

Abstract
Mechanism-inspired drug repurposing that augments standard treatments offers a cost-effective and a rapid route toward addressing the burgeoning problem of plateauing of effective therapeutics for drug-resistant micrometastases. We show that the antibiotic minocycline, by its ability to minimize DNA repair via reduced expression of tyrosyl-DNA phosphodiesterase-1 (Tdp1), removes a key process attenuating the efficacy of irinotecan, a frequently used chemotherapeutic against metastatic disease. Moreover, minocycline and irinotecan cooperatively mitigate each other’s undesired cytokine inductions of VEGF and IL-8 respectively, thereby reinforcing the benefits of each modality. These mechanistic interactions result in synergistic enhancement of irinotecan-induced platinum-resistant epithelial ovarian cancer cell death, reduced micrometastases in the omenta and mesentery by >75%, and an extended overall survival by 50% in a late-stage peritoneal carcinomatosis mouse model. Economic incentives and easy translatability make the repurposing of minocycline as a reinforcer of the topoisomerase class of chemotherapeutics extremely valuable and merits further investigations.
Keywords: Tetracyclines, Nanoliposomal irinotecan, Tyrosyl-DNA phosphodiesterase-1, Ovarian cancer, Drug repurposing
Introduction
Treatment-resistance is one of the major driving forces of mortality in cancer patients despite advances in targeted therapies, chemoradiotherapy, and their cocktails(1,2). Moreover, the high cost associated with developing marginally effective therapies is becoming unsustainable. Therefore, cost-effective, mechanism-based approaches are urgently needed to break the plateau in therapeutic advances for treatment-refractory cancers.
Here, based on an understanding of molecular mechanisms, we repurpose an FDA-approved antibiotic minocycline to overcome chemoresistance pathways and enhance the antitumor activity of irinotecan, a frequently used chemotherapeutic against treatment-refractory peritoneal carcinomatosis and several other cancers. This unique re-tasking of minocycline as a cost-effective component within a combination anti-cancer approach offers several advantages: (i) At the DNA level, minocycline, when combined with irinotecan, simultaneously reduced the expression of a DNA repair enzyme, tyrosyl-DNA phosphodiesterase 1 (Tdp1), and induces double-stranded DNA breaks to synergistically enhance the anticancer activity against platinum-resistant EOC cells; (ii) At the molecular level, minocycline and irinotecan cooperatively overcome each other’s resistance pathways, through mitigation of secretion of tumor growth-promoting cytokines, vascular endothelial growth factor (VEGF) and pro-inflammatory cytokine Interleukin-8 (IL-8), to reinforce the benefits of each modality; and (iii) Minocycline, with a distinct molecular mechanism of action, enhances chemotherapeutic efficacy with no additional off-target toxicities in vivo. We provide evidence that minocycline realizes these complementary interactions to significantly potentiate irinotecan efficacy, reduce the metastatic burden and enhance survival outcomes in challenging peritoneal carcinomatosis animal models.
An ongoing challenge for treating drug resistant, disseminated tumors such as peritoneal carcinomatoses of gastrointestinal and gynecological malignancies is the lack of therapeutic options that are effective, safe, tolerable, and low-cost in patients(3). These obstacles are especially problematic in the treatment of platinum-refractory epithelial ovarian cancer (EOC) that has often already spread within the abdomen and formed numerous nodules studding peritoneal surfaces(4,5). A significant fraction (70–80%) of advanced stage EOC patients will experience disease relapse after first-line treatment that consists mainly of debulking surgery followed by platinum-based chemotherapy(6,7). In contrast to the steady increase in survival for most cancer, advances have been slow for EOC, for which the 5-year related survival has only inched up from 36% in 1975 to around 46% today(8). Recurrent EOC with intrinsic or acquired resistance is considered incurable primarily due to the poor response rate (6–30%) of salvage therapies, such as irinotecan, topotecan, etoposide, and bevacizumab(9–11). Salvage chemotherapy with irinotecan provides a median overall survival (OS) of 10.1 months in patients with metastatic drug-resistant EOC(12). Recently, there has been much excitement with the combination of irinotecan and bevacizumab (or etoposide) showing activity and a modest improvement of OS to 13.8–19 months(10,13,14) in heavily pre-treated patients with recurrent ovarian EOC. However, the highly cytotoxic chemotherapy cocktails oftentimes require dose de-escalation due to intolerable toxicity in patients, and thus have limited use(10,15). The rapid embrace of these modalities despite their toxicities, and modest impact on survival, is a testimony to the dismal state of recurrent EOC treatment and a desperate need for a conceptual shift to new combinatorial approaches beyond traditional chemotherapy and biological cocktails. Moreover, the ability of tumors to adapt to alternative pathways of survival and growth also suggests that combination treatments will need to attack several targets, perhaps simultaneously, and ideally counterstriking treatment resistance pathways.
Irinotecan is a camptothecin derivative that inhibits topoisomerase I (Top1) by trapping Top1-DNA cleavage complexes (Top1cc), which ultimately causes double-stranded DNA breaks and promotes cell death(16). Its action is limited by one of the repair enzymes of Top1cc, tyrosyl-DNA phosphodiesterase 1 (Tdp1), which resolves the Top1cc adduct by cleaving the 3’(5′)-tyrosyl-DNA bond between Top1 and DNA in cells, allowing DNA religation(17,18) and cell proliferation (Fig. 1A). Therefore, in the context of a Top1 inhibitor, it may be desirable to inhibit Tdp1 in order to reduce the degree of Topo1cc repair and increase the DNA damage(19,20). Tdp1 is an emerging anticancer target found to be upregulated in drug-resistant variants of human EOC and other cancers(21,22). Several Tdp1 inhibitors have been identified through drug discovery projects(20,23,24). However, these are largely in preclinical research with no in vivo evaluations confirming the survival benefit of Tdp1 inhibitors or clinical trials in progress. We demonstrate that this barrier may be overcome by the use of a well-known tetracycline antibiotic minocycline without any added normal tissue toxicity yet increased therapeutic efficacy.
Fig. 1. The combination of minocycline and irinotecan simultaneously reduced Tpd1 expression and induced DNA damage in cancer cells.
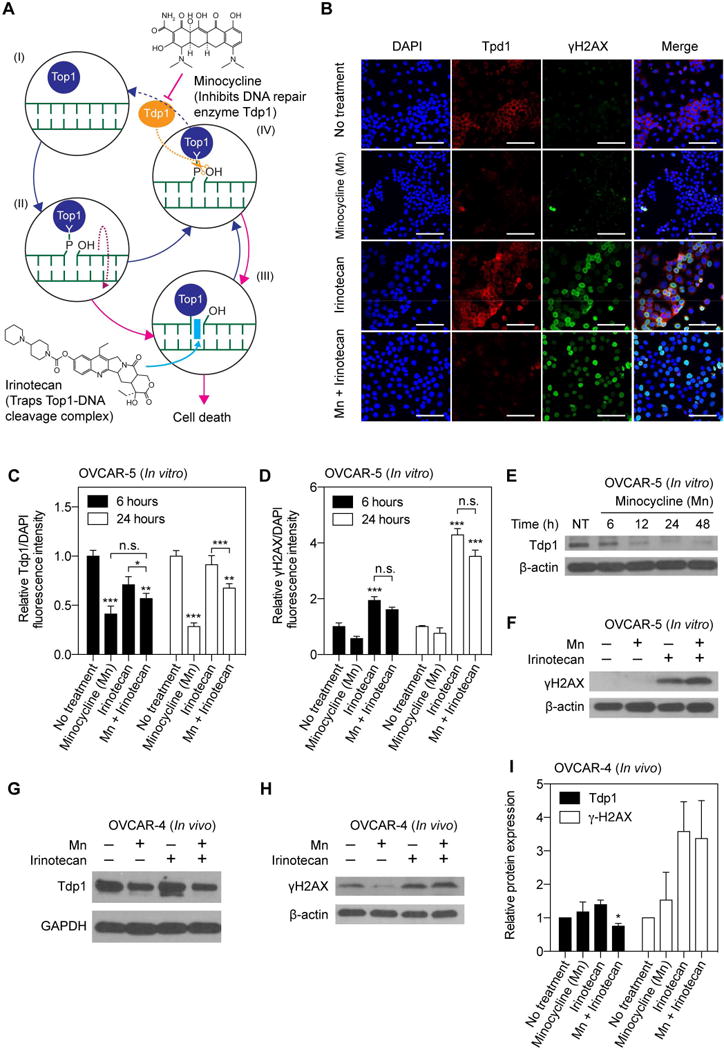
(A) Schematic representation of minocycline (Mn) inhibiting tyrosyl-DNA phosphodiesterase 1 (Tdp1), removing a barrier attenuating irinotecan action against topoisomerase I (Top1). (I-II) Top1 is a protein with enzymatic activity that relaxes supercoiled double-strand DNA (green), thereby permitting DNA replication and RNA transcription. (III) Irinotecan (purple) blocks DNA religation by trapping Top1-DNA cleavage complexes (Top1cc) and causes double-stranded DNA breaks, which ultimately promotes cell death. (IV) Mn inhibits Tdp1, which resolves the Top1cc adduct by cleaving the 3’(5′)-tyrosyl-DNA bond between Top1 and DNA in cells, preventing DNA repair. (B) Monolayer cultures of human OVCAR-5 cells were incubated with Mn, irinotecan or their combination for 6, 24, and 48 hours. Representative immunofluorescence imaging of Tdp1 (red fluorescence) and γH2AX (green fluorescence) in OVCAR-5 cells subjected to (i) No-treatment (NT); (ii) Mn ; (iii) Irinotecan; and (iv) Combination of Mn and irinotecan for 24 hours. (C, D) To quantify immunofluorescence intensities, at least 6 images, evenly distributed across the entire monolayer culture dish, were collected from at least 3 samples for each condition. At 6 and 24 hours post-treatment, Tdp1 and γH2AX fluorescence intensities were normalized to DAPI area per image. Relative Tdp1 levels were found to be significantly lower in the Mn treatment group compared to the NT and irinotecan alone groups. The γH2AX signal was significantly higher in both the combination and the irinotecan alone groups, compared to the NT and irinotecan alone groups. (*P<0.05, **P<0.01, ***P<0.001, Kruskal–Wallis one-way ANOVA with Dunn’s post hoc test) Asterisks denote significance compared to the NT group or amongst the indicated groups at each time point. Results are mean ± standard error of the mean. (E) Representative immunoblotting showing a time-dependent reduction of Tdp1 expression in OVCAR-5 cells at 6, 24, and 48 hours after Mn incubation. (F) Immunoblot analysis of γH2AX in OVCAR-5 cells at 24 hours post-treatment. (G, H) Representative immunoblotting and (I) quantification of in vivo tumors at 72 hours after treatment termination (i.e. two weeks after treatment initiation; see methods for in vivo treatment schedule) showing the combination of Mn and irinotecan significantly reduced Tdp1 expression and induced γH2AX levels.
