

Abstract
The mineralocorticoid receptor (MR) and its downstream signaling play an important role in hypertensive renal injury. The interaction of advanced glycation end products (AGE) with their receptor (RAGE) is involved in the progression of renal disease. However, the pathological crosstalk between AGE–RAGE axis and MR system in kidney derangement remains unclear. We screened DNA-aptamer directed against RAGE (RAGE-apt) in vitro and examined its effects on renal injury in uninephrectomized deoxycorticosterone acetate (DOCA)/salt-induced hypertensive mice. RAGE, GTP-bound Rac-1 (Rac1), and MR were co-localized in the podocytes of DOCA mice. The deletion of RAGE gene significantly inhibited mesangial matrix expansion and tubulointerstitial fibrosis in DOCA mice, which was associated with the reduction of glomerular oxidative stress, MR, Rac1, and urinary albumin excretion (UAE) levels. RAGE-apt attenuated the increase in carboxymethyllysine (CML), RAGE, nitrotyrosine, Rac1, and MR levels in the kidneys and reduced UAE in DOCA mice. Aldosterone (Aldo) increased nitrotyrosine, CML, and RAGE gene expression in murine podocytes, whereas CML stimulated MR and Rac1 levels, which were blocked by RAGE-apt. The present study indicates the crosstalk between the AGE–RAGE axis and Aldo–MR system, suggesting that RAGE-apt may be a novel therapeutic tool for the treatment of MR-associated renal diseases.
Introduction
Hypertensive nephropathy (HN) is one of the common causes of end-stage renal disease requiring renal replacement therapy in industrialized countries1,2. Chronic hypertension induces arteriosclerosis in the renal afferent and efferent arterioles and subsequently causes ischemic changes in the glomeruli via the intrarenal renin–angiotensin–aldosterone (Aldo) system (RAAS) activation, thereby leading to renal damage, including podocyte loss, glomerular sclerosis, and tubulointerstitial fibrosis3–5. Furthermore, mineralocorticoid receptor (MR) activation in podocyte has been reported to accelerate the development and progression of HN6. Since the inhibition of MR activation attenuates Aldo-induced podocyte injury in an animal model of salt-sensitive HN7, the blockade of MR activation may be a promising therapeutic strategy for podocyte damage in HN. Indeed, several blood-lowering agents, such as MR antagonists and/or angiotensin II type 1 receptor blockers, show beneficial effects on HN8,9. However, the effects of these agents on HN are modest and partial; hence, pathways other than RAAS may also be involved in the progression of HN10.
Advanced glycation end products (AGE) are a heterogeneous group of molecules formed by a non-enzymatic reaction between reducing sugars and amino acids11. AGE modification of proteins causes not only structural alteration but also interacts with the receptor for AGE (RAGE), contributing to the development of numerous age-related devastating disorders, such as chronic kidney disease, cardiovascular diseases, and cancer12–14. Furthermore, since AGE activate Aldo–MR pathway through the interaction with RAGE on MRC-5 fibroblasts15, it is conceivable that the AGE–RAGE system may accelerate MR downstream pathways, thereby showing involvement in renal injury.
Aptamers are short, single-stranded DNA or RNA molecules that can bind with high affinity and specificity to a wide range of target proteins16. Recently, we reported that DNA-aptamer (DNA-apt) directed against AGE inhibited the binding of AGE to RAGE and attenuated renal injury in obese type 2 diabetic mice17,18. These findings suggest that aptamers may be a therapeutic tool in the prevention of AGE–RAGE-related disorders. Therefore, in this study, we screened a high-affinity DNA-apt directed against RAGE (RAGE-apt) using a combinatorial chemistry in vitro, and we examined its effects on renal injury in uninephrectomized deoxycorticosterone acetate (DOCA)/salt-induced hypertensive mice (DOCA mice). Furthermore, we explored how the AGE–RAGE axis activated MR downstream pathways using immortalized murine podocyte cells (MPC).
Results
Distribution of RAGE, GTP-Bound Rac1, and MR in the Glomeruli of DOCA Mice
First, we investigated the co-localization and distribution of RAGE, MR, and Rac1 in the glomeruli of uninephrectomized DOCA/salt-induced hypertensive mice (DOCA mice) by an immunofluorescence double staining with RAGE, MR, GTP-bound Rac1, and Wilms’ tumor 1 (WT-1), a marker of podocytes. RAGE, GTP-bound Rac1, and MR were merged with WT-1 (Fig. 1a). Further, immunofluorescence double staining demonstrated that RAGE, GTP-bound Rac1, and MR were co-localized with each other (Fig. 1b) in podocytes in the kidneys of DOCA mice.
Figure 1.

RAGE, GTP-bound Rac1, and MR were co-localized in podocytes of DOCA/salt-induced hypertensive mice. (a) Sections of kidney tissue from DOCA mice showing glomeruli co-stained for RAGE, GTP-bound Rac1, and MR (green), the podocyte marker WT1 (red), and overlaid images (yellow). (b) Sections of kidney tissue from DOCA mice co-stained for RAGE, GTP-bound Rac1, and MR and their merged images. RAGE, GTP-bound Rac1, and MR were together co-localized in podocytes from DOCA mice (arrows). All sections are 4-μm thin and evaluated by confocal microscopy. Bars = 20 μm. RAGE, receptor for advanced glycation end products; WT1, Wilms’ tumor 1; MR, mineralocorticoid receptor.
Renal RAGE Expression in DOCA Mice
Renal RAGE expression was significantly upregulated in DOCA mice compared with uninephrectomized salt-administrated mice (Cont mice) (Fig. 2a). We confirmed that RAGE was strongly expressed in the lung of C57BL/6 J (wild type, WT) mice, which was completely deleted in RAGE-knockout (RAGE-KO) mice (Fig. 2b).
Figure 2.

