies). Reactions were monitored by TLC, and visualized using Ultraviolet (UV) light at 254 nm. Melting points were recorded on a Stuart Melting Point apparatus (model - SMP20) and are uncorrected. Bruker 400 Ultrashield™ spectrometer (1H at 400 MHz and 13C at 100 MHz) equipped with a z-axis gradient probe was used to record NMR experiments. 1H NMR and 13C NMR chemical shifts were reported downfield from tetramethylsilane (TMS, internal standard) in parts per million (δ ppm). The 1H NMR data are presented as follows: chemical shift (multiplicity {s (singlet), bs (broad singlet), d (doublet), t (triplet), dd (doublet of doublets), m (multiplet) and oct (octet)}, number of protons, coupling constant). The 13C NMR (proton decoupled, fluorine coupled) data are presented as follows: chemical shift (multiplicity {d (doublet), t (triplet)}). Column chromatography purifications were performed with silica gel (40–63 μm) obtained from Silicycle Inc. (Quebec City, Canada) and flash chromatography was performed using Reveleris® X2 flash chromatography system (BÜCHI Corporation, New Castle, DE). Preparative TLC was performed using Silica Gel GF 1000 μm 20×20cm glass backed plates from Analtech (Miles Scientific, Newark, DE). Target compounds purity analysis was accomplished utilizing Agilent 1100 HPLC system with an autosampler (Agilent, Santa Clara, CA) eluting a C-18 reverse phase column (Agilent Eclipse plus C18, 3.5 μm, 4.6 × 100 mm) with an isocratic mobile phase flow (1.0 mL/min), and samples monitored under UV light at 254 nm. All target compounds were established to be ≥95% pure (major peak area/total combined area of peaks). Mass analyses were carried out on Agilent 1260 infinity series liquid chromatography (LC) system (C18 column, Agilent InfinityLab poroshell 120, EC-C18, 2.7 μm, 4.6 × 50 mm) connected with Agilent 6120 quadrupole mass spectrometer (MS). The elemental analyses (C, H, and N) were performed by Atlantic Microlabs, Inc., (Norcross, GA) and the observed values were within ±0.4% of calculated values.
Synthesis
Compounds 2,31 619 and 1020 were synthesized according to the procedures as described in our previous reports.
Method A: Typical Procedure for Peptide Coupling Reactions
A homogenous solution/suspension of carboxylic acid (1 eq) in either dry N, N-dimethylacetamide (DMA; 10 mL) or dry dichloromethane (DCM; 15 mL), under nitrogen atmosphere, was brought to 0°C and then added with diisopropylethylamine (DIEA; 1.5 eq), HCTU (1.5 eq), and HOBt (1.5 eq). The resulting mixture was stirred for 10–15 min at 0°C before the addition of cold solution/suspension of amine (1.2 eq) in appropriate solvent (DMA or DCM; ~1–2 mL). It was then stirred overnight at rt, after which the resulting solution was evaporated. Ethyl acetate was added to a crude product mass and washed with 1N aqueous potassium bisulfate (KHSO4) solution. The acidic aqueous layer was then extracted with 2X ethyl acetate. Combined ethyl acetate extracts were partitioned with saturated aqueous solution of sodium carbonate (Na2CO3). Ethyl acetate extract was collected, dried over anhydrous magnesium sulfate (MgSO4) and evaporated. The remaining mass was purified by chromatography (preparative TLC or flash chromatography) using n-hexanes/ethyl acetate as mobile phase to obtain coupled product as a colorless oil. This oil was triturated with ethyl acetate and n-hexanes to provide a pure solid.
Method B: General Procedure for the N-Boc Deprotection
N-Boc protected amine (1 eq) dissolved in DCM (20 mL) was slowly added to trifluoroacetic acid (TFA; 10 eq) at 0°C. A resulting reaction mixture was kept at 0°C for 10 min and then it was stirred at rt for an appropriate period (4–12 h). The volatiles were evaporated to leave viscous mass, which was dissolved in water. It was partitioned with ethyl acetate and the acidic aqueous layer was collected. The water layer was carefully neutralized using saturated aqueous solution of Na2CO3 and then extracted with ethyl acetate. The ethyl acetate extract was passed over anhydrous MgSO4 and evaporated to obtain free amine, which was used without further purification.
Method C: Procedure for the Synthesis of Carboxamides by Mixed-Anhydride Method
Appropriate carboxylic acid (1 eq) in anhydrous THF (50 mL) under nitrogen was cooled to −20°C. Then, isobutyl chloroformate (1.2 eq) and N-methylmorpholine (NMM; 1.2 eq) were added to the reaction mixture and stirred at −20°C for 1–2 h. To this suspension was added excess (25 mL) of aqueous ammonia (30%), which produced a clear biphasic solution that was allowed to stir for an additional 30 min – 2 h at rt. After concentrating the reaction mixture, it was extracted with 2X ethyl acetate. The combined ethyl acetate extract was partitioned with aqueous 1N KHSO4, collected, dried over anhydrous MgSO4 and evaporated to provide desired carboxamide as a solid.
Method D: General Procedure for the Synthesis of Thioamides
To a solution of carboxamide (1 eq) in anhydrous THF (40 mL) was added Lawesson’s reagent (0.57 eq) under nitrogen atmosphere. The suspension was stirred for 8 h at rt. At this time, entire solid material in the reaction mixture dissolved to produce a clear yellow solution. Subsequently, saturated aqueous NaHCO3 solution was slowly added to the reaction flask until effervescence ceases. Ethyl acetate (15 mL) was added to the quenched mixture and stirred for an additional hour. The TLC analysis showed two spots with near Rf values of which the upper faint spot disappeared from the organic layer after extraction workup. The ethyl acetate extract was dried (anhydrous MgSO4), concentrated and then purified through column chromatography using n-hexanes/ethyl acetate as eluent giving pure thioamide as a thick oil.
Method E: General Procedure for the Construction of Thiazole Scaffold
The thioamide (1 eq) in 40 mL anhydrous ethanol under inert condition was cooled using a sodium chloride/ice-bath (−20°C). To it, was added calcium carbonate (CaCO3; 2.5 eq) and the suspension was stirred at −20°C for 15 min and subsequently ethyl bromopyruvate (1.1 eq) was added. The dark mixture was further stirred at rt for 8 h. Afterwards, the solid was filtered off and the filtrates were concentrated to furnish crude product, which was purified using column chromatography with n-hexanes/ethyl acetate as eluent.
Method F: General Procedure for Peptide Coupling with Acyl Chlorides
Diisopropylethylamine (DIEA, 2 eq) and appropriate acyl chloride (1.2 eq) were sequentially added to a solution of amine (1 eq) in anhydrous THF (20 mL) at 0°C and stirred overnight at rt under nitrogen atmosphere. Ethyl acetate was added to the reaction mixture and it was then successively washed with 2N HCl and saturated aqueous Na2CO3 solution. The organic layers were collected, dried (anhydrous MgSO4) and concentrated to a crude residue, which was purified by flash chromatography providing the coupled product.
Method G: General Procedure for the Thiazole Ester Hydrolysis
The thiazole ester in a 20 mL of THF/methanol/water (10/2/3) solvent mixture at 0°C was added sodium hydroxide (10 eq). The reaction mixture was brought to rt and allowed to stir overnight after which it was concentrated using rotary vacuum evaporation. The residual liquid was added with more water and partitioned with ethyl acetate. The aqueous layer was collected, acidified using 2N HCl and partitioned with ethyl acetate (2X). The organic extracts were collected, dried over anhydrous MgSO4 and evaporated to provide crude acid that was used without further purification.
Tert-butyl (S)-(2-methyl-1-(4-((3,4,5-trimethoxyphenyl)carbamoyl)thiazol-2-yl)propyl) carbamate (3)
Compound 3 was obtained from acid 2 (475 mg, 1.58 mmol) and 3,4,5-trimethoxyaniline (350 mg, 1.91 mmol) using method A as a yellow solid (544 mg, 74%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 9.96 (s, 1H), 8.29 (s, 1H), 7.75 (d, 1H, J = 8.2 Hz), 7.30 (s, 2H), 4.77-4.74 (m, 1H), 3.77 (s, 6H), 3.64 (s, 3H), 2.37-2.29 (m, 1H), 1.42 (s, 9H), 0.90 (d, 6H, J = 6.5 Hz).
(S)-2-(1-Amino-2-methylpropyl)-N-(3,4,5-trimethoxyphenyl)thiazole-4-carboxamide (4)
Amine 4 was obtained from compound 3 using method B (12 h) as a yellow oil. 1H NMR (400 MHz; CDCl3; TMS) δ 9.08 (s, 1H), 8.09 (s, 1H), 7.02 (s, 2H), 4.21-4.18 (m, 1H), 3.87 (s, 6H), 3.81 (s, 3H), 2.31-2.23 (m, 1H), 1.01 (d, 3H, J = 6.5 Hz), 0.92 (d, 3H, J = 6.5 Hz).
(S)-2-(1-(2-Benzoylbenzamido)-2-methylpropyl)-N-(3,4,5-trimethoxyphenyl)thiazole-4-carboxamide (5)
Compound 5 was obtained from 2-benzoyl benzoic acid (50 mg, 0.22 mmol) and amine 4 (97 mg, 0.27 mmol) using method A as a white solid (42 mg, 33%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 8.79 (s, 1H), 8.16 (s, 1H), 7.79 (d, 1H, J = 6.8 Hz), 7.62-7.55 (m, 2H), 7.48 (s, 1H), 7.26 (d, 1H, J = 7.0 Hz), 7.13 (s, 2H), 7.06-6.93 (m, 5H), 4.13 (d, 1H, J = 10.9 Hz), 3.81 (s, 6H), 3.65 (s, 3H), 3.19-3.11 (m, 1H), 1.06 (d, 3H, J = 6.5), 0.71 (d, 3H, J = 6.5 Hz). m/z (ESI-MS) 574.3 (C31H31N3O6S requires 574.19, [M + H]+). HPLC tR (Acetonitrile/water 60/40) = 3.2 min, purity 98%.
(S)-2-(2-methyl-1-(3,4,5-trimethoxybenzamido)propyl)thiazole-4-carboxylic acid (6)
The synthesis and 1H NMR details for compound 6 are reported elsewhere.19 13C NMR (100 MHz; DMSO-D6; TMS) δ 172.98, 166.27, 162.58, 153.03, 147.12, 140.71, 129.41, 129.05, 105.64, 60.55, 57.93, 56.51, 32.19, 20.31, 19.74.
(S)-N-(3-Benzoylphenyl)-2-(2-methyl-1-(3,4,5-trimethoxybenzamido)propyl)thiazole-4-carboxamide (7)
Compound 7 was obtained from acid 6 (50 mg, 0.13 mmol) and 3-aminobenzophenone (30 mg, 0.15 mmol) using method A as a white solid (30 mg, 41%). 1H NMR (400 MHz; CDCl3; TMS) δ 9.22 (s, 1H), 8.17-8.15 (m, 2H), 7.94-7.93 (m, 1H), 7.84-7.82 (m, 2H), 7.61 (d, 1H, J = 7.3 Hz), 7.55-7.47 (m, 4H), 7.05 (s, 2H), 6.67 (d, 1H, J = 8.6 Hz), 5.39 (dd, 1H, J = 8.8 Hz, 6.7 Hz), 3.90 (s, 6H), 3.88 (s, 3H), 2.56 (oct, 1H, J = 6.7 Hz), 1.10 (d, 3H, J = 6.8 Hz), 1.06 (d, 3H, J = 6.8 Hz). m/z (ESI-MS) 574.2 (C31H31N3O6S requires 574.19, [M + H]+). HPLC tR (Acetonitrile/water 60/40) = 5.4 min, purity 99%.
2-Amino-N-phenylbenzamide (8).32
A diethyl ether solution (70 mL) of anthranilic acid (500 mg, 3.65 mmol) was added dropwise to thionyl chloride (5 mL) and refluxed for 2 h. The reaction mixture was then co-evaporated with DCM for three times. The remaining mass was taken up in ether and added to a stirring solution containing aniline (0.4 mL, 4.38 mmol) in pyridine (15 mL) and ether (40 mL), and heated at refluxing temperatures for 3 h. The dark solution was evaporated and the crude product was purified using flash chromatography (n-hexanes/ethyl acetate: 100/0% to 0/100%; gradient) as an off-white solid (8; 250 mg, 32%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 9.98 (s, 1H), 7.70 (d, 2H, J = 8.2 Hz), 7.61 (d, 1H, J = 7.9 Hz), 7.32 (t, 2H, J = 7.5 Hz), 7.19 (t, 1H, J = 7.5 Hz), 7.07 (t, 1H, J = 7.3 Hz), 6.75 (d, 1H, J = 8.0 Hz), 6.58 (t, 1H, J = 7.4 Hz), 6.31 (bs, 2H).
N-(2-Aminophenyl)benzamide (9).33
2-nitroaniline (300 mg, 2.17 mmol) in anhydrous THF (20 mL) was stirred overnight with pyridine (0.26 mL, 3.25 mmol) and benzoyl chloride (0.24 mL, 2.06 mmol) after which the mixture was concentrated. The crude product was dissolved in ethyl acetate and washed sequentially with 2N HCl and saturated aqueous solution of NaHCO3. The organic layer was dried (anhydrous MgSO4) and evaporated to obtain the coupled product as a yellow oil, which was dissolved in methanol and subjected to palladium catalyzed hydrogenation using pressure vessel (60 psi) at rt for overnight. The solution was then passed through celite and evaporated to furnish 9 (330 mg, 75%) as a light brown solid. 1H NMR (400 MHz; CDCl3; TMS) δ 11.39 (s, 1H), 9.04 (d, 1H, J = 8.5 Hz), 8.31 (d, 1H, J = 8.5 Hz), 8.03 (d, 2H, J = 7.9 Hz), 7.75 (t, 1H, J = 7.8 Hz), 7.65-7.62 (m, 1H), 7.59-7.55 (m, 2H), 7.25 (t, 1H, J = 7.8 Hz).
(2-Aminophenyl)(4-bromophenyl)methanol (11)
2-Amino-4′-bromobenzophenone (300 mg, 1.09 mmol) in combination with sodium borohydride (41 mg, 1.09 mmol) was refluxed in ethanol (15 mL) for 2 h and then the solution was concentrated. The crude remaining was dissolved in ethyl acetate and partitioned with water. The organic extract was dried (anhydrous MgSO4) and concentrated to yield a brown solid (11, 236 mg, 78%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 7.48 (d, 2H, J = 8.2 Hz), 7.31 (d, 2H, J = 7.9 Hz), 7.03 (d, 1H, J = 7.6 Hz), 6.95 (t, 1H, J = 7.2 Hz), 6.59 (d, 1H, J = 8.0 Hz), 6.52 (t, 1H, J = 7.3 Hz), 5.96 (d, 1H, J = 3.8 Hz), 5.71 (d, 1H, 3.8 Hz), 4.97 (s, 2H).
3-((Tert-butoxycarbonyl)amino)benzoic acid (12).34
Compound 12 was obtained by reacting 3-aminobenzoic acid (1.20 g, 8.75 mmol) with di-tert-butyl dicarbonate (2.86 g, 13.12 mmol) and triethylamine (1.83 mL, 13.12 mmol) in a solvent mixture of dioxane (20 mL) and water (20 mL) at rt for 24 h. The reaction mixture was concentrated, acidified with 3N HCl and extracted with ethyl acetate. The organic layer was dried over anhydrous MgSO4 and evaporated to produce a white solid (1.91 g, 92%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 9.38 (s, 1H), 8.15 (s, 1H), 7.63 (d, 1H, J = 6.3 Hz), 7.47 (d, 1H, J = 7.8 Hz), 7.21 (t, 1H, J = 7.8 Hz), 1.47 (s, 9H).
2-Azidobenzoic acid (13)
To anthranilic acid (500 mg, 3.65 mmol) dissolved in a cooled (0°C) aq. solution of 10% HCl was slowly added a 1.5 mL of aqueous sodium nitrite (302 mg, 4.38 mmol) solution and allowed to stir for 15 min at rt. Then, 1 mL of aqueous sodium azide (285 mg, 4.38 mmol) solution was added carefully resulting in vigorous precipitation. The precipitates were filtered and washed with water to obtain 13 as an orange solid in quantitative yield. 1H NMR (400 MHz; DMSO-d6; TMS) δ 13.21 (s, 1H), 7.77-7.26 (m, 4H).
3-Azidobenzoic acid (14).35
Compound 14 was prepared from 3-aminobenzoic acid using the same procedure as that for azide 13 as a cream colored solid. 1H NMR (400 MHz; DMSO-d6; TMS) δ 13.28 (s, 1H), 7.73-7.33 (m, 4H).
4-Azidobenzoic acid (15).35
Compound 15 was prepared from 4-aminobenzoic acid using the same procedure as that for azide 13 as a cream colored solid. 1H NMR (400 MHz; DMSO-d6; TMS) δ 13.03 (bs, 1H), 7.95 (d, 2H, J = 7.4 Hz), 7.18 (d, 2H, J = 7.4 Hz).
Tert-butyl (1-benzylpiperidin-4-yl)carbamate (16).36
Tert-butyl piperidin-4-ylcarbamate (1.00 g, 4.99 mmol) was alkylated by heating for overnight with benzyl chloride (0.86 mL, 7.49 mmol) and triethylamine (1.39 mL, 9.98 mmol) at refluxing temperature in THF (20 mL). Afterwards, ethyl acetate was used to dilute the reaction mixture and then washed with aqueous 1N KHSO4. The ethyl acetate layer was passed over anhydrous MgSO4 and evaporated. The resulting crude mass was purified using flash chromatography to furnish 16 (1.11 g, 76%) as a yellow solid. 1H NMR (400 MHz; CDCl3; TMS) δ 7.75 (d, 1H, J = 7.4 Hz), 7.42-7.31 (m, 5H), 3.49 (s, 2H), 3.07-2.93 (m, 1H), 2.82-2.80 (m, 2H), 2.12-2.07 (m, 2H), 1.93-1.81 (m, 4H), 1.45 (s, 9H).