VEGF and IL-8 are implicated in the peritoneal metastasis of EOC and development of malignant ascites(25,26). VEGF is a key regulator of angiogenesis and was identified to induce peritoneal vascular hyperpermeability, promote the pathogenesis of malignant ascites, and contribute to the carcinogenesis of EOC(25). IL-8 is known to maintain the mesenchymal phenotype of cancer cells and to facilitate metastatic carcinoma progression through mitogenic and angiogenic effects(27). Secretions of VEGF and IL-8 have been correlated with increased tumorigenicity, ascites formation, and poor prognosis in EOC patients(28–30) and in vivo models(31). Huang et al. showed that inhibiting the expression of VEGF and IL-8 via blockade of nuclear factor-κB signaling inhibited angiogenesis, reduced malignant ascites and prolonged survival in mice(32). While a number of potent therapeutic agents targeting angiogenesis, cancer-related inflammation, and DNA repair defects have been explored in combination with chemotherapy for EOC, only few have yield survival benefits in the clinic(33). Efforts to understand VEGF and IL-8 secretion in response to conventional chemotherapy and to develop gentler and cost-effective strategies remain a high priority.
At a time where the average cost to bring a drug to market is over $2.5 billion, drug repurposing allows a more efficient development at lower costs and potentially with fewer safety concerns as preclinical and clinical safety data already exist. Thus far, several advancements in cancer treatment have been made by repurposing long-standing drugs as monotherapies or as part of combinations, such as chloroquine(34), metformin(35), vitamin D(36), itraconazole(37). Repurposing of minocycline, a broad-spectrum tetracycline antibiotic, has proven to be effective for gynecologic tumors in preclinical studies(38,39). Studies have reported the cytostatic and cytotoxic effects of minocycline against EOC—via G0-G1 cell cycle arrest, and modulation of vascular endothelial growth factor (VEGF), pro-inflammatory cytokine Interleukin-6 (IL-6) and its receptor(38,39), which play pivotal roles in the development of peritoneal carcinomatosis, the potent proliferative and metastatic capacity of EOC, and also correlate with poor prognosis in patients(40,41). Moreover, through a quantitative high-throughput screening assay for inhibitors of human Tdp1, Yves Pommier identified that minocycline hydrochloride (SID: 144206734), at micromolar concentrations, could be involved in the inhibition of Tdp1-mediated repair pathway in chicken Tdp1-transfected DT40 cells (BioAssay AID: 686978 and 686979).
In view of the poor prognosis for patients with treatment-resistant cancers and the rising costs required for developing a brand-new therapeutics, this is the first in vivo study to show that the combination of minocycline and irinotecan, at a clinically relevant dose and schedule, can effectively reduce metastatic burden in the omenta and mesenteries by more than 75%, significantly extending survival outcomes in a challenging mouse model for peritoneal metastatic EOC. Our findings also offer a platform for designing new tetracycline-based approaches that provide the dual advantage of overcoming DNA repair mechanisms while mitigating cytokine secretion for the enhanced antitumor efficacy of topoisomerase inhibitors, without additional side effects.
Materials and Methods
Cell lines and culture
Human epithelial ovarian carcinoma cell lines OVCAR-5 (derived from the ascitic fluid of EOC patient without prior treatment; tumor characterized to be platinum-resistant) and OVCAR-4 (derived from the ascitic fluid of platinum-refractory EOC patient) were used to assess the combinatorial effects of minocycline and irinotecan. Authenticated OVCAR-5 and OVCAR-4 cells were obtained from ATCC in 2011 and Fox Chase Cancer Center in 2015, respectively, tested free of mycoplasma contamination using commercially available kits (MycoAlert™, Lonza; Latest date: April 2017), and cultured according to the supplier’s instruction. Authenticated OVCAR-5 and OVCAR-4 were propagated for less than 30 and 4 passages after resuscitation, respectively. No further authentication of the cell line was done by the authors. OVCAR-5 cells were grown in RPMI-1640 medium (with L-glutamine, Cellgro) supplemented with 10 % heat-inactivated fetal calf serum (Gibco), 100 U/mL penicillin and 100 μg/mL streptomycin. OVCAR-4 cells were cultured in equivalent conditions, with the exception of supplementing 0.2 U/mL bovine insulin in the medium, as recommended by the supplier. Both cells lines were maintained in an incubator at 37 °C in an atmosphere of 5% CO2, passaged and plated at required cell concentrations under sterile conditions.
In vitro treatment response
OVCAR-5 and OVCAR-4 cells were plated in sterile 35 mm cell culture dishes (BD Biosciences) at a density of 105,000 cells in 2 mL media and allowed to grow for 24 hours. To establish dose response in monolayer, cultures were treated with irinotecan (Selleckchem), minocycline hydrochloride (Sigma-Aldrich), or the combination of the two agents at concentrations ranging from 0 to 200 μM. Cytotoxicity was assessed via MTT assay following a 48-hour drug-incubation and all readouts were normalized to untreated controls. Non-linear regression trends were generated with GraphPad (Prism) software to calculate the IC50, which represents the concentration of the tested drug that is required for 50% inhibition of the cell viability. All IC50 values were validated using the CompuSyn software (ComboSyn, Inc, Paramus, NJ). In vitro combination effects were evaluated at a constant drug ratio. Each drug and their combination were used at a concentration approximately equal to its IC50, and concentrations within 2.5-fold increments below and above the IC50 value. Combination index (CI) values, based on the Loewe additivity model, were calculated using the CompuSyn software to determine whether the drug-drug interactions are synergistic (CI<1), additive (CI=1), or antagonistic (CI>1). Isobolograms at effect levels of 50% and 75% fraction affected (Fa; inhibition of cancer cell viability) were created to determine the dose-depended interaction of minocycline and irinotecan.
Molecular characterization of DNA damage and repair
Human epithelial ovarian carcinoma cells on glass bottom 24-well plates were incubated for 6 or 24 hours with media containing irinotecan, minocycline, or their combination at concentrations ranging from 0 to 100 μM. For immunofluorescence, cells were washed with room temperature (RT) PBS and immediately fixed and permeabilized in 1:1 acetone:methanol (-20 °C) for 1 minute, remove acetone:methanol carefully wash with RT PBS, followed by blocking (Dako, Protein Block Serum-free) for 30 minutes at RT. Cells were then incubated at 37 °C (4°C) with 100 μL of primary antibodies, anti-Tdp1 (Abcam) and anti-phospho-histone H2A.X SER 139 (EMD Millipore) at 5 μg/mL (dilute in DAKO diluent at 4 °C). After 2 hours (or overnight), cells were washed three times with RT PBS (critical) and incubated for 1 hour with fluorescently labeled secondary antibodies (Donkey Anti-Rabbit IgG Alexa Fluor 647, Goat Anti-Mouse IgG Alexa Fluor 488, Abcam) at 37 °C followed by three more washes with PBS. Forty microliters of SlowFade Gold Antifade Mountant with DAPI was added to cells prior to the placement of a glass coverslip. Fluorescence images were obtained immediately on an Olympus FV-1000 confocal microscope.
Quantification of interleukin-8 (IL-8) and vascular endothelial growth factor (VEGF) Secretions
Monolayer OVCAR-5 cultures were treated with irinotecan, minocycline hydrochloride, or the combination of the two agents at concentrations ranging from 0 to 100 μM. Supernatant from OVCAR-5 cells was collected at various incubation time points (24 and 48 hours) after treatment. IL-8 and VEGF secretion into the supernatant were measured using respective ELISAs (VEGF, R&D Systems; IL-8, BioLegend). Assays were performed according to manufacturer guidelines with appropriate standard curves. All results are plotted as fold change with respect to untreated controls collected at the same time points.
