

Abstract
Objectives
Oxidative stress plays a major role in the onset and progression of involutional osteoporosis. However, classical antioxidants fail to restore osteoblast function. Interestingly, the bone anabolism of parathyroid hormone (PTH) has been shown to be associated with its ability to counteract oxidative stress in osteoblasts. The PTH counterpart in bone, which is the PTH-related protein (PTHrP), displays osteogenic actions through both its N-terminal PTH-like region and the C-terminal domain.
Methods
We examined and compared the antioxidant capacity of PTHrP (1-37) with the C-terminal PTHrP domain comprising the 107-111 epitope (osteostatin) in both murine osteoblastic MC3T3-E1 cells and primary human osteoblastic cells.
Results
We showed that both N- and C-terminal PTHrP peptides at 100 nM decreased reactive oxygen species production and forkhead box protein O activation following hydrogen peroxide (H2O2)-induced oxidation, which was related to decreased lipid oxidative damage and caspase-3 activation in these cells. This was associated with their ability to restore the deleterious effects of H2O2 on cell growth and alkaline phosphatase activity, as well as on the expression of various osteoblast differentiation genes. The addition of Rp-cyclic 3′,5′-hydrogen phosphorothioate adenosine triethylammonium salt (a cyclic 3',5'-adenosine monophosphate antagonist) and calphostin C (a protein kinase C inhibitor), or a PTH type 1 receptor antagonist, abrogated the effects of N-terminal PTHrP, whereas protein phosphatase 1 (an Src kinase activity inhibitor), SU1498 (a vascular endothelial growth factor receptor 2 inhibitor), or an anti osteostatin antiserum, inhibited the effects of C-terminal PTHrP.
Conclusion
These findings indicate that the antioxidant properties of PTHrP act through its N- and C-terminal domains and provide novel insights into the osteogenic action of PTHrP.
Cite this article: S. Portal-Núñez, J. A. Ardura, D. Lozano, I. Martínez de Toda, M. De la Fuente, G. Herrero-Beaumont, R. Largo, P. Esbrit. Parathyroid hormone-related protein exhibits antioxidant features in osteoblastic cells through its N-terminal and osteostatin domains. Bone Joint Res 2018;7:58–68. DOI: 10.1302/2046-3758.71.BJR-2016-0242.R2.
Keywords: Parathyroid hormone-related protein, Osteostatin, Antioxidant activity
Article focus
Oxidative stress is associated with bone loss.
We assessed in vitro the putative antioxidant properties of the osteogenic parathyroid hormone-related protein (PTHrP) and their ability to modulate osteoblastic function.
Key messages
PTHrP displays antioxidative stress activity in mouse and human osteoblastic cells.
PTHrP (1-37) and PTHrP (107-111) (osteostatin) counteract the decrease of osteoblastic function triggered by oxidative stress.
N- and C-terminal domains of PTHrP exhibit similar antioxidative stress features through different signalling mechanisms in osteoblastic cells.
Strengths and limitations
PTHrP preserves osteoblastic function in an in vitro oxidative environment that further supports the putative use of PTHrP peptides as osteoporosis therapy.
Caution should be taken when translating these in vitro results to an in vivo scenario.
Introduction
It is widely accepted that bone mass in humans starts to decline between the second and third decades of life, and this has been attributed to an increase in oxidative stress.1,2 Compelling evidence indicates that oxidative stress is a major cause of the ageing process, and that it occurs as the result of an imbalance between the production of reactive oxygen species (ROS) and the molecular defence mechanisms that control ROS, such as antioxidant enzymes catalase (CAT), superoxide dismutase (SOD), glutathione peroxidase (GPx), and antioxidant compounds (glutathione, among others).3,4 It has been reported that age-related conditions that are associated with bone derangement, such as post-menopausal oestrogen depletion and diabetes mellitus, produce an increase of ROS.5,6 The expression of ROS inhibiting molecules, namely the enzymes SOD7 and CAT,8 as well as factors that induce cell cycle arrest in response to excessive oxidative stress (such as growth arrest DNA damage 45 protein),9 can be regulated by forkhead box protein O (FOXO) transcription factors, which are essential to bone integrity.10 In fact, it has recently been demonstrated that knocking down SOD results in bone deterioration in mice.11 In humans, an augmented oxidative stress has been traced to decreased bone mineral density.12,13 The mechanisms by which oxidative stress may impair bone formation are complex and involve DNA damage,14 lipid peroxidation,15 protein carbonylation,16 inhibition of Wnt pathway activation,17 decreased osteoblast proliferation18 and differentiation,19,20 and increased osteoblast apoptosis.21
Parathyroid hormone (PTH)-related protein (PTHrP) is abundantly expressed in bone, where it performs an important role in bone formation and remodelling; similarly to PTH, intermittent administration of PTHrP displays anabolic actions in rodents and/or humans.22 In fact, the N-terminal region of PTHrP shows homology with the same region of PTH and interacts with the common PTH type 1 receptor (PTH1R), which is a class II G protein-coupled receptor. PTH binding to PTH1R produces the activation of protein kinase A (PKA) by cyclic 3',5'-adenosine monophosphate (cAMP) and phospholipase C.23 On the other hand, the PTH-unrelated C-terminal tail of PTHrP contains the 107-111 epitope (named osteostatin), which does not interact with the parathyroid hormone 1 receptor (PTHR1) but can apparently signal through opening voltage-sensitive calcium channels and protein kinase C (PKC) activation in osteoblastic cells.24 It has recently been shown that osteostatin may transactivate vascular endothelial growth factor receptor 2 (VEGFR2) to promote osteoblastic function through an Src kinase-mediated mechanism.25
Recent data have disclosed the impact of the antioxidative stress properties of PTH on its bone anabolic action in aged mice.26 In this regard, we recently reported that PTHrP (1-36) and PTHrP (107-139) were able to prevent the inhibitory effect of oxidative stress on the Wnt pathway activation in mouse mesenchymal C3H10T1/2 cells, suggesting their antioxidant potential.27 The aim of the present study was to determine the antioxidant properties of both N- and C-terminal domains of PTHrP in murine osteoblastic MC3T3-E1 cells and human osteoblastic cells (hOBs). Regarding the latter domain, we tested both the native fragment PTHrP (107-139) and osteostatin, both of which have a simple structure that would make them attractive agents for promoting bone formation and bone regeneration.28-32 The impact of the antioxidant properties of these PTHrP peptides on their ability to affect murine and hOB function has also been explored in this study.
