

Abstract
Strict maternal inheritance renders the mitochondrial genome susceptible to accumulating mutations that harm males, but are otherwise benign or beneficial for females. This ‘mother's curse’ effect can degrade male survival and fertility if unopposed by counteracting evolutionary processes. Coadaptation between nuclear and mitochondrial genomes—with nuclear genes evolving to compensate for male-harming mitochondrial substitutions—may ultimately resolve mother's curse. However, males are still expected to incur a transient fitness cost during mito-nuclear coevolution, and it remains unclear how severe such costs should be. We present a population genetic analysis of mito-nuclear coadaptation to resolve mother's curse effects, and show that the magnitude of the ‘male mitochondrial load’—the negative impact of mitochondrial substitutions on male fitness components—may be large, even when genetic variation for compensatory evolution is abundant. We also find that the male load is surprisingly sensitive to population size: male fitness costs of mito-nuclear coevolution are particularly pronounced in both small and large populations, and minimized in populations of intermediate size. Our results reveal complex interactions between demography and genetic constraints during the resolution of mother's curse, suggesting potentially widespread species differences in susceptibility to mother's curse effects.
Keywords: epistasis, sex-specific selection, sexual conflict, adaptation, compensatory evolution
1. Introduction
With rare exceptions, mitochondria are maternally inherited, with little to no transmission from fathers to offspring [1]. This unique inheritance pattern may lead to unusual evolutionary dynamics of mitochondrial genes, compared with genes that are encoded within the nuclear genome [2–4]. Whereas strict maternal transmission ensures that the mitochondrial genome is evolutionarily responsive to natural selection in females, it inhibits evolutionary responses to selection in males. Mitochondrial DNA (mtDNA) mutations that reduce the survival, mating success or fertility of males may therefore accumulate within a population, as long as these mutations do not reduce female fitness (i.e. they may be benign or beneficial for females [2–6]). The evolutionary accumulation of mtDNA mutations with male-harming effects, attributable to the maternal inheritance of mitochondria, has been dubbed ‘mother's curse’ [4].
Several observations suggest that mother's curse influences the evolution of mitochondrial genomes, the genetic basis of female and male adaptations and the manifestation of disease. In plants, cytoplasmic male sterility (CMS) is widespread, typically has a mitochondrial genetic basis, and is important in both the evolution of plant breeding systems and production of hybrid seeds in commercial agriculture [2,7,8]. Sex-specific fitness effects of mitochondrial genetic variation have received less attention in animal systems; even so, some mtDNA mutations are known to have male-biased effects on animal fertility (e.g. [9–12]) and neurodegenerative disease (e.g. [13,14]). Studies in Drosophila have identified signals of mother's curse in patterns of (i) male-biased effects of mitochondrial haplotypes on the transcription of nuclear-encoded genes [15] (but see [16,17]), (ii) male-biased genetic variance in ageing [18,19] and (iii) male-limited genetic variation for sterility [11,20]. Overall, the pervasiveness of mother's curse effects among species, populations and environmental contexts remains unclear (e.g. [21,22]). Nevertheless, current examples suggest that at least a fraction of mtDNA mutations have male-biased or male-limited fitness costs (e.g. [6,11,23]). The evolutionary logic of the mother's curse hypothesis predicts that such mutations are likely to accumulate within populations and depress male fitness components, unless they are countered by evolutionary mechanisms that oppose their spread, or compensate for their negative effects in males [3,24,25].
Two general evolutionary scenarios can potentially resolve mother's curse, and rescue male fitness declines caused by mtDNA mutation accumulation. First, some forms of inbreeding, kin selection, assortative mating and paternal mitochondrial transmission generate direct purifying selection against mtDNA mutations with male-limited costs [26–30]. These processes provide scope for the elimination of male-harming mtDNA mutations, although purifying selection through males is likely to remain inefficient [28,31], particularly when male-harming alleles have small fitness effects [26,27]. Second, changes to the nuclear genome may compensate for the fitness costs of mtDNA alleles by masking their male fitness consequences [6,24,32]. Mitochondrial and nuclear genomes are functionally coupled through their shared roles in encoding the cellular machinery of energy production [33–35], and this close coupling provides a mechanistic basis for mito-nuclear genetic interactions. Mito-nuclear interactions for fitness—including sex-specific fitness components—have been reported in several animal systems (e.g. [24,34,36–44]). Likewise, in plant systems that harbour mtDNA alleles that cause CMS, nuclear genes play central roles in restoring male fertility [8].