Nanoliposomal irinotecan preparation and characterization
Nanoliposomes are well-established pharmaceutical carriers that have been shown to decrease systemic toxicity and improve the pharmacokinetics and intratumoral accumulation of irinotecan and it’s active metabolite, SN-38(42). Motivated by the recent clinical success of nanoliposomal irinotecan (ONIVYDE®) for patients with metastatic cancer(42,43), a similar laboratory formulation of nanoliposomal irinotecan (nal-IRI) was prepared following our established protocol(44) and used for in vivo studies. Briefly, dipalmitoylphosphatidylcholine (DPPC), cholesterol, distearoylphosphatidylethanolamine-methoxy polyethylene glycol (DSPE-PEG), and dioleoyltrimethylammoniumpropane (DOTAP) (Avanti Polar Lipids) were mixed in chloroform at 20:10:1:2.5 molar ratio. Chloroform was removed by rotary evaporation overnight to afford a thin lipid film. The resulting lipid film was rehydrated with 1 mL 7 mM of irinotecan hydrochloride trihydrate in phosphate-buffered saline (PBS) at irinotecan-to-total lipid ratio of 17 mol%, and subjected to freeze-thaw cycles (4–45 °C) for 2 hours. The dispersion was then extruded 10 times through two stacked polycarbonate membranes (0.1 μm pore size; Nuclepore, Whatman) at 42 °C using a mini-extruder system (Avanti Polar Lipids) to form unilamellar vesicles. Un-encapsulated drugs were removed by dialysis (Spectra/Por, MWCO 300kD, Spectrum Laboratories) against PBS. Irinotecan concentration was determined by UV-Vis spectroscopy with appropriate standard curves. Characterizations of the nal-IRI, including size, stability, shelf life, drug release, in vivo pharmacokinetics and biodistribution, were reported previously.
Mouse model of peritoneal carcinomatosis and in vivo treatment response
All animal experiments were conducted in accordance with the Massachusetts General Hospital (MGH) Institutional Animal Care and Use Committee (IACUC) guidelines. An orthotopic xenograft mouse model of human ovarian carcinomatosis was established with gynecologic oncologists and characterized in our laboratory. Female athymic (nu/nu) Swiss mice (20–25 g and 4–6 weeks old; Cox Breeding Laboratories) were injected intraperitoneally with 31.5 × 106 OVCAR-5 cells suspended in 2 mL of PBS. Treatments were initiated at 7 (or 14 days) post-implantation. Tumor-bearing mice were randomized into the following groups: (i) no treatment; (ii) minocycline; (iii) nanoliposomal irinotecan (nal-IRI); and (iv) combination of minocycline and nal-IRI treatment. For nal-IRI treatment, animals received three-cycles of 20 mg·kg−1 body weight intravenous injection of nal-IRI on days 11, 15, 18 (or days 18, 22, 25) post-implantation. The selected nal-IRI mouse dose of 20 mg/kg is equivalent to human dose ~60 mg/m2 of and similar to the clinical (irinotecan liposome injection) of ~70 mg/m2. Minocycline antibiotics were administered by intraperitoneal injection in 1 mL of sterile PBS. A single bolus intraperitoneal injection of Mn at 50 mg·kg−1 body weight was administered on day 7 (or day 14), which is followed by eight more cycles of Mn at 25 mg·kg−1 body weight on the following days post-implantation: 8, 9, 10, 11, 14, 15, 17, 18 (or days 15, 16, 17, 18, 21, 24, 25). The route of intraperitoneal administration for minocycline was chosen to allow direct access for the drug to the peritoneal cavity, where the ovarian cancer has spread. Furthermore, previous study have suggested that the intraperitoneal injections of minocycline in mice in our study leads to a serum minocycline level (Cmax, 5–10 μg/mL) comparable to that obtained after oral dosing of 200 mg in humans (Cmax, 3–4 μg/mL)(45,46). At 20 days post-implantation, animals were euthanized and necropsied to assess acute tumor burden following treatment. The following tissues were resected and weighed at the time of necropsy: subgastric omentum, pelvic omentum, bowel mesentery, uterus, and diaphragm. Corresponding tissue from healthy, non-tumored mice were also collected, averaged, and subtracted from the tissue weight in tumored animals. The weights of the collected tissue were also summed for the total burden per animal. For survival studies, mice were observed daily and removed from the study once the humane endpoints were reached. Moribundity and animal mobility (i.e. unable to move) were used as the endpoint with proper justification and special approval by MGH IACUC. Mouse body weight was monitored before and longitudinally after treatment as a metric of toxicity in a subcutaneous mouse model of human ovarian cancer.
Western Blot
Cells were harvested by scraping in cold PBS and followed by centrifugation (1000 rpm, 5 min). The cell pellet was re-suspended in 2× packed cell volume (PCV) of RIPA lysis buffer (Thermo) supplemented with 1% phosphatase and protease inhibitor cocktail respectively (Sigma, Millipore). The mixture was incubated on a shaker (1h, 4 °C) and sonicated (2 min). Subsequent to centrifugation (14 000 rcf, 10 min, 4 °C), supernatants (control, drug-treated samples) were collected and protein concentrations were determined using the BCA assay (Bio-Rad). Protein expression was analyzed using a standard Western protocol (Bio-Rad). As briefly described, protein lysates were separated on 4–20% precast polyacrylamide gel (Mini-PROTEAN TGX, Bio-Rad) and transferred onto a polyvinylidene difluoride (PVDF) membrane (Thermo). Subsequent to blocking with 5% BSA/TBST solution, proteins were further detected using antibodies against γ-H2AX (Millipore) and Tdp1 (Santa Cruz). Anti β-actin antibodies (Cell Signaling) were used for loading control. Ramos whole cell lysate (Santa Cruz) was used as positive control of Tdp1 primary antibody. Visualization of protein bands was developed by chemiluminescence (ECL, Bio-Rad) with exposure to X-ray film (Thermo). The quantitative analysis of protein expression was done using ImageJ software. Western blot analyses of the target proteins were repeated at least for three times.
Statistical analyses
Results are mean ± standard error of the mean (SEM). Statistical tests were carried out using GraphPad Prism (GraphPad Software). Specific tests are indicated in the figure captions. The synergy between irinotecan and minocycline was determined based on Drug Combination Analyses (CompuSyn). No exclusion criteria were used, and no data points or animals were excluded from analysis. Survival curves were created using the method of Kaplan and Meier. The log-rank test was used to test if the difference between survival times between two groups is statistically significant or not. Investigators were blinded to experimental groups during tumor volume monitoring and survival studies unless noted otherwise.
Results
Subhead 1: Combination minocycline and irinotecan simultaneously reduce Tdp1 expression and induce DNA double-strand breaks in ovarian carcinoma cells
The impacts of minocycline, irinotecan, or their combination on DNA repair enzyme Tdp1 and DNA damage were assessed by confocal microscope imaging of Tdp1 and double-strand DNA break marker γH2AX in monolayer OVCAR5 cells (Fig. 1A,B). Representative immunofluorescence images (Fig. 1B) showed that minocycline treatment for 24-hours decreased the expression of Tdp1, but did not significantly modulate the γH2AX expression in OVCAR-5 cells. In contrast, irinotecan treatment for 24-hours up-regulated the expression of γH2AX, but did not significantly alter Tdp1 expression in OVCAR-5 cells. Only in the presence of both minocycline and irinotecan, was both a significant loss in Tdp1 signal and gain in γH2AX expression observed in cancer cells. Quantitative analyses of Tdp1 and γH2AX immunofluorescence intensity revealed that, at 6 and 24-hours post-incubation, minocycline significantly reduced the Tdp1 immunofluorescence intensity by 60-70% (P < 0.001). Whereas, irinotecan did not significantly modulate Tdp1 levels (Fig. 1B). However, irinotecan induced up to 2 and 4-fold increase in γH2AX immunofluorescence signal at 6 and 24-hours post-treatment, respectively (P < 0.001; Fig. 1C) suggesting the presence of DNA double-strand breaks in OVCAR5 cells. Minocycline alone did not increase γH2AX expressions. Importantly, cells treated with the combination of minocycline and irinotecan showed both a decrease in the Tdp1 signal by ~32% and an increase in the γH2AX intensity up to 3.2-fold, compared to the untreated cells (Fig. 1C,D). Immunoblotting confirmed that minocycline incubation for up to 48-hours resulted in a time-dependent reduction in Tdp1 protein levels in OVCAR-5 cells (Fig. 1E). The γH2AX protein expression levels in OVCAR-5 cells treated with irinotecan alone and the combination of minocycline and irinotecan were significantly higher than in cells that received no-treatment or minocycline alone (Fig. 1F). We also evaluated the Tdp1 and γH2AX expressions in OVCAR-4 tumors ex vivo at 72 hours after treatment termination (see in vivo methods). We observed that the combination of minocycline and irinotecan reduce tumoral Tdp1 expression by 25 percent and induce γH2AX levels by over 3 fold, compared to the no-treatment tumors (Fig. 1G–I). While Mn significantly reduced Tdp1 levels in OVCAR-5 cells in vitro (Fig. 1C–F), Mn did not attenuate the Tdp1 level in OVCAR-4 tumors (Fig. 1G–I). The discrepancy of results between the two cell lines is presumably due to different etiologies of the two lines, contrasting cellular Tdp1 baseline levels (Fig. S1), different model systems (in vitro v.s. in vivo), and dissimilar drug exposure conditions.