Materials and Methods
Cells and reagents
Previously well-characterized osteoblastic MC3T3-E1-subclon 4 cells (CRL-2593; American Type Culture Collection, Manassas, Virginia), showing high mineralization capacity under differentiation conditions, were used. These cells have been used in previous studies evaluating several osteogenic actions of PTH and PTHrP.28,29,33,34 Primary hOBs were obtained from trabecular bone explants from two osteoarthritic patients undergoing total knee arthroplasty surgery, as previously reported.22,28,29,35-37 Briefly, hOBs were cultured from bone slices with Dulbecco’s modified Eagle medium (MEM; Thermo Fisher Scientific, Waltham, Massachusetts) containing 20% heat-inactivated fetal bovine serum (FBS), 2 mM glutamine and 100 U/ml of penicillin/streptomycin, and experiments were carried out using cells at the third passage. The Ethics Committee for Clinical Research at Instituto de Investigación Sanitaria-Fundación Jiménéz Díaz, Madrid, Spain approved the protocol, and written informed consent from the patients was granted.
MC3T3-E1 cells were grown in α-MEM with 10% FBS and antibiotics were made up of 1% penicillin/streptomycin (standard medium) in a humidified atmosphere of 5% CO2 at 37°C. When present, PTH (1-34) (Sigma-Aldrich, St Louis, Missouri), PTHrP (1-37), PTHrP (107-139), and osteostatin (Bachem AG, Bubendorf, Switzerland), depending on the type of experiment (detailed in each case), were added at 100 nM to the cell culture. This dose has demonstrated osteogenic capacity in previous studies using PTHrP (1-37) and osteostatin.27,29,34,38 In order to evaluate gene expression changes as described below, the PTH1R antagonist (Asn10, Leu11, D-Trp12) PTHrP (7-34) amide (PTHrP 7-34), at 100 nM, or neutralizing rabbit polyclonal antiserum C7 recognizing the osteostatin epitope in the entire PTHrP molecule, at 1:100 dilution,39 were added together with PTHrP (1-37) or osteostatin, respectively. CAT (Sigma-Aldrich), at 200 U/ml, was used as an antioxidant control. In alkaline phosphatase (ALP) activity and viability assays, we used the PKA inhibitor, adenosine 3’,5’-cyclic monophosphorothioate, and Rp-isomer (Rp-cyclic 3’,5’-hydrogen phosphorothioate adenosine triethylammonium salt (Rp-cAMPs)) (Santa Cruz Biotechnology Inc., Santa Cruz, California), at 25 µM; the PKC inhibitor, calphostin C (CalpC) (Calbiochem Corp., La Jolla, California), at 250 nM; or the Src kinase inhibitor PP-1 (Calbiochem Corp.) and VEGFR2 phosphorylation inhibitor SU1498 (Sigma-Aldrich), each at 10 µM, all of them added 30 minutes prior to the tested peptides.
ROS level determination
ROS were measured by evaluating the fluorescence increment of 2’,7’-dichlorofluorescein diacetate (DCF) dye reacting with strong oxidants such as cell peroxides, as previously described.40,41 Briefly, a final concentration of 10 µM DCF was added to subconfluent cells (10 000 cells/cm2 to 20 000 cells/cm2) in a FBS-depleted medium for 15 minutes at 37ºC. After extensive washing with phosphate-buffered saline (PBS), pH 7.4, cells were incubated for another 15 minutes, followed by treatment with the tested peptides at 100 nM for ten minutes in standard medium. After removing this medium, cells were washed with PBS followed by an addition of 200 µM hydrogen peroxide (H2O2) for ten minutes in a FBS-depleted medium and then tripsinized, measuring the increment of fluorescence in a FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, New Jersey).
Confocal experiments were also performed following the same protocol described above for DCF loading and peptide and H2O2 treatments. Cells were subsequently fixed with methanol including the DNA fluorescent dye 4’,6-diamidino-2-phenylindole dihydrochloride (1 mg/ml) for ten minutes, mounted in FluorSave reagent (Calbiochem Corp.) and examined using a Leica DM IRB confocal microscope (Leica, Wetzlar, Germany).
FOXO-luciferase (luc) assay
Subconfluent MC3T3-E1 cells were transfected with a mixture of 1 µg of FOXO-luc reporter plasmid and 5 ng of a renilla coding plasmid (Promega Corp., Fitchburg, Wisconsin) (transfection control) using X-tremeGENE 9 DNA Transfection Reagent (Roche Diagnostics, Indianapolis, Indiana), following the manufacturer’s instructions. After transfection, cells were treated with the peptides (each at 100 nM) for one hour in standard medium and then washed with PBS before the addition of 100 µM H2O2, followed by an overnight incubation in 1% FBS-containing medium at 37ºC. Thereafter, cells were homogenized with a lysis buffer, and luciferase and renilla activities were quantified with a dual luciferase kit (Promega Corp.), following the manufacturer’s instructions, using the FB12 tube luminometer (Berthold Technologies GmbH & Co. KG, Bad Wildbad, Germany). Renilla activity values were used to normalize the luciferase values.