1-Benzylpiperidin-4-amine (17)
Compound 17 was obtained from N-Boc protected 16 using method B in a period of 4 h as a colorless oil. 1H NMR (400 MHz; CDCl3; TMS) δ 7.33-7.23 (m, 5H), 3.51 (s, 2H), 2.85 (d, 1H, J = 11.7 Hz), 2.71-2.64 (m, 2H), 2.03 (t, 2H, J = 11.3 Hz), 1.82-1.74 (m, 4H), 1.46-1.37 (m, 2H).
(S)-2-(2-Methyl-1-(3,4,5-trimethoxybenzamido)propyl)-N-(2-(phenylcarbamoyl)phenyl)thiazole-4-carboxamide (18)
Compound 18 was obtained from acid 6 (100 mg, 0.25 mmol) and aniline 8 (65 mg, 0.30) using method A as a white solid (10 mg, 7%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 12.06 (s, 1H), 10.55 (s, 1H), 9.02 (d, 1H, J = 8.4 Hz), 8.61 (d, 1H, J = 8.4 Hz), 8.39 (s, 1H), 7.90 (d, 1H, J = 7.8 Hz), 7.85-7.80 (m, 2H), 7.62 (t, 1H, J = 7.8 Hz), 7.37 (t, 2H, J = 7.8 Hz), 7.31-7.23 (m, 3H), 7.14 (t, 1H, 7.4 Hz), 5.21-5.17 (m, 1H), 3.83 (s, 6H), 3.71 (s, 3H), 2.69-2.60 (m, 1H), 1.09-1.05 (m, 6H). m/z (ESI-MS) 589.3 (C31H32N4O6S requires 589.20, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 13.8 min, purity 95%.
(S)-N-(2-Benzamidophenyl)-2-(2-methyl-1-(3,4,5-trimethoxybenzamido)propyl)thiazole-4-carboxamide (19)
Compound 19 was obtained from acid 6 (100 mg, 0.25 mmol) and aniline 9 (65 mg, 0.30) using method A as a white solid (10 mg, 7%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 10.45 (s, 1H), 9.89 (s, 1H), 8.90 (d, 1H, J = 8.5 Hz), 8.36 (s, 1H), 8.13-8.07 (m, 3H), 7.64 (t, 1H, J = 6.9 Hz), 7.57 (t, 2H, J = 6.9 Hz), 7.42-7.35 (m, 2H), 7.27-7.22 (m, 3H), 5.07-5.03 (m, 1H), 3.83 (s, 6H), 3.71 (s, 3H), 2.34-2.24 (m, 1H), 0.81 (t, 3H, J = 6.5 Hz), 0.66 (d, 3H, J = 6.5 Hz). m/z (ESI-MS) 589.3 (C31H32N4O6S requires 589.20, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 7.3 min, purity 95%.
(S)-2-(2-Methyl-1-(3,4,5-trimethoxybenzamido)propyl)-N-(2-phenoxyphenyl)thiazole-4-carboxamide (20)
Compound 20 was obtained from acid 6 (100 mg, 0.25 mmol) and 2-phenoxyaniline (56 mg, 0.30) using method A as a white solid (30 mg, 21%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 9.79 (s, 1H), 8.97 (d, 1H, J = 8.2 Hz), 8.42 (d, 1H, J = 8.0 Hz), 8.39 (s, 1H), 7.39 (t, 2H, J = 7.3 Hz), 7.28-7.12 (m, 5H), 7.09-7.04 (m, 3H), 5.11-5.07 (m, 1H), 3.83 (s, 6H), 3.71 (s, 3H), 2.40-2.33 (m, 1H), 0.99 (d, 3H, J = 6.6 Hz), 0.87 (d, 3H, J = 6.6 Hz). m/z (ESI-MS) 562.20 (C30H31N3O6S requires 562.19, [M + H]+). HPLC tR (acetonitrile/water 60/40) = 9.6 min, purity 100%.
(S)-N-(2-Benzylphenyl)-2-(2-methyl-1-(3,4,5-trimethoxybenzamido)propyl)thiazole-4-carboxamide (21)
Compound 21 was obtained from acid 6 (100 mg, 0.25 mmol) and 2-benzylamine (56 mg, 0.30) using method A as a white solid (22 mg, 16%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 9.53 (s, 1H), 8.95 (d, 1H, J = 8.2 Hz), 8.33 (s, 1H), 7.84 (d, 1H, J = 7.6 Hz), 7.35-7.17 (m, 10H), 5.23-5.19 (m, 1H), 4.09-4.00 (m, 2H), 3.84 (s, 6H), 3.71 (s, 3H), 2.47-2.41 (m, 1H), 1.06 (d, 3H, J = 6.3 Hz), 0.95 (d, 3H, J = 6.3 Hz). m/z (ESI-MS) 560.3 (C31H33N3O5S requires 560.21, [M + H]+). HPLC tR (acetonitrile/water 60/40) = 7.6 min, purity 99%.
Tert-butyl ((1S)-1-(4-((2-(hydroxy(phenyl)methyl)phenyl)carbamoyl)thiazol-2-yl)-2-methylpropyl)carbamate (22)
Compound 22 was obtained from acid 2 (500 mg, 1.66 mmol) and 2-aminobenzhydrol 1020 (398 mg, 2.00 mmol) using method A as a white solid (552 mg, 69%). 1H NMR (400 MHz; CDCl3; TMS) δ 10.47 (s, 1H), 8.30 (d, 1H, J = 8.2 Hz), 8.11 (s, 1H), 8.04 (d, 1H, J = 3.4 Hz), 7.44-7.03 (m, 8H), 6.02-6.01 (m, 1H), 5.42-5.40 (m, 1H), 4.94-4.91 (m, 1H), 2.40-2.31 (m, 1H), 1.47 (s, 9H), 1.00-0.98 (m, 6H).
Tert-butyl ((1S)-1-(4-((2-((4-bromophenyl)(hydroxy)methyl)phenyl)carbamoyl)thiazol-2-yl)-2-methylpropyl)carbamate (23)
Compound 23 was obtained from acid 2 and benzhydrol 11, using method A as a dark brown oil. 1H NMR (400 MHz; CDCl3; TMS) δ 8.10-6.38 (m, 11H), 5.44-5.39 (m, 1H), 4.91-4.88 (m, 1H), 2.40-2.32 (m, 1H), 1.45 (s, 9H), 0.98 (d, 3H, J = 6.7 Hz), 0.92 (d, 3H, J = 6.7 Hz).
Tert-butyl (S)-(1-(4-((2-benzoylphenyl)carbamoyl)thiazol-2-yl)-2-methylpropyl) carbamate (24)
To a solution of benzhydrol derivative 22 (500 mg, 1.04 mmol) in dry THF (15 mL), Dess–Martin periodinane (662 mg, 1.56 mmol) was added and stirred at rt until the TLC indicated reaction completion (~30 min). Subsequently saturated NaHCO3 and sodium thiosulfate (Na2S2O3) aqueous solutions were added for quenching the reaction. The mixture was partitioned with ethyl acetate and the organic layer was collected, dried (anhydrous MgSO4) and evaporated to give a crude mass, which was purified by column chromatography (n-hexane/ethyl acetate 80/20) to obtain compound 24 (478 mg, 96%) as a white solid. 13C NMR (100 MHz; CDCl3; TMS) δ 198.47, 171.96, 159.82, 155.55, 150.22, 139.76, 138.69, 133.81, 133.26, 132.33, 129.98, 128.23, 124.48, 123.85, 122.41, 121.57, 79.91, 57.83, 33.38, 28.32, 19.25, 17.60. 1H NMR (400 MHz; CDCl3; TMS) δ 12.25 (s, 1H), 8.81 (d, 1H, J = 8.4 Hz), 8.14 (s, 1H), 7.78 (d, 2H, J = 7.8 Hz), 7.65-7.59 (m, 3H), 7.52-7.48 (m, 2H), 7.15 (t, 1H, J = 7.4 Hz), 5.53-5.51 (m, 1H), 5.00-4.96 (m, 1H), 2.49 (oct, 1H, J = 6.5 Hz), 1.46 (s, 9H), 1.05 (d, 3H, J = 6.7 Hz), 1.00 (d, 3H, J = 6.7 Hz). m/z (ESI-MS) 480.2 (C26H29N3O4S requires 480.19, [M + H]+).
Tert-butyl (S)-(1-(4-((2-(4-bromobenzoyl)phenyl)carbamoyl)thiazol-2-yl)-2-methylpropyl) carbamate (25)
Compound 25 was prepared following the same procedure used for the synthesis of 24 except compound 23 was used as starting material. 1H NMR (400 MHz; DMSO-d6; TMS) δ 8.06-8.03 (m, 1H), 7.99-7.96 (m, 1H), 7.90-7.88 (m, 1H,), 7.79-7.58 (m, 3H), 7.42-7.38 (m, 2H), 7.17-7.12 (m, 2H), 5.71-5.50 (m, 1H), 2.37-2.34 (m, 1H), 1.45 (s, 9H), 0.98-0.92 (m, 6H).
(S)-2-(1-Amino-2-methylpropyl)-N-(2-benzoylphenyl)thiazole-4-carboxamide (26)
Amine 26 was obtained from N-Boc protected 24 using method B (12 h) as a colorless oil and it was used without additional purification.
(S)-2-(1-Amino-2-methylpropyl)-N-(2-(4-bromobenzoyl)phenyl)thiazole-4-carboxamide (27)
Amine 27 was obtained from N-Boc protected 25 using method B (12 h) as a colorless oil, which was carried forward without additional purification.
(S)-N-(2-(4-Bromobenzoyl)phenyl)-2-(2-methyl-1-(3,4,5-trimethoxybenzamido)propyl) thiazole-4-carboxamide (28)
Compound 28 was obtained from 3,4,5-trimethoxybenzoyl chloride (122 mg, 0.53 mmol) and amine 27 (200 mg, 0.44 mmol) using method F as a white solid (159 mg, 56%). 1H NMR (400 MHz; CDCl3; TMS) δ 12.39 (s, 1H), 8.83 (d, 1H, J = 8.4 Hz), 8.19 (s, 1H), 7.68-7.62 (m, 3H), 7.55 (d, 1H, J = 7.8 Hz), 7.51-7.49 (m, 2H), 7.34 (d, 1H, J = 8.4 Hz), 7.19-7.15 (m, 3H), 5.54 (dd, 1H, J = 8.7 Hz, 5.7 Hz), 3.92 (s, 3H), 3.85 (s, 6H), 2.54–2.46 (m, 1H), 1.10 (d, 3H, J = 6.8 Hz), 1.06 (d, 3H, J = 6.8 Hz). m/z (ESI-MS) 652.2 (C31H30BrN3O6S requires 652.11, [M + H]+). HPLC tR (acetonitrile/water 60/40) = 14.1 min, purity 98%.
(S)-N-(2-(4-Azidobenzoyl)phenyl)-2-(2-methyl-1-(3,4,5-trimethoxybenzamido)propyl) thiazole-4-carboxamide (29)
The 4′-bromo-derivative 28 (120 mg, 0.18 mmol) was suspended in a 30 mL mixture of ethanol:water (7:3) under nitrogen atmosphere. Sodium hydroxide (2 mg, 0.05 mmol), copper iodide (4 mg, 0.02 mmol), (S)-proline (6 mg, 0.05 mmol) and sodium azide (23 mg, 0.36 mmol) were added to the solution and refluxed for 24 h. Afterwards, the reaction solution was cooled, concentrated and partitioned with ethyl acetate. The ethyl acetate layer was dried over anhydrous MgSO4 and concentrated to give crude mass, which was purified by preparative TLC (n-hexane/ethyl acetate - 70/30) to yield compound 29 (30 mg, 27%) as a white solid. 1H NMR (400 MHz; CDCl3; TMS) δ 12.35 (d, 1H, J = 33.9 Hz), 8.81 (dd, 1H, J = 12.8 Hz, 8.4 Hz), 8.19 (d, 1H, J = 2.6 Hz), 7.68-7.61 (m, 3H), 7.55 (dd, 1H, J = 7.8 Hz, 4.4 Hz), 7.50 (d, 1H, J = 8.3 Hz), 7.37-7.32 (m, 1H), 7.19 (d, 2H, J = 4.1 Hz), 7.17-7.14 (m, 1H), 7.10 (d, 1H, J = 8.6 Hz), 5.56-5.52 (m, 1H), 3.92 (s, 3H), 3.85-3.84 (m, 6H), 2.49 (oct, 1H, J = 6.4 Hz), 1.10 (d, 3H, J = 6.8 Hz), 1.06 (d, 3H, J = 6.8 Hz). m/z (ESI-MS) 615.3 (C31H30N6O6S requires 615.19, [M + H]+). HPLC tR (acetonitrile/water 60/40) = 18.0 min, purity 98%.
Tert-butyl (S)-(3-((1-(4-((2-benzoylphenyl)carbamoyl)thiazol-2-yl)-2-methylpropyl) carbamoyl)phenyl)carbamate (30)
Compound 30 was obtained from acid 12 (100 mg, 0.42 mmol) and amine 26 (190 mg, 0.50 mmol) using method A as a white solid (110 mg, 44%). 1H NMR (400 MHz; CDCl3; TMS) δ 12.44 (s, 1H), 8.82 (d, 1H, J = 8.2 Hz), 8.19 (s, 1H), 7.81 (bs, 1H), 7.75-7.70 (m, 5H), 7.60-7.57 (m, 3H), 7.38-7.34 (m, 2H), 7.14 (t, 1H, J = 7.5 Hz), 7.03 (t, 1H, J = 8.0 Hz), 6.98 (d, 1H, J = 7.4 Hz), 5.53 (dd, 1H, J = 8.5 Hz, 5.3 Hz), 2.44 (oct, 1H, J = 6.5 Hz), 1.44 (s, 9H), 1.04 (d, 3H, J = 6.8 Hz), 1.01 (d, 3H, J = 6.8 Hz).
(S)-2-(1-(3-Aminobenzamido)-2-methylpropyl)-N-(2-benzoylphenyl)thiazole-4-carboxamide (31)
Compound 31 was obtained from N-Boc compound 30 (40 mg, 0.07 mmol) in 4 h using method B as a white solid (30 mg, 90%). mp 146–150 °C. 1H NMR (400 MHz; CDCl3; TMS) δ 12.60 (s, 1H), 8.89 (d, 1H, J = 8.6 Hz), 8.18 (s, 1H), 7.76 (d, 2H, J = 7.3 Hz), 7.68-7.62 (m, 3H), 7.54-7.50 (m, 3H), 7.38-7.34 (m, 2H), 7.16 (t, 1H, J = 7.7 Hz), 6.95 (t, 1H, J = 7.8 Hz), 6.75 (dd, 1H, J = 7.9 Hz, 2.1 Hz), 5.56 (dd, 1H, J = 8.6 Hz, 5.3 Hz), 3.69-3.66 (m, 2H), 2.45 (oct, 1H, J = 6.3 Hz), 1.08 (d, 3H, J = 6.8 Hz), 1.05 (d, 3H, J = 6.8 Hz). 13C NMR (100 MHz; CDCl3; TMS) δ 199.28, 169.50, 166.87, 159.68, 149.89, 140.14, 138.94, 134.92, 134.29, 133.75, 132.32, 129.93, 129.43, 128.36, 124.05, 123.90, 122.41, 121.38, 118.24, 117.15, 116.09, 114.21, 56.22, 34.38, 18.91, 18.19. m/z (ESI-MS) 499.2 (C28H26N4O3S requires 499.18, [M + H]+). HPLC tR (acetonitrile/water 60/40) = 5.6 min, purity 99%. Anal. Calcd for C28H26N4O3S·0.3 H2O: C, 66.73; H, 5.32; N, 11.12. Found: C, 67.02; H, 4.99; N, 10.75.
(S)-2-(1-(2-Azidobenzamido)-2-methylpropyl)-N-(2-benzoylphenyl)thiazole-4-carboxamide (32)
Compound 32 was obtained from acid 13 (50 mg, 0.31 mmol) and amine 26 (139 mg, 0.37) using method A as a white solid (5 mg, 3%). 1H NMR (400 MHz; CDCl3; TMS) δ 12.32 (s, 1H), 8.80 (d, 1H, J = 8.4 Hz), 8.48 (d, 1H, J = 8.4 Hz), 8.17-8.15 (m, 2H), 7.67-7.44 (m, 9H), 7.14-7.10 (m, 2H), 5.55 (dd, 1H, J = 8.6 Hz, 5.3 Hz), 2.61-2.54 (m, 1H), 1.09 (d, 6H, J = 6.6 Hz). m/z (ESI-MS) 525.2 (C28H24N6O3S requires 525.16, [M + H]+). HPLC tR (acetonitrile/water 60/40) = 15.8 min, purity 98%.