Subhead 2: Mutually reinforcing cooperation between minocycline and irinotecan reduces secretion of cytokines
Preclinical and clinical studies have identified IL-8 and VEGF as key contributors to EOC progression and predictors of poor prognosis in patients(26,30,47,48). We, therefore, measured the secretion of these cytokines in the surrounding media of EOC cells treated with minocycline, irinotecan, and their combination using ELISA kits. Minocycline at 100 μM reduced IL-8 secretion by ~26% in OVCAR-5 cells at 24-hours, compared to the baseline IL-8 level of 849±75 pg/mL in the no-treatment cells (P < 0.05; Fig. 2A). In contrast, a significant dose-dependent increase in the IL-8 secretion by 20-50% was observed 24-hours after irinotecan chemotherapy (50–100 μM; P < 0.01; Fig. 2A). A combination of minocycline and irinotecan at the micromolar range (10, 50 and 100 μM) effectively suppressed IL-8 secretion at a baseline level (890±56 pg/mL). Furthermore, it was found that minocycline (50 and 100 μM) significantly increased the levels of VEGF secretion by ~2.7-fold to 1356±130 pg/mL, compared to the VEGF levels in the no-treatment control (500±52 pg/mL) (P < 0.01; Fig. 2B). However, the combination was able to suppress this increase and effectively maintained the VEGF secretion at the baseline level (442±48 pg/mL), similar to that of the no-treatment and irinotecan control groups. Together, these results suggest a mutually reinforcing cooperation between minocycline and irinotecan in which minocycline mitigates the irinotecan-stimulated secretion of IL-8, while irinotecan reduces the minocycline-induced secretion of VEGF (Fig. 2C).
Fig. 2. A mechanistic cooperation between minocycline and irinotecan that reduces the secretion of IL-8 and VEGF cytokines.
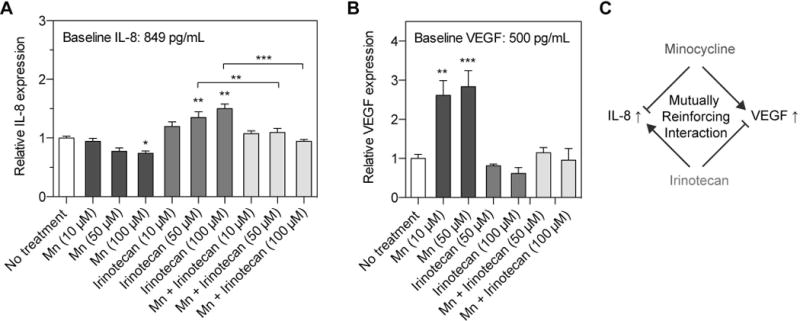
Monolayer cultures of human OVCAR-5 cells were incubated with minocycline (Mn), irinotecan or their combination at various concentrations (10, 50, and 100 μM). Secretion of IL-8 and VEGF into the supernatant was measured using respective ELISA kits. (A) Minocycline at 100 μM reduced the IL-8 secretion by ~26% at 24-hours post-treatment. In contrary, a dose-dependent increase in IL-8 secretion up to 50% was observed in irinotecan-treated cells, compared the no-treatment group. A combination of Mn and irinotecan maintained the IL-8 secretion at a baseline level, similar to that of the no-treatment (NT) group. (B) A surge of VEGF secretion by over 200% was observed after 48 hours of Mn treatment, while both the combination and irinotecan treatments maintained VEGF secretion around the (NT) baseline level. Together, these results suggest that Mn could effectively mitigate the surge of IL-8 secretion induced by irinotecan treatment. On the other hand, irinotecan abrogates the increase of VEGF secretion triggered by Mn treatment. (n = 4-7; *P<0.05, **P<0.01, ***P<0.001, Kruskal–Wallis one-way ANOVA with Dunn’s post hoc test) Asterisks denote significance compared to the NT group or amongst the indicated groups at each time point. Results are mean ± standard error of the mean. (C) A mutually reinforcing interaction between Mn and irinotecan.
Subhead 3: Combination of minocycline and irinotecan synergize in ovarian carcinoma cells
We assessed the effects of minocycline, irinotecan, and their combination on the viability of platinum-resistant human EOC cells lines of OVCAR-5 and OVCAR-4 (both expressing Tdp1, Fig. S1). Cells were treated with minocycline, irinotecan or their combination in a serial dilution from 0–100 μM (Fig. 3A,B). Cell viability was analyzed 48 hours later by MTT tetrazolium assay. Our results show that minocycline treatment for 48 hours reduced the viability of OVCAR-5 and OVCAR-4 in a dose-dependent manner with the half maximal inhibitory concentration (IC50) of 62.8 and 57.8 μM, respectively (Fig. 3A,B). Similarly, a 48-hour incubation of irinotecan reduced viability of both OVCAR-5 and OVCAR-4 cells in a dose-dependent manner with an IC50 of 76.2 and 100.6 μM, respectively (Fig. 3A,B). The IC50 values of minocycline and irinotecan for OVCAR-5 and OVCAR-4 cells are within the micromolar range, similar to data published by others with EOC and other cancer lines(38,49,50). A fixed drug ratio method based on Loewe’s concept of additivity was employed to test if minocycline and irinotecan can enhance each other’s anticancer activity(51,52). Using CompuSyn software and robust regression fits of the dose-response curve trend lines (R2=0.978-0.993), combination index (CI) values were calculated to determine if the dug-drug interaction is synergistic (CI<1), additive (CI=1), or antagonistic (CI>1)(51,52). In both EOC lines, synergy was observed at higher dose levels and at higher inhibitory activities of the cancer cell viability (>50% fraction affected; Fig. 3C,D).
Fig. 3. Minocycline synergizes with irinotecan in Tdp1-expressing human EOC cells.
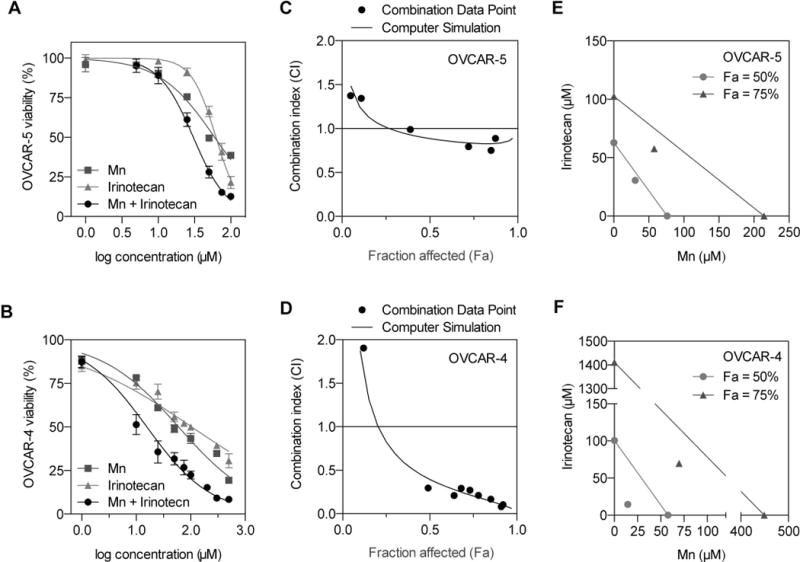
Monolayer cultures of human OVCAR-5 and OVCAR-4 cells (105,000 cells per 35-mm dish) were incubated with minocycline (Mn), irinotecan or their combination (ranging from 0–200 μM) for 48 hours. MTT assay was used to assess cytotoxicity, and all readouts were normalized to untreated controls. (A, B) Graphs of percentage cell viability vs. log concentration showing the combination of Mn and irinotecan (black circles) resulted in lower cell viability, compared to Mn alone and irinotecan alone. Results are mean ± standard error of the mean (n = 6-8). (C, D) Using the CompuSyn software and robust regression fits of the dose-response curve trend lines (R2=0.978-0.993), the combination index (CI) values were calculated to determine the Mn-irinotecan interactions: synergistic (CI<1), additive (CI=1), or antagonistic (CI>1). A modest synergy was observed in OVCAR-5 cells, whereas a strong synergy was observed in the OVCAR-4 cells. (E, F) To determine dose-dependent interaction of Mn and irinotecan, isobolograms at effect levels of 50% and 75% fraction affected (Fa: inhibition of cancer cell viability) were created. The combination data points at both 50% and 75% effect levels lie well below their additive isoboles, suggesting a synergy between Mn and irinotecan.