Malondialdehyde (MDA) assay
Lipid peroxidation levels were determined by measuring the formation of MDA using a commercial kit (BioVision, Inc., Mountain View, California) per the manufacturer’s instructions. Briefly, MC3T3-E1 cells and hOBs (2 × 106 cells) were pretreated with PTHrP (1-37), PTHrP (107-139), osteostatin or PTH (1-34), each at 100 nM, for ten minutes in a serum-depleted medium. Cells were then washed three times with PBS and treated with 200 µM H2O2 for 24 hours, followed by centrifugation at 500 ×g for five minutes. The pellets were then washed three times with PBS. Pellets were subsequently resuspended in 300 μl of lysis buffer, as supplied by the kit, with 3 μl butylated hydroxytoluene (0.1 mM), sonicated, and centrifuged (13 000 ×g for ten minutes). The supernatants (200 μl) from each sample were added to 600 μl of thiobarbituric acid and incubated in a water bath at 95°C for 60 minutes. Samples were then ice-cooled for ten minutes and 200 μl (from each 800 μl of reaction mixture) was placed into a 96-well microplate for measuring absorbance at 532 nm. The MDA supplied in the kit was used as standard and MDA levels were determined by comparing the absorbance of samples with that of the standard controls. The protein concentration of the samples was measured with bicinchoninic acid protein assay (Sigma-Aldrich), using bovine serum albumin as standard. Results were expressed as nmols of MDA per mg of protein.
Caspase-3 assay
Subconfluent cells were serum-depleted overnight prior to incubation with the different peptides tested (each at 100 nM) for one hour in FBS-depleted medium. After washing the cells with PBS, 100 µM H2O2 was added for two, six, and 24 hours in the latter medium. Caspase-3 activity was analyzed using the CaspACE Assay System (Promega) following the manufacturer’s instructions. In brief, cell extracts obtained in lysis buffer were submitted to three freeze-and-thaw cycles and then centrifuged at 15 000 g at 4ºC. Supernatants were then incubated with the substrate Ac-DEVD-p-nitroaniline (Sigma-Aldrich) for four hours at 37ºC, and caspase-3 activity was determined by measuring absorbance at 405 nm. Caspase activity was expressed as pmol p-nitroaniline/h/µg protein (measured as described above).
Cell proliferation
Cells were seeded at 3000 cells/cm2 in 96-well plates. After 24 hours, cells were treated with each tested peptide, at 100 nM, or CAT (an antioxidant control) at 200 U/ml, for one hour in standard medium. After washing with PBS, 200 µM H2O2 was added for one hour in FBS-depleted medium. The medium was then replaced with the standard medium. This procedure was repeated after 48 hours and 24 hours after this repetition we proceeded to measure cell proliferation (a total five days since assay onset). At this time, cell proliferation was determined by the addition of resazurin salt solution42 (Sigma-Aldrich), at 10 µg/ml. After four hours, absorbance was measured at both 540 nm and 630 nm. The percentage difference in cell growth between treated and control cells was calculated using the following equation: [(O2 × A1) - (O1 × A2) / (O2 × P1) - (O1 × P2)] × 100, where O1 and O2 are the molar extinction coefficients of oxidized resazurin salt at 540 nm and 630 nm, respectively; A1 and A2 are the absorbance of treated cells at 540 nm and 630 nm, respectively; and P1 and P2 correspond to the absorbance of control cells (cells without treatment) at 540 nm and 630 nm, respectively.
Cell viability
Subclonfluent MC3T3-E1 cells were pre-treated for 30 minutes with the inhibitors prior to the addition of PTHrP peptides for 30 minutes in standard medium. Controls were not pretreated. The cells were then washed with PBS and incubated with 200 µM H2O2 for six hours in either serum-depleted or standard medium (to control for endogenous growth factors in FBS). After treatment, nonadherent cells were collected and pooled with adherent cells (following trypsinization), and cell viability was assessed by trypan blue exclusion, as previously described.36,43 Nonviable cells exhibiting intracellular trypan blue staining and total cell numbers were counted to calculate the percentage of cell viability in each case.
ALP activity assay
Subconfluent MC3T3-E1 cells and hOBs were preincubated with the different inhibitors for 30 minutes, subsequently followed by the addition of the tested PTHrP peptides, each at 100 nM for one hour in standard medium and 200 µM of H2O2 for two hours in FBS-depleted medium. The FBS-depleted medium was then replaced with standard medium and cells were incubated for a further 24 hours. ALP activity was measured in cell extracts obtained with PBS and 0.1% Triton X-100 (Sigma-Aldrich) using p-nitrophenyl phosphate as substrate, as previously described.28 ALP activity was normalized to cell protein content determined as described in MDA and Caspase assays.
Quantitative real-time polymerase chain reaction (PCR)
Subconfluent MC3T3-E1 cells were treated as described for the ALP activity assay. Following treatment, cell total RNA was extracted with TRIzol (Thermo Fisher Scientific, Waltham, Massachusetts). Synthesis of complementary DNA (cDNA) was performed using the high-capacity cDNA reverse transcription kit following the manufacturer’s instructions (Applied Biosystems, Grand Island, New York). A quantitative real-time PCR was performed in an ABI PRISM 7500 system (Applied Biosystems). TaqMan minor groove binder probes (Applied Biosystems, Assay-by-Design) were obtained for amplification of the following genes: runt-related transcription factor 2 (Runx2), osterix (Osx) and ALP using Premix ex-Taq polymerase (Takara Bio Inc., Otsu, Japan). The messenger RNA (mRNA) copy numbers were calculated for each sample using the cycle threshold value and normalized against 18S ribosomal RNA, as previously reported.44,45 Results were expressed as n-fold mRNA values versus corresponding values in nontreated control cells.
Statistical analysis
Results are expressed as mean ± standard error of the mean (sem) throughout the text. Differences among groups were analyzed by the Kruskal-Wallis test followed by Dunn’s post hoc test and p < 0.05 was considered significant. Statistical analysis was performed with SPSS v21 software (IBM Corp., Armonk, New York).