(S)-2-(1-(3-Azidobenzamido)-2-methylpropyl)-N-(2-benzoylphenyl)thiazole-4-carboxamide (33)
Compound 33 was obtained from acid 14 (50 mg, 0.31 mmol) and amine 26 (139 mg, 0.37) using method A as a white solid (15 mg, 9%). 1H NMR (400 MHz; CDCl3; TMS) δ 12.79 (s, 1H), 8.90 (d, 1H, J = 8.4 Hz), 8.20-8.18 (m, 1H), 7.78 (d, 1H, J = 7.4 Hz), 7.72-7.62 (m, 7H), 7.52 (t, 2H, J = 7.4 Hz), 7.16 (t, 1H, J = 7.6 Hz), 7.07 (d, 1H, J = 7.8 Hz), 7.00 (d, 1H, J = 7.6 Hz), 5.57 (dd, 1H, J = 8.6 Hz, 5.1 Hz), 2.44 (oct, 1H, J = 6.3 Hz), 1.09 (d, 3H, J = 6.8 Hz), 1.05 (d, 3H, J = 6.8 Hz). m/z (ESI-MS) 525.2 (C28H24N6O3S requires 525.16, [M + H]+). HPLC tR (acetonitrile/water 60/40) = 16.9 min, purity 98%.
(S)-2-(1-(4-Azidobenzamido)-2-methylpropyl)-N-(2-benzoylphenyl)thiazole-4-carboxamide (34)
Compound 34 was obtained from acid 15 (50 mg, 0.31 mmol) and amine 26 (139 mg, 0.37) using method A as a white solid (50 mg, 31%). 1H NMR (400 MHz; CDCl3; TMS) δ 12.96 (s, 1H), 8.94 (d, 1H, J = 8.3 Hz), 8.19 (s, 1H), 8.03 (d, 2H, J = 8.6 Hz), 7.75-7.64 (m, 6H), 7.57 (t, 2H, J = 7.5 Hz), 7.15 (t, 1H, J = 7.6 Hz), 6.69 (d, 2H, J = 8.6 Hz), 5.59 (dd, 1H, J = 8.5 Hz, 5.1 Hz), 2.41 (oct, 1H, J = 6.8 Hz), 1.10 (d, 3H, J = 6.8 Hz), 1.06 (d, 3H, J = 6.8 Hz). m/z (ESI-MS) 525.2 (C28H24N6O3S requires 525.16, [M + H]+). HPLC tR (acetonitrile/water 60/40) = 15.1 min, purity 98%.
(S)-2-(1-(4-Azidobenzamido)-2-methylpropyl)-N-(2-(4-bromobenzoyl)phenyl)thiazole-4-carboxamide (35)
Compound 35 was obtained from acid 15 (50 mg, 0.31 mmol) and amine 27 (170 mg, 0.37) using method A as a white solid (40 mg, 22%). 1H NMR (400 MHz; CDCl3; TMS) δ 12.71 (s, 1H), 8.89 (d, 1H, J = 8.4 Hz), 8.18 (s, 1H), 8.00 (d, 2H, J = 8.6 Hz), 7.68-7.63 (m, 4H), 7.58-7.55 (m, 3H), 7.14 (t, 1H, J = 7.6 Hz), 6.80 (d, 2H, J = 8.5 Hz), 5.55 (dd, 1H, J = 8.4 Hz, 5.1 Hz), 2.41 (oct, 1H, J = 6.2 Hz), 1.07 (d, 3H, J = 6.8 Hz), 1.03 (d, 3H, J = 6.8 Hz). m/z (ESI-MS) 603.1 (C28H23BrN6O3S requires 603.07, [M + H]+). HPLC tR (acetonitrile/water 70/30) = 12.0 min, purity 98%.
(S)-N-Cyclopropyl-2-(2-methyl-1-(3,4,5-trimethoxybenzamido)propyl)thiazole-4-carboxamide (36)
Compound 36 was obtained from acid 6 (100 mg, 0.25 mmol) and cyclopropanamine (17 mg, 0.30) using method A as a white solid (6 mg, 5%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 8.90 (d, 1H, J = 7.9 Hz), 8.22 (d, 1H, J = 3.9 Hz), 8.16 (s, 1H), 7.21 (s, 2H), 5.15-5.11 (m, 1H), 3.84 (s, 6H), 3.71 (s, 3H), 2.47-2.41 (m, 1H), 1.28-1.22 (m, 1H), 1.03 (d, 3H, J = 6.5 Hz), 0.91 (d, 3H, J = 6.5 Hz), 0.70-0.60 (m, 4H). m/z (ESI-MS) 434.2 (C21H27N3O5S requires 434.17, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 2.3 min, purity 96%.
(S)-N-Cyclobutyl-2-(2-methyl-1-(3,4,5-trimethoxybenzamido)propyl)thiazole-4-carboxamide (37)
Compound 37 was obtained from acid 6 (100 mg, 0.25 mmol) and cyclobutanamine (22 mg, 0.30 mmol) using method A as a white solid (15 mg, 13%). 1H NMR (400 MHz; CDCl3; TMS) δ 8.04 (s, 1H), 7.35 (d, 1H, J = 7.7 Hz), 7.06 (s, 2H), 6.61 (d, 1H, J = 8.7 Hz), 5.40 (dd, 1H, J = 8.8 Hz, 6.3 Hz), 4.59 (oct, 1H, J = 8.6 Hz), 3.96 (s, 6H), 3.92 (s, 3H), 2.55-2.40 (m, 3H), 2.09-1.98 (m, 2H), 1.83-1.77 (m, 2H), 1.09-1.06 (m, 6H). m/z (ESI-MS) 448.3 (C22H29N3O5S requires 448.19, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 3.4 min, purity 100%.
(S)-N-Cyclopentyl-2-(2-methyl-1-(3,4,5-trimethoxybenzamido)propyl)thiazole-4-carboxamide (38)
Compound 38 was obtained from acid 6 (100 mg, 0.25 mmol) and cyclopentanamine (26 mg, 0.30 mmol) using method A as a white solid (22 mg, 19%). 1H NMR (400 MHz; CDCl3; TMS) δ 8.04 (s, 1H), 7.17 (d, 1H, J = 7.6 Hz), 7.05 (s, 2H), 6.60 (d, 1H, J = 8.8 Hz), 5.40 (dd, 1H, J = 8.8 Hz, 6.5 Hz), 4.40 (oct, 1H, J = 7.2 Hz), 3.95 (s, 6H), 3.92 (s, 3H), 2.56-2.44 (m, 1H), 2.15-2.06 (m, 2H), 1.79-1.64 (m, 4H), 1.57-1.50 (m, 2H), 1.08-1.05 (m, 6H). m/z (ESI-MS) 462.2 (C23H31N3O5S requires 462.20, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 4.7 min, purity 96%.
(S)-N-Cyclohexyl-2-(2-methyl-1-(3,4,5-trimethoxybenzamido)propyl)thiazole-4-carboxamide (39)
Compound 39 was obtained from acid 6 (100 mg, 0.25 mmol) and cyclohexanamine (30 mg, 0.30 mmol) using method A as a white solid (30 mg, 25%). mp 87–89 °C. 1H NMR (400 MHz; DMSO-d6; TMS) δ 8.94 (d, 1H, J = 8.3 Hz), 8.17 (s, 1H), 7.88 (d, 1H, J = 8.4 Hz), 7.22 (s, 2H), 5.13 (t, 1H, J = 8.4 Hz), 3.85 (s, 6H), 3.77 (bs, 1H), 3.71 (s, 3H), 2.50–2.40 (m, 1H), 1.81-1.78 (m, 2H), 1.73-1.70 (m, 2H), 1.61-1.58 (m, 1H), 1.42-1.25 (m, 4H), 1.19-1.09 (m, 1H), 1.04 (d, 3H, J = 6.8 Hz), 0.92 (d, 3H, J = 6.8 Hz). 13C NMR (100 MHz; CDCl3; TMS) δ 171.49, 167.02, 159.84, 153.33, 149.65, 141.46, 129.25, 123.04, 104.75, 60.94, 56.89, 56.45, 48.40, 33.29, 33.04, 25.50, 24.87, 19.60, 18.31. m/z (ESI-MS) 476.3 (C24H33N3O5S requires 476.21, [M + H]+). HPLC tR (acetonitrile/water 60/40) = 3.0 min, purity 99%. Anal. Calcd for C24H33N3O5S·0.45 H2O: C, 59.59; H, 7.06; N, 8.69. Found: C, 59.75; H, 7.04; N, 8.48.
(S)-N-(Adamantan-1-yl)-2-(2-methyl-1-(3,4,5-trimethoxybenzamido)propyl)thiazole-4-carboxamide (40)
Compound 40 was obtained from acid 6 (100 mg, 0.25 mmol) and adamantan-1-amine (46 mg, 0.30 mmol) using method A as a white solid (40 mg, 30%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 8.92 (d, 1H, J = 8.2 Hz), 8.11 (s, 1H),7.21 (s, 3H), 5.11-5.07 (m, 1H), 3.83 (s, 6H), 3.70 (s, 3H), 2.45-2.38 (m, 1H), 2.05 (s, 9H), 1.66 (s, 6H), 1.04 (d, 3H, J = 6.5 Hz), 0.92 (d, 3H, J = 6.5 Hz). m/z (ESI-MS) 528.3 (C28H37N3O5S requires 528.25, [M + H]+). HPLC tR (acetonitrile/water 60/40) = 8.4 min, purity 99%.
Tert-butyl (S)-4-(2-(2-methyl-1-(3,4,5-trimethoxybenzamido)propyl)thiazole-4-carboxamido)piperidine-1-carboxylate (41)
Compound 41 was obtained from acid 6 (200 mg, 0.51 mmol) and tert-butyl 4-aminopiperidine-1-carboxylate (122 mg, 0.61 mmol) using method A as a white solid (50 mg, 34%). 1H NMR (400 MHz; CDCl3; TMS) δ 7.78 (s, 1H), 7.06 (s, 2H), 6.79 (d, 1H, J = 8.8 Hz), 5.43 (dd, 1H, J = 8.8 Hz, 6.2 Hz), 4.63-4.52 (m, 2H), 4.30-4.23 (m, 1H), 3.94 (s, 6H), 3.91 (s, 3H), 3.76-3.66 (m, 2H), 3.21-3.17 (m, 1H), 3.00-2.95 (m, 1H), 2.50-2.46 (m, 1H), 2.06-1.93 (m, 3H), 1.46 (s, 9H), 1.06-1.03 (m, 6H). m/z (ESI-MS) 577.3 (C28H40N4O7S requires 577.26, [M + H]+). HPLC tR (acetonitrile/water 60/40) = 3.1 min, purity 95%.
(S)-2-(2-Methyl-1-(3,4,5-trimethoxybenzamido)propyl)-N-(piperidin-4-yl)thiazole-4-carboxamide (42)
Compound 42 was obtained from N-Boc compound 41 (30 mg, 0.05 mmol) using method B (12 h) as a white solid (24 mg, 97%). 1H NMR (400 MHz; CDCl3; TMS) δ 8.04 (s, 1H), 7.16 (d, 1H, J = 8.4 Hz), 7.06 (s, 2H), 6.65 (d, 1H, J = 8.7 Hz), 5.39 (dd, 1H, J = 8.7 Hz, 6.6 Hz), 4.12-4.00 (m, 1H), 3.95 (s, 6H), 3.92 (s, 3H), 3.13 (d, 2H, J = 11.8 Hz), 2.77 (t, 2H, J = 11.8 Hz), 2.57-2.47 (m, 1H), 2.07-2.06 (m, 2H), 1.53-1.41 (m, 2H), 1.08 (d, 3H, J = 6.8 Hz), 1.06 (d, 3H, J = 6.8 Hz). m/z (ESI-MS) 477.3 (C23H32N4O5S requires 477.21, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 3.3 min, purity 99%.
(S)-2-(2-Methyl-1-(3,4,5-trimethoxybenzamido)propyl)-N-(tetrahydro-2H-pyran-4-yl) thiazole-4-carboxamide (43)
Compound 43 was obtained from acid 6 (200 mg, 0.51 mmol) and tetrahydro-2H-pyran-4-amine (62 mg, 0.61 mmol) using method A as a white solid (100 mg, 41%). 1H NMR (400 MHz; CDCl3; TMS) δ 8.05 (s, 1H), 7.16 (d, 1H, J = 8.2 Hz), 7.05 (s, 2H), 6.61 (d, 1H, J = 8.8 Hz), 5.39 (dd, 1H, J = 8.8 Hz, 6.5 Hz), 4.24-4.14 (m, 1H), 4.02 (d, 2H, J = 11.9 Hz), 3.95 (s, 6H), 3.92 (s, 3H), 3.56 (t, 2H, J = 11.9 Hz), 2.56-2.46 (m, 1H), 2.02-1.99 (m, 2H), 1.64-1.57 (m, 2H), 1.08 (d, 3H, J = 6.8 Hz), 1.06 (d, 3H, J = 6.8 Hz). m/z (ESI-MS) 478.3 (C23H31N3O6S requires 478.19, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 1.9 min, purity 99%.
(S)-3,4,5-Trimethoxy-N-(2-methyl-1-(4-(4-oxopiperidine-1-carbonyl)thiazol-2-yl)propyl) benzamide (44)
Compound 44 was obtained from acid 6 (100 mg, 0.25 mmol) and 4-piperidone monohydrate hydrochloride (47 mg, 0.30 mmol) using method A as a white solid (40 mg, 33%). 1H NMR (400 MHz; CDCl3; TMS) δ 7.93 (s, 1H), 7.02 (s, 2H), 6.65 (d, 1H, J = 8.7 Hz), 5.42 (dd, 1H, J = 8.6 Hz, 6.1 Hz), 4.09-4.03 (m, 4H), 3.92 (s, 6H), 3.89 (s, 3H), 2.57-2.45 (m, 5H), 1.05 (dd, 6H, J = 6.6 Hz, 1.3 Hz). m/z (ESI-MS) 476.2 (C23H29N3O6S requires 476.18, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 1.8 min, purity 99%.
Tert-butyl (S)-4-((2-(2-methyl-1-(3,4,5-trimethoxybenzamido)propyl)thiazole-4-carboxamido)methyl)piperidine-1-carboxylate (45)
Compound 45 was obtained from acid 6 (200 mg, 0.51 mmol) and tert-butyl 4-(aminomethyl)piperidine-1-carboxylate (130 mg, 0.61 mmol) using method A as a white solid (70 mg, 23%). 1H NMR (400 MHz; CDCl3; TMS) δ 8.05 (s, 1H), 7.38-7.35 (m, 1H), 7.05 (s, 2H), 6.56 (d, 1H, J = 8.8 Hz), 5.40 (dd, 1H, J = 8.8 Hz, 6.4 Hz), 3.95 (s, 3H), 3.93 (s, 6H), 3.37-3.33 (m, 4H), 2.74-2.67 (m, 2H), 2.54-2.51 (m, 1H), 1.77-1.74 (m, 3H), 1.61 (s, 9H), 1.27-1.17 (m, 2H), 1.08-1.05 (m, 6H).
(S)-2-(2-Methyl-1-(3,4,5-trimethoxybenzamido)propyl)-N-(piperidin-4-ylmethyl)thiazole-4-carboxamide (46)
Compound 46 was obtained from N-Boc compound 45 (70 mg, 0.12 mmol) using method B (12 h) as a white solid (55 mg, 95%). 1H NMR (400 MHz; CDCl3; TMS) δ 8.04 (s, 1H), 7.37-7.34 (m, 1H), 7.05 (s, 2H), 6.65 (d, 1H, J = 8.8 Hz), 5.39 (dd, 1H, J = 8.8 Hz, 6.4 Hz), 3.94 (s, 6H), 3.91 (s, 3H), 3.37-3.33 (m, 2H), 3.11 (d, 2H, J = 11.9 Hz), 2.61 (t, 2H, J = 11.9 Hz), 2.51 (oct, 1H, J = 6.7 Hz), 1.78-1.74 (m, 3H), 1.29-1.19 (m, 2H), 1.08-1.05 (m, 6H). m/z (ESI-MS) 491.3 (C24H34N4O5S requires 491.22, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 3.8 min, purity 99%.
Tert-butyl (S)-(1-(2-(2-methyl-1-(3,4,5-trimethoxybenzamido)propyl)thiazole-4-carbonyl)piperidin-4-yl)carbamate (47)
Compound 47 was obtained from acid 6 (200 mg, 0.51 mmol) and tert-butyl piperidin-4-ylcarbamate (122 mg, 0.61 mmol) using method A as a white solid (30 mg, 10%). The coupled product was carried forward without characterization.
(S)-N-(1-(4-(4-Aminopiperidine-1-carbonyl)thiazol-2-yl)-2-methylpropyl)-3,4,5-trimethoxybenzamide (48)
Compound 48 was obtained from N-Boc compound 47 (20 mg, 0.04 mmol) using method B (12 h) as a white solid (16 mg, 97%). 1H NMR (400 MHz; CDCl3; TMS) δ 7.76 (s, 1H), 7.06 (s, 2H), 6.84 (d, 1H, J = 8.7 Hz), 5.43 (dd, 1H, J = 8.8 Hz, 6.2 Hz), 4.61-4.57 (m, 1H), 4.24-4.20 (m, 1H), 3.94 (s, 6H), 3.91 (s, 3H), 3.18-3.11 (m, 1H), 3.03-2.94 (m, 2H), 2.49 (oct, 1H, J = 6.7 Hz), 1.98-1.77 (m, 2H), 1.46-1.36 (m, 2H), 1.06-1.03 (m, 6H). m/z (ESI-MS) 477.3 (C23H32N4O5S requires 477.21, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 2.3 min, purity 99%.
Tert-butyl ((1S,4R)-4-((2-((S)-2-methyl-1-(3,4,5-trimethoxybenzamido)propyl)thiazole-4-carboxamido)methyl)cyclohexyl)carbamate (49)
Compound 49 was obtained from acid 6 (200 mg, 0.51 mmol) and trans-tert-butyl (4-(aminomethyl)cyclohexyl)carbamate (139 mg, 0.61 mmol) using method A as a white solid (25 mg, 8%). 1H NMR (400 MHz; CDCl3; TMS) δ 8.04 (s, 1H), 7.36-7.33 (m, 1H), 7.04 (s, 2H), 6.57 (d, 1H, J = 8.8 Hz), 5.40 (dd, 1H, J = 8.8 Hz, 6.4 Hz), 3.95 (s, 6H), 3.93 (s, 3H), 3.41-3.28 (m, 5H), 2.55-2.45 (m, 1H), 2.06-2.03 (m, 4H), 1.59 (s, 9H), 1.30-1.26 (m, 4H), 1.09-1.05 (m, 6H).