Isobolograms were generated to visualize the synergy read-outs at the doses required for each drug to provide 50% and 75% of cancer cell viability inhibition (Fig. 3E,F). We reported the 50% and 75% effect levels as IC50, which is frequently used to assess the potency of anticancer drugs. Additionally, it is critical to show synergy at a high inhibitory activity for oncology applications, as traditional anti-cancer drugs are used at or near the maximum tolerated dose. In the OVCAR-5 cell line, the combination data point at the 50% effect level is only slightly below the additive isobole indicating minimal synergy (Fig. 3E). On the other hand, the combination at the 75% effect level is further below the additive isobole suggesting a stronger synergetic effect. When minocycline is added to irinotecan, the dose requirement for irinotecan to achieve 50% OVCAR-5 viability inhibition reduced by almost 2-fold. For OVCAR-4, the combination data points at both 50% and 75% effect levels lie well below their additive isoboles, showing strong synergy between minocycline and irinotecan (Fig. 3F). The dose requirement to achieve 50% inhibition for irinotecan reduced by 7-fold to ~14 μM in the combination setting relative to irinotecan alone. This reduction in irinotecan dose level was even more pronounced at 75% inhibition at which the dose requirements were reduced by ~20-fold (irinotecan) and 6-fold (minocycline) as compared to each monotherapy.
Subhead 4: Combination of minocycline and nanoliposomal irinotecan reduce metastatic burden in peritoneal carcinomatosis mouse model of EOC
The vast majority (>75%) of EOC patients have intra-abdominal metastatic disease with nodules studding the peritoneal surface at the time of diagnosis. Therefore, we evaluated the impact of combining minocycline with a nanoliposomal formulation irinotecan (nal-IRI, Fig. S2, Table S1) on metastasis control in a mouse model for intraperitoneally disseminated EOC (Fig. 4A, see methods). Treatments were initiated seven days following orthotopic implantation of human OVCAR-5 cells in mice. For minocycline treatment, a single bolus intraperitoneal injection of minocycline (50 mg/kg) was administered on day 7, which was then followed by eight more minocycline injections (25 mg/kg) to allow direct access for the drug to the peritoneal cavity, where EOC has spread (Fig. 4A; see Methods). For nal-IRI chemotherapy, three intravenous injections of nal-IRI (20 mg/kg irinotecan hydrochloride salt) were administered over two weeks. Tumor weights from the pelvic omentum, bowel mesentery, subgastric omentum, uterus, diaphragm, were measured individually and were summed to reflect the site-specific and overall disease burdens. At 20-days post-implantation (i.e. 2-days after treatment termination), the combination of minocycline and nal-IRI dramatically reduced the tumor burden by 76±3% to an overall tumor weight of 311±33 mg, compared to 1278±159 mg in no-treatment mice (P < 0.01; Fig. 4B). Both minocycline monotherapy and nal-IRI alone only modestly reduced the overall tumor burden by 40±15% to a tumor weight of 743±52 mg, but this was not statistically different from the no-treatment animals. None of the monotherapies (minocycline nor nal-IRI) significantly reduced the site-specific disease burden. In contrast, combined minocycline and nal-IRI effectively reduced the tumor burden at the pelvic omentum, bowel mesentery, and subgastric omentum sites by 88%, 72%, and 58%, respectively, compared to the no-treatment group (P < 0.05; Fig. 4C–E). Tumor burden in the uterus and diaphragm were not significantly reduced by the combination treatment (Fig. 4F,G).
Fig. 4. Combined minocycline antibiotic and nal-IRI enhanced the anti-tumor effects in a peritoneal carcinomatosis mouse model.
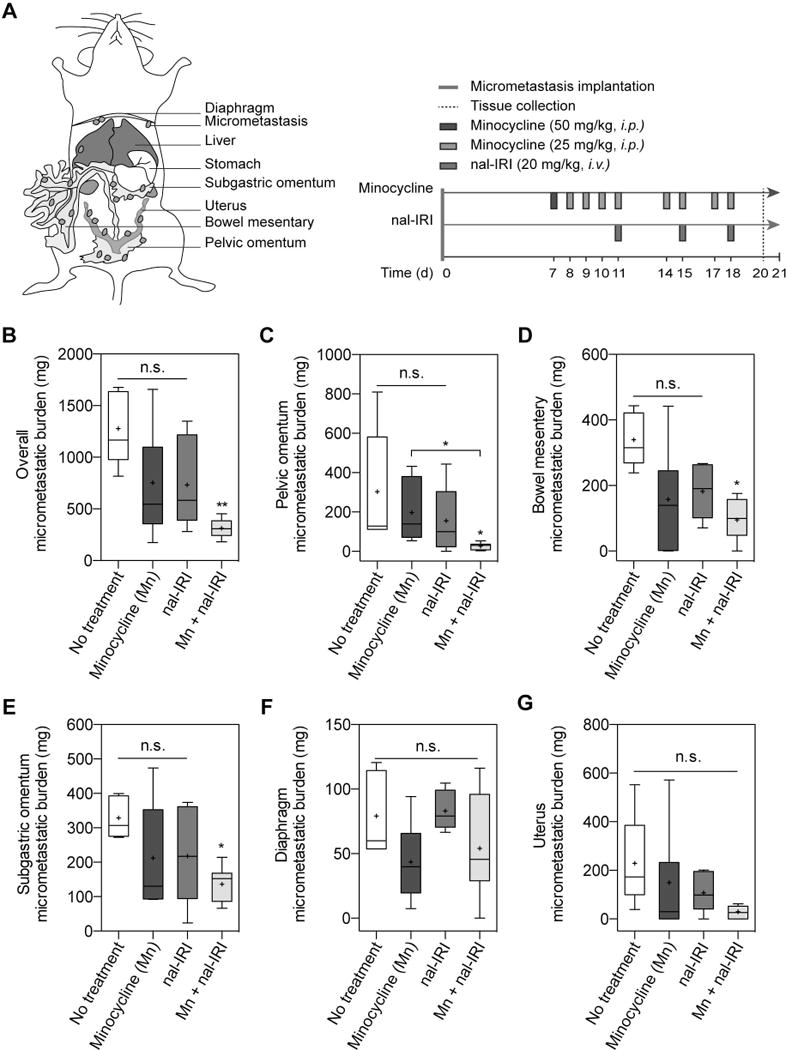
(A) To assess the efficacy of minocycline (Mn) and nal-IRI in controlling peritoneal carcinomatosis, treatments were initiated seven days after OVCAR-5 tumor implantation in mice randomized to the following groups: (i) no-treatment (NT); (ii) Mn; (iii) nal-IRI (three doses at 20 mg/kg each, on days 11, 15 and 18); and (iv) Combination of Mn and nal-IRI. On day 21 post-implantation, pelvic omentum, bowel mesentery, subgastric omentum, diaphragm, and uterus were resected and weighed at the time of necropsy. Corresponding tissue from healthy (non-tumored) mice were also collected, averaged, and subtracted from the tissue weight in tumored animals. The weights of the collected tissue were also summed for the total burden per animal. (B) Combination treatment of Mn and irinotecan significantly reduced the overall peritoneal carcinomatosis burden by >75%, compared to the NT control. (C-E) Tumor burdens at (C) pelvic omentum, (D) bowel mesentery, and (E) subgastric omentum were significantly lower after the combination treatment, compared to the NT control. Disease burden at (F) diaphragm and (G) uterus were not significantly altered after the treatments. (n = 5-7 animals per group; *P<0.05, **P<0.01, ***P<0.001, Kruskal–Wallis one-way ANOVA with Dunn’s post hoc test).
Subhead 5: Combining minocycline and nanoliposomal irinotecan prolong animal survival in peritoneal carcinomatosis mouse model of EOC
Most patients diagnosed with Stage IV EOC have a five-year survival rate of ~17% and often rapidly succumb to their disease. It was, therefore, critical to determine if the significant improvement in micrometastases control provided by combination minocycline and nal-IRI translated to a durable survival enhancement (Fig. 5A). Using moribundity (defined as inability to ambulate) as the endpoint, the no treatment group in the same mouse model for intraperitoneal metastatic EOC used in the previous section demonstrated a median overall survival (OS) time of only 21 days. As in the tumor burden studies, treatments for the survival studies were initiated 7 days after intraperitoneal tumor implantation. Minocycline combined with nal-IRI significantly prolonged the median OS by 50% to 32.5 days, compared to 21 and 22 days with nal-IRI alone and minocycline alone, respectively (P <0.01; Fig. 5B). All animals in the no-treatment, Mn alone, and nal-IRI alone groups were dead at days 27 and 32, and 34, respectively. Our second treatment timeline served to imitate a late intervention for late stage ovarian carcinomatosis. In this group, treatments were initiated 14 days after OVCAR-5 implantation (when animal death first occurs). Despite only 60% of animals completing the entire treatment course, the combination of minocycline and nal-IRI continued to extend the median OS to 30.5 days (P < 0.01), compared to the no-treatment animals (median OS 22 days) (P <0.01; Fig. 5C). Both minocycline alone and nal-IRI monotherapy showed no survival benefits compared to the no-treatment. These results, in two treatment schedules, suggest that minocycline repurposing offers a significant advantage to achieving significant and durable survival benefits with nal-IRI.
Fig. 5. A combination of minocycline and nal-IRI achieve significant survival enhancement in a peritoneal carcinomatosis mouse model.