Results
Both N- and C-terminal PTHrP peptides counteract H2O2-induced oxidative stress in osteoblastic cells
First, we aimed to explore whether N- and C-terminal PTHrP peptides might be able to prevent ROS production by the prominent oxidative stress-related agent H2O2 in osteoblastic cells. Short exposure (ten minutes) of MC3T3-E1 cells to 200 µM H2O2 produced a significant (p < 0.01) increase in intracellular ROS (Figs 1 and 2). However, this increase was prevented by each N- or C-terminal PTHrP peptide tested or PTH (1-34) (a positive control) (each at 100 nM). Further confirmation of the capacity of these peptides to impair oxidative stress was obtained by using confocal microscopy to assess H2O2-induced DCF fluorescence changes in MC3T3-E1 cells and in hOBs (Figs 3 and 4).

Oxidative stress induced by hydrogen peroxide (H2O2) can be prevented by N- and C-terminal parathyroid hormone (PTH) and PTH-related protein (PTHrP) peptides in osteoblastic cells. Cells were exposed to 200 μM H2O2 following pretreatment with each PTHrP peptide or PTH (1-34) (100 nM) or control. Reactive oxygen species levels were measured by 2’,7’dichloro fluorescein diacetate (DFC) fluorescence. Representative flow cytometry images for a) control, b) H2O2, c) PTH + H2O2, d) PTHrP (1-37) + H2O2, e) PTHrP (107-139) + H2O2, and f) osteostatin + H2O2. M1 and M2 denote the two areas used to quantify increment in fluorescence.
Fig. 2.

Representative flow cytometry quantification. †p < 0.01 versus control; ‡p < 0.05 versus H2O2 treatment; §p < 0.01 versus H2O2 treatment. Data are represented by mean ± standard error of the mean (sem). Kruskal–Wallis test followed by Dunn’s post hoc test. N-ter(1-37), C-ter(107-139), and Ost denote parathyroid hormone-related protein (1-37), parathyroid hormone-related protein (107-139), and osteostatin, respectively.

Confocal microscopy images of MC3T3-E1 cells for a) control, b) H2O2, c) parathyroid hormone (PTH) + H2O2, d) PTH-related protein (1-37) + H2O2, e) PTH-related protein (107-139) + H2O2, and f) osteostatin + H2O. The white bar denotes 75 μm.

Confocal microscopy images of human osteoblastic cells for a) control, b) H2O2, c) parathyroid hormone (PTH) + H2O2, d) PTH-related protein (1-37) + H2O2, e) PTH-related protein (107-139) + H2O2, and f) osteostatin + H2O. The white bar denotes 20 μm.
It has been demonstrated that increased cell ROS lead to an augmented FOXO transcriptional activity.46,47 Thus, we next examined the changes in this activity elicited by H2O2 and the possible modulatory effects of the different peptides evaluated in MC3T3-E1 cells. We found that preincubation with either PTHrP peptide or PTH (1-34) abrogated the oxidant-related effect of H2O2 in these cells (Fig. 5).
Fig. 5.
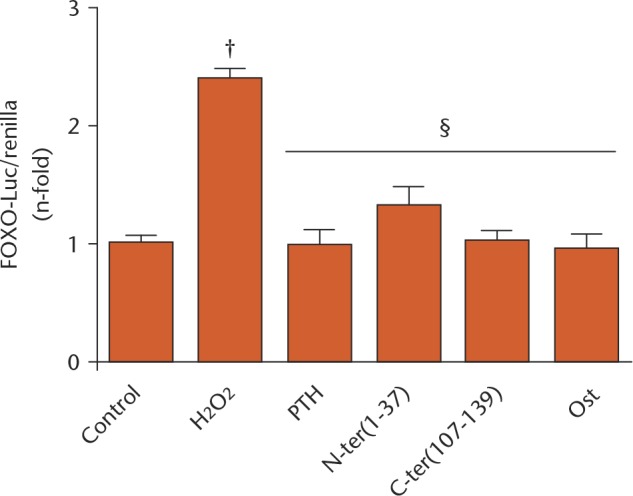
Forkhead box protein O (FOXO) activity in MC3T3-E1 cells was measured with a FOXO-luciferase plasmid. Bars represent the mean ± standard error of the mean (sem) of three independent experiments performed in triplicate. †p < 0.01 versus control; §p < 0.01 versus H2O2 treatment. Kruskal–Wallis test followed by Dunn’s post hoc test. N-ter(1-37), C-ter(107-139), and Ost denote parathyroid hormone-related protein (1-37), parathyroid hormone-related protein (107-139), and osteostatin, respectively.
Oxidative stress induces different kinds of cellular damage, which impairs regular cell function.1 Thus, we next analyzed the changes in MDA levels induced by H2O2 as an oxidative stress damage marker and the possible modulatory action of the different PTHrP peptides. As expected, this oxidative agent increased MDA levels, which were normalized by pretreating MC3T3-E1 cells and hOBS with the PTHrP peptides or PTH (1-34) (Figs 6a and b). It has been demonstrated that oxidative stress triggers an apoptotic cascade resulting in caspase activation.21 We found that caspase-3 activity increased significantly after 24 hours of exposure to H2O2, and all of the peptides tested were similarly effective in normalizing this activity in these cells (Fig. 6c).

Parathyroid hormone (PTH) and both N- and C-terminal PTH-related protein (PTHrP) peptides prevented the increase in malondialdehyde (MDA) levels and caspase-3 activation induced by H2O2 in osteoblastic cells. Cells were pretreated with the different peptides at 100 nM followed by H2O2 (200 μM) for a) and b) ten minutes, or c) two to 24 hours. Changes in MDA levels in a) MC3T3-E1 cells and b) human osteoblastic cells, and in c) caspase-3 activity in MC3T3-E1 cells, are shown. Data are mean ± standard error of the mean (sem) of three independent experiments performed at least in triplicate. *p < 0.05 versus corresponding time control; †p < 0.01 versus corresponding time control; ‡p < 0.05 versus H2O2 at the corresponding time; §p < 0.01 versus H2O2 at the corresponding time. Kruskal–Wallis test followed by Dunn’s post hoc test. N-ter(1-37), C-ter(107-139), and Ost denote PTHrP (1-37), PTHrP (107-139), and osteostatin, respectively.