N-(((1R,4S)-4-Aminocyclohexyl)methyl)-2-((S)-2-methyl-1-(3,4,5-trimethoxybenzamido) propyl)thiazole-4-carboxamide (50)
Compound 50 was obtained from N-Boc compound 49 (25 mg, 0.04 mmol) using method B (12 h) as a white solid (20 mg, 96%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 8.97 (d, 1H, J = 8.3 Hz), 8.32 (t, 1H, J = 6.3 Hz), 8.16 (s, 1H), 7.82-7.72 (m, 2H), 7.22 (s, 2H), 5.15-5.11 (m, 1H), 3.84 (s, 6H), 3.71 (s, 3H), 3.21-3.04 (m, 2H), 2.97-2.90 (m, 1H), 2.46-2.39 (m, 1H), 1.93 (d, 2H, J = 11.6 Hz), 1.75 (d, 2H, J = 11.6 Hz), 1.55-1.47 (m, 2H), 1.30-1.20 (m, 3H), 1.05 (d, 3H, J = 6.7 Hz), 0.92 (d, 3H, J = 6.7 Hz). m/z (ESI-MS) 505.3 (C25H36N4O5S requires 505.24, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 2.5 min, purity 99%.
(S)-N-(1,1-Dioxidotetrahydro-2H-thiopyran-4-yl)-2-(2-methyl-1-(3,4,5-trimethoxy benzamido)propyl)thiazole-4-carboxamide (51)
Compound 51 was obtained from acid 6 (100 mg, 0.25 mmol) and 4-aminotetrahydro-2H-thiopyran-1,1-dioxide hydrochloride (56 mg, 0.30 mmol) using method A as a white solid (12 mg, 9%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 8.93 (d, 1H, J = 8.3 Hz), 8.39 (d, 1H, J = 8.3 Hz), 8.20 (s, 1H), 7.21 (s, 2H), 5.16-5.12 (m, 1H), 4.23-4.15 (m, 1H), 3.84 (s, 6H), 3.70 (s, 3H), 3.40-3.27 (m, 2H), 3.09-3.05 (m, 2H), 2.47-2.44 (m, 1H), 2.23-2.04 (m, 4H), 1.03 (d, 3H, J = 6.7 Hz), 0.93 (d, 3H, J = 6.7 Hz). m/z (ESI-MS) 526.2 (C23H31N3O7S2 requires 526.15, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 1.7 min, purity 96%.
(S)-2-(2-Methyl-1-(3,4,5-trimethoxybenzamido)propyl)-N-(4-oxocyclohexyl)thiazole-4-carboxamide (52)
Compound 52 was obtained from acid 6 (100 mg, 0.25 mmol) and 4-aminocyclohexan-1-one (34 mg, 0.30 mmol) using method A as a white solid (15 mg, 12%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 8.94 (d, 1H, J = 8.3 Hz), 8.21 (s, 1H), 8.15 (d, 1H, J = 8.3 Hz), 7.21 (s, 2H), 5.15-5.11 (m, 1H), 4.36-4.26 (m, 1H), 3.83 (s, 6H), 3.70 (s, 3H), 2.60-2.53 (m, 2H), 2.47-2.40 (m, 1H), 2.24-2.21 (m, 2H), 2.08-2.03 (m, 2H), 1.92-1.82 (m, 2H), 1.04 (d, 3H, J = 6.7 Hz), 0.91 (d, 3H, J = 6.7 Hz). m/z (ESI-MS) 490.3 (C24H31N3O6S requires 490.19, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 2.0 min, purity 96%.
(S)-N-(4,4-Difluorocyclohexyl)-2-(2-methyl-1-(3,4,5-trimethoxybenzamido)propyl) thiazole-4-carboxamide (53)
Compound 53 was obtained from acid 6 (150 mg, 0.38 mmol) and 4,4-difluorocyclohexan-1-amine (62 mg, 0.46 mmol) using method A as a white solid (85 mg, 44%). mp 183–185 °C. 1H NMR (400 MHz; CDCl3; TMS) δ 8.06 (s, 1H), 7.16 (d, 1H, J = 8.2 Hz), 7.04 (s, 2H), 6.59 (d, 1H, J = 8.8 Hz), 5.38 (dd, 1H, J = 8.8 Hz, 6.4 Hz), 4.13-4.05 (m, 1H), 3.94 (s, 6H), 3.91 (s, 3H), 2.51 (oct, 1H, J = 6.6 Hz), 2.18-2.09 (m, 4H), 2.02-1.85 (m, 2H), 1.74-1.62 (m, 2H), 1.08 (t, 3H, J = 6.8 Hz), 1.06 (d, 3H, J = 6.8 Hz). 13C NMR (100 MHz; CDCl3; TMS) δ 171.56, 166.96, 160.24, 153.39, 149.77, 141.60, 129.30, 123.19, 104.75, 60.97, 56.84, 56.52, 46.19, 33.36, 32.25 (t, J = 24.7 Hz), 28.70 (d, J = 8.5 Hz), 19.61, 18.18. m/z (ESI-MS) 512.2 (C24H31F2N3O5S requires 512.19, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 4.6 min, purity 99%. Anal. Calcd for C24H31F2N3O5S: C, 56.35; H, 6.11; N, 8.21. Found: C, 56.18; H, 5.95; N, 7.95.
(S)-N-(4,4-Dimethylcyclohexyl)-2-(2-methyl-1-(3,4,5-trimethoxybenzamido)propyl) thiazole-4-carboxamide (54)
Compound 54 was obtained from acid 6 (100 mg, 0.25 mmol) and 4,4-dimethylcyclohexan-1-amine (39 mg, 0.30 mmol) using method A as a white solid (5 mg, 4%). 1H NMR (400 MHz; CDCl3; TMS) δ 8.02 (s, 1H), 7.11 (d, 1H, J = 8.2 Hz), 7.03 (s, 2H), 6.58 (d, 1H, J = 8.9 Hz), 5.38 (dd, 1H, J = 8.8 Hz, 6.4 Hz), 3.93 (s, 6H), 3.89 (s, 3H), 2.52-2.42 (m, 1H), 1.89-1.83 (m, 2H), 1.45-1.35 (m, 7H), 1.06-1.03 (m, 6H), 0.94 (s, 6H). m/z (ESI-MS) 504.3 (C26H37N3O5S requires 504.25, [M + H]+). HPLC tR (acetonitrile/water 60/40) = 6.3 min, purity 99%.
(S)-N, N-Dicyclohexyl-2-(2-methyl-1-(3,4,5-trimethoxybenzamido)propyl)thiazole-4-carboxamide (55)
Compound 55 was obtained from acid 6 (100 mg, 0.25 mmol) and dicyclohexylamine (55 mg, 0.30 mmol) using method A as a white solid (84 mg, 59%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 7.59 (s, 1H), 7.05 (s, 2H), 7.00 (d, 1H, J = 8.6 Hz), 5.40 (dd, 1H, J = 8.9 Hz, 6.3 Hz), 3.90 (s, 6H), 3.88 (s, 3H), 3.83-3.69 (m, 1H), 3.08-2.99 (m, 1H), 2.56-2.40 (m, 3H), 1.80-1.51 (m, 18H), 1.04 (d, 3H, J = 6.8 Hz), 1.01 (d, 3H, J = 6.8 Hz). m/z (ESI-MS) 558.3 (C30H43N3O5S requires 558.29, [M + H]+). HPLC tR (acetonitrile/water 60/40) = 14.5 min, purity 99%.
(S)-N-(1-(4-([1,4′-Bipiperidine]-1′-carbonyl)thiazol-2-yl)-2-methylpropyl)-3,4,5-trimethoxybenzamide (56)
Compound 56 was obtained from acid 6 (100 mg, 0.25 mmol) and 1,4′-bipiperidine (51 mg, 0.30 mmol) using method A as a white solid (13 mg, 9%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 8.95 (d, 1H, J = 8.3 Hz), 7.96 (s, 1H), 7.23 (s, 2H), 5.11-5.07 (m, 1H), 4.48-4.45 (m, 1H), 4.12-4.03 (m, 1H), 3.83 (s, 6H), 3.70 (s, 3H), 3.05-2.97 (m, 1H), 2.73-2.67 (m, 1H), 2.46-2.40 (m, 6H), 1.81-1.76 (m, 1H), 1.70-1.59 (m, 1H), 1.46-1.36 (m, 8H), 1.04 (d, 3H, J = 6.7 Hz), 0.93 (d, 3H, J = 6.7 Hz). m/z (ESI-MS) 545.3 (C28H40N4O5S requires 545.27, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 2.7 min, purity 99%.
(S)-N-(1-Benzylpiperidin-4-yl)-2-(2-methyl-1-(3,4,5-trimethoxybenzamido)propyl) thiazole-4-carboxamide (57)
Compound 57 was obtained from acid 6 (200 mg, 0.51 mmol) and 1-benzylpiperidin-4-amine (17, 116 mg, 0.61 mmol) using method A as a white solid (142 mg, 49%). 1H NMR (400 MHz; CDCl3; TMS) δ 8.02 (s, 1H), 7.34-7.33 (m, 5H), 7.14 (d, 1H, J = 8.4 Hz), 7.05 (s, 2H), 6.67 (d, 1H, J = 8.8 Hz), 5.38 (dd, 1H, J = 8.8 Hz, 6.4 Hz), 4.03-3.96 (m, 1H), 3.94 (s, 6H), 3.91 (s, 3H), 3.53 (s, 2H), 2.90-2.86 (m, 2H), 2.49 (oct, 1H, J = 6.6 Hz), 2.18 (t, 2H, J = 11.09 Hz), 2.04-1.99 (m, 2H), 1.66-1.55 (m, 2H), 1.07 (d, 3H, J = 6.8 Hz), 1.05 (d, 3H, J = 6.8 Hz). m/z (ESI-MS) 567.3 (C30H38N4O5S requires 567.26, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 1.8 min, purity 99%.
(S)-2-(2-Methyl-1-(3,4,5-trimethoxybenzamido)propyl)-N-(1-(pyrimidin-2-yl)piperidin-4-yl)thiazole-4-carboxamide (58)
Compound 58 was obtained from acid 6 (100 mg, 0.25 mmol) and 1-(pyrimidin-2-yl)piperidin-4-amine (54 mg, 0.30 mmol) using method A as a white solid (55 mg, 39%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 8.92 (d, 1H, J = 8.3), 8.36 (d, 2H, J = 4.7 Hz), 8.18 (s, 1H), 8.12 (d, 1H, J = 8.4 Hz), 7.20 (s, 2H), 6.61 (t, 1H, J = 4.5 Hz), 5.15-5.10 (m, 1H), 4.67 (d, 2H, J = 12.8 Hz), 4.14-4.06 (m, 1H), 3.83 (s, 6H), 3.70 (s, 3H), 2.99 (t, 2H, J = 12.9 Hz), 2.47-2.40 (m, 1H), 1.84-1.81 (m, 2H), 1.61-1.52 (m, 2H), 1.02 (d, 3H, J = 6.7 Hz), 0.91 (d, 3H, J = 6.7 Hz). m/z (ESI-MS) 555.3 (C27H34N6O5S requires 555.23, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 3.1min, purity 98%.
2-((S)-2-Methyl-1-(3,4,5-trimethoxybenzamido)propyl)-N-((S)-1,2,3,4-tetrahydronaphthalen-1-yl)thiazole-4-carboxamide (59)
Compound 59 was obtained from acid 6 (100 mg, 0.25 mmol) and (S)-1,2,3,4-tetrahydronaphthalen-1-amine (45 mg, 0.30 mmol) using method A as a white solid (8 mg, 6%). 1H NMR (400 MHz; CDCl3; TMS) δ 8.10 (s, 1H), 7.47 (d, 1H, J = 8.8 Hz), 7.33-7.32 (m, 1H), 7.22-7.12 (m, 3H), 6.96 (s, 2H), 6.55 (d, 1H, J = 8.8 Hz), 5.42-5.36 (m, 1H), 5.33 (dd, 1H, J = 8.8 Hz, 6.6 Hz), 3.87 (s, 3H), 3.84 (s, 6H), 2.89-2.78 (m, 2H), 2.42 (oct, 1H, J = 6.6 Hz), 2.21-2.14 (m, 1H), 1.94-1.88 (m, 3H), 1.02 (d, 3H, J = 6.7 Hz), 0.99 (d, 3H, J = 6.7 Hz). m/z (ESI-MS) 524.3 (C28H33N3O5S requires 524.21, [M + H]+). HPLC tR (acetonitrile/water 60/40) = 4.9 min, purity 99%.
2-((S)-2-Methyl-1-(3,4,5-trimethoxybenzamido)propyl)-N-((R)-1,2,3,4-tetrahydronaphthalen-1-yl)thiazole-4-carboxamide (60)
Compound 60 was obtained from acid 6 (100 mg, 0.25 mmol) and (R)-1,2,3,4-tetrahydronaphthalen-1-amine (45 mg, 0.30 mmol) using method A as a white solid (22 mg, 17%). mp 97–100 °C. 1H NMR (400 MHz; CDCl3; TMS) δ 8.10 (s, 1H), 7.47 (d, 1H, J = 8.8 Hz), 7.37-7.34 (m, 1H), 7.22-7.19 (m, 2H), 7.15-7.13 (m, 1H), 6.99 (m, 2H), 6.56 (d, 1H, J = 8.8 Hz), 5.40-5.32 (m, 2H), 3.88 (bs, 9H), 2.85-2.81 (m, 2H), 2.42 (oct, 1H, J = 6.8 Hz), 2.20-2.14 (m, 1H), 1.91-1.84 (m, 3H), 1.02-0.99 (m, 6H). 13C NMR (100 MHz; CDCl3; TMS) δ 170.96, 166.88, 160.39, 153.35, 150.16, 141.41, 137.69, 136.63, 129.26, 128.72, 127.38, 126.39, 125.30, 123.15, 104.56, 60.96, 56.68, 56.40, 47.52, 33.50, 30.44, 29.32, 20.33, 19.56, 18.08. m/z (ESI-MS) 524.3 (C28H33N3O5S requires 524.21, [M + H]+). HPLC tR (acetonitrile/water 60/40) = 5.1 min, purity 99%. Anal. Calcd for C28H33N3O5S·0.7 H2O: C, 62.71; H, 6.47; N, 7.84. Found: C, 62.78; H, 6.07; N, 7.44.
(S)-N-(1-(4-(4,4-Difluoropiperidine-1-carbonyl)thiazol-2-yl)-2-methylpropyl)-3,4,5-trimethoxybenzamide (61)
Compound 61 was obtained from acid 6 (100 mg, 0.25 mmol) and 4,4-difluoropiperidine hydrochloride (48 mg, 0.30 mmol) using method A as a white solid (30 mg, 24%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 8.96 (d, 1H, J = 8.3 Hz), 8.08 (s, 1H), 7.22 (s, 2H), 5.13-5.09 (m, 1H), 3.83 (s, 6H), 3.77-3.73 (m, 4H), 3.70 (s, 3H), 2.47-2.40 (m, 1H), 2.09-1.99 (m, 4H), 1.04 (d, 3H, J = 6.7 Hz), 0.93 (d, 3H, J = 6.7 Hz). m/z (ESI-MS) 498.3 (C23H29F2N3O5S requires 498.18, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 4.0 min, purity 98%.
(S)-3,4,5-Trimethoxy-N-(2-methyl-1-(4-(4-(trifluoromethyl)piperidine-1-carbonyl)thiazol-2-yl)propyl)benzamide (62)
Compound 62 was obtained from acid 6 (100 mg, 0.25 mmol) and 4-(trifluoromethyl)piperidine hydrochloride (58 mg, 0.30 mmol) using method A as a white solid (30 mg, 22%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 8.96 (d, 1H, J = 8.3 Hz), 8.02 (s, 1H), 7.23 (s, 2H), 5.13-5.08 (m, 1H), 4.57-4.44 (m, 1H), 4.25-4.15 (m, 1H), 3.84 (s, 6H), 3.70 (s, 3H), 3.13-3.02 (m, 1H), 2.84-2.65 (m, 2H), 2.47-2.42 (m, 1H), 1.92-1.74 (m, 2H), 1.47-1.36 (m, 2H), 1.05 (d, 3H, J = 6.6 Hz), 0.94 (d, 3H, J = 6.6 Hz). m/z (ESI-MS) 530.2 (C24H30F3N3O5S requires 530.19, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 3.1 min, purity 99%.
(S)-N-((4,4-Difluorocyclohexyl)methyl)-2-(2-methyl-1-(3,4,5-trimethoxybenzamido) propyl)thiazole-4-carboxamide (63)
Compound 63 was obtained from acid 6 (100 mg, 0.25 mmol) and (4,4-difluorocyclohexyl)methanamine hydrochloride (56 mg, 0.30 mmol) using method A as a white solid (25 mg, 19%). 1H NMR (400 MHz; CDCl3; TMS) δ 8.05 (s, 1H), 7.40-7.37 (m, 1H), 7.04 (s, 2H), 6.55 (d, 1H, J = 8.8 Hz), 5.41 (dd, 1H, J = 8.8 Hz, 6.4 Hz), 3.95 (s, 6H), 3.92 (s, 3H), 3.44-3.31 (m, 2H), 2.51 (oct, 1H, J = 6.7 Hz), 2.18-2.09 (m, 2H), 1.89-1.65 (m, 5H), 1.44-1.34 (m, 2H), 1.09-1.06 (m, 6H). m/z (ESI-MS) 526.3 (C25H33F2N3O5S requires 526.21, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 6.1 min, purity 99%.