(a) RAGE protein expression in the kidney of Cont and DOCA mice (n = 3, respectively), (b) RAGE protein expression in lung of WT and RAGE-KO mice, (c) SBP levels of Cont, DOCA, and DOCA–RAGE-KO mice (n = 5–8 per group), §p < 0.05 vs Cont, (d) UAE levels of Cont, DOCA, and DOCA–RAGE-KO mice (n = 5–6 per group), (e and f) mesangial matrix expansion and tubulointerstitial fibrosis assessed by periodic acid-Schiff and Masson’s trichrome stains, respectively, in Cont, DOCA, and DOCA–RAGE-KO mice (n = 5 per group), (g–i) immunohistochemical analysis for glomerular 8-OHdG, GTP-bound Rac1, and MR levels in Cont, DOCA, and DOCA–RAGE KO mice (n = 5 per group), (j) glomerular CML staining by immunohistochemistry (n = 4–5 per group), (k) podocin expression (n = 4–5 per group), (l) cortical mRNA expression levels of MR, SGK1, and TGF-β in Cont, DOCA, and DOCA–RAGE-KO mice (n = 5 per group). Data are presented as the mean ± SEM. #p < 0.05, ##p < 0.01 vs. DOCA mice. All sections were 4-μm thin. Bars = 20 μm. RAGE, receptor for advanced glycation end products; Cont, control; WT, wild type; KO, knockout; 8-OHdG, 8-hydroxy-2ʹ-deoxyguanosine; MR, mineralocorticoid receptor; SGK1, serum/glucocorticoid regulated kinase1; TGF-β, transforming growth factor-β.
DOCA-Induced Renal Injury was Attenuated in RAGE-KO Mice
We next studied whether activation of MR by DOCA/salt-induced renal damage, which was attenuated in RAGE KO mice. Systolic blood pressure (SBP), blood urea nitrogen (BUN), and creatinine (Cr) levels significantly elevated in DOCA and DOCA–RAGE-KO mice compared with the Cont mice. Although there were no significant differences in terms of SBP (Fig. 2c) and plasma Cr level (Table 1) between the DOCA mice and DOCA–RAGE-KO mice, plasma BUN level tended to be attenuated in DOCA–RAGE-KO mice compared to DOCA mice (p = 0.065) (Table 1). The increases in urinary albumin excretion (UAE), mesangial matrix expansion, and tubulointerstitial fibrosis in DOCA mice were significantly reduced in DOCA–RAGE-KO mice (UAE; 184.3 ± 29.4 vs 47.7 ± 24.2 μg/mg Cr) (Fig. 2d–f). 8-hydroxy-2′-deoxyguanosine (8-OHdG), glomerular CML deposition, Rac1, and MR levels were increased in the kidneys of DOCA mice, all of which were attenuated in DOCA–RAGE-KO mice (Fig. 2g–j). Podocin expression was decreased in the glomeruli of DOCA mice, which was prevented by the genetic deletion of RAGE in DOCA-RAGE-KO mice (Fig. 2k), suggesting that RAGE could be involved in the progression of podocyte injury in MR-activated kidney injury. In addition, enhanced renal gene expression of MR, serine/threonine-protein kinase 1 (SGK1), and transforming growth factor β (TGF-β) were significantly ameliorated in DOCA-RAGE-KO mice (Fig. 2l).
Table 1.
Clinical characteristics of mice in experiment design.
| Parameters | cont | DOCA | DOCA-RAGE-KO | DOCA | |
|---|---|---|---|---|---|
| Ctrl-apt | RAGE-apt | ||||
| Body weight (g) | 23.3 ± 0.6 | 20.3 ± 0.9* | 21.3 ± 0.5* | 20.4 ± 0.2 | 21.6 ± 0.7 |
| KW/BW ratio (mg/g) | 9.54 ± 0.5 | 11.9 ± 0.4* | 12.3 ± 0.6* | 11.7 ± 0.5 | 10.1 ± 0.3# |
| Serum BUN (mg/dL) | 30.7 ± 0.7 | 57.9 ± 3.1* | 48.6 ± 3.8*‡ | 60.0 ± 4.5 | 63.8 ± 3.6 |
| Serum Cr (mg/dL) | 0.10 ± 0.01 | 0.18 ± 0.02* | 0.18 ± 0.01* | 0.17 ± 0.02 | 0.20 ± 0.01 |
Data are mean ± SEM. *p < 0.05 vs. Ctrl, #p < 0.05 vs DOCA-Ctrl-apt mice, ‡p = 0.065 vs DOCA.
RAGE: receptor for advanced glycation end products, KO: knockout, Ctrl: control, apt: aptamer, KW: kidney weight, BW: body weight, BUN: blood urea nitrogen, Cr: creatinine.
Characterization of RAGE-aptamers
We selected 7 RAGE-apts by a Systematic Evolution of Ligands using the EXponential enrichment (SELEX) method as previously described18. We selected #2 RAGE-apt and used it in the following experiments because of its strongest antagonistic ability for RAGE. #2 RAGE-apt has double bulge loops (Fig. 3a), which may be important for binding to the v-domain of the human RAGE (v-RAGE). A sensitive 27-MHz quartz crystal microbalance (QCM) also revealed that #2 RAGE-apt bound to v-RAGE with a dissociation constant (Kd) of 0.1 nM; RAGE-apt markedly blocked the binding of CML-BSA to v-RAGE (Fig. 3b). We also found that CML-BSA bound to v-RAGE with a Kd of 28.3 nM.
Figure 3.
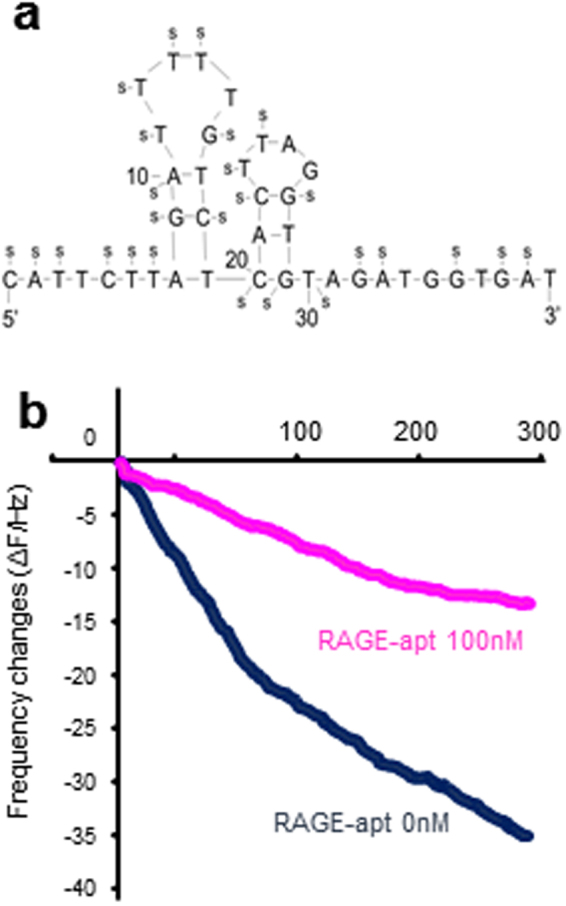
(a) Structure of #2 RAGE-apt, (b) RAGE-apt (100 nM) blocks the binding of CML to vRAGE assessed by QCM method. RAGE, receptor for advanced glycation end products; RAGE-apt, RAGE-aptamer; Ctrl-apt, control-aptamer; ELISA, enzyme-linked immunosorbent assay; CML-BSA, carboxymethyllysine-bovine serum albumin; QCM, quartz crystal microbalance.