Protective effects of both N- and C-terminal PTHrP peptides against the deleterious effects of H2O2 on osteoblastic cell function
Osteoblast growth has been shown to be altered negatively by oxidative stress.18,19 This prompted us to examine whether PTHrP would prevent the negative effects of a short exposure (one hour) to H2O2 during MC3T3-E1 cell growth for five days. Indeed, we showed a diminished cell proliferation with H2O2 treatment, which was similarly compensated for by both N- and C-terminal PTHrP peptides (Fig. 7a). Of note, CAT (used as an antioxidant control) produced a dramatic stimulation for cell proliferation, suggesting that basal H2O2 levels keep proliferation under control in these cells. In addition, we found that either the N- or C-terminal PTHrP domain could counteract the inhibitory effect of acute exposure of MC3T3-E1 cells to H2O2 in the presence or absence of FBS on cell viability (Fig. 7b).

Parathyroid hormone (PTH) and both N- and C-terminal PTH-related protein (PTHrP) peptides counteract the deleterious effects of H2O2 on MC3T3-E1 cell growth. Cells were preincubated with different inhibitors for 30 minutes: Rp-cyclic 3’,5’-hydrogen phosphorothioate adenosine triethylammonium salt (Rp-cAMPS; 25 µM), protein phosphatase 1 and Su1498 (5 µM) or calphostin C (CalpC) (250 nM), and then treated with PTHrP peptides (100 nM) before exposure to H2O2. Control cells were not treated. a) For calculation of cell proliferation, cell growth percentage values in treated samples were expressed as n-fold versus control (normalized to 1). Catalase (200 U/ml) was used as an antioxidant control. b) Cell viability was expressed as the percentage of viable cells over total cells. Data are mean ± standard error of the mean (sem) of three independent experiments performed at least in triplicate. *p < 0.05 versus control; †p < 0.01 versus control; ‡p < 0.05 versus H2O2; §p < 0.01 versus H2O2; probability associated with comparison A (Ap) < 0.05 versus H2O2 + N-ter (1-37); probability associated with comparison B (Bp) < 0.05 versus H2O2 + Ost. Kruskal–Wallis test followed by Dunn’s post hoc test. N-ter(1-37), C-ter(107-139) and Ost denote PTHrP (1-37), PTHrP (107-139), and osteostatin, respectively. FBS, fetal bovine serum.
We also aimed to explore the putative changes elicited by short exposure to H2O2 on various osteoblast differentiation markers and the modulatory effect of PTHrP peptides in MC3T3-E1 cells and hOBs. This oxidant was found to reduce ALP activity in both osteoblastic cell types (Figs 8a and b), as well as the gene expression of ALP, Runx2, and Osx in MC3T3-E1 cells (Fig. 8c); these deleterious effects were prevented by pretreating these cells with each PTHrP peptide tested (Figs 8a to c).

Parathyroid hormone (PTH) and both N- and C-terminal PTH-related protein (PTHrP) peptides counteract the negative effects of H2O2 on osteoblastic cell differentiation. a) MC3T3-E1 cells and b) human osteoblastic cells were preincubated or not with different inhibitors, as described in the legend for Figure 7, and then treated or not (control) with the PTHrP peptides (100 nM) before exposure to H2O2. Alkaline phosphatase (ALP) activity was expressed as n-fold over control (mean ± standard error of the mean (sem): 2.6 nmoles/min/mg (sem 0.5) and 4.3 nmoles/min/mg (sem 0.22) protein for MC3T3-E1 cells and human osteoblastic cells, respectively). c) MC3T3-E1 cells were pre treated with the PTHrP peptides in the presence or absence of the antagonist PTHrP (7-34) or the anti-osteostatin antiserum C7 before addition of H2O2. Changes in gene expression of ALP, runt-related transcription factor 2 (Runx2) and osterix (Osx) were analyzed in cell total RNA isolates by quantitative polymerase chain reaction. †p < 0.01 versus control; ‡p < 0.05 versus H2O2;§p < 0.01 versus H2O2; probability associated with comparison A (Ap) < 0.05; probability associated with comparison AA (AAp) < 0.01 versus H2O2 + N-ter(1-37); probability associated with comparison B (Bp) < 0.05; comparison associated with comparison BB (BBp) < 0.01 versus H2O2 + Ost. N-ter(1-37), C-ter(107-139) and Ost denote PTHrP (1-37), PTHrP (107-139) and osteostatin, respectively. Rp-cAMPS, Rp-cyclic 3’,5’-hydrogen phosphorothioate adenosine triethylammonium salt; CalpC, calphostin C; mRNA, messenger RNA.
As a specificity control of each PTHrP peptide action in this setting, we used several inhibitors or antagonists. Thus, we found that Rp-cAMPS (a PKA inhibitor) inhibited the ability of PTHrP (1-37) to preserve cell viability and ALP activity upon exposure to H2O2 in MC3T3-E1 cells, whereas CalpC (a PKC inhibitor), protein phosphatase 1 (Src activation inhibitor) or SU1498 (VEGFR2 activation inhibitor) abrogated such ability of osteostatin to restore ALP activity upon oxidant treatment in these cells (Figs 7b, 8a, and 8b). In addition, PTHrP (7-34) (a PTHR1 antagonist) and anti-osteostatin antiserum C7 inhibited the positive effects of PTHrP (1-37) and osteostatin, respectively, on osteoblast differentiation-related genes in these cells following H2O2 treatment (Fig. 8c).