2-((S)-2-methyl-1-(3,4,5-trimethoxybenzamido)propyl)-N-((1R,4S)-4-(trifluoromethyl) cyclohexyl)thiazole-4-carboxamide (64)
Compound 64 was obtained from acid 6 (100 mg, 0.25 mmol) and 4-(trifluoromethyl)cyclohexan-1-amine (51 mg, 0.30 mmol) using method A as a white solid (20 mg, 15%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 8.94 (d, 1H, J = 8.3 Hz), 8.17 (s, 1H), 7.99 (d, 1H, J = 8.3 Hz), 7.21 (s, 2H), 5.15-5.10 (m, 1H), 3.83 (s, 6H), 3.70 (s, 3H), 2.50-2.40 (m, 1H), 2.23-2.20 (m, 1H), 1.91 (d, 4H, J = 10.9 Hz), 1.53-1.33 (m, 4H), 1.26-1.23 (m, 1H), 1.03 (d, 3H, J = 6.7 Hz), 0.91 (d, 3H, J = 6.7 Hz). m/z (ESI-MS) 544.3 (C25H32F3N3O5S requires 544.20, [M + H]+). HPLC tR (acetonitrile/water 60/40) = 4.1 min, purity 100%.
(S)-N-(4-Fluorophenyl)-2-(2-methyl-1-(3,4,5-trimethoxybenzamido)propyl)thiazole-4-carboxamide (65)
Compound 65 was obtained from acid 6 (100 mg, 0.25 mmol) and 4-fluoroaniline (34 mg, 0.30 mmol) using method A as a white solid (118 mg, 95%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 10.2 (s, 1H), 8.97 (d, 1H, J = 7.7 Hz), 8.38 (s, 1H), 7.85 (s, 2H), 7.24-7.19 (m, 4H), 5.25-5.21 (m, 1H), 3.85 (s, 6H), 3.71 (s, 3H), 2.51-2.48 (m, 1H), 1.06 (d, 3H, J = 5.8 Hz), 0.96 (d, 3H, J = 5.8 Hz). m/z (ESI-MS) 488.2 (C24H26FN3O5S requires 488.16, [M + H]+). HPLC tR (acetonitrile/water 60/40) = 3.7 min, purity 99%.
(S)-2-(2-Methyl-1-(3,4,5-trimethoxybenzamido)propyl)-N-(3,4,5-trifluorophenyl)thiazole-4-carboxamide (66)
Compound 66 was obtained from acid 6 (100 mg, 0.25 mmol) and 3,4,5-trifluoroaniline (45 mg, 0.30 mmol) using method A as a white solid (100 mg, 75%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 10.56 (s, 1H), 8.97 (d, 1H, J = 8.3 Hz), 8.44 (s, 1H), 7.88 (dd, 2H, J = 10.5 Hz, 6.7 Hz), 7.23 (s, 2H), 5.25-5.21 (m, 1H), 3.85 (s, 6H), 3.71 (s, 3H), 2.49-2.44 (m, 1H), 1.06 (d, 3H, J = 6.7 Hz), 0.95 (d, 3H, J = 6.7 Hz). m/z (ESI-MS) 524.2 (C24H24F3N3O5S requires 524.14, [M + H]+). HPLC tR (acetonitrile/water 60/40) = 6.6 min, purity 100%.
(S)-2-(2-Methyl-1-(3,4,5-trimethoxybenzamido)propyl)-N-(4,4,4-trifluorobutyl)thiazole-4-carboxamide (67)
Compound 67 was obtained from acid 6 (200 mg, 0.61 mmol) and 4,4,4-trifluorobutan-1-amine hydrochloride (100 mg, 0.46 mmol) using method A as a white solid (145 mg, 57%). 1H NMR (400 MHz; CDCl3; TMS) δ 8.04 (s, 1H), 7.37 (t, 1H, J = 5.7 Hz), 7.05 (s, 2H), 6.65 (d, 1H, J = 8.7 Hz), 5.39 (dd, 1H, J = 8.8 Hz, 6.7 Hz), 3.93 (s, 6H), 3.91 (s, 3H), 3.53 (q, 2H, J = 6.7 Hz), 2.49 (oct, 1H, J = 6.7 Hz), 2.27-2.15 (m, 2H), 1.91 (quin, 2H, J = 7.5 Hz), 1.08-1.04 (m, 6H). m/z (ESI-MS) 504.2 (C22H28F3N3O5S requires 504.17, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 4.8 min, purity 95%.
Tert-butyl (S)-(1-(4-((4,4-difluorocyclohexyl)carbamoyl)thiazol-2-yl)-2-methylpropyl)carbamate (68)
Compound 68 was obtained from acid 2 (500 mg, 1.66 mmol) and 4,4-difluorocyclohexan-1-amine (270 mg, 2.00 mmol) using method A as a white solid (464 mg, 67%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 8.17 (d, 1H, J = 8.6 Hz), 8.14 (s, 1H), 7.69 (d, 1H, J = 8.6 Hz), 4.69-4.66 (m, 1H), 3.99-3.94 (m, 1H), 2.29-2.20 (m, 1H), 2.04-2.01 (m, 2H), 1.91-1.70 (m, 6H), 1.40 (s, 9H), 0.89-0.86 (m, 6H).
(S)-N-(4,4-Difluorocyclohexyl)-2-(2-methyl-1-(thiazole-4-carboxamido)propyl)thiazole-4-carboxamide (69)
Compound 69 was obtained from thiazole-4-carboxylic acid (100 mg, 0.77 mmol) and amine 77 (295 mg, 0.93 mmol) using method A as a yellow solid (20 mg, 6%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 9.24-9.23 (m, 1H), 8.89 (d, 1H, J = 8.9 Hz), 8.40-8.39 (m, 1H), 8.23 (d, 1H, J = 8.4 Hz), 8.17 (s, 1H), 5.16-5.12 (m, 1H), 4.00-3.93 (m, 1H), 2.49-2.42 (m, 1H), 2.02-1.67 (m, 8H), 0.97 (d, 3H, J = 6.7 Hz), 0.87 (d, 3H, J = 6.7 Hz). m/z (ESI-MS) 429.2 (C18H22F2N4O2S2 requires 429.12, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 3.5 min, purity 97%.
(S)-N-(4,4-Difluorocyclohexyl)-2-(2-methyl-1-(pyrimidine-5-carboxamido)propyl) thiazole-4-carboxamide (70)
Compound 70 was obtained from pyrimidine-5-carboxylic acid (100 mg, 0.81 mmol) and amine 77 (307 mg, 0.97 mmol) using method A as a gray solid (10 mg, 3%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 9.39 (d, 1H, J = 8.1 Hz), 9.35 (s, 1H), 9.19 (s, 2H), 8.21-8.19 (m, 2H), 5.22-5.18 (m, 1H), 3.98-3.96 (m, 1H), 2.47-2.39 (m, 1H), 2.03-1.67 (m, 8H), 1.03 (d, 3H, J = 6.7 Hz), 0.93 (d, 3H, J = 6.7 Hz). m/z (ESI-MS) 424.2 (C19H23F2N5O2S requires 424.15, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 1.9 min, purity 98%.
(S)-N-(4,4-Difluorocyclohexyl)-2-(2-methyl-1-(quinoline-3-carboxamido)propyl)thiazole-4-carboxamide (71)
Compound 71 was obtained from quinolone-3-carboxylic acid (100 mg, 0.58 mmol) and amine 77 (220 mg, 0.69 mmol) using method A as a white solid (60 mg, 22%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 9.38 (d, 1H, J = 8.2 Hz), 9.28 (d, 1H, J = 2.1 Hz), 8.89 (s, 1H), 8.23-8.21 (m, 2H), 8.14 (d, 1H, J = 8.0 Hz), 8.11 (d, 1H, J = 8.3 Hz), 7.89 (t, 1H, J = 7.5 Hz), 7.72 (t, 1H, J = 7.5 Hz), 5.28-5.24 (m, 1H), 4.02-3.95 (m, 1H), 2.49-2.44 (m, 1H), 2.06-1.69 (m, 8H), 1.07 (d, 3H, J = 6.7 Hz), 0.96 (d, 3H, J = 6.7 Hz). m/z (ESI-MS) 473.3 (C24H26F2N4O2S requires 473.17, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 4.3 min, purity 97%.
(S)-N-(4,4-Difluorocyclohexyl)-2-(2-methyl-1-(tetrahydro-2H-pyran-4-carboxamido) propyl)thiazole-4-carboxamide (72)
Compound 72 was obtained from tetrahydro-2H-pyran-4-carboxylic acid (100 mg, 0.77 mmol) and amine 77 (293 mg, 0.92 mmol) using method A as a yellow solid (35 mg, 11%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 8.45 (d, 1H, J = 8.3 Hz), 8.17 (d, 1H, J = 8.2 Hz), 8.14 (s, 1H), 4.99 (dd, 1H, J = 8.8 Hz, 6.5 Hz), 3.97-3.86 (m, 3H), 3.31-3.28 (m, 1H), 2.64-2.56 (m, 1H), 2.33 (oct, 1H, J = 6.8 Hz), 2.03-1.55 (m, 13H), 0.89 (dd, 6H, J =6.9 Hz, 2.0 Hz). m/z (ESI-MS) 430.2 (C20H29F2N3O3S requires 430.19, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 2.2 min, purity 97%.
(S)-2-(1-(4,4-Difluorocyclohexane-1-carboxamido)-2-methylpropyl)-N-(4,4-difluorocyclohexyl)thiazole-4-carboxamide (73)
Compound 73 was obtained from 4,4-difluorocyclohexane carboxylic acid (100 mg, 0.61 mmol) and amine 77 (232 mg, 0.73 mmol) using method A as a white solid (100 mg, 30%). 1H NMR (400 MHz; CDCl3; TMS) δ 8.03 (s, 1H), 7.12 (d, 1H, J = 8.2 Hz), 6.02 (d, 1H, J = 8.8 Hz), 5.21 (dd, 1H, J = 8.8 Hz, 6.1 Hz), 4.13-4.06 (m, 1H), 2.42-2.30 (m, 2H), 2.26-2.10 (m, 6H), 2.01-1.65 (m, 10H), 0.98 (dd, 6H, J = 6.8 Hz, 5.3 Hz). m/z (ESI-MS) 464.2 (C21H29F4N3O2S requires 464.19, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 5.3 min, purity 95%.
(S)-N-(4,4-Difluorocyclohexyl)-2-(1-(3,4-dimethoxybenzamido)-2-methylpropyl)thiazole-4-carboxamide (74)
Compound 74 was obtained from 3,4-dimethoxybenzoyl chloride (76 mg, 0.38 mmol) and amine 77 (100 mg, 0.32 mmol) using method F as a white solid (16 mg, 11%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 8.84 (d, 1H, J = 8.3 Hz), 8.19-8.17 (m, 2H), 7.56 (d, 1H, J = 8.6 Hz), 7.45 (s, 1H), 7.04 (d, 1H, J = 8.6 Hz), 5.16-5.12 (m, 1H), 4.02-3.93 (m, 1H), 3.82 (s, 3H, 3.81 (s, 3H), 2.47-2.40 (m, 1H), 2.04-1.68 (m, 8H), 1.02 (d, 3H, J = 6.6 Hz), 0.91 (d, 3H, J = 6.6 Hz). m/z (ESI-MS) 482.2 (C23H29F2N3O4S requires 482.18, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 3.9 min, purity 99%.
(S)-N-(4,4-Difluorocyclohexyl)-2-(1-(4-methoxybenzamido)-2-methylpropyl)thiazole-4-carboxamide (75)
Compound 75 was obtained from 4-methoxybenzoyl chloride (0.05 mL, 0.38 mmol) and amine 77 (100 mg, 0.32 mmol) using method F as a white solid (30 mg, 21%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 8.84 (d, 1H, J = 8.3 Hz), 8.19-8.16 (m, 2H), 7.88 (d, 2H, J = 8.7 Hz), 7.02 (d, 2H, J = 8.7 Hz), 5.16-5.12 (m, 1H), 4.01-3.92 (m, 1H), 3.82 (s, 3H), 2.45-2.38 (m, 1H), 2.03-1.67 (m, 8H), 1.01 (d, 3H, J = 6.5 Hz), 0.91 (d, 3H, J = 6.5 Hz). m/z (ESI-MS) 452.2 (C22H27F2N3O3S requires 452.17, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 5.3 min, purity 98%.
(S)-2-(1-Benzamido-2-methylpropyl)-N-(4,4-difluorocyclohexyl)thiazole-4-carboxamide (76)
Compound 76 was obtained from benzoyl chloride (0.04 mL, 0.38 mmol) and amine 77 (100 mg, 0.32 mmol) using method F as a white solid (30 mg, 23%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 9.01 (d, 1H, J = 8.3 Hz), 8.20-8.18 (m, 2H), 7.88 (d, 2H, J = 7.3), 7.58-7.48 (m, 3H), 5.18-5.14 (m, 1H), 3.99-3.96 (m, 1H), 2.43 (oct, 1H, J = 6.8 Hz), 2.03-1.68 (m, 8H), 1.02 (d, 3H, J = 6.5 Hz), 0.92 (d, 3H, J = 6.5 Hz). m/z (ESI-MS) 422.2 (C21H25F2N3O2S requires 422.16, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 5.2 min, purity 98%.
(S)-2-(1-Amino-2-methylpropyl)-N-(4,4-difluorocyclohexyl)thiazole-4-carboxamide (77)
Compound 77 was obtained from N-Boc compound 68 (240 mg, 0.57 mmol) using method B (12 h) as a white solid (174 mg, 95%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 8.13 (d, 1H, J = 8.3 Hz), 8.09 (s, 1H), 4.00-3.92 (m, 2H), 2.26-2.15 (m, 3H), 2.02-1.66 (m, 8H), 0.94 (d, 3H, J = 6.9 Hz), 0.78 (d, 3H, J = 6.9 Hz). 13C NMR (100 MHz; DMSO-d6; TMS) δ 179.81, 160.72, 150.12, 123.63, 58.88, 46.05, 34.31, 32.39 (t, J = 24.0 Hz), 28.36 (d, J = 9.5 Hz), 19.80, 16.87. m/z (ESI-MS) 318.2 (C14H21F2N3OS requires 318.14, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 3.3 min, purity 99%.
(S)-N-(4,4-Difluorocyclohexyl)-2-(2-methyl-1-(3,4,5-trifluorobenzamido)propyl)thiazole-4-carboxamide (78)
Compound 78 was obtained from 3,4,5-trifluorobenzoyl chloride (0.05 mL, 0.38 mmol) and amine 77 (100 mg, 0.32 mmol) using method F as a white solid (14 mg, 9%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 9.17 (d, 1H, J = 8.3 Hz), 8.20-8.18 (m, 2H), 7.88 (t, 2H, J = 7.6 Hz), 5.16-5.12 (m, 1H), 4.02-3.94 (m, 1H), 2.45-2.39 (m, 1H), 2.04-1.67 (m, 8H), 1.02 (d, 3H, J = 6.7 Hz), 0.91 (d, 3H, J = 6.7 Hz). m/z (ESI-MS) 476.3 (C21H22F5N3O2S requires 476.14, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 12.1 min, purity 95%.
(S)-N-(4,4-Difluorocyclohexyl)-2-(2-methyl-1-(2-(3,4,5-trimethoxyphenyl)acetamido) propyl)thiazole-4-carboxamide (79)
Compound 79 was obtained from 2-(3,4,5-trimethoxyphenyl)acetic acid (100 mg, 0.44 mmol) and amine 77 (168 mg, 0.53 mmol) using method A as a white solid (100 mg, 43%). 1H NMR (400 MHz; CDCl3; TMS) δ 8.73 (d, 1H, J = 8.7 Hz), 8.19-8.16 (m, 2H), 6.59 (s, 2H), 4.99 (dd, 1H, J = 8.7 Hz, 6.5 Hz), 4.00-3.91 (m, 1H), 3.73 (s, 6H), 3.62 (s, 3H), 3.49 (s, 2H), 2.35 (oct, 1H, J = 6.7 Hz), 2.02-1.66 (m, 8H), 0.92 (d, 3H, J = 6.7 Hz), 0.87 (d, 3H, J = 6.7 Hz). m/z (ESI-MS) 526.3 (C25H33F2N3O5S requires 526.21, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 3.5 min, purity 97%.
(S)-N-(4,4-Difluorocyclohexyl)-2-(2-methyl-1-(2-oxo-2-(3,4,5-trimethoxyphenyl) acetamido)propyl)thiazole-4-carboxamide (80)
Compound 80 was obtained from 2-oxo-2-(3,4,5-trimethoxyphenyl)acetic acid (100 mg, 0.42 mmol) and amine 77 (159 mg, 0.50 mmol) using method A as a light orange solid (34 mg, 15%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 9.68 (d, 1H, J = 8.6 Hz), 8.22 (s, 1H), 8.20 (d, 1H, J = 8.5 Hz), 7.16 (s, 2H), 5.19 (dd, 1H, J = 8.8 Hz, 7.1 Hz), 4.01-3.92 (m, 1H), 3.79-3.78 (s, 9H), 2.44 (oct, 1H, J = 6.7 Hz), 2.03-1.65 (m, 8H), 0.96 (d, 6H, J = 6.6 Hz). m/z (ESI-MS) 540.2 (C25H31F2N3O6S requires 540.19, [M + H]+). HPLC tR (acetonitrile/water 60/40) = 3.4 min, purity 100%.