Effects of RAGE-aptamer on Renal Injury in DOCA mice
Although the continuous administration of RAGE-apt did not affect SBP in DOCA mice, it reduced the increase in UAE (Fig. 4a–c). As with DOCA-RAGE-KO mice, podocin expression decreased in DOCA-Ctrl-apt–treated mice, which was restored in RAGE-apt–treated DOCA mice (Fig. 4d). Nitrotyrosine (NT) was co-localized with CML in the podocytes of DOCA-Ctrl-apt mice, and their levels increased in DOCA-Ctrl-apt–treated mice, both of which were suppressed by RAGE-apt treatment (Fig. 4e). The increases in glomerular CML, RAGE, MR, and Rac1 expressions as well as plasma CML and cortical RAGE levels in DOCA-Ctrl-apt–treated mice were also significantly reduced by RAGE-apt or spironolactone (Spiro) (Fig. 4f–k).
Figure 4.

RAGE-apt ameliorates the progression of MR-associated podocyte injury through the inhibition of oxidative stress production and Rac1 activation. (a and b) SBP levels in Cont, DOCA-Ctrl-apt, DOCA–RAGE-apt, and DOCA-Spiro mice (n = 4–12 per group), †p < 0.05 vs Cont, $p < 0.05 vs DOCA-Spiro, (c) UAE levels (n = 4–12 per group), (d) podocin expression (n = 4–5 per group), (e) double staining for nitrotyrosine and CML by immunofluorescence (n = 4–5 per group), (f) glomerular CML staining by immunohistochemistry (n = 4–5 per group), (g) plasma CML levels (n = 4–12 per group), (h) glomerular RAGE expression (n = 4–5 per group), (i) cortical RAGE protein expression by Western blot (n = 4 per group), (j and k) glomerular GTP-bound Rac1 and MR expression (n = 4–5 per group). Data are presented as mean ± SEM. #p < 0.05, ##p < 0.01 vs DOCA-Ctrl-apt mice, †p < 0.05, ††p < 0.01 vs DOCA-Spiro mice. All kidney sections are 4-μm thin. Bars = 20 μm. RAGE, receptor for advanced glycation end products; MR, mineralocorticoid receptor; SBP, systolic blood pressure; Cont, control; Ctrl-apt, control aptamer; RAGE-apt, RAGE aptamer; UAE, urinary albumin excretion; Spiro, spironolactone; CML, carboxymethyllysine.
Aldosterone Induces CML, ROS, and RAGE Production in Murine Podocyte Cells (MPC)
Aldo increased NT levels, which was suppressed by pretreatment with diphenylene iodonium (DPI), an inhibitor of NADPH oxidase, or Spiro (Fig. 5a and b). Furthermore, Aldo increased intracellular CML levels and RAGE gene expression in MPC, both of which were reduced by the anti-oxidant manganese (III) tetrakis (4-benzoic acid) porphyrin chloride (MnTBAP) (Fig. 5c–e)19,20.
Figure 5.

Aldo–MR system enhances nitrotyrosine and CML production through oxidative stress generation in MPC. (a) MPC were incubated with Aldo (1 µM) for 5, 15, and 30 min, and nitrotyrosine expression, a marker of peroxynitrite, was measured by Western blot (n = 3). (b) MPC were incubated with Aldo with or without DPI or Spiro for 15 min, and nitrotyrosine was measured by Western blot in MPC (n = 6). (c and d) MPC were incubated with Aldo for 30 min, then intracellular CML (ELISA) and CML (Western blot) expression were determined with or without MnTBAP, an anti-oxidant, in MPC (n = 6). (e) Aldo was administrated for 4 h, and RAGE mRNA expression were measured with or without MnTBAP by RT-PCR in MPC (n = 6). Data are presented as mean ± SEM. *p < 0.05 vs 0 min, #p < 0.05, ##p < 0.01 vs Aldo. Aldo, aldosterone; MR, mineralocorticoid receptor; CML, carboxymethyllysine; MPC, mouse podocyte cells; DPI, diphenyleneiodonium; MnTBAP, manganese (III) tetrakis (4-benzoic acid) porphyrin chloride; RAGE, receptor for advanced glycation end products; RT-PCR, real-time polymerase chain reaction.
RAGE-aptamer Improves CML-BSA-elicited MR and GTP-bound Rac1 Expressions in MPC
CML-BSA increased MR levels in a dose- and time-dependent manner in MPC. RAGE-apt, NSC23766, an inhibitor of Rac1, but not Spiro, significantly blocked the increase in MR expression in MPC (Fig. 6a–c). In addition, CML-BSA enhanced the activation of Rac1 in MPC (Fig. 6d). Taken together, these data indicate that inhibition of AGE-RAGE axis can block podocyte injury induced by MR activation (Fig. 7).
Figure 6.