Discussion
Oestrogen depletion has traditionally been considered the hallmark of the pathogenesis of involutional osteoporosis,48 but age-related factors intrinsic to bone, mainly oxidative stress, appear also to contribute to bone loss in this condition.1,2 In this regard, the recent finding that intermittent PTH, currently the only bone anabolic therapy, may function, as least partially, through reducing oxidative stress in aged bone26 provides insight into the mechanisms and possible treatment options for bone loss. However, the use of antioxidants alone or in combination with PTH presents several drawbacks. Thus, common antioxidants such as N-acetyl cysteine and CAT produce a decrease in ROS levels, but can also decrease osteoblastogenesis by blocking Wnt pathway activation,49 thereby inhibiting β-catenin-dependent transcription of osteogenic genes.26 In addition, antioxidants have anti-osteoclastic features that would inhibit bone remodelling.26
Previous studies have shown the ability of N- and C-terminal PTHrP peptides to promote cell growth and osteoblast differentiation in MC3T3-E1 cells and other osteoblastic cell preparations. Studies on these proteins have also reported their osteogenic properties in vivo in high oxidative stress-related conditions, namely diabetes mellitus and ovariectomized mice.24,25,27-29,32,34-36,39 Very recently, the osteoregenerative features of both PTHrP (1-37) and osteostatin have been reported in vivo in an ageing rat model with diabetes mellitus, although only indirect evidence for the antioxidant capacities of these peptides was provided.50
In this in vitro study, we disclose the antioxidant capacity of both N- and C-terminal domains of PTHrP in well-characterized osteoblastic cells. To induce oxidative stress, we used H2O2 at concentrations widely used in previous studies.18,38,51 ROS elevation can impair bone formation by interaction with the Wnt/β-catenin pathway.17 Thus, ROS-mediated activation of FOXO transcription factors diverts β-catenin from the promoter regions of genes involved in proliferation and differentiation of cells of the osteoblastic lineage, impairing these cellular programmes.52 Our data show that these PTHrP peptides were able to prevent the H2O2-induced generation of cell ROS in both murine osteoblastic cells and hOBs, and accordingly, the increase in FOXO transcriptional activity as demonstrated by a FOXO-luc reporter assay in the former cells. This is consistent with recent results showing the ability of PTHrP to increase Wnt/β-catenin signalling in osteoblastic cells exposed to high glucose concentrations, another oxidative stress-related scenario.33 Oxidative stress also triggers cellular damage and apoptosis through caspase activation.21,53 Here we show that pretreatment with these peptides decreased lipid peroxidation and blocked caspase-3 activation, thus protecting MC3T3-E1 cells from apoptosis in both osteoblastic cell types used.
It has been reported previously that oxidative stress impairs osteoblast function.5,17-19,21,51 In the present study, we report that short exposure (one to six hours) of MC3T3-E1 cells to H2O2 significantly decreased cell proliferation and viability by approximately 30%. This decrease is moderate compared with previous data in the same osteoblast cell line, although previous studies used prolonged exposure to the oxidant in a FBS-containing medium and/or higher oxidant concentration than that used herein.18,19 In addition, ALP enzymatic activity and mRNA levels, as well as gene expression of other well-known osteoblast differentiation markers, were all diminished by acute exposure to H2O2, and this was also prevented by pretreatment with both PTHrP peptides in MC3T3-E1 cells and hOBs.
Previous reports have shown the important role of the cAMP/PKA pathway in the anabolic actions elicited by the homologous N-terminal domains of PTH and PTHrP.22,24,25,35,36 This fact was confirmed by using a PKA inhibitor in the present oxidative stress scenario. In addition, specific inhibitors of PKC and Src-dependent VEGFR2 activation inhibited the protective/restoring role of osteostatin in viability and ALP activity, supporting the notion that both pathways play a major role in its osteogenic actions.24,25,36 Our results thus further emphasize the involvement of distinct signalling pathways triggered by both the N- and C-terminal PTHrP domains in osteoblasts.
Taken together, the present data indicate that both N- and C-terminal domains of PTHrP display various antioxidant features in both murine osteoblastic cells and hOBs. However, we understand that this is an in vitro study and caution should be taken when translating our results to an in vivo scenario. Even so, these findings provide novel insights that add support to the generally accepted use of PTHrP peptides as osteoporosis therapy.
Footnotes
Author Contribution: S. Portal-Núñez: Designing the study, Data acquisition, Statistical analysis, Writing the manuscript, Final approval of the manuscript.
J. A. Ardura: Designing the study, Data acquisition, Writing the manuscript, Statistical analysis.
D. Lozano: Data acquisition, Writing the manucript, Statistical analysis.
I. Martínez de Toda: Data acquisition.
M. De la Fuente: Designing the study, Revising the manuscript, Final approval of the manuscript.
G. Herrero-Beaumont: Revising the manuscript, Financial support.
R. Largo: Revising the manuscript, Financial support.
P. Esbrit: Study direction, Designing the study, Writing and revising the manuscript, Financial support.
Conflicts of Interest Statement: None declared
Follow us @BoneJointRes
Funding Statement
This work has been funded by grants from the Fundación para la Investigación Ósea y Metabolismo Mineral-FEIOMM and the Instituto de Salud Carlos III (PI11/00449, PI15/00340, PI1600065, RD12/0043/0029, RD12/0043/0008 and RD12/0043/0018,). J. A. Ardua, D. Lozano, and S. Portal-Núñez are recipients of postdoctoral contracts from the Ministerio de Economía y Competitividad, Juan de la Cierva program JCI-2011-09548, FPDI-2013-17268, and RETICEF [FEDER “una manera de hacer Europa” (RD12/0043/0008)].