(S)-N-Cyclohexyl-2-(2-methyl-1-(3,4,5-trimethoxyphenylthioamido)propyl)thiazole-4-carbothioamide (81) and (S)-N-(1-(4-(cyclohexylcarbamothioyl)thiazol-2-yl)-2-methyl propyl)-3,4,5-trimethoxybenzamide (82)
Compounds 81 and 82 were obtained from carboxamide 39 (200 mg, 0.42 mmol) using method D (except using 1.1 eq of Lawesson’s reagent and preparative TLC; n-hexane/ethyl acetate – 50/50; for purification) as yellow solids.
Dithioamide 81: Yield - 20 mg, 9%; 1H NMR (400 MHz; DMSO-d6; TMS) δ 10.6 (d, 1H, J = 8.1 Hz), 9.74 (d, 1H, J = 8.3 Hz), 8.34 (s, 1H), 7.05 (s, 2H), 5.88-5.83 (m, 1H), 4.48-4.40 (m, 1H), 3.84 (s, 6H), 3.71 (s, 3H), 2.62 (oct, 1H, J = 6.8 Hz), 1.94 (t, 2H, J = 13.2 Hz), 1.75 (d, 2H, J = 12.2 Hz), 1.63 (d, 1H, J = 12.2 Hz), 1.49 (q, 2H, J = 11.5 Hz), 1.33 (q, 2H, J = 11.5 Hz), 1.20 (t, 1H, J = 12.2 Hz), 1.09 (d, 3H, J = 6.7 Hz), 0.97 (d, 3H, J = 6.7 Hz). m/z (ESI-MS) 508.2 (C24H33N3O3S3 requires 508.17, [M + H]+). HPLC tR (acetonitrile/water 70/30) = 9.4 min, purity 99%.
Monothioamide 82: Yield - 8 mg, 4%; 1H NMR (400 MHz; DMSO-d6; TMS) δ 9.79 (d, 1H, J = 8.3 Hz), 8.92 (d, 1H, J = 8.3), 8.32 (s, 1H), 7.21 (s, 2H), 5.17-5.13 (m, 1H), 4.48-4.39 (m, 1H), 3.84 (s, 6H), 3.71 (s, 3H), 2.45 (oct, 1H, J = 6.8 Hz), 1.93 (t, 2H, J = 11.2 Hz), 1.75 (d, 2H, J = 12.3 Hz), 1.63 (d, 1H, J = 12.3 Hz), 1.56-1.46 (m, 2H), 1.33 (q, 2H, J = 12.3 Hz), 1.20 (t, 1H, J = 12.3 Hz), 1.04 (d, 3H, J = 6.7 Hz, 3.7 Hz), 0.94 (d, 3H, J = 6.7 Hz, 3.7 Hz). m/z (ESI-MS) 492.2 (C24H33N3O4S2 requires 492.19, [M + H]+). HPLC tR (acetonitrile/water 70/30) = 4.7 min, purity 99%.
(S)-N-(4,4-Difluorocyclohexyl)-2-(2-methyl-1-(3,4,5-trimethoxyphenylthioamido)propyl) thiazole-4-carbothioamide (83) and (S)-N-(1-(4-((4,4-difluorocyclohexyl)carbamothioyl) thiazol-2-yl)-2-methylpropyl)-3,4,5-trimethoxybenzamide (84)
Compounds 83 and 84 were obtained from carboxamide 53 (100 mg, 0.20 mmol) using method D (except using 1.1 eq of Lawesson’s reagent and preparative TLC; n-hexane/ethyl acetate - 50/50; for purification) as yellow solids,
Dithioamide 83: Yield - 3 mg, 3%; 1H NMR (400 MHz; DMSO-d6; TMS) δ 10.65 (d, 1H, J = 8.3 Hz), 9.96 (d, 1H, J = 8.3 Hz), 8.35 (s, 1H), 7.05 (s, 2H), 5.89-5.84 (m, 1H), 4.66-4.59 (m, 1H), 3.84 (s, 6H), 3.71 (s, 3H), 2.67-2.59 (m, 1H), 2.12-1.81 (m, 8H), 1.09 (d, 3H, J = 6.7 Hz), 0.98 (d, 3H, J = 6.7 Hz). m/z (ESI-MS) 544.2 (C24H31F2N3O3S3 requires 544.15, [M + H]+). HPLC tR (acetonitrile/water 70/30) = 5.9 min, purity 99%.
Monothioamide 84: Yield - 3 mg, 3%; 1H NMR (400 MHz; DMSO-d6; TMS) δ 10.02 (d, 1H, J = 8.3 Hz), 8.93 (d, 1H, J = 8.3 Hz), 8.34 (s, 1H), 7.21 (s, 2H), 5.18-5.14 (m, 1H), 4.67-4.60 (m, 1H), 3.84 (s, 6H), 3.71 (s, 3H), 2.47-2.43 (m, 1H), 2.10-1.82 (m, 8H), 1.04 (d, 3H, J = 6.7 Hz), 0.94 (d, 3H, J = 6.7 Hz). m/z (ESI-MS) 528.2 (C24H31F2N3O4S2 requires 528.17, [M + H]+). HPLC tR (acetonitrile/water 60/40) = 6.9 min, purity 97%.
Tert-butyl (2-amino-2-oxoethyl)carbamate (85)
Compound 85 was obtained from N-Boc glycine (5 g, 28.54 mmol) using method C as a light yellow solid (4.62 g, 93%). 1H NMR (400 MHz; CDCl3; TMS) δ 6.64-6.50 (m, 2H), 5.70-5.65 (m, 1H), 3.80-3.76 (m, 2H), 1.40 (s, 9H).
Tert-butyl (S)-(1-amino-4-methyl-1-oxopentan-2-yl)carbamate (86)
Compound 86 was obtained from N-Boc (S)-leucine (7 g, 30.27 mmol) using method C as a white solid (6.94 g, 99%). 1H NMR (400 MHz; CDCl3; TMS) δ 6.47 (s, 1H), 5.91 (s, 1H), 5.09 (s, 1H), 4.18 (s, 1H), 1.74-1.60 (m, 2H), 1.54-1.49 (m, 1H), 1.44 (s, 9H), 0.96-0.93 (m, 6H).
Tert-butyl (S)-(1-amino-1-oxo-3-phenylpropan-2-yl)carbamate (87)
Compound 87 was obtained from N-Boc (S)-phenylalanine (10 g, 37.69 mmol) using method C as a white solid (9.74 g, 98%). 1H NMR (400 MHz; CDCl3; TMS) δ 7.32-7.28 (m, 2H), 7.26-7.22 (m, 3H), 6.24-6.14 (m, 1H), 5.95-5.84 (m, 1H), 5.27-5.22 (m, 1H), 4.44-4.40 (m, 1H), 3.06 (s, 2H), 1.40 (s, 9H).
Tert-butyl (S)-(1-amino-3-(4-hydroxyphenyl)-1-oxopropan-2-yl)carbamate (88)
Compound 88 was obtained from N-Boc (S)-tyrosine (10 g, 35.54 mmol) using method C as a white solid (9.45 g, 95%). 1H NMR (400 MHz; CDCl3; TMS) δ 8.00 (s, 1H), 7.01 (d, 2H, J = 8.0 Hz), 6.74 (d, 2H, J = 8.0 Hz), 6.30 (s, 1H), 6.02 (s, 1H), 5.38 (d, 1H, J = 8.0 Hz), 4.36-4.30 (m, 1H), 2.88 (s, 2H), 1.39 (s, 9H).
(S)-(9H-Fluoren-9-yl)methyl 2-carbamoylpyrrolidine-1-carboxylate (89)
Compound 89 was obtained from Fmoc (S)-proline (10 g, 29.64 mmol) using method C as a white solid (9.21 g, 92%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 7.89 (t, 2H, J = 6.8 Hz), 7.68 (t, 2H, J = 6.8 Hz), 7.44-7.33 (m, 4H), 7.14-6.97 (m, 2H), 4.31-4.07 (m, 4H), 3.51-3.45 (m, 1H), 3.35-3.31 (m, 1H), 2.26-1.79 (m, 4H).
Tert-butyl (2-amino-2-thioxoethyl)carbamate (90)
Compound 90 was obtained from carboxamide 85 (4.5 g, 25.83 mmol) using method D as a colorless oil (3.44 g, 70%). 1H NMR (400 MHz; CDCl3; TMS) δ 8.06 (s, 2H), 5.54 (s, 1H), 3.91-3.82 (m, 2H), 1.45 (s, 9H).
Tert-butyl (S)-(1-amino-4-methyl-1-thioxopentan-2-yl)carbamate (91)
Compound 91 was obtained from carboxamide 86 (6 g, 26.05 mmol) using method D as a yellow oil (5.25 g, 82%). 1H NMR (400 MHz; CDCl3; TMS) δ 8.39 (s, 1H), 7.81 (s, 1H), 5.25 (s, 1H), 4.55-4.52 (m, 1H), 1.76-1.63 (m, 3H), 1.44 (s, 9H), 0.97-0.94 (m, 6H).
Tert-butyl (S)-(1-amino-3-phenyl-1-thioxopropan-2-yl)carbamate (92)
Compound 92 was obtained from carboxamide 87 (9 g, 34.05 mmol) using method D as a yellow oil (7.44 g, 78%). 1H NMR (400 MHz; CDCl3; TMS) δ 8.23 (s, 1H), 8.05 (s, 1H), 7.25-7.19 (m, 5H), 5.61 (s, 1H), 4.77-4.75 (m, 1H), 3.11-2.99 (m, 2H), 1.33 (s, 9H).
Tert-butyl (S)-(1-amino-3-(4-hydroxyphenyl)-1-thioxopropan-2-yl)carbamate (93)
Compound 93 was obtained from carboxamide 88 (9 g, 32.10 mmol) using method D as a yellow oil (8.17 g, 86%). 1H NMR (400 MHz; CDCl3; TMS) δ 7.83 (s, 1H), 7.75 (s, 1H), 7.04 (d, 2H, J = 8.0 Hz), 6.72 (d, 2H, J = 8.0 Hz), 5.59 (s, 1H), 4.58 (q, 1H, J = 7.2 Hz), 2.99 (s, 2H), 1.40 (s, 9H).
(S)-(9H-Fluoren-9-yl)methyl-2-carbamothioylpyrrolidine-1-carboxylate (94)
Compound 94 was obtained from carboxamide 89 (9 g, 26.75 mmol) using method D as a yellow oil (6.78 g, 72%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 9.78-9.60 (m, 1H), 9.33-9.18 (m, 1H), 7.90 (t, 2H, J = 7.6 Hz), 7.73-7.68 (m, 2H), 7.44-7.40 (m, 2H), 7.37-7.31 (m, 2H), 4.73-4.47 (m, 1H), 4.27-4.15 (m, 3H), 3.62-3.55 (m, 1H), 3.43-3.39 (m, 1H), 2.36-2.15 (m, 1H),1.97-1.80 (m, 3H).
Ethyl 2-(((tert-butoxycarbonyl)amino)methyl)thiazole-4-carboxylate (95)
Compound 95 was obtained from thioamide 90 (3 g, 15.77 mmol) using method E as a dark red oil (1.89 g, 42%). 1H NMR (400 MHz; CDCl3; TMS) δ 8.12 (s, 1H), 5.61 (s, 1H), 4.65 (d, 2H, J = 6.2 Hz), 4.41 (q, 2H, J = 6.9 Hz), 1.46 (s, 9H), 1.40 (t, 3H, J = 7.1 Hz).
Ethyl-2-(1-((tert-butoxycarbonyl)amino)-3-methylbutyl)thiazole-4-carboxylate (96)
Compound 96 was obtained from thioamide 91 (5 g, 20.29 mmol) using method E as a light brown solid (3.96 g, 57%). [α]D25= −11.84° (c 0.1, CHCl3) (literature, [α]D25 = −44.0°);40 1H NMR (400 MHz; CDCl3; TMS) δ 8.08 (s, 1H), 5.17-5.13 (m, 1H), 4.44-4.38 (m, 3H), 1.96-1.68 (m, 3H), 1.44 (s, 9H), 1.32-1.28 (m, 3H), 0.98-0.95 (m, 6H).
Ethyl-2-(1-((tert-butoxycarbonyl)amino)-2-phenylethyl)thiazole-4-carboxylate (97)
Compound 97 was obtained from thioamide 92 (7.2 g, 25.68 mmol) using method E as a yellow solid (5.99 g, 62%). [α]D25= −1.34° (c 0.1, CHCl3) (literature, [α]D25= −20.1°);40 1H NMR (400 MHz; CDCl3; TMS) δ 8.05 (s, 1H), 7.28-7.21 (m, 3H), 7.12 (d, 2H, J = 6.9 Hz), 5.47-5.46 (m, 1H), 5.31-5.28 (m, 1H), 4.45-4.40 (m, 2H), 3.35-3.39 (m, 2H), 1.39 (s, 9H), 1.27-1.23 (m, 3H).
Ethyl-2-(1-((tert-butoxycarbonyl)amino)-2-(4-hydroxyphenyl)ethyl)thiazole-4-carboxylate (98)
Compound 98 was obtained from thioamide 93 (7.5 g, 25.30 mmol) using method E as a yellow solid (5.36 g, 54%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 8.42 (s, 1H), 7.79 (d, 1H, J = 8.3 Hz), 7.07 (d, 2H, J = 8.0 Hz), 6.66 (d, 2H, J = 8.0 Hz), 4.90-4.85 (m, 1H), 4.34-4.28 (m, 3H), 3.18-3.14 (m, 2H), 1.32 (s, 9H), 1.23-1.20 (m, 3H).
Ethyl-2-(1-(((9H-fluoren-9-yl)methoxy)carbonyl)pyrrolidin-2-yl)thiazole-4-carboxylate (99)
Compound 99 was obtained from thioamide 94 (6.5 g, 18.44 mmol) using method E as a reddish brown oil (3.89 g, 47%). 1H NMR (400 MHz; DMSO-d6; TMS) δ 8.37 (d, 1H, J = 8.0 Hz), 7.88 (d, 1H, J = 7.4 Hz), 7.76 (d, 1H, J = 6.4 Hz), 7.66 (d, 1H, J = 6.4 Hz), 7.40-7.11 (m, 5H), 5.12 (s, 1H), 4.40-4.27 (m, 5H), 3.52-3.40 (m, 2H), 2.34-2.26 (m, 1H), 2.02-1.82 (m, 3H), 1.33-1.26 (m, 3H).
Ethyl 2-(3,4,5-trimethoxybenzamido)thiazole-4-carboxylate (100)
Compound 100 was obtained from 3,4,5-trimethoxybenzoyl chloride (1.61 g, 6.97 mmol) and ethyl 2-amino thiazole-4-carboxylate (1 g, 5.81 mmol) using method F as a white solid (1.48 g, 70%). 1H NMR (400 MHz; CDCl3; TMS) δ 11.43 (s, 1H), 7.78 (s, 1H), 7.14 (s, 2H), 4.08 (q, 2H, J = 7.0 Hz), 3.80 (s, 3H), 3.72 (s, 6H), 1.18-1.16 (m, 3H).
N-(4,4-Difluorocyclohexyl)-2-(3,4,5-trimethoxybenzamido)thiazole-4-carboxamide (101)
Ester 100 (120 mg, 0.33 mmol) was hydrolyzed using method G providing corresponding acid, which was reacted further with 4,4-difluorocyclohexylamine (54 mg, 0.40 mmol) using method A to obtain compound 101 (80 mg, 54% over two steps) as a white solid. 1H NMR (400 MHz; DMSO-d6; TMS) δ 12.68 (bs, 1H), 7.85 (s, 1H), 7.82 (d, 1H, J = 8.0 Hz), 7.48 (s, 2H), 3.98-3.91 (m, 1H), 3.87 (s, 6H), 3.74 (s, 3H), 2.05-1.89 (m, 6H), 1.65-1.55 (m, 2H). m/z (ESI-MS) 456.2 (C20H23F2N3O5S requires 456.13, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 3.7 min, purity 98%.
Ethyl 2-((3,4,5-trimethoxybenzamido)methyl)thiazole-4-carboxylate (102)
Compound 95 (1 g, 3.49 mmol) was hydrolyzed using method B to obtain free amine as an oil, which was reacted with 3,4,5-trimethoxybenzoyl chloride (0.97 g, 4.19 mmol) using method F to furnish compound 102 as a brown solid (0.62 g, 47% over two steps). 1H NMR (400 MHz; CDCl3; TMS) δ 8.41 (t, 1H, J = 5.8 Hz), 8.03 (s, 1H), 7.08 (s, 2H), 4.79 (d, 2H, J = 5.9 Hz), 4.27 (q, 2H, J = 7.1 Hz), 3.76 (s, 3H), 3.71 (s, 6H), 1.26 (t, 3H, J = 7.0 Hz).
Ethyl-2-(3-methyl-1-(3,4,5-trimethoxybenzamido)butyl)thiazole-4-carboxylate (103)
Compound 96 (1 g, 2.92 mmol) was hydrolyzed using method B to obtain free amine as an oil, which was reacted with 3,4,5-trimethoxybenzoyl chloride (0.79 g, 3.40 mmol) using method F to give compound 103 as a light yellow solid (0.86 g, 68% over two steps). 1H NMR (400 MHz; CDCl3; TMS) δ 8.09 (s, 1H), 7.04 (s, 2H), 6.87 (d, 1H, J = 8.5 Hz), 5.62 (q, 1H, J = 7.9 Hz), 4.41 (q, 2H, J = 7.1 Hz), 3.91 (s, 6H), 3.87 (s, 3H), 2.03-2.00 (m, 2H), 1.75-1.65 (m, 1H), 1.39 (t, 3H, J = 6.9 Hz), 1.00 (dd, 6H, J = 6.7 Hz, 2.1 Hz).