CML-BSA increases MR expression through RAGE–Rac1 axis in MPC. (a) MPC were incubated with CML-BSA (50, 100, and 200 μg/ml) for 24 h, and MR expression was determined by Western blot (n = 4, respectively). (b) CML-BSA (200 μg/ml) was administrated for 6, 12, and 24 h, and MR expression was determined (n = 4, respectively). (c) MPC were co-incubated with CML-BSA or BSA (200 μg/ml) with or without Ctrl-apt (100 nM), RAGE-apt (100 nM), NSC23766 (100 μM), a Rac1 inhibitor, or Spiro (10 μM) for 24 h, and MR expression was evaluated by Western blot (n = 5). (d) CML-BSA or BSA (200 μg/ml) was co-incubated with Ctrl- or RAGE-apt (100 nM) for 5 min in MPC, and Rac1 activation was determined (n = 4). Data are presented as the mean ± SEM. #p < 0.05, ##p < 0.01 vs CML-BSA. CML-BSA, carboxymethyllysine-bovine serum albumin; MR, mineralocorticoid receptor; RAGE, receptor for advanced glycation end products; MPC, mouse podocyte cells; BSA, bovine serum albumin; Ctrl-apt, control aptamer; RAGE-apt, RAGE aptamer; Rac1, GTP-bound Rac1; Spiro, spironolactone.
Figure 7.
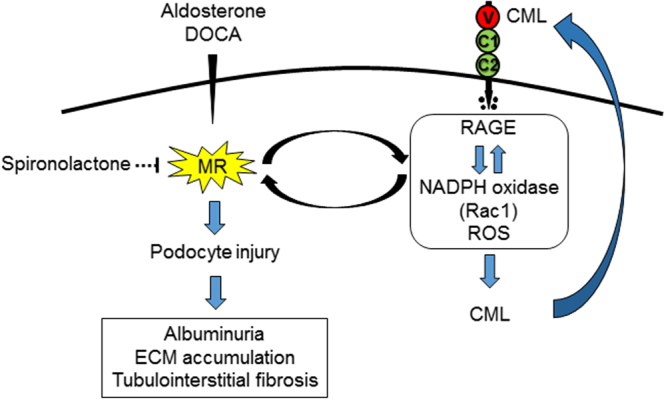
Hypothetic pathways of Aldo–MR system and AGE–RAGE axis-mediated podocyte injury in hypertensive nephropathy. MR, mineralocorticoid receptor; ECM, extracellular matrix; CML, carboxymethyllysine; RAGE, receptor for advanced glycation end products; ROS, reactive oxygen species.
Discussion
We showed for the first time that although the administration of RAGE-apt for 3 weeks did not affect blood pressure levels, it reduced UAE, restored podocin expression, and inhibited the increases in glomerular CML, RAGE, MR, and Rac1 levels in DOCA mice. CML and NT were co-localized and increased in the podocytes of the glomeruli of DOCA mice, which was also prevented by RAGE-apt. Since Spiro mimicked the effects of RAGE-apt, the present study suggests that DOCA could stimulate the AGE–RAGE axis in podocytes of the glomeruli through the interaction with MR via the Rac1 pathway, thereby leading to podocyte damage and increased UAE in DOCA mice.
Podocyte dysfunction and loss are one of the characteristic features of several renal diseases21–23. AGE and RAGE are reported to be strongly expressed in glomerular sclerosis lesions in patients with hypertension24. Further, RAGE is dominantly overexpressed in podocyte24, thus suggesting the clinical relevance of AGE–RAGE axis in podocyte injury in hypertensive renal disease. Recently, we have found that RAGE-neutralizing antibody inhibits the AGE-induced upregulation of RAGE mRNA levels, oxidative stress generation, and apoptosis in podocytes25. Therefore, the interaction of AGE with RAGE per se could modulate RAGE expression via the production of intracellular reactive oxygen species (ROS) as a second messenger. Since oxidative stress is generated by MR-mediated Rac1 overexpression and ROS stimulates intracellular AGE production in podocytes26,27, DOCA-elicited MR activation may enhance ROS generation and subsequently promote intracellular AGE production, which could elicit podocyte injury through interaction with RAGE. Nitric oxide is shown to rapidly react with superoxide anion radical derived from NADPH oxidase, forming ONOO- in kidney cells28. Indeed, AGE induced the production of inducible nitric oxide synthase (iNOS) and accelerated ONOO- production through the interaction with RAGE in diabetic condition29,30. That could be the reason why RAGE-apt inhibited the expression levels of nitrotyrosine in the podocytes of DOCA mice. Therefore, we hypothesize that nitric oxide may play a crucial role for ONOO- production, leading to podocyte injury through the crosstalk with RAGE in DOCA-induced kidney injury.
In the present study, genetic RAGE deletion also significantly suppressed DOCA-induced increase in MR production, Rac1 activation, CML deposition, and the downstream signaling of SGK1 and TGF-β. These findings are in accordance with the observations of RAGE-apt experiments showing that RAGE-apt could prevent DOCA-elicited renal injury via the Rac1–ROS–AGE pathway. Further, we observed that Aldo significantly increased NT and CML expressions in association with increased RAGE gene expression in MPC. Since CML accumulation in atherosclerotic plaques was diminished in RAGE-deleted ApoE-KO diabetic mice31, it is conceivable that engagement of Aldo-elicited CML to RAGE stimulates ROS production, which in turn promotes the further production of CML, thus forming a positive feedback loop between RAGE downstream signaling and CML generation. Moreover, CML increased MR expression and Rac1 activation, which were blocked by the RAGE-apt in MPC. These findings suggest that MR expression and AGE–RAGE system are closely correlated with each other, leading to podocyte injury (Fig. 7). DOCA-induced mesangial matrix expansion and tubulointerstitial fibrosis were restored in RAGE-KO mice (Fig. 2e–f). In addition, glomerular matrix accumulation was ameliorated by the treatment with RAGE-apt (Supplementary Figure S3). Since DOCA-elicited SGK1 and TGF-β expression were ameliorated in RAGE-KO mice, RAGE-apt might restore the MR-induced glomerular matrix accumulation through the suppression of SGK1 and TGF-β expression, thereby protecting damages in several renal cells including podocytes, mesangial cells, and proximal tubular cells.