References
- 1. Manolagas SC. From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis. Endocr Rev 2010;31:266-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Khosla S, Melton LJ, III, Riggs BL. The unitary model for estrogen deficiency and the pathogenesis of osteoporosis: is a revision needed? J Bone Miner Res 2011;26:441-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. De la, Fuente M, Miquel J. An update of the oxidation-inflammation theory of aging: the involvement of the immune system in oxi-inflamm-aging. Curr Pharm Des 2009;15:3003-3026. [DOI] [PubMed] [Google Scholar]
- 4. Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol 1956;11:298-300. [DOI] [PubMed] [Google Scholar]
- 5. Almeida M, Han L, Martin-Millan M, et al. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J Biol Chem 2007;282:27285-27297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hamada Y, Kitazawa S, Kitazawa R, et al. Histomorphometric analysis of diabetic osteopenia in streptozotocin-induced diabetic mice: a possible role of oxidative stress. Bone 2007;40:1408-1414. [DOI] [PubMed] [Google Scholar]
- 7. Kops GJPL, Dansen TB, Polderman PE, et al. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 2002;419:316-321. [DOI] [PubMed] [Google Scholar]
- 8. Nemoto S, Finkel T. Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science 2002;295:2450-2452. [DOI] [PubMed] [Google Scholar]
- 9. Tran H, Brunet A, Grenier JM, et al. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science 2002;296:530-534. [DOI] [PubMed] [Google Scholar]
- 10. Ambrogini E, Almeida M, Martin-Millan M, et al. FoxO-mediated defense against oxidative stress in osteoblasts is indispensable for skeletal homeostasis in mice. Cell Metab 2010;11:136-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nojiri H, Saita Y, Morikawa D, et al. Cytoplasmic superoxide causes bone fragility owing to low-turnover osteoporosis and impaired collagen cross-linking. J Bone Miner Res 2011;26:2682-2694. [DOI] [PubMed] [Google Scholar]
- 12. Cervellati C, Bonaccorsi G, Cremonini E, et al. Oxidative stress and bone resorption interplay as a possible trigger for postmenopausal osteoporosis. Biomed Res Int 2014;2014:569563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Basu S, Michaëlsson K, Olofsson H, Johansson S, Melhus H. Association between oxidative stress and bone mineral density. Biochem Biophys Res Commun 2001;288:275-279. [DOI] [PubMed] [Google Scholar]
- 14. Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J 2003;17:1195-1214. [DOI] [PubMed] [Google Scholar]
- 15. Jacob KD, Noren Hooten N, Trzeciak AR, Evans MK. Markers of oxidant stress that are clinically relevant in aging and age-related disease. Mech Ageing Dev 2013;134:139-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stadtman ER, Levine RL. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids 2003;25:207-218. [DOI] [PubMed] [Google Scholar]
- 17. Almeida M, Han L, Martin-Millan M, O’Brien CA, Manolagas SC. Oxidative stress antagonizes Wnt signaling in osteoblast precursors by diverting beta-catenin from T cell factor- to forkhead box O-mediated transcription. J Biol Chem 2007;282:27298-27305. [DOI] [PubMed] [Google Scholar]
- 18. Li M, Zhao L, Liu J, et al. Hydrogen peroxide induces G2 cell cycle arrest and inhibits cell proliferation in osteoblasts. Anat Rec (Hoboken) 2009;292:1107-1113. [DOI] [PubMed] [Google Scholar]
- 19. Mody N, Parhami F, Sarafian TA, Demer LL. Oxidative stress modulates osteoblastic differentiation of vascular and bone cells. Free Radic Biol Med 2001;31:509-519. [DOI] [PubMed] [Google Scholar]
- 20. Bai XC, Lu D, Bai J, et al. Oxidative stress inhibits osteoblastic differentiation of bone cells by ERK and NF-kappaB. Biochem Biophys Res Commun 2004;314:197-207. [DOI] [PubMed] [Google Scholar]
- 21. Almeida M, Han L, Ambrogini E, Bartell SM, Manolagas SC. Oxidative stress stimulates apoptosis and activates NF-kappaB in osteoblastic cells via a PKCbeta/p66shc signaling cascade: counter regulation by estrogens or androgens. Mol Endocrinol 2010;24:2030-2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Esbrit P, Alcaraz MJ. Current perspectives on parathyroid hormone (PTH) and PTH-related protein (PTHrP) as bone anabolic therapies. Biochem Pharmacol 2013;85:1417-1423. [DOI] [PubMed] [Google Scholar]
- 23. Sneddon WB, Magyar CE, Willick GE, et al. Ligand-selective dissociation of activation and internalization of the parathyroid hormone (PTH) receptor: conditional efficacy of PTH peptide fragments. Endocrinology 2004;145:2815-2823. [DOI] [PubMed] [Google Scholar]
- 24. Valín A, Guillén C, Esbrit P. C-terminal parathyroid hormone-related protein (PTHrP) (107-139) stimulates intracellular Ca(2+) through a receptor different from the type 1 PTH/PTHrP receptor in osteoblastic osteosarcoma UMR 106 cells. Endocrinology 2001;142:2752-2759. [DOI] [PubMed] [Google Scholar]
- 25. García-Martín A, Acitores A, Maycas M, Villanueva-Peñacarrillo ML, Esbrit P. Src kinases mediate VEGFR2 transactivation by the osteostatin domain of PTHrP to modulate osteoblastic function. J Cell Biochem 2013;114:1404-1413. [DOI] [PubMed] [Google Scholar]
- 26. Jilka RL, Almeida M, Ambrogini E, et al. Decreased oxidative stress and greater bone anabolism in the aged, when compared to the young, murine skeleton with parathyroid hormone administration. Aging Cell 2010;9:851-867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. de Castro LF, Lozano D, Portal-Núñez S, et al. Comparison of the skeletal effects induced by daily administration of PTHrP (1-36) and PTHrP (107-139) to ovariectomized mice. J Cell Physiol 2012;227:1752-1760. [DOI] [PubMed] [Google Scholar]
- 28. Lozano D, Sánchez-Salcedo S, Portal-Núñez S, et al. Parathyroid hormone-related protein (107-111) improves the bone regeneration potential of gelatin-glutaraldehyde biopolymer-coated hydroxyapatite. Acta Biomater 2014;10:3307-3316. [DOI] [PubMed] [Google Scholar]