Ethyl-2-(2-phenyl-1-(3,4,5-trimethoxybenzamido)ethyl)thiazole-4-carboxylate (104)
Compound 97 (1.5 g, 3.98 mmol) was hydrolyzed using method B to obtain free amine as an oil, which was reacted with 3,4,5-trimethoxybenzoyl chloride (1.10 g, 4.78 mmol) using method F to yield compound 104 as a light yellow solid (1.53 g, 82% over two steps). 1H NMR (400 MHz; CDCl3; TMS) δ 8.05 (s, 1H), 7.29-7.21 (m, 3H), 7.16 (d, 2H, J = 7.1 Hz), 7.03-7.00 (m, 1H), 6.93 (s, 2H), 5.77 (q, 1H, J = 7.3 Hz), 4.42 (q, 2H, 7.1 Hz), 3.92 (s, 3H), 3.87 (m, 6H), 3.52-3.40 (m, 2H), 1.41 (t, 3H, J = 7.1 Hz). 13C NMR (100 MHz; CDCl3; TMS) δ 171.29, 166.62, 161.17, 153.19, 146.98, 141.17, 136.24, 129.41, 128.98, 128.75, 127.49, 127.21, 104.42, 61.59, 60.92, 56.29, 52.73, 41.26, 14.37.
Ethyl-2-(2-(4-hydroxyphenyl)-1-(3,4,5-trimethoxybenzamido)ethyl)thiazole-4-carboxylate (105)
Compound 98 (1.5 g, 3.82 mmol) was hydrolyzed using method B to obtain free amine as an oil, which was reacted with 3,4,5-trimethoxybenzoyl chloride (1.06 g, 4.59 mmol) using method F to obtain compound 105 as a white solid (1.37 g, 74% over two steps). 1H NMR (400 MHz; DMSO-d6; TMS) δ 9.22 (s, 1H), 9.13 (d, 1H, J = 8.1 Hz), 8.45 (s, 1H), 7.50 (d, 1H, J = 7.8 Hz), 7.20-7.12 (m, 4H), 6.65 (d, 1H, J = 8.0 Hz), 5.63-5.42 (m, 1H), 4.35-4.29 (m, 2H), 3.85 (m, 2H), 3.82 (s, 6H), 3.69 (s, 3H), 1.34-1.29 (m, 3H).
Ethyl-2-(1-(3,4,5-trimethoxybenzoyl)pyrrolidin-2-yl)thiazole-4-carboxylate (106)
Compound 99 (1.5 g, 3.34 mmol) was stirred with a solution of 5% piperidine in DMF (10 mL) for 2 h. It was further diluted with ethyl acetate and partitioned with 2N HCl solution. Aqueous layer was collected and basified with aqueous saturated solution of Na2CO3. It was then extracted 2× with ethyl acetate. The combined organic layer was dried (anhydrous MgSO4) and evaporated to obtain free amine as an oil, which was reacted further with 3,4,5-trimethoxybenzoyl chloride (1.00 g, 4.34 mmol) using method F to prepare compound 106 as a white solid (0.75 g, 53% over two steps). 1H NMR (400 MHz; DMSO-d6; TMS) δ 8.45 (s, 1H), 6,83 (s, 2H), 5.47-5.43 (m, 1H), 4.32-4.27 (m, 2H), 3.82 (s, 6H), 3.70 (s, 3H), 3.63-3.58 (m, 2H), 2.44-2.37 (m, 1H), 2.17-2.11 (m, 1H), 1.98-1.89 (m, 2H), 1.32-1.28 (m, 3H).
N-(4,4-Difluorocyclohexyl)-2-((3,4,5-trimethoxybenzamido)methyl)thiazole-4-carboxamide (107)
Ester 102 (130 mg, 0.34 mmol) was hydrolyzed using method G providing acid, which was further reacted with 4,4-difluorocyclohexan-1-amine (56 mg, 0.41 mmol) using method A to obtain compound 107 (11 mg, 7% over two steps) as a cream colored solid. 1H NMR (400 MHz; DMSO-d6; TMS) δ 9.43 (t, 1H, J = 5.9 Hz), 8.30 (d, 1H, J = 8.3 Hz), 8.17 (s, 1H), 7.25 (s, 2H), 4.77 (d, 2H, J = 5.8 Hz), 4.01-3.92 (m, 1H), 3.83 (s, 6H), 3.71 (s, 3H), 2.04-1.69 (m, 8H). m/z (ESI-MS) 470.2 (C21H25F2N3O5S requires 470.15, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 2.2 min, purity 95%.
N-(4,4-Difluorocyclohexyl)-2-(3-methyl-1-(3,4,5-trimethoxybenzamido)butyl)thiazole-4-carboxamide (108)
Ester 103 (123 mg, 0.28 mmol) was hydrolyzed using method G providing acid, which was further reacted with 4,4-difluorocyclohexan-1-amine (46 mg, 0.34 mmol) using method A to furnish compound 108 (115 mg, 78% over two steps) as a white solid. 1H NMR (400 MHz; DMSO-d6; TMS) δ 9.07 (d, 1H, J = 8.2 Hz), 8.21 (d, 1H, J = 8.3 Hz), 8.15 (s, 1H), 7.27 (s, 2H), 5.48-5.42 (m, 1H), 4.02-3.94 (m, 1H), 3.84 (s, 6H), 3.71 (s, 3H), 2.04-1.70 (m, 11H), 0.98 (d, 3H, J = 6.5 Hz), 0.94 (d, 3H, J = 6.5 Hz). m/z (ESI-MS) 526.3 (C25H33F2N3O5S requires 526.21, [M + H]+). HPLC tR (acetonitrile/water 60/40) = 3.6 min, purity 97%.
N-(4,4-Difluorocyclohexyl)-2-(2-phenyl-1-(3,4,5-trimethoxybenzamido)ethyl)thiazole-4-carboxamide (109)
Ester 104 (125 mg, 0.27 mmol) was hydrolyzed using method G providing acid, which was further reacted with 4,4-difluorocyclohexan-1-amine (43 mg, 0.32 mmol) using method A to provide compound 109 (100 mg, 67% over two steps) as a white solid. mp 188-191 °C. 1H NMR (400 MHz; DMSO-d6; TMS) δ 9.15 (d, 1H, J = 8.3 Hz), 8.23 (d, 1H, J = 8.4 Hz), 8.19 (s, 1H), 7.40 (d, 2H, J = 7.5 Hz), 7.30 (t, 2H, J = 7.5 Hz), 7.20 (t, 1H, J = 7.3 Hz), 7.11 (s, 2H), 5.60-5.54 (m, 1H), 4.05-3.96 (m, 1H), 3.81 (s, 6H), 3.69 (s, 3H), 3.61 (dd, 1H, J = 13.8 Hz, 4.0 Hz), 3.29 (dd, 1H, J = 13.8 Hz, 11.0 Hz), 2.06-1.72 (m, 8H). 13C NMR (100 MHz; CDCl3; TMS) δ 172.08, 166.79, 159.98, 153.31, 149.29, 141.43, 135.81, 129.38, 128.87, 127.49, 123.73, 104.48, 60.95, 56.35, 52.42, 46.28, 41.08, 32.18 (t, J = 24.7 Hz), 28.63 (d, J = 6.4 Hz). m/z (ESI-MS) 560.3 (C28H31F2N3O5S requires 560.20, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 7.3 min, purity 95%. Anal. Calcd for C28H31F2N3O5S: C, 60.09; H, 5.58; N, 7.51. Found: C, 59.89; H, 5.58; N, 7.51.
N-(4,4-Difluorocyclohexyl)-2-(2-(4-hydroxyphenyl)-1-(3,4,5-trimethoxybenzamido)ethyl) thiazole-4-carboxamide (110)
Ester 105 (120 mg, 0.25 mmol) was hydrolyzed using method G providing acid, which was reacted further with 4,4-difluorocyclohexan-1-amine (40 mg, 0.30 mmol) using method A to obtain compound 110 (50 mg, 35% over two steps) as a white solid. 1H NMR (400 MHz; DMSO-d6; TMS) δ 9.23 (s, 1H), 9.07 (d, 1H, J = 8.3 Hz), 8.23 (d, 1H, J = 8.3 Hz), 8.17 (s, 1H), 7.17 (d, 2H, J = 8.4 Hz), 7.11 (s, 2H), 6.66 (d, 2H, J = 8.4 Hz), 5.49-5.44 (m, 1H), 4.04-3.95 (m, 1H), 3.82 (s, 6H), 3.69 (s, 3H), 3.47 (dd, 1H, J = 13,9 Hz, 4.3 Hz), 3.15 (dd, 1H, J = 13.9 Hz, 10.8 Hz), 2.06-1.69 (m, 8H). m/z (ESI-MS) 576.2 (C28H31F2N3O6S requires 576.19, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 2.8 min, purity 97%.
N-(4,4-Difluorocyclohexyl)-2-(1-(3,4,5-trimethoxybenzoyl)pyrrolidin-2-yl)thiazole-4-carboxamide (111)
Ester 106 (120 mg, 0.29 mmol) was hydrolyzed using method G providing acid, which was further reacted with 4,4-difluorocyclohexan-1-amine (47 mg, 0.35 mmol) using method A to furnish compound 111 (56 mg, 39% over two steps) as a white solid. 1H NMR (400 MHz; DMSO-d6; TMS) δ 8.27 (d, 1H, J = 8.0 Hz), 8.16 (s, 1H), 6.82 (s, 2H), 5.48-5.44 (m, 1H), 4.01-3.94 (m, 1H), 3.82 (s, 6H), 3.70 (s, 3H), 3.62-3.55 (m, 2H), 2.45-2.36 (m, 1H), 2.22-2.16 (m, 1H), 2.03-1.69 (m, 10H). m/z (ESI-MS) 510.2 (C24H29F2N3O5S requires 510.18, [M + H]+). HPLC tR (acetonitrile/water 50/50) = 3.0 min, purity 99%.
Bioassay Procedures
Chemicals
Dulbecco’s Modified Eagle’s Medium (DMEM), fetal bovine serum (FBS), penicillin/streptomycin, and Trypsin 0.25% EDTA were products of Hyclone (GE Healthcare Bio-Sciences, Pittsburgh, PA), Thermo Fisher Scientific (Waltham, MA). Phosphate buffered saline (PBS) 20× concentrate (pH 7.5) was purchased from AMRESCO (Solon, OH). Paclitaxel, verapamil, zosuquidar, G418, 3-(4,5-dimethylthiazol-yl)-2,5-diphenyltetrazolium bromide (MTT), dimethyl sulfoxide (DMSO), and other chemicals were obtained from Sigma Chemical Co. (St. Louis, MO). The lyophilized compounds were solubilized in DMSO at 10 mM stock concentrations and kept at −20°C until experimentation. Those stocks were further diluted in media on the same day, prior to experimentation to the respective working concentrations for reversal assay. Microplates were read and analyzed with Multiskan™ GO Microplate Spectrophotometer using SkanIt™ Software Research Addition purchased from Thermo Scientific (Waltham, MA). Vivid CYP3A4 green screening kit was purchased from Thermo Fisher Scientific (Waltham, MA).
Cell Lines and Cell Culture
HEK 293-pcDNA 3.1 and HEK-ABCB1 cells were generated by transfecting the HEK293 cells respectively with the control pcDNA3.1 vector and ABCB1 expression vector. All transfected cells were cultured in DMEM medium with 2 mg/mL of G418. These cell lines were maintained in T-75 culture flasks, using DMEM media, supplemented with 10% Fetal Bovine Serum and 1% penicillin/streptomycin and incubated in 5% CO2 at 37°C. The cells were frequently examined under a microscope and only used for assay when both cell lines were growing well. The cells were washed with PBS and grown as adherent monolayers, maintained on drug free media at least 14 days prior to experimentation. Cells were checked on under the microscope throughout the assay to ensure even plate seeding and growth. The untreated row of cells in each 96-well plate confirmed the growth over the course of the experiment.
ATPase Activity of P-gp
High Five insect cell membrane vesicles prepared from P-gp expressing insect cells were used to measure the vanadate-sensitive ATPase activity of P-gp in the presence of indicated concentrations of synthesized compounds. All compounds were dissolved in dimethyl sulfoxide (DMSO) and 100× concentrated working stocks of compounds were prepared. In the ATPase assay, the final concentration of DMSO was kept at 1% (v/v). The basal ATPase activity was measured in the absence of synthesized compounds but, in the presence of same percentage volume of DMSO solvent. The drug-modulated activity was measured in the presence of indicated compounds. Total membrane protein (10 μg/100 μL), was pre-incubated with the synthesized compound or DMSO control at 37 °C for 5 min in a 50 mM MES-Tris buffer (pH 6.8) containing 50 mM KCl, 10 mM MgCl2, 5 mM NaN3, 1 mM EGTA, 1 mM ouabain, and 2 mM DTT in the presence and absence of sodium orthovanadate (0.3 mM). The reaction was initiated by the addition of 5 mM ATP and terminated with addition of 0.1 mL of 5% (w/v) sodium dodecyl sulfate solution (final concentration, 2.5%); the extent of inorganic phosphate liberated over 20 minutes at 37 °C was measured using a colorimetric method as described previously.53 The results reported as mean ± SD are obtained from three independent experiments performed in duplicates. For some compounds, mean values from two independent experiments in triplicates are reported.
[125I]-Iodoarylazidoprazosin (IAAP) photoaffinity Labeling of human P-gp
Increasing concentrations of compound 53 (0–5 μM) or compound 109 (0–10 μM) were incubated with P-gp expressing High Five insect cell membrane vesicles (65–75 μg protein/100 μL) in 50 mM MES-Tris buffer (pH 6.8) and 150 mM sodium chloride at 37 °C for 10 min and then the samples were displaced to 4 °C bath. At that time, 4–5 nM [125I]-IAAP (2200 Ci/mmol; PerkinElmer Lifesciences Corp.) was added under subdued light. Photo-cross-linking with [125I]-IAAP was performed by exposing the samples to 366 nm UV light for 10 min and the incorporation of IAAP in P-gp after SDS-PAGE was quantified as described previously.54 [125I]-IAAP incorporation into P-gp band in the absence of compound (only DMSO) was taken as 100%.
MDR Reversal (Cell Killing) Cytotoxicity Assay55
The chemo-sensitivity was determined using cell viability colorimetric assay, which was designed and performed using 96-well plates in triplicate. Parental HEK 293-pcDNA 3.1 cells and P-gp overexpressing (ABCB1 transfected) cells evenly seeded at 5000 cells per well in 160 μL of culture medium were incubated under 5% CO2 at 37°C. After 24 h incubation, the cells were dosed with designated treatments. Combination treatments received 20 μL of the compounds at a non-toxic concentration of 10 μM and incubated for 1 hour prior to the chemotherapy. Verapamil and zosuquidar were used as positive control reversal agents at 10 μM and 0.25 μM, respectively. ABCB1 substrate chemotherapy drug paclitaxel was serially diluted and dosed to respective cell lines. HEK 293-pcDNA 3.1 and HEK-ABCB1 received paclitaxel to up to 10 μM. The cells under treatment were then incubated under 5% CO2 at 37°C for 72 h. Following the 3 days, each well received 20 μL of MTT solution (4 mg/mL) followed by incubation of the plates for 4 h at 37°C to ensure the complete formation of the formazan crystallization. After aspirating the culture media MTT solution, the resulting formazan was dissolved in 100 μL of dimethyl sulfoxide. Finally, plates were shaken for 10 min and absorbances were measured at 570 nm, using a microplate reader, and analyzed.
Cytochrome P4503A4 inhibition assay
The CYP3A4 inhibition assay was conducted by following the manufacturer’s guidelines (vivid® CYP450 green screening kit; Thermofisher Scientific, Waltham, MA) and as reported in Wang and colleagues.20
Molecular descriptors calculation
The molecular descriptors were calculated using the calculate property functionality in Maestro v10.1 (AlogP, HBA, HBD, Polar SA and Number of rotatable bonds; Schrödinger, LLC, New York, NY, 2015), Qikprop (Mol. Wt. and No. of N and O; QikProp, Schrödinger, LLC, New York, NY, 2015), ACD/Chemsketch (Molar refractivity and Molar volume; ACD/Labs 2017.1.2, Advanced Chemistry Development Inc. Toronto, Ontario, Canada) and Open Notebook Science Challenge (A and B; AbrahamModel001; http://showme.physics.drexel.edu/onsc/models/AbrahamDescriptorsModel001.php).
In silico PAINS analysis
A program KNIME (v3.4.1, KNIME GmbH, Konstanz, Germany) was used to run the workflow (http://myexperiment.org/workflows/1841) for PAINS analysis.56, 57 Target compounds were manually input into the workflow in the form of SMILES. The output file for the run listed the predicted PAINS.
Molecular Modeling
All docking experiments were performed on Mac Pro 6-core Intel Xenon X5 processor with Macintosh Operating System (OS X El Capitan) using Schrodinger 2015-1 (Schrödinger, LLC, New York, NY, 2015) software. Ligand preparations of the thiazole compounds, homology modeling (except using template as PDB: 4Q9H),51 and protein preparation of the homology model were essentially performed following our reported protocols.19 Homology model validation was performed based on Ramachandran plot and root-mean-square deviation (RMSD) of Cα atoms of the residues of the generated model and experimental structure. The Ramachandran plot analysis revealed >94% residues in the favored region, ~4% residues in the allowed regions, and ~1.2% residues (all glycine and Ala82) in the disallowed regions. The RMSD was found to be 0.061 Å suggesting good alignment. The grid was generated by selecting residues at 4 Å from bound inhibitors in homology modeled proteins (template PDBs: 4Q9I, 4Q9J, 4Q9K, 4Q9L). These residues are as follows: 61, 64, 65, 68, 69, 72, 118, 125, 222, 299, 303, 306, 307, 310, 336, 339, 340, 342, 343, 721, 725, 728, 729, 732, 770, 841, 842, 870, 871, 872, 942, 945, 949, 953, 957, 975, 978, 979, 982, 983, 984, 985, 986, 987, 990, and 991. Default protocol was used to perform induced-fit docking except the number of poses were reduced to 10. The surface representation of the bound ligand is generated by using ‘Create binding site surfaces’ tool in Maestro with default parameters and residue-type color scheme.