In the present study, we observed that RAGE-apt did not affect plasma BUN and Cr levels when compared to DOCA-Ctrl-apt mice. On the basis of the result showing that genetic deletion of RAGE tended to improve the plasma BUN level (p = 0.065, Table 1), the pharmacokinetics of RAGE-apt such as bioavailability and dissociation constant might be associated with the failure to attenuate plasma BUN and Cr levels in DOCA-mice. Secondly, we believe the polyuria associated with the DOCA model may mask some protective effects as measured by serum markers of kidney function. It is generally accepted that urine volume was increased in DOCA/salt-treated mice, which was reduced by normalization of blood pressure using hydralazine, a direct acting vasodilator without affecting any urine electrolytes32, suggesting that increased blood pressure could be strongly associated with urine volume and subsequent dehydration-induced renal function. In the present study, we observed that the administration with Spiro could reduce the elevated systolic blood pressure in DOCA mice (Fig. 4b) and significantly decrease their urine volume (DOCA-Ctrl-apt vs DOCA-Spiro; 2.47 ± 0.27 vs 1.37 ± 0.12 (mL), p < 0.01). On the other hand, the pharmacological deletion of RAGE using RAGE-apt did not affect systolic blood pressure (Fig. 4b) and the amount of urine volume (DOCA-Ctrl-apt vs DOCA-RAGE-apt; 2.47 ± 0.27 vs 2.71 ± 0.21 (mL), p = 0.51). We hypothesize that polyuria could lead to dehydration, minimizing the apparent protective effect of RAGE-apt as measured by serum markers of kidney function in spite of their structural improvement. In addition, histological changes are sometimes more sensitive than markers of kidney function for detecting kidney injury in early phase of chronic kidney disease. Therefore, we have concluded that RAGE-apt could be a promising therapeutic agent against MR-activated kidney injury.
There is a growing body of evidence that MR antagonism using Spiro or eplerenone can reduce urinary albumin excretion and retard the progression of chronic kidney disease in several clinical studies33,34. We hypothesize that Spiro is an excellent combination therapy to use in combination with RAGE-apt in MR-activated kidney injury. Our in vitro data suggested that aldosterone-induced nitrotyrosine production was reduced by the treatment with Spiro (Fig. 5b), but it does not completely block the CML-induced MR activation (Fig. 6c). In addition, we found that the CML deposition in the kidney tissue was partially, but significantly, improved by the treatment with Spiro in vivo, though not to the same extent as RAGE-apt (Fig. 4f). These findings suggest that RAGE-apt and Spiro target independent pathways in the progression of podocyte injury, thereby the combination therapy of Spiro plus RAGE-apt can be promising treatment against MR-activated kidney injury.
Several therapeutic agents targeting RAGE have been reported12,35,36. Administration of soluble RAGE suppressed the progression of diabetic atherosclerosis in ApoE-KO mice35. Long-term treatment with neutralizing RAGE antibody ameliorated renal injury in obese type 2 diabetic mice model36. However, due to several limitations, these agents have not been utilized in clinical setting. On the contrary, RAGE-apts have several advantages over soluble RAGE protein or RAGE antibody. First, the synthesis of aptamers does not rely on animal systems; therefore, they can be easily selected from oligonucleotide library in vitro. Second, aptamers are quite thermally stable, whereas proteins are sensitive to temperature. Third, aptamers do not have immunogenicity over soluble proteins or antibodies. Finally, a small size allows for more efficient entry of RAGE-apt into various organs37,38. Recently, we found that AGE-DNA-apt significantly inhibited the progression of experimental diabetic nephropathy17. However, since AGEs are composed of complex and heterogeneous compounds11, targeting one type of AGEs may not sufficiently block the AGE–RAGE system. Therefore, RAGE-apt may be a more favorable tool for blockade of the AGE–RAGE axis. Although some aptamers targeting receptor may have an agonistic function39, our RAGE-apt did not show any agonistic activity of RAGE because RAGE-apt alone did not affect the gene expression of RAGE, connective tissue growth factor, and monocyte chemokine protein-1 (Supplementary Figure S4). Therefore, our RAGE-apt could be safe and effective for the treatment of several renal injury, including HN12.
Concise Methods
Animal Preparation
Eight-week-old male WT mice were purchased from CLEA Japan (Tokyo, Japan). The RAGE-KO mouse strains on C57BL/6J background were created by Professor Arnold (Deutsches Krebsforschungszentrum Stiftung des öffentlichen Rechts, Heidelberg, Germany) and Professor Nawroth (University Clinical Centre of Heidelberg) and were kindly given from Dr. Bierhaus (Deutsches Krebsforschungszentrum Stiftung des öffentlichen Rechts, Heidelberg, Germany)40,41.
Hypertension was induced in C57BL/6J and RAGE-KO mice by uninephrectomy with the administration of DOCA (50 mg, a 21-day continuous-release, Innovation Research, USA) and 4% salt diet, with the following categories: 4% salt diet, Cont; 4% salt diet with DOCA, DOCA; RAGE-KO mice treated with DOCA/salt, DOCA–RAGE-KO. RAGE-apt (2.0 × 10−4 μg/day) or Ctrl-apt (2.0 × 10−4 μg/day) were administrated in DOCA mice subcutaneously by an osmotic pump (model 2006; ALZET, Cupertino, USA). Spiro (30 mg/kg per day) was administrated by a feeding needle. All experimental procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the ethics committee of Kurume University School of Medicine.
Measurement of Clinical Variables
Albuminuria was determined with a commercially available ELISA kit (Exocell, Philadelphia, USA). Blood was collected, and the plasma was stored at −40 °C. BUN and Cr levels in the plasma were measured by an enzymatic method using an auto-analyzer (LABOSPECT 008; Hitachi, Tokyo, Japan).
Preparation of CML-BSA
CML-BSA was prepared by a reaction with glyoxylic acid with BSA in the presence of NaBH3CN, as previously described42.
Screening and Blocking Capacity of RAGE–DNA Aptamer
RAGE-apt was obtained by an in vitro selection process, that is, SELEX methods, from a pool of ~1015 different nucleic acid sequences as previously described18. Seven types of sequences directed against RAGE were obtained in this study and sequences of selected RAGE-apts and Ctrl-apt are displayed in Supplementary Figure S5a. These aptamers were modified with phosphorothioate for protection from degradation by nuclease43. We examined the blocking capacity of RAGE-apts between CML-BSA and v-RAGE by ELISA. v-RAGE (residues 23–121) was prepared as previously described44,45. In brief, CML-BSA was immobilized on the bottom of 96 well-ELISA plate, and v-RAGE was added with RAGE- or Ctrl-apts. CML-BSA-bound v-RAGE was captured by horseradish peroxidase-conjugated antibody against v-RAGE and then detected via an enzyme-catalyzed color reaction. Clone #2 showed the highest binding affinity to CML-BSA (Supplementary Figure S5b). Next, to confirm the inhibitory capacity of #2 RAGE-apt against the interaction of CML-BSA with RAGE, we performed sensitive 27-MHz QCM (Ainix Q, Intium, Tokyo, Japan)46. In brief, v-RAGE was immobilized on an avidin-bound QCM surface. After adding CML-BSA to a reaction vessel with or without RAGE-apt (100 nM), the time course of the frequency decrease on the QCM was monitored.