- 29. Lozano D, Feito MJ, Portal-Núñez S, et al. Osteostatin improves the osteogenic activity of fibroblast growth factor-2 immobilized in Si-doped hydroxyapatite in osteoblastic cells. Acta Biomater 2012;8:2770-2777. [DOI] [PubMed] [Google Scholar]
- 30. Trejo CG, Lozano D, Manzano M, et al. The osteoinductive properties of mesoporous silicate coated with osteostatin in a rabbit femur cavity defect model. Biomaterials 2010;31:8564-8573. [DOI] [PubMed] [Google Scholar]
- 31. Manzano M, Lozano D, Arcos D, et al. Comparison of the osteoblastic activity conferred on Si-doped hydroxyapatite scaffolds by different osteostatin coatings. Acta Biomater 2011;7:3555-3562. [DOI] [PubMed] [Google Scholar]
- 32. Lozano D, Manzano M, Doadrio JC, et al. Osteostatin-loaded bioceramics stimulate osteoblastic growth and differentiation. Acta Biomater 2010;6:797-803. [DOI] [PubMed] [Google Scholar]
- 33. López-Herradón A, Portal-Núñez S, García-Martín A, et al. Inhibition of the canonical Wnt pathway by high glucose can be reversed by parathyroid hormone-related protein in osteoblastic cells. J Cell Biochem 2013;114:1908-1916. [DOI] [PubMed] [Google Scholar]
- 34. Lozano D, de Castro LF, Dapía S, et al. Role of parathyroid hormone-related protein in the decreased osteoblast function in diabetes-related osteopenia. Endocrinology 2009;150:2027-2035. [DOI] [PubMed] [Google Scholar]
- 35. de Gortázar AR, Alonso V, Alvarez-Arroyo MV, Esbrit P. Transient exposure to PTHrP (107-139) exerts anabolic effects through vascular endothelial growth factor receptor 2 in human osteoblastic cells in vitro. Calcif Tissue Int 2006;79:360-369. [DOI] [PubMed] [Google Scholar]
- 36. Alonso V, de Gortázar AR, Ardura JA, et al. Parathyroid hormone-related protein (107-139) increases human osteoblastic cell survival by activation of vascular endothelial growth factor receptor-2. J Cell Physiol 2008;217:717-727. [DOI] [PubMed] [Google Scholar]
- 37. Villalvilla A, García-Martín A, Largo R, et al. The adipokine lipocalin-2 in the context of the osteoarthritic osteochondral junction. Sci Rep 2016;6:29243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ardura JA, Portal-Núñez S, Castelbón-Calvo I, et al. Parathyroid hormone-related protein protects osteoblastic cells from oxidative stress by activation of MKP1 phosphatase. J Cell Physiol 2017;232:785-796. [DOI] [PubMed] [Google Scholar]
- 39. Lozano D, Fernández-de-Castro L, Portal-Núñez S, et al. The C-terminal fragment of parathyroid hormone-related peptide promotes bone formation in diabetic mice with low-turnover osteopaenia. Br J Pharmacol 2011;162:1424-1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Peiró C, Lafuente N, Matesanz N, et al. High glucose induces cell death of cultured human aortic smooth muscle cells through the formation of hydrogen peroxide. Br J Pharmacol 2001;133:967-974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Huang X, Frenkel K, Klein CB, Costa M. Nickel induces increased oxidants in intact cultured mammalian cells as detected by dichlorofluorescein fluorescence. Toxicol Appl Pharmacol 1993;120:29-36. [DOI] [PubMed] [Google Scholar]
- 42. O’Brien J, Wilson I, Orton T, Pognan F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur J Biochem 2000;267:5421-5426. [DOI] [PubMed] [Google Scholar]
- 43. Alonso V, Pérez-Martínez FC, Calahorra FJ, Esbrit P. Phytoestrogen modulation of bone-related cytokines and its impact on cell viability in human prostate cancer cells. Life Sci 2009;85:421-430. [DOI] [PubMed] [Google Scholar]
- 44. Portal-Núñez S, Lozano D, de Castro LF, et al. Alterations of the Wnt/beta-catenin pathway and its target genes for the N- and C-terminal domains of parathyroid hormone-related protein in bone from diabetic mice. FEBS Lett 2010;584:3095-3100. [DOI] [PubMed] [Google Scholar]
- 45. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001;25:402-408. [DOI] [PubMed] [Google Scholar]
- 46. van der Horst A, de Vries-Smits AMM, Brenkman AB, et al. FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat Cell Biol 2006;8:1064-1073. [DOI] [PubMed] [Google Scholar]
- 47. Furuyama T, Nakazawa T, Nakano I, Mori N. Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF-16 homologues. Biochem J 2000;349:629-634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Riggs BL, Khosla S, Melton LJ., III A unitary model for involutional osteoporosis: estrogen deficiency causes both type I and type II osteoporosis in postmenopausal women and contributes to bone loss in aging men. J Bone Miner Res 1998;13:763-773. [DOI] [PubMed] [Google Scholar]
- 49. Funato Y, Michiue T, Asashima M, Miki H. The thioredoxin-related redox-regulating protein nucleoredoxin inhibits Wnt-beta-catenin signalling through dishevelled. Nat Cell Biol 2006;8:501-508. [DOI] [PubMed] [Google Scholar]
- 50. Ardura JA, Portal-Núñez S, Lozano D, et al. Local delivery of parathyroid hormone-related protein-derived peptides coated onto a hydroxyapatite-based implant enhances bone regeneration in old and diabetic rats. J Biomed Mater Res A 2016;104:2060-2070. [DOI] [PubMed] [Google Scholar]
- 51. Fatokun AA, Stone TW, Smith RA. Responses of differentiated MC3T3-E1 osteoblast-like cells to reactive oxygen species. Eur J Pharmacol 2008;587:35-41. [DOI] [PubMed] [Google Scholar]
- 52. Hoogeboom D, Essers MAG, Polderman PE, et al. Interaction of FOXO with beta-catenin inhibits beta-catenin/T cell factor activity. J Biol Chem 2008;283:9224-9230. [DOI] [PubMed] [Google Scholar]
- 53. Almeida M, Ambrogini E, Han L, Manolagas SC, Jilka RL. Increased lipid oxidation causes oxidative stress, increased peroxisome proliferator-activated receptor-gamma expression, and diminished pro-osteogenic Wnt signaling in the skeleton. J Biol Chem 2009;284:27438-27448. [DOI] [PMC free article] [PubMed] [Google Scholar]