Supplementary Material
Acknowledgments
This research was supported by the Department of Pharmaceutical Sciences of St. John’s University. Drs. BA and SVA were supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
ABBREVIATIONS
- Cryo-EM
Cryo-Electron Microscopy
- DTT
Dithiothreitol
- EGTA
Ethylene glycol-bis(2-aminoethylether)-N,N,N,N-tetraacetic acid
- HCTU
O-(1H-6-Chlorobenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- HOBt
1-Hydroxybenzotriazole
- IAAP
Iodoarylazidoprazocin
- MES
2-(N-morpholino)ethanesulfonic acid
- PAINS
Pan assay interference compounds
- P-gp
P-glycoprotein.
Authors will release the atomic coordinates and experimental data upon article publication.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: Human P-gp homology model (PDB) generated using mouse P-gp (4Q9H) X-ray structure.51
Molecular formula strings (CSV).
1H-NMR spectrums for compounds 39, 54, 77 and 81–84.
Calculated molecular descriptors for target compounds.
References
- 1.Longley DB, Johnston PG. Molecular mechanisms of drug resistance. J Pathol. 2005;205:275–292. doi: 10.1002/path.1706. [DOI] [PubMed] [Google Scholar]
- 2.Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 3.Sharom FJ. ABC multidrug transporters: structure, function and role in chemoresistance. Pharmacogenomics. 2008;9:105–127. doi: 10.2217/14622416.9.1.105. [DOI] [PubMed] [Google Scholar]
- 4.Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta. 1976;455:152–162. doi: 10.1016/0005-2736(76)90160-7. [DOI] [PubMed] [Google Scholar]
- 5.Gottesman MM, Pastan I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu Rev Biochem. 1993;62:385–427. doi: 10.1146/annurev.bi.62.070193.002125. [DOI] [PubMed] [Google Scholar]
- 6.Dey S, Ramachandra M, Pastan I, Gottesman MM, Ambudkar SV. Evidence for two nonidentical drug-interaction sites in the human P-glycoprotein. Proc Natl Acad Sci U S A. 1997;94:10594–10599. doi: 10.1073/pnas.94.20.10594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orlowski S, Mir LM, Belehradek J, Jr, Garrigos M. Effects of steroids and verapamil on P-glycoprotein ATPase activity: progesterone, desoxycorticosterone, corticosterone and verapamil are mutually non-exclusive modulators. Biochem J. 1996;317(Pt 2):515–522. doi: 10.1042/bj3170515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chufan EE, Kapoor K, Sim HM, Singh S, Talele TT, Durell SR, Ambudkar SV. Multiple transport-active binding sites are available for a single substrate on human P-glycoprotein (ABCB1) PLoS One. 2013;8:e82463. doi: 10.1371/journal.pone.0082463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Higgins CF. Multiple molecular mechanisms for multidrug resistance transporters. Nature. 2007;446:749–757. doi: 10.1038/nature05630. [DOI] [PubMed] [Google Scholar]
- 10.Frank GA, Shukla S, Rao P, Borgnia MJ, Bartesaghi A, Merk A, Mobin A, Esser L, Earl LA, Gottesman MM, Xia D, Ambudkar SV, Subramaniam S. Cryo-EM analysis of the conformational landscape of human P-glycoprotein (ABCB1) during its catalytic cycle. Mol Pharmacol. 2016;90:35–41. doi: 10.1124/mol.116.104190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Palmeira A, Sousa E, Vasconcelos MH, Pinto MM. Three decades of P-gp inhibitors: skimming through several generations and scaffolds. Curr Med Chem. 2012;19:1946–2025. doi: 10.2174/092986712800167392. [DOI] [PubMed] [Google Scholar]
- 12.Robert J, Jarry C. Multidrug resistance reversal agents. J Med Chem. 2003;46:4805–4817. doi: 10.1021/jm030183a. [DOI] [PubMed] [Google Scholar]
- 13.Wandel C, Kim RB, Kajiji S, Guengerich P, Wilkinson GR, Wood AJ. P-glycoprotein and cytochrome P-450 3A inhibition: dissociation of inhibitory potencies. Cancer Res. 1999;59:3944–3948. [PubMed] [Google Scholar]
- 14.Bohme M, Buchler M, Muller M, Keppler D. Differential inhibition by cyclosporins of primary-active ATP-dependent transporters in the hepatocyte canalicular membrane. FEBS Lett. 1993;333:193–196. doi: 10.1016/0014-5793(93)80403-h. [DOI] [PubMed] [Google Scholar]
- 15.Shukla S, Ohnuma S, Ambudkar SV. Improving cancer chemotherapy with modulators of ABC drug transporters. Curr Drug Targets. 2011;12:621–630. doi: 10.2174/138945011795378540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buchman AL, Paine MF, Wallin A, Ludington SS. A higher dose requirement of tacrolimus in active crohn’s disease may be related to a high intestinal P-glycoprotein content. Dig Dis Sci. 2005;50:2312–2315. doi: 10.1007/s10620-005-3053-3. [DOI] [PubMed] [Google Scholar]
- 17.Englund G, Jacobson A, Rorsman F, Artursson P, Kindmark A, Rönnblom A. Efflux transporters in ulcerative colitis: Decreased expression of BCRP (ABCG2) and Pgp (ABCB1) Inflamm Bowel Dis. 2007;13:291–297. doi: 10.1002/ibd.20030. [DOI] [PubMed] [Google Scholar]
- 18.Binkhathlan Z, Lavasanifar A. P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: current status and future perspectives. Curr Cancer Drug Targets. 2013;13:326–346. doi: 10.2174/15680096113139990076. [DOI] [PubMed] [Google Scholar]
- 19.Singh S, Prasad NR, Chufan EE, Patel BA, Wang YJ, Chen ZS, Ambudkar SV, Talele TT. Design and synthesis of human ABCB1 (P-glycoprotein) inhibitors by peptide coupling of diverse chemical scaffolds on carboxyl and amino termini of (S)-valine-derived thiazole amino acid. J Med Chem. 2014;57:4058–4072. doi: 10.1021/jm401966m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang YJ, Patel BA, Anreddy N, Zhang YK, Zhang GN, Alqahtani S, Singh S, Shukla S, Kaddoumi A, Ambudkar SV, Talele TT, Chen ZS. Thiazole-valine peptidomimetic (TTT-28) antagonizes multidrug resistance in vitro and in vivo by selectively inhibiting the efflux activity of ABCB1. Sci Rep. 2017;7:42106. doi: 10.1038/srep42106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Azzaria M, Schurr E, Gros P. Discrete mutations introduced in the predicted nucleotide-binding sites of the mdr1 gene abolish its ability to confer multidrug resistance. Mol Cell Biol. 1989;9:5289–5297. doi: 10.1128/mcb.9.12.5289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loo TW, Clarke DM. Covalent modification of human P-glycoprotein mutants containing a single cysteine in either nucleotide-binding fold abolishes drug-stimulated ATPase activity. J Biol Chem. 1995;270:22957–22961. doi: 10.1074/jbc.270.39.22957. [DOI] [PubMed] [Google Scholar]
- 23.Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu Rev Pharmacol Toxicol. 1999;39:361–398. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- 24.Loo TW, Clarke DM. P-glycoprotein ATPase activity requires lipids to activate a switch at the first transmission interface. Biochem Biophys Res Commun. 2016;472:379–383. doi: 10.1016/j.bbrc.2016.02.124. [DOI] [PubMed] [Google Scholar]
- 25.Mistry P, Stewart AJ, Dangerfield W, Okiji S, Liddle C, Bootle D, Plumb JA, Templeton D, Charlton P. In vitro and in vivo reversal of P-glycoprotein-mediated multidrug resistance by a novel potent modulator, XR9576. Cancer Res. 2001;61:749–758. [PubMed] [Google Scholar]
- 26.Dantzig AH, Law KL, Cao J, Starling JJ. Reversal of multidrug resistance by the P-glycoprotein modulator, LY335979, from the bench to the clinic. Curr Med Chem. 2001;8:39–50. doi: 10.2174/0929867013373903. [DOI] [PubMed] [Google Scholar]
- 27.Qiu Q, Shi W, Li Z, Zhang B, Pan M, Cui J, Dai Y, Huang W, Qian H. Exploration of 2-((Pyridin-4-ylmethyl) amino) nicotinamide derivatives as potent reversal agents against P-glycoprotein-mediated multidrug resistance. J Med Chem. 2017;60:2930–2943. doi: 10.1021/acs.jmedchem.6b01879. [DOI] [PubMed] [Google Scholar]
- 28.Polli JW, Wring SA, Humphreys JE, Huang L, Morgan JB, Webster LO, Serabjit-Singh CS. Rational use of in vitro P-glycoprotein assays in drug discovery. J Pharmacol Exp Ther. 2001;299:620–628. [PubMed] [Google Scholar]
- 29.Chufan EE, Kapoor K, Ambudkar SV. Drug-protein hydrogen bonds govern the inhibition of the ATP hydrolysis of the multidrug transporter P-glycoprotein. Biochem Pharmacol. 2016;101:40–53. doi: 10.1016/j.bcp.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schinkel AH, Jonker JW. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv Drug Deliv Rev. 2003;55:3–29. doi: 10.1016/s0169-409x(02)00169-2. [DOI] [PubMed] [Google Scholar]
- 31.Singh S, Prasad NR, Kapoor K, Chufan EE, Patel BA, Ambudkar SV, Talele TT. Design, synthesis, and biological evaluation of (S)-valine thiazole-derived cyclic and noncyclic peptidomimetic oligomers as modulators of human P-glycoprotein (ABCB1) ChemBioChem. 2014;15:157–169. doi: 10.1002/cbic.201300565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liang Y, Su B, Zhao J, Sun W. The synthesis of new asymmetric double schiff bases containing a new o-amino benzoic acid derivative. Synth Commun. 2004;34:3235–3242. [Google Scholar]
- 33.Dobrotǎ C, Graeupner J, Dumitru I, Matache M, Paraschivescu CC. Expedient access to fused quinoxalines via Dess–Martin periodinane-mediated cyclization of unsymmetrical phenylenediamide derivatives. Tetrahedron Lett. 2010;51:1262–1264. [Google Scholar]
- 34.Mu F, Coffing SL, Riese DJ, 2nd, Geahlen RL, Verdier-Pinard P, Hamel TE, Johnson J, Cushman M. Design, synthesis, and biological evaluation of a series of lavendustin A analogues that inhibit EGFR and Syk tyrosine kinases, as well as tubulin polymerization. J Med Chem. 2001;44:441–452. doi: 10.1021/jm000387g. [DOI] [PubMed] [Google Scholar]
- 35.DeMuth DR, Luzzio FA. Anti-Biofilm Compounds. 9,167,820 B2. US Patent. 2015
- 36.Nguyen T, Sakasegawa Y, Doh-Ura K, Go ML. Anti-prion activities and drug-like potential of functionalized quinacrine analogs with basic phenyl residues at the 9-amino position. Eur J Med Chem. 2011;46:2917–2929. doi: 10.1016/j.ejmech.2011.04.016. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Q, Takacs JM. Click-connected ligand scaffolds: macrocyclic chelates for asymmetric hydrogenation. Org Lett. 2008;10:545–548. doi: 10.1021/ol702890s. [DOI] [PubMed] [Google Scholar]
- 38.Bruno P, Pena S, Just-Baringo X, Albericio F, Alvarez M. Total synthesis of aeruginazole A. Org Lett. 2011;13:4648–4651. doi: 10.1021/ol2018592. [DOI] [PubMed] [Google Scholar]
- 39.Yao Y, Tu Z, Liao C, Wang Z, Li S, Yao H, Li Z, Jiang S. Discovery of novel class I histone deacetylase inhibitors with promising in vitro and in vivo antitumor activities. J Med Chem. 2015;58:7672–7680. doi: 10.1021/acs.jmedchem.5b01044. [DOI] [PubMed] [Google Scholar]
- 40.Bredenkamp MW, Holzapfel CW, van Zyl WJ. The chiral synthesis of thiazole amino acid enantiomers. Synth Commun. 1990;20:2235–2249. [Google Scholar]
- 41.Holzapfel CW, Pettit GR. Antineoplastic agents. Par 108. Structural biochemistry. Part 23. Synthesis of the dolastatin thiazole amino acid component (gln)Thz. J Org Chem. 1985;50:2323–2327. [Google Scholar]
- 42.Kelly RC, Gebhard I, Wicnienski N. Synthesis of (R)- and (S)-(glu)thz and the corresponding bisthiazole dipeptide of dolastatin 3. J Org Chem. 1986;51:4590–4594. [Google Scholar]
- 43.Dean BM, Mijovi MPV, Walker J. Chemistry of micrococcin P. Part VI. Racemisation of 2-(l-amino-2-methylpropyl)thiazole-4-carboxylic acid, and related studies. J Chem Soc. 1961;0:3394–3400. [Google Scholar]
- 44.Daniels RN, Melancon BJ, Wang EA, Crews BC, Marnett LJ, Sulikowski GA, Lindsley CW. Progress toward the total synthesis of lucentamycin A: total synthesis and biological evaluation of 8-epi-lucentamycin A. J Org Chem. 2009;74:8852–8855. doi: 10.1021/jo902115s. [DOI] [PubMed] [Google Scholar]
- 45.Didziapetris R, Japertas P, Avdeef A, Petrauskas A. Classification analysis of P-glycoprotein substrate specificity. J Drug Target. 2003;11:391–406. doi: 10.1080/10611860310001648248. [DOI] [PubMed] [Google Scholar]
- 46.Jabeen I, Pleban K, Rinner U, Chiba P, Ecker GF. Structure-activity relationships, ligand efficiency, and lipophilic efficiency profiles of benzophenone-type inhibitors of the multidrug transporter P-glycoprotein. J Med Chem. 2012;55:3261–3273. doi: 10.1021/jm201705f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klopman G, Shi LM, Ramu A. Quantitative structure-activity relationship of multidrug resistance reversal agents. Mol Pharmacol. 1997;52:323–334. doi: 10.1124/mol.52.2.323. [DOI] [PubMed] [Google Scholar]
- 48.Crivori P, Reinach B, Pezzetta D, Poggesi I. Computational models for identifying potential P-glycoprotein substrates and inhibitors. Mol Pharmaceut. 2006;3:33–44. doi: 10.1021/mp050071a. [DOI] [PubMed] [Google Scholar]
- 49.Pajeva IK, Globisch C, Wiese M. Combined pharmacophore modeling, docking, and 3D QSAR studies of ABCB1 and ABCC1 transporter inhibitors. ChemMedChem. 2009;4:1883–1896. doi: 10.1002/cmdc.200900282. [DOI] [PubMed] [Google Scholar]
- 50.Baell JB, Holloway GA. New substructure filters for removal of Pan Assay Interference Compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 51.Szewczyk P, Tao H, McGrath AP, Villaluz M, Rees SD, Lee SC, Doshi R, Urbatsch IL, Zhang Q, Chang G. Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein. Acta Crystallogr D Biol Crystallogr. 2015;71:732–741. doi: 10.1107/S1399004715000978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wüthrich K, Grathwohl C, Schwyzer R. Cis, trans, and nonplanar peptide bonds in oligopeptides: 13C NMR studies. In: Blout ER, Bovey FA, Goodman M, Lotan N, editors. Peptides, Polypeptides and Proteins; Wiley; New York: 1974. pp. 300–307. [Google Scholar]
- 53.Ambudkar SV. Drug-stimulatable ATPase activity in crude membranes of human MDR1-transfected mammalian cells. Methods Enzymol. 1998;292:504–514. doi: 10.1016/s0076-6879(98)92039-0. [DOI] [PubMed] [Google Scholar]
- 54.Sauna ZE, Ambudkar SV. Evidence for a requirement for ATP hydrolysis at two distinct steps during a single turnover of the catalytic cycle of human P-glycoprotein. Proc Natl Acad Sci U S A. 2000;97:2515–2520. doi: 10.1073/pnas.97.6.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carmichael J, DeGraff WG, Gazdar AF, Minna JD, Mitchell JB. Evaluation of a Tetrazolium-based semiautomated colorimetric assay: assessment of chemosensitivity testing. Cancer Res. 1987;47:936–942. [PubMed] [Google Scholar]
- 56.Berthold MR, Cebron N, Dill F, Gabriel TR, Kötter T, Meinl T, Ohl P, Sieb C, Thiel K, Wiswedel B. KNIME: The Konstanz Information Miner. In: Preisach C, Burkhardt H, Schmidt-Thieme L, Decker R, editors. Data analysis, machine learning and applications; Springer; Heidelberg, Berlin: 2008. pp. 319–326. [Google Scholar]
- 57.Saubern S, Guha R, Baell JB. KNIME workflow to assess PAINS filters in SMARTS format. Comparison of RDKit and Indigo cheminformatics libraries. Mol Inf. 2011;30:847–850. doi: 10.1002/minf.201100076. [DOI] [PubMed] [Google Scholar]
- 58.Trubetskoy O, Marks B, Zielinski T, Yueh M, Raucy J. A simultaneous assessment of CYP3A4 metabolism and induction in the DPX-2 cell line. AAPS J. 2005;7:E6–E13. doi: 10.1208/aapsj070102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.