Immunohistochemical Analysis
Specimens of kidney cortex were fixed with 4% paraformaldehyde, embedded in paraffin, sectioned at 4-µm intervals, and mounted on glass slides. The sections were incubated with antibodies (Abs) raised against MR (1:100), RAGE (1:100) (kindly provided by Professor Yamamoto Y, Kanazawa University Graduate School of Medical Science, Kanazawa, Japan), CML (1:1000), 8-OHdG (1:1000), and Rac1 (1:100) after pretreatment with blocking agent (Supplementary Figure S1). The sections were incubated with Envision HRP-labeled polymer anti-rabbit and mouse (ready-to-use) (Dako, cat# K536111–2). Immunoreactivity in 20 different fields (×600) in each sample was evaluated by image analysis software (version 6.57; Optimas, Media Cybernetics, Silver Spring, MD).
Immunofluorescence Study by Confocal Microscopy
To determine the localization of RAGE, MR, and Rac1 in the glomeruli of DOCA mice, we performed an immunofluorescence staining with Abs raised against RAGE (1:100), MR (1:100), Rac1 (1:100), and WT-1 (1:1000) as a primary Ab (Supplementary Figure S1). Goat anti-rabbit IgG Alexa 488 and anti-mouse IgG Alexa 594 (1:1000; Molecular Probes, Eugene, OR, USA) were used as secondary Abs. The sections were analyzed under a confocal laser microscope, FV 1000 (OLYMPUS, Tokyo, Japan). Double staining of CML and NT was also performed using anti-CML and anti-NT Abs as a primary antibody. Podocin was evaluated by immunofluorescence study with anti-podocin Ab (1:1000, kindly provided by Dr. Asanuma K) as a primary Ab and goat anti-mouse IgG Alexa 488 (1:1000) as a secondary Ab to investigate podocyte dysfunction.
Morphological Analysis
Four-µm paraffin sections were stained with periodic acid-Schiff and Masson’s trichrome for light microscopic analysis47.
Measurement of CML in Plasma and Cell Lysate
CML levels in plasma and cell lysate were measured using a competitive ELISA according to the manufacturer’s instructions (Cell Biolabs, Inc. cat #STA-816).
Cells
Conditionally immortalized MPC were kindly provided by Dr. Asanuma K, and cultured as previously described25,48. MPC was treated with Aldo (1 µM), CML-BSA (50–200 µg/ml) or non-glycated BSA (200 µg/ml) with RAGE-apt (100 nM), Ctrl-apt (100 nM), NSC23766 (100 µM), Spiro (10 µM), and MnTBAP (10 µM) in a medium containing 1% fetal bovine serum.
Western Blot
Kidney, lung, and MPC were lysed, and lysates were prepared as previously described49. Membranes were incubated with the following primary Abs: RAGE, NT, CML, MR, SGK1, and GAPDH (1:1000, respectively) and β-actin (1:2000) during overnight (Supplementary Figure S1). Horseradish peroxidase-conjugated anti-mouse and anti-rabbit secondary antibodies (1:2000) were applied. Protein expressions were visualized by Signal Enhancer HIKARI (Nacalai Tesque, Inc., Kyoto, Japan).
Real-Time RT-PCR
Total RNAs were extracted from each kidney cortex and MPC with TRIzol reagent (Invitrogen, USA), and isolated RNAs were used to synthesize cDNA with Rever Tra Ace qPCR RT Master Mix with gDNA Remover (Toyobo, Tokyo, Japan). Quantitative real-time RT-PCR was performed using Assay-on-Demand and Taqman 5 fluorogenic nuclease chemistry (Applied Biosystems, USA). Information of primers and probes are shown in Supplementary Figure S2. The relative mRNA expressions of each gene were calculated with ∆∆Ct method.
Rac1 Activity by G-LISA
For evaluation of GTP-bound Rac1 activity, the Rac1 G-LISA Activation Assay Biochem Kit (Cytoskeleton, Denver, CO) was performed according to the manufacture’s instruction. In brief, MPC were placed on ice and washed with ice-cold PBS after the incubation with CML-BSA (200 μg/mL) or BSA (200 μg/mL) for 5 min in the presence or absence of RAGE-apt. MPC were harvested with ice-cold lysis buffer, then lysates were added to the wells of the Rac1-GTPase binding plate. After addition of primary and horseradish peroxidase-conjugated secondary Abs, signals were measured using a microplate luminometer (2030 ARVO x3, Perkin Elmer).
Statistical Analyses
All data are presented as mean ± SEM. One-way ANOVA and following unpaired t-test were used to assess the differences among groups when appropriate. All statistical analyses were performed using GraphPad Prism 7 Software (GraphPad Software, San Diego, CA). p < 0.05 was considered statistically significant.
Electronic supplementary material
Acknowledgements
This work was supported, in part, by Grants-in-Aid for Welfare and Scientific Research (C) (no. 22590904) (K.F.) and from the Ministry of Education, Culture, Sports, Science and Technology of Japan. I thank Prof. Yamamoto Y for providing an excellent antibody against RAGE.