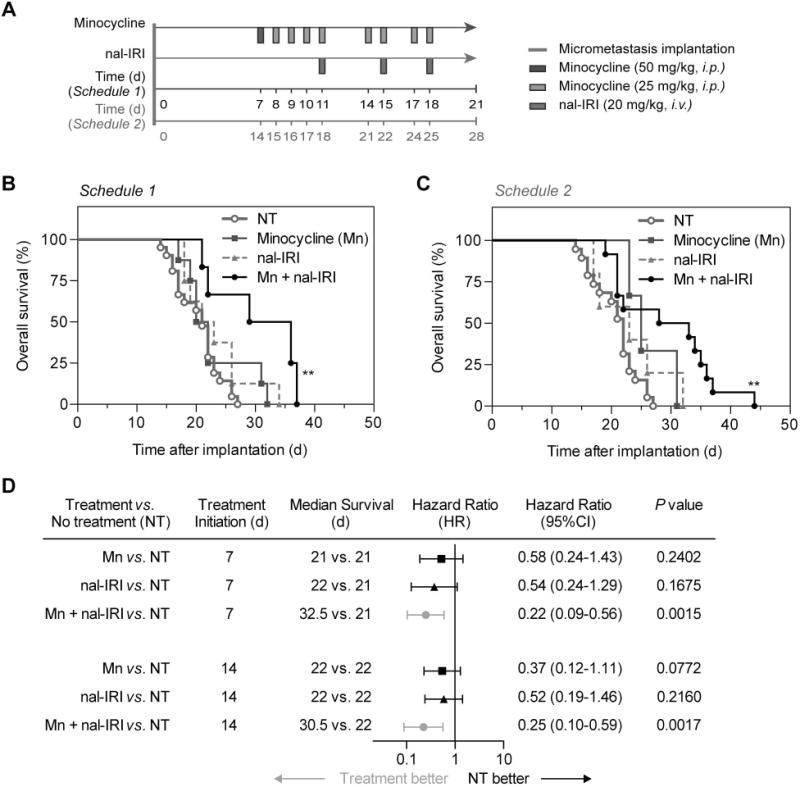
(A) Swiss nude mice were intraperitoneally inoculated with OVCAR-5 cells, divided into four groups, and subjected to (i) no-treatment (NT); (ii) Minocycline (Mn); (iii) nal-IRI (three doses at 20 mg/kg each); and (iv) Combination of Mn and nal-IRI. Treatments were initiated at 7 days (schedule 1) or 14 post-implantation (schedule 2). Moribundity and animal mobility (i.e. unable to move) were used as the endpoint with proper justification and special approval by MGH IACUC. (B, C) Kaplan-Meier plot of animal overall survival in the OVCAR-5 model. (n = 3-19 animals per group; P < 0.01, log-rank test between the NT and combination group). (D) A global test demonstrated a difference exists among the groups. Median survival time, hazard ratio forest plot, and differences in survival were evaluated by the log-rank test. Specifically, pairwise comparisons were performed to evaluate the advantage of treatment over no-treatment. Animals treated with the combination of Mn and nal-IRI were found to be significantly less likely to die by the next time point (hazard ratio < 1). No advantages to monotherapies (compared to NT) were observed.
The forest plot (Fig. 5D) summarizes the hazard ratio data across all conditions. Here, the hazard ratio is defined as the ratio of the probability of death in the treatment arm to the probability in the no-treatment (control) arm, and represents the instantaneous risk over the study time period. A hazard ratio of less than 0.25 (P <0.05) observed in the combination groups, suggests that animals treated with both minocycline and nal-IRI at any given time point were four times more likely to survive by the next time point compared to the no-treatment group (Fig. 5D). In contrast, all other treatment groups cross the 1.0 value, indicating the hazard ratio is not significant and there is no clear advantage for minocycline alone or nal-IRI alone compared to the no-treatment arm. Furthermore, mouse body weight was longitudinally monitored before and after treatment as a metric of side effects. In the combination group, the change in mouse weight was consistent with the monotherapies, indicating that co-administration of minocycline and nal-IRI does not appreciably add to the in vivo systemic toxicity (Fig. S3).
Discussion
Platinum-refractory and other resistant tumors represent a major problem in cancer management. Second-line therapies are critically needed to delay the time to symptomatic disease progression and prolong overall survival, but their effects are often mitigated by intrinsic and acquired resistance pathways. Therefore, it would be extremely useful to identify agents that can mechanistically counteract the natural defenses of cancer cells against therapeutic agents. These effects would be maximally enhanced if the counteracting entity synergistically interacted with the therapeutic agent in multiple ways. In this report, we present exactly such an example to treat metastatic EOC in two cell lines and in two orthotopic models in vivo.
Camptothecin derivatives, irinotecan and topotecan, are the only FDA-approved topoisomerase I inhibitors routinely prescribed for several cancers including ovarian cancer and gastrointestinal malignancies. Considering the consistently poor prognosis for platinum-refractory patients with peritoneal carcinomatosis, as well as the skyrocketing costs and extended development phase required to create new breakthrough medications, our findings offer mechanism-inspired prospects to repurpose antibiotic minocycline for enhancing the class of camptothecin derivatives with multiple advantages: (i) From a biological perspective, the ability of minocycline to affect the tdp1 pathway of DNA repair removes a barrier mitigating the effect of topoisomerase inhibitors. In addition, minocycline effectively suppresses of both constitutive and stimulated interleukin cytokines that are major contributing factors in cancer growth and progression; (ii) From the clinical and translational angle, antibiotics are already commonly used—on an as-needed basis—to reduce the risk of infection-related death after chemotherapy(53). Moreover, the adverse events of minocycline are milder or non-overlapping with the major side effects of irinotecan; and (iii) Given the high cost of new drug development, repurposing of minocycline to potentiate irinotecan makes economical sense if the combination becomes available for cancer patients.
This report shows that minocycline reduces Tdp1 expression level in platinum-resistant cancer cells in a treatment time-dependent manner. These findings confirmed the earlier hypothesis and reports by Pommier and colleagues(16,19,23) and furthermore implements the potential application. We also demonstrated that the combination of minocycline and irinotecan is highly synergistic (CI<0.3) in OVCAR-4 cells expressing high levels of Tdp1, while only an additive-to-mild synergy was observed in OVCAR-5 cells that express lower levels of Tdp1 (Fig. S1). These observations suggest a crucial role of Tdp1 levels in the combination of minocycline and irinotecan, which warrants further study. So far, there are no reported structural-activity relationships providing insights into minocycline’s inhibitory effect against Tdp1. Based on data in the literature, we suggest that the reported minocycline inhibition of PARP-1(54) may interfere PARP1–Tdp1 coupling, which is essential for the repair of trapped Top1cc by Tdp1(55), thereby, removing a barrier mitigating the effect of topoisomerase inhibitors. In addition to minocycline reduction of Tdp1 levels, this study presents a mutually reinforcing mechanistic cooperation between minocycline and irinotecan to simultaneously suppress VEGF and IL-8 secretion in vitro. Further in vivo validation to determine IL-8 and VEGF levels in serum is necessary to explore the impacts of minocycline and irinotecan combination therapy on the activity of innate immune cells and ascites formation.
The above mechanistic findings motivated us to evaluate the efficacy of combined minocycline and irinotecan in vivo. Previous studies by Pourgholami et al. have shown that daily minocycline alone treatment at 10–30 mg/kg via drinking water is well-tolerated by mice and effectively controlled the tumor growth in a subcutaneous model of OVCAR-3(38). Here, using a more challenging mouse model of disseminated ovarian micrometastases, we demonstrate that routine intraperitoneal injection of minocycline (25 mg/kg, see Methods) alone have negligible antitumor effects and no survival benefits (Fig. 4–5). Only minocycline combined with a clinically relevant low-dose irinotecan in a nanoliposomal formulation (20 mg/kg irinotecan hydrochloride salt; Table S1) achieved a meaningful survival improvement, without appreciable addition to the in vivo systemic toxicity. Clinically, the common adverse events that may occur within minocycline (e.g. allergic reaction, nausea, rash, and mild diarrhea)(56) are milder or non-overlapping with the major side effects of irinotecan (e.g. neutropenia, diarrhea)(15), thus affording another compelling rationale to repurpose minocycline for patients receiving irinotecan. In addition, antibiotic prophylaxis in afebrile neutropenic patients is already clinically feasible and beneficial in reducing the risk of infection-related death(53). Specifically, minocycline has shown promises in preventing neurotoxicity(57), one of the side effects that limit the use of platinum-based drugs, and several clinical trials are already ongoing to investigate the value of minocycline in reducing the side effects for cancer patients who received chemo-radiation therapy and surgery (Clinicaltrials.gov: NCT01636934, NCT02055963, NCT01173692, NCT01693523).
Clinically, oral or intravenous administration of minocycline at 200 mg is safe and results in a peak plasma concentration (Cmax) of 3–4 μg/mL(45,46,56). In our animal study, minocycline was intraperitoneally administered using an established in vivo dosing schedule (Table S2) to achieve a similar minocycline Cmax of 5–10 μg/mL in mice(45,46,56). In light of the recent FDA-approved liposomal irinotecan injection (ONIVYDE®) with favorable pharmacokinetic and safety profiles(43), a similar laboratory formulation of nal-IRI, established and well characterized by us, was used intravenously in this animal study(44) (Fig. S2, Table S2). Further investigations of intraperitoneal co-injection of minocycline and irinotecan (or its more potent metabolite SN-38) could potentially result in superior outcomes in vivo. Because simultaneously increasing the rate of DNA nicking (by irinotecan arresting Top1cc) and reducing the rate of DNA religation (by minocycline reduction of Tdp1 level) is crucial to ensure the steady state concentration of Top1cc complexes remains high in cancer cells. Looking forward, advances of multi-agent nanoliposomal formulations coupled with targeted delivery capability hold high potential to simultaneously co-delivery both agents to further improve therapeutic outcomes in the future(58).