Whether evolutionary changes in the nuclear genome can compensate for male-harming mtDNA mutants is not controversial. However, the speed and efficiency with which nuclear genome evolution resolves mother's curse remains an open question [32]. Several models have considered the coevolutionary dynamics of male-harming mitochondrial mutations and compensatory alleles in the nuclear genome (e.g. [2,32,36,45–47]). Theory of mito-nuclear coevolution is particularly well developed in contexts of CMS, where conditions for the evolutionary invasion, maintenance and fixation of male-sterility genotypes are well characterized (reviewed by Jacobs & Wade [48]). On the other hand, prior theory largely focuses on the deterministic dynamics and long-term evolutionary equilibria of mitochondrial and nuclear genes. Much less attention has been given to non-equilibrium dynamics of mito-nuclear coevolution, where mito-nuclear dynamics should generate transient reductions in male fitness components prior to the ultimate resolution of mother's curse. There is currently no clear expectation for the severity of male fitness costs that arise from bouts of mito-nuclear coevolution, nor expectations for the impacts of genetic drift and population size on the manifestation of mother's curse effects.
Here, we present a theoretical population genetic analysis of mito-nuclear coevolution in the context of mother's curse. We specifically quantify the ‘male mitochondrial load’—the reduction in male fitness components during coevolutionary cycles between male-harming mtDNA substitutions and nuclear compensatory substitutions that restore the fitness components. Our formulation of load is conceptually similar to ‘substitution loads’ or ‘lag loads’ that arise during adaptation to a new environment (e.g. [49–51]), with mtDNA divergence serving as an analogue of environmental change. The theoretical framework also parallels a long tradition of compensatory evolution models in population genetics [52–54]. Our results consider male-harming mtDNA substitutions that are fixed by positive selection in females (i.e. sexually antagonistic mutations), as well as substitutions that are neutral in females and fixed by genetic drift. We show that the magnitude of the male mitochondrial load can be large relative to classical genetic loads. We also demonstrate that the load is highly sensitive to the effective population size of the species: the male mitochondrial load exhibits a non-monotonic relation with population size, which is minimized in intermediate-sized populations. These results shed new light on the contexts of selection and demography that are likely to elevate or reduce the impact of mother's curse effects.
2. Model
(a). Model structure and assumptions
Our baseline model follows the evolution of a pair of interacting mito-nuclear loci that affect male fitness. Derived (mutant) alleles at the mitochondrial locus are assumed to reduce male fitness relative to the ancestral genotype; these alleles may be neutral or beneficial for females. For simplicity, we assume that the nuclear (i.e. compensatory) locus is male-limited in expression (alleles are neutral in females); we later revisit this assumption. Generations are discrete with a life cycle of (i) birth, (ii) selection, (iii) meiosis and mutation, followed by random mating, and (iv) death of the adults.
Interacting loci have two alleles each. The mitochondrial locus has alleles M and m, and the nuclear locus has alleles A and a. M and A represent the ancestral (wild-type) alleles, whereas m and a are derived alleles. We assume that mitochondrial inheritance is strictly maternal (there is no paternal transmission). The nuclear locus is diploid, with biparental inheritance. Following prior models [3,26,27,30], we assume that each individual in the population is homoplasmic for one of the two mitochondrial alleles. This simplifying assumption is reasonable under strict maternal inheritance, with mitochondria strongly bottlenecked during oogenesis; we return to this issue in the Discussion. Following the tradition of compensatory evolution models (e.g. [54]), we assume that derived mutations are individually deleterious (fitness costs of sm and sa are associated with the m and a alleles, respectively), but beneficial for males in combination (table 1). Thus, AA/M and aa/m genotypes yield maximal fitness, and the remaining genotypic combinations impose a cost to males.
Table 1.
Relative fitness as a function of the mito-nuclear genotype.
| nuclear genotype |
|||
|---|---|---|---|
| AA | Aa | aa | |
| male relative fitness | |||
| M cytotype | 1 | 1 − sa/2 | 1 − sa |
| m cytotype | 1 − sm | 1 − sm/2 | 1 |
| female relative fitness | |||
| M cytotype | 1 | 1 | 1 |
| m cytotype | 1 + sf | 1 + sf | 1 + sf |
Each evolutionary cycle between a mitochondrial and nuclear locus initiates with the invasion—from a single, initial copy—of a derived, male-harming mitochondrial allele, m. Allele m evolves under positive selection in females or genetic drift until it is eventually fixed in the population. The nuclear locus evolves under recurrent mutation and selection in males. The population is initially fixed for the ancestral allele, A, which is beneficial for males in combination with the ancestral mitochondrial allele, M. For simplicity, we assume that mutations from A to a are unidirectional, with a rate of v, per meiosis; introduction of back-mutation is trivial, and simply allows for the maintenance of rare A alleles in populations that become fixed for the m cytotype. The evolutionary cycle completes when both derived alleles (a and m) are fixed.
(b). Quantifying the male mitochondrial load
Each cycle of mito-nuclear coevolution contributes to the male genetic load, as follows. Let t (t ≥ 0) represent the number of generations following the initial appearance of a mitochondrial allele destined to eventually fix. Following the standard genetic load definition, the male load at generation t is
| 2.1 |
where  is the mean of relative male fitness at generation t, and wmax is the relative fitness of the best genotype for males (i.e. AAM and aam from table 1, where wmax is scaled to one in our model). The cumulative contribution of the coevolutionary cycle to the male load is
is the mean of relative male fitness at generation t, and wmax is the relative fitness of the best genotype for males (i.e. AAM and aam from table 1, where wmax is scaled to one in our model). The cumulative contribution of the coevolutionary cycle to the male load is 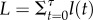 , where τ is the duration, in generations, of the cycle (i.e. the time between the origin of the male-harming allele, m, to the eventual fixation of the derived mito-nuclear combination, am). From this, the average contribution of a single coevolutionary cycle to the male load is
, where τ is the duration, in generations, of the cycle (i.e. the time between the origin of the male-harming allele, m, to the eventual fixation of the derived mito-nuclear combination, am). From this, the average contribution of a single coevolutionary cycle to the male load is
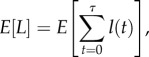 |
2.2 |
where E[x] denotes the expectation. Biologically, (N/2)E[L] represents the average number of males that are selectively eliminated during a typical bout of mito-nuclear coevolution, where N/2 is the number of males in each generation (see [50] and [55], pp. 81–82). This concept of the male load parallels other forms of genetic load that arise during adaptation, particularly Haldane's [49] ‘cost of selection’ and Maynard Smith's [51] ‘lag load’ (see [56], p. 169). We return to this comparison further below.
Equation (2.2) quantifies the cumulative contribution of single mito-nuclear coevolutionary cycles to the male load, but it does not take into account the tempo of male-harming mtDNA substitutions that initiate each cycle. To quantify the fitness cost to males, per generation, we scale the cumulative load per coevolutionary cycle by the average number of generations between cycles of mito-nuclear coevolution (denoted as TM):
| 2.3 |
([55], pp. 81–82), where K = 1/TM is the rate of substitution of male-harming alleles in the mitochondrial genome. Equation (2.3) applies when the time scale for resolution of mother's curse is faster than the tempo of male-harming mtDNA substitution. Otherwise mitochondrial function in males will systematically degenerate over time—a situation more closely aligned with models of mutational meltdown (e.g. [57,58]).
(c). Model analysis
Our analytical results assume that selection coefficients are small (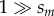 , sa > 0), and population-scaled selection is strong (Nsm,
, sa > 0), and population-scaled selection is strong (Nsm, 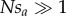 , where N is the size of a Wright–Fisher population). Under random mating, deviations from Hardy–Weinberg equilibrium and mito-nuclear linkage disequilibrium are negligible. We assume throughout that the effective size of a