Author Contributions
S.Y., Y.H., and K.F. conceived the original idea of this study. K.T., M.Y., S.I., G.K., Y.K., Y.N., R.A., N.O., and T.M. carried out in vivo and vitro experiments. K.A., S.U., and S.O. supervised on these experiments. K.T. and K.F. wrote most of the initial draft of the manuscript and C.B edited the manuscript. All co-authors read and approved the final manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-21176-5.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Walker WG. Hypertension-related renal injury: a major contributor to end-stage renal disease. Am J Kidney Dis. 1993;22:164–173. doi: 10.1016/S0272-6386(12)70183-X. [DOI] [PubMed] [Google Scholar]
- 2.Rutkowski B, Tylicki L, Manitius J, Lysiak-Szydlowska W. Hypertensive nephropathy - an increasing clinical problem. Miner Electrolyte Metab. 1999;25:65–68. doi: 10.1159/000057422. [DOI] [PubMed] [Google Scholar]
- 3.Durvasula RV, Shankland SJ. The renin-angiotensin system in glomerular podocytes: mediator of glomerulosclerosis and link to hypertensive nephropathy. Curr Hypertens Rep. 2006;8:132–138. doi: 10.1007/s11906-006-0009-8. [DOI] [PubMed] [Google Scholar]
- 4.Seccia TM, Caroccia B, Calò LA. Hypertensive nephropathy. Moving from classic to emerging pathogenetic mechanisms. J Hypertens. 2017;35:205–212. doi: 10.1097/HJH.0000000000001170. [DOI] [PubMed] [Google Scholar]
- 5.Wei SY, et al. Multiple Mechanisms are Involved in Salt-Sensitive Hypertension-Induced Renal Injury and Interstitial Fibrosis. Sci Rep. 2017;7:45952. doi: 10.1038/srep45952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nagase M. Recent topics on podocytes and aldosterone. J Ren Nutr. 2015;25:201–204. doi: 10.1053/j.jrn.2014.10.016. [DOI] [PubMed] [Google Scholar]
- 7.Nagase M, Matsui H, Shibata S, Gotoda T, Fujita T. Salt-induced nephropathy in obese spontaneously hypertensive rats via paradoxical activation of the mineralocorticoid receptor: role of oxidative stress. Hypertension. 2007;50:877–883. doi: 10.1161/HYPERTENSIONAHA.107.091058. [DOI] [PubMed] [Google Scholar]
- 8.Epstein M. Aldosterone as a mediator of progressive renal disease: pathogenetic and clinical implications. Am J Kidney Dis. 2001;37:677–688. doi: 10.1016/S0272-6386(01)80115-3. [DOI] [PubMed] [Google Scholar]
- 9.Ishimitsu T, et al. Effects of valsartan on the progression of chronic renal insufficiency in patients with nondiabetic renal diseases. Hypertens Res. 2005;28:865–870. doi: 10.1291/hypres.28.865. [DOI] [PubMed] [Google Scholar]
- 10.Shibata S, et al. Rac1 GTPase in rodent kidneys is essential for salt-sensitive hypertension via a mineralocorticoid receptor-dependent pathway. J Clin Invest. 2011;121:3233–3243. doi: 10.1172/JCI43124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation. 2006;114:597–605. doi: 10.1161/CIRCULATIONAHA.106.621854. [DOI] [PubMed] [Google Scholar]
- 12.Fukami K, Taguchi K, Yamagishi S, Okuda S. Receptor for advanced glycation endproducts and progressive kidney disease. Curr Opin Nephrol Hypertens. 2015;24:54–60. doi: 10.1097/MNH.0000000000000091. [DOI] [PubMed] [Google Scholar]
- 13.Yamagishi S, Fukami K, Matsui T. Evaluation of tissue accumulation levels of advanced glycation end products by skin autofluorescence: A novel marker of vascular complications in high-risk patients for cardiovascular disease. Int J Cardiol. 2015;185:263–268. doi: 10.1016/j.ijcard.2015.03.167. [DOI] [PubMed] [Google Scholar]
- 14.Yamagishi S, Matsui T, Fukami K. Role of receptor for advanced glycation end products (RAGE) and its ligands in cancer risk. Rejuvenation Res. 2015;18:48–56. doi: 10.1089/rej.2014.1625. [DOI] [PubMed] [Google Scholar]
- 15.Matsui T, Takeuchi M, Yamagishi S. Nifedipine, a calcium channel blocker, inhibits inflammatory and fibrogenic gene expressions in advanced glycation end product (AGE)-exposed fibroblasts via mineralocorticoid receptor antagonistic activity. Biochem Biophys Res Commun. 2010;396:566–570. doi: 10.1016/j.bbrc.2010.04.149. [DOI] [PubMed] [Google Scholar]
- 16.Zhou J, Rossi J. Aptamers as targeted therapeutics: current potential and challenges. Nat Rev Drug Discov. 2017;16:181–202. doi: 10.1038/nrd.2016.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaida Y, et al. DNA aptamer raised against AGEs blocks the progression of experimental diabetic nephropathy. Diabetes. 2013;62:3241–3250. doi: 10.2337/db12-1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Higashimoto Y, et al. In vitro selection of DNA aptamers that block toxic effects of AGE on cultured retinal pericytes. Microvasc Res. 2007;74:65–69. doi: 10.1016/j.mvr.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 19.Mollace V, et al. The effect of peroxynitrite decomposition catalyst MnTBAP on aldehyde dehydrogenase-2 nitration by organic nitrates: role in nitrate tolerance. Pharmacol Res. 2014;89:29–35. doi: 10.1016/j.phrs.2014.07.007. [DOI] [PubMed] [Google Scholar]
- 20.Batinić-Haberle I, et al. Pure MnTBAP selectively scavenges peroxynitrite over superoxide: comparison of pure and commercial MnTBAP samples to MnTE-2-PyP in two models of oxidative stress injury, an SOD-specific Escherichia coli model and carrageenan-induced pleurisy. Free Radic Biol Med. 2009;46:192–201. doi: 10.1016/j.freeradbiomed.2008.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang G, et al. Podocyte loss in human hypertensive nephrosclerosis. Am J Hypertens. 2009;22:300–306. doi: 10.1038/ajh.2008.360. [DOI] [PubMed] [Google Scholar]
- 22.Burger D, et al. Urinary podocyte microparticles identify prealbuminuric diabetic glomerular injury. J Am Soc Nephrol. 2014;25:1401–1407. doi: 10.1681/ASN.2013070763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wiggins RC. The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int. 2007;71:1205–1214. doi: 10.1038/sj.ki.5002222. [DOI] [PubMed] [Google Scholar]