The average financial burden of health care for advanced ovarian cancer patients is above $65,000 during the initial treatment period(59). A preliminary cost analysis of including minocycline to irinotecan chemotherapy regimen indicates significant drug cost savings compared to other irinotecan-based combination therapies (Table S3 and Table S4). While the combination of etoposide and irinotecan in a phase II study has low costs comparable to the proposed combination of minocycline and irinotecan(14), serious side effects, particularly myelosuppression, were common despite a significant reduction in irinotecan dose. Conversely, the combination of bevacizumab and irinotecan regimen resulted in significantly lower toxicity due to a dose reduction of both agents(13), but lead to a much higher overall cost due to the bevacizumab.
Given the high cost and modest impact of new oncology medicine, this study makes a strong case for mechanism-based mutually reinforcing combinations such as minocycline (Tdp1 inhibitor) and irinotecan (Top1 intercalator) provide cost-effective translatable repurposing of drugs and warrant further clinical investigation as an effective treatment for treatment-refractory cancers.
Supplementary Material
Acknowledgments
The authors thank Mr. Zachary Silber for help with discussions and experimental repeats.
Financial support: This work was supported by National Institutes of Health Grants P01CA084203 (T. Hasan), R01CA156177 (T. Hasan), R01CA160998 (T. Hasan), K99CA194269 (H-C. Huang), K99CA175292 (I. Rizvi), and R00CA175292 (I. Rizvi).
Abbreviations list
- CI
Combination index
- DOTAP
Dioleoyltrimethylammoniumpropane
- DPPC
Dipalmitoylphosphatidylcholine
- DSPE-PEG
Distearoylphosphatidylethanolamine-methoxy polyethylene glycol
- EOC
Epithelial ovarian cancer
- IC50
Half maximal inhibitory concentration
- IACUC
Institutional Animal Care and Use Committee
- IL-6
Interleukin-6
- IL-8
Interleukin-8
- MGH
Massachusetts General Hospital
- nal-IRI
Nanoliposomal irinotecan
- Cmax
Peak plasma concentration
- PBS
Phosphate-buffered saline
- RT
Room temperature
- SEM
Standard error of the mean
- Top1cc
Top1-DNA cleavage complexes
- Top1
Topoisomerase I
- Tdp1
Tyrosyl-DNA phosphodiesterase-1
- VEGF
Vascular endothelial growth factor
Footnotes
Competing interests: The authors declare no potential conflicts of interest.
References
- 1.Steeg PS. Targeting metastasis. Nat Rev Cancer. 2016;16:201–18. doi: 10.1038/nrc.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ceelen WP, Flessner MF. Intraperitoneal therapy for peritoneal tumors: biophysics and clinical evidence. Nature reviews Clinical oncology. 2010;7:108–15. doi: 10.1038/nrclinonc.2009.217. [DOI] [PubMed] [Google Scholar]
- 3.Lambert LA. Looking up: Recent advances in understanding and treating peritoneal carcinomatosis. CA Cancer J Clin. 2015;65:284–98. doi: 10.3322/caac.21277. [DOI] [PubMed] [Google Scholar]
- 4.Vaughan S, Coward JI, Bast RC, Berchuck A, Berek JS, Brenton JD, et al. Rethinking ovarian cancer: recommendations for improving outcomes. Nat Rev Cancer. 2011;11:719–25. doi: 10.1038/nrc3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lengyel E. Ovarian cancer development and metastasis. The American journal of pathology. 2010;177:1053–64. doi: 10.2353/ajpath.2010.100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bowtell DD, Bohm S, Ahmed AA, Aspuria PJ, Bast RC, Jr, Beral V, et al. Rethinking ovarian cancer II: reducing mortality from high-grade serous ovarian cancer. Nat Rev Cancer. 2015;15:668–79. doi: 10.1038/nrc4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Narod S. Can advanced-stage ovarian cancer be cured? Nature reviews Clinical oncology. 2016;13:255–61. doi: 10.1038/nrclinonc.2015.224. [DOI] [PubMed] [Google Scholar]
- 8.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 9.Coleman RL, Monk BJ, Sood AK, Herzog TJ. Latest research and treatment of advanced-stage epithelial ovarian cancer. Nature reviews Clinical oncology. 2013;10:211–24. doi: 10.1038/nrclinonc.2013.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Musa F, Pothuri B, Blank SV, Ling HT, Speyer JL, Curtin J, et al. Phase II study of irinotecan in combination with bevacizumab in recurrent ovarian cancer. Gynecologic Oncology. 2017;144:279–84. doi: 10.1016/j.ygyno.2016.11.043. [DOI] [PubMed] [Google Scholar]
- 11.Gershenson DM. Irinotecan in epithelial ovarian cancer. Oncology (Williston Park, NY) 2002;16:29–31. [PubMed] [Google Scholar]
- 12.Bodurka DC, Levenback C, Wolf JK, Gano J, Wharton JT, Kavanagh JJ, et al. Phase II Trial of Irinotecan in Patients With Metastatic Epithelial Ovarian Cancer or Peritoneal Cancer. Journal of Clinical Oncology. 2003;21:291–7. doi: 10.1200/JCO.2003.02.091. [DOI] [PubMed] [Google Scholar]
- 13.Liu Y, Ren Z, Xu S, Bai H, Ma N, Wang F. Low-dose-intensity bevacizumab with weekly irinotecan for platinum- and taxanes-resistant epithelial ovarian cancer. Cancer Chemotherapy and Pharmacology. 2015;75:645–51. doi: 10.1007/s00280-015-2680-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shoji T, Takatori E, Omi H, Kumagai S, Yoshizaki A, Yokoyama Y, et al. Phase II clinical study of the combination chemotherapy regimen of irinotecan plus oral etoposide for the treatment of recurrent ovarian cancer (Tohoku Gynecologic Cancer Unit 101 Group Study) International journal of gynecological cancer : official journal of the International Gynecological Cancer Society. 2011;21:44–50. doi: 10.1097/IGC.0b013e3181ffbe9f. [DOI] [PubMed] [Google Scholar]
- 15.Ko AH, Tempero MA, Shan YS, Su WC, Lin YL, Dito E, et al. A multinational phase 2 study of nanoliposomal irinotecan sucrosofate (PEP02, MM-398) for patients with gemcitabine-refractory metastatic pancreatic cancer. British Journal of Cancer. 2013;109:920–5. doi: 10.1038/bjc.2013.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 17.Pouliot JJ, Yao KC, Robertson CA, Nash HA. Yeast gene for a Tyr-DNA phosphodiesterase that repairs topoisomerase I complexes. Science. 1999;286:552–5. doi: 10.1126/science.286.5439.552. [DOI] [PubMed] [Google Scholar]
- 18.Pommier Y, Huang S-yN, Gao R, Das BB, Murai J, Marchand C. Tyrosyl-DNA-phosphodiesterases (TDP1 and TDP2) DNA repair. 2014;19:114–29. doi: 10.1016/j.dnarep.2014.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang SN, Pommier Y, Marchand C. Tyrosyl-DNA Phosphodiesterase 1 (Tdp1) inhibitors. Expert opinion on therapeutic patents. 2011;21:1285–92. doi: 10.1517/13543776.2011.604314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zakharenko A, Luzina O, Koval O, Nilov D, Gushchina I, Dyrkheeva N, et al. Tyrosyl-DNA Phosphodiesterase 1 Inhibitors: Usnic Acid Enamines Enhance the Cytotoxic Effect of Camptothecin. Journal of natural products. 2016;79:2961–7. doi: 10.1021/acs.jnatprod.6b00979. [DOI] [PubMed] [Google Scholar]
- 21.Gao R, Das BB, Chatterjee R, Abaan O, Agama K, Matuo R, et al. Epigenetic and genetic inactivation of tyrosyl-DNA-phosphodiesterase 1 (TDP1) in human lung cancer cells from the NCI-60 panel. DNA repair. 2014;13:1–9. doi: 10.1016/j.dnarep.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meisenberg C, Ward SE, Schmid P, El-Khamisy SF. TDP1/TOP1 Ratio as a Promising Indicator for the Response of Small Cell Lung Cancer to Topotecan. Journal of cancer science & therapy. 2014;6:258–67. doi: 10.4172/1948-5956.1000280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marchand C, Lea WA, Jadhav A, Dexheimer TS, Austin CP, Inglese J, et al. Identification of phosphotyrosine mimetic inhibitors of human tyrosyl-DNA phosphodiesterase I by a novel AlphaScreen high-throughput assay. Molecular cancer therapeutics. 2009;8:240–8. doi: 10.1158/1535-7163.MCT-08-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weidlich IE, Dexheimer T, Marchand C, Antony S, Pommier Y, Nicklaus MC. Inhibitors of human tyrosyl-DNA phospodiesterase (hTdp1) developed by virtual screening using ligand-based pharmacophores. Bioorganic & medicinal chemistry. 2010;18:182–9. doi: 10.1016/j.bmc.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kipps E, Tan DSP, Kaye SB. Meeting the challenge of ascites in ovarian cancer: new avenues for therapy and research. Nat Rev Cancer. 2013;13:273–82. doi: 10.1038/nrc3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Masoumi Moghaddam S, Amini A, Morris DL, Pourgholami MH. Significance of vascular endothelial growth factor in growth and peritoneal dissemination of ovarian cancer. Cancer Metastasis Reviews. 2012;31:143–62. doi: 10.1007/s10555-011-9337-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008;14:6735–41. doi: 10.1158/1078-0432.CCR-07-4843. [DOI] [PubMed] [Google Scholar]
- 28.Santin AD, Hermonat PL, Ravaggi A, Cannon MJ, Pecorelli S, Parham GP. Secretion of vascular endothelial growth factor in ovarian cancer. European journal of gynaecological oncology. 1999;20:177–81. [PubMed] [Google Scholar]
- 29.Bamias A, Koutsoukou V, Terpos E, Tsiatas ML, Liakos C, Tsitsilonis O, et al. Correlation of NK T-like CD3+CD56+ cells and CD4+CD25+(hi) regulatory T cells with VEGF and TNFα in ascites from advanced ovarian cancer: Association with platinum resistance and prognosis in patients receiving first-line, platinum-based chemotherapy. Gynecologic Oncology. 2008;108:421–7. doi: 10.1016/j.ygyno.2007.10.018. [DOI] [PubMed] [Google Scholar]
- 30.Kassim SK, El-Salahy EM, Fayed ST, Helal SA, Helal T, Azzam EE-d, et al. Vascular endothelial growth factor and interleukin-8 are associated with poor prognosis in epithelial ovarian cancer patients. Clinical Biochemistry. 2004;37:363–9. doi: 10.1016/j.clinbiochem.2004.01.014. [DOI] [PubMed] [Google Scholar]
- 31.Shahzad MM, Arevalo JM, Armaiz-Pena GN, Lu C, Stone RL, Moreno-Smith M, et al. Stress effects on FosB- and interleukin-8 (IL8)-driven ovarian cancer growth and metastasis. The Journal of biological chemistry. 2010;285:35462–70. doi: 10.1074/jbc.M110.109579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang S, Robinson JB, Deguzman A, Bucana CD, Fidler IJ. Blockade of nuclear factor-kappaB signaling inhibits angiogenesis and tumorigenicity of human ovarian cancer cells by suppressing expression of vascular endothelial growth factor and interleukin 8. Cancer Res. 2000;60:5334–9. [PubMed] [Google Scholar]
- 33.Coward JIG, Middleton K, Murphy F. New perspectives on targeted therapy in ovarian cancer. International Journal of Women's Health. 2015;7:189–203. doi: 10.2147/IJWH.S52379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kimura T, Takabatake Y, Takahashi A, Isaka Y. Chloroquine in cancer therapy: a double-edged sword of autophagy. Cancer Res. 2013;73:3–7. doi: 10.1158/0008-5472.CAN-12-2464. [DOI] [PubMed] [Google Scholar]
- 35.Kasznicki J, Sliwinska A, Drzewoski J. Metformin in cancer prevention and therapy. Annals of Translational Medicine. 2014;2:57. doi: 10.3978/j.issn.2305-5839.2014.06.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. 2014;159:80–93. doi: 10.1016/j.cell.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pantziarka P, Sukhatme V, Bouche G, Meheus L, Sukhatme VP. Repurposing Drugs in Oncology (ReDO)—itraconazole as an anti-cancer agent. ecancermedicalscience. 2015;9:521. doi: 10.3332/ecancer.2015.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pourgholami MH, Mekkawy AH, Badar S, Morris DL. Minocycline inhibits growth of epithelial ovarian cancer. Gynecol Oncol. 2012;125:433–40. doi: 10.1016/j.ygyno.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 39.Ataie-Kachoie P, Morris DL, Pourgholami MH. Minocycline suppresses interleukine-6, its receptor system and signaling pathways and impairs migration, invasion and adhesion capacity of ovarian cancer cells: in vitro and in vivo studies. PLoS One. 2013;8:e60817. doi: 10.1371/journal.pone.0060817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coward J, Kulbe H, Chakravarty P, Leader D, Vassileva V, Leinster DA, et al. Interleukin-6 as a Therapeutic Target in Human Ovarian Cancer. Clinical Cancer Research. 2011;17:6083–96. doi: 10.1158/1078-0432.CCR-11-0945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duncan TJ, Al-Attar A, Rolland P, Scott IV, Deen S, Liu DT, et al. Vascular endothelial growth factor expression in ovarian cancer: a model for targeted use of novel therapies? Clin Cancer Res. 2008;14:3030–5. doi: 10.1158/1078-0432.CCR-07-1888. [DOI] [PubMed] [Google Scholar]
- 42.Drummond DC, Noble CO, Guo Z, Hong K, Park JW, Kirpotin DB. Development of a highly active nanoliposomal irinotecan using a novel intraliposomal stabilization strategy. Cancer Res. 2006;66:3271–7. doi: 10.1158/0008-5472.CAN-05-4007. [DOI] [PubMed] [Google Scholar]
- 43.Carnevale J, Ko AH. MM-398 (nanoliposomal irinotecan): emergence of a novel therapy for the treatment of advanced pancreatic cancer. Future oncology (London, England) 2016;12:453–64. doi: 10.2217/fon.15.333. [DOI] [PubMed] [Google Scholar]
- 44.Huang HC, Mallidi S, Liu J, Chiang CT, Mai Z, Goldschmidt R, et al. Photodynamic Therapy Synergizes with Irinotecan to Overcome Compensatory Mechanisms and Improve Treatment Outcomes in Pancreatic Cancer. Cancer Res. 2016;76:1066–77. doi: 10.1158/0008-5472.CAN-15-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wells JE, Hurlbert RJ, Fehlings MG, Yong VW. Neuroprotection by minocycline facilitates significant recovery from spinal cord injury in mice. Brain : a journal of neurology. 2003;126:1628–37. doi: 10.1093/brain/awg178. [DOI] [PubMed] [Google Scholar]
- 46.Agwuh KN, MacGowan A. Pharmacokinetics and pharmacodynamics of the tetracyclines including glycylcyclines. The Journal of antimicrobial chemotherapy. 2006;58:256–65. doi: 10.1093/jac/dkl224. [DOI] [PubMed] [Google Scholar]
- 47.Stronach EA, Cunnea P, Turner C, Guney T, Aiyappa R, Jeyapalan S, et al. The role of interleukin-8 (IL-8) and IL-8 receptors in platinum response in high grade serous ovarian carcinoma. Oncotarget. 2015;6:31593–603. doi: 10.18632/oncotarget.3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mayerhofer K, Bodner K, Bodner-Adler B, Schindl M, Kaider A, Hefler L, et al. Interleukin-8 serum level shift in patients with ovarian carcinoma undergoing paclitaxel-containing chemotherapy. Cancer. 2001;91:388–93. [PubMed] [Google Scholar]
- 49.O'Meara AT, Sevin B-U. In VitroSensitivity of Fresh Ovarian Carcinoma Specimens to CPT-11 (Irinotecan) Gynecologic Oncology. 1999;72:143–7. doi: 10.1006/gyno.1998.5241. [DOI] [PubMed] [Google Scholar]
- 50.HOFMANN C, BUTTENSCHOEN K, STRAETER J, HENNE-BRUNS D, KORNMANN M. Pre-clinical Evaluation of the Activity of Irinotecan as a Basis for Regional Chemotherapy. Anticancer Research. 2005;25:795–804. [PubMed] [Google Scholar]
- 51.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacological reviews. 2006;58:621–81. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 52.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Advances in enzyme regulation. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 53.Gafter-Gvili A, Fraser A, Paul M, Vidal L, Lawrie TA, van de Wetering MD, et al. Antibiotic prophylaxis for bacterial infections in afebrile neutropenic patients following chemotherapy. The Cochrane database of systematic reviews. 2012;1:Cd004386. doi: 10.1002/14651858.CD004386.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alano CC, Kauppinen TM, Valls AV, Swanson RA. Minocycline inhibits poly(ADP-ribose) polymerase-1 at nanomolar concentrations. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:9685–90. doi: 10.1073/pnas.0600554103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Das BB, Huang S-yN, Murai J, Rehman I, Amé J-C, Sengupta S, et al. PARP1–TDP1 coupling for the repair of topoisomerase I–induced DNA damage. Nucleic Acids Research. 2014;42:4435–49. doi: 10.1093/nar/gku088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goulden V, Glass D, Cunliffe WJ. Safety of long-term high-dose minocycline in the treatment of acne. The British journal of dermatology. 1996;134:693–5. doi: 10.1111/j.1365-2133.1996.tb06972.x. [DOI] [PubMed] [Google Scholar]
- 57.Plane JM, Shen Y, Pleasure DE, Deng W. Prospects for Minocycline Neuroprotection. Archives of neurology. 2010;67:1442–8. doi: 10.1001/archneurol.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.H HC, H T. The “Nano” World in Photodynamic Therapy Austin. J Nanomed Nanotechnol. 2014;2:1020. [PMC free article] [PubMed] [Google Scholar]
- 59.Urban RR, He H, Alfonso-Cristancho R, Hardesty MM, Goff BA. The Cost of Initial Care for Medicare Patients With Advanced Ovarian Cancer. Journal of the National Comprehensive Cancer Network : JNCCN. 2016;14:429–37. doi: 10.6004/jnccn.2016.0049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.