- 24.Wendt T, et al. Glucose, glycation, and RAGE: implications for amplification of cellular dysfunction in diabetic nephropathy. J Am Soc Nephrol. 2003;14:1383–1395. doi: 10.1097/01.ASN.0000065100.17349.CA. [DOI] [PubMed] [Google Scholar]
- 25.Ishibashi Y, et al. PEDF inhibits AGE-induced podocyte apoptosis via PPAR-gamma activation. Microvasc Res. 2013;85:54–58. doi: 10.1016/j.mvr.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 26.Nagase M, Fujita T. Role of Rac1-mineralocorticoid-receptor signalling in renal and cardiac disease. Nat Rev Nephrol. 2013;9:86–98. doi: 10.1038/nrneph.2012.282. [DOI] [PubMed] [Google Scholar]
- 27.Nishikawa T, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 28.Pfeiffer S, Mayer B. Lack of tyrosine nitration by peroxynitrite generated at physiological pH. J Biol Chem. 1998;273:27280–27285. doi: 10.1074/jbc.273.42.27280. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y, et al. The alternative crosstalk between RAGE and nitrative thioredoxin inactivation during diabetic myocardial ischemia-reperfusion injury. Am J Physiol Endocrinol Metab. 2012;303:E841–852. doi: 10.1152/ajpendo.00075.2012. [DOI] [PubMed] [Google Scholar]
- 30.San Martin A, et al. Nox1-based NADPH oxidase-derived superoxide is required for VSMC activation by advanced glycation end-products. Free Radic Biol Med. 2007;42:1671–1679. doi: 10.1016/j.freeradbiomed.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 31.Soro-Paavonen A, et al. Receptor for advanced glycation end products (RAGE) deficiency attenuates the development of atherosclerosis in diabetes. Diabetes. 2008;57:2461–2469. doi: 10.2337/db07-1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grobe JL, et al. Angiotensinergic signaling in the brain mediates metabolic effects of deoxycorticosterone (DOCA)-salt in C57 mice. Hypertension. 2011;57:600–607. doi: 10.1161/HYPERTENSIONAHA.110.165829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ando K, et al. Anti-albuminuric effect of the aldosterone blocker eplerenone in non-diabetic hypertensive patients with albuminuria: a double-blind, randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2014;2:944–953. doi: 10.1016/S2213-8587(14)70194-9. [DOI] [PubMed] [Google Scholar]
- 34.Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int. 2006;70:2116–2123. doi: 10.1038/sj.ki.5001854. [DOI] [PubMed] [Google Scholar]
- 35.Park L, et al. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat Med. 1998;4:1025–1031. doi: 10.1038/2012. [DOI] [PubMed] [Google Scholar]
- 36.Flyvbjerg A, et al. Long-term renal effects of a neutralizing RAGE antibody in obese type 2 diabetic mice. Diabetes. 2004;53:166–172. doi: 10.2337/diabetes.53.1.166. [DOI] [PubMed] [Google Scholar]
- 37.Osborne SE, Matsumura I, Ellington AD. Aptamers as therapeutic and diagnostic reagents: problems and prospects. Curr Opin Chem Biol. 1997;1:5–9. doi: 10.1016/S1367-5931(97)80102-0. [DOI] [PubMed] [Google Scholar]
- 38.Famulok M, Hartig JS, Mayer G. Functional aptamers and aptazymes in biotechnology, diagnostics, and therapy. Chem Rev. 2007;107:3715–3743. doi: 10.1021/cr0306743. [DOI] [PubMed] [Google Scholar]
- 39.Yunn NO, et al. Agonistic aptamer to the insulin receptor leads to biased signaling and functional selectivity through allosteric modulation. Nucleic Acids Res. 2015;43:7688–7701. doi: 10.1093/nar/gkv767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Constien R, et al. Characterization of a novel EGFP reporter mouse to monitor Cre recombination as demonstrated by a Tie2 Cre mouse line. Genesis. 2001;30:36–44. doi: 10.1002/gene.1030. [DOI] [PubMed] [Google Scholar]
- 41.Liliensiek B, et al. Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J Clin Invest. 2004;113:1641–1650. doi: 10.1172/JCI200418704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reddy S, Bichler J, Wells-Knecht KJ, Thorpe SR, Baynes JW. N epsilon-(carboxymethyl)lysine is a dominant advanced glycation end product (AGE) antigen in tissue proteins. Biochemistry. 1995;34:10872–10878. doi: 10.1021/bi00034a021. [DOI] [PubMed] [Google Scholar]
- 43.King DJ, Ventura DA, Brasier AR, Gorenstein DG. Novel combinatorial selection of phosphorothioate oligonucleotide aptamers. Biochemistry. 1998;37:16489–16493. doi: 10.1021/bi981780f. [DOI] [PubMed] [Google Scholar]
- 44.Yamagishi S, et al. Pigment epithelium-derived factor protects cultured retinal pericytes from advanced glycation end product-induced injury through its antioxidative properties. Biochem Biophys Res Commun. 2002;296:877–882. doi: 10.1016/S0006-291X(02)00940-3. [DOI] [PubMed] [Google Scholar]
- 45.Suga T, et al. Activation of receptor for advanced glycation end products induces osteogenic differentiation of vascular smooth muscle cells. J Atheroscler Thromb. 2011;18:670–683. doi: 10.5551/jat.7120. [DOI] [PubMed] [Google Scholar]
- 46.Okahata Y, Niikura K, Sugiura Y, Sawada M, Morii T. Kinetic studies of sequence-specific binding of GCN4-bZIP peptides to DNA strands immobilized on a 27-MHz quartz-crystal microbalance. Biochemistry. 1998;37:5666–5672. doi: 10.1021/bi980037k. [DOI] [PubMed] [Google Scholar]
- 47.Takamiya Y, et al. Experimental diabetic nephropathy is accelerated in matrix metalloproteinase-2 knockout mice. Nephrol Dial Transplant. 2013;28:55–62. doi: 10.1093/ndt/gfs387. [DOI] [PubMed] [Google Scholar]
- 48.Mundel P, et al. Synaptopodin: an actin-associated protein in telencephalic dendrites and renal podocytes. J Cell Biol. 1997;139:193–204. doi: 10.1083/jcb.139.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakayama Y, et al. Asymmetric dimethylarginine accumulates in the kidney during ischemia/reperfusion injury. Kidney Int. 2014;85:570–578. doi: 10.1038/ki.2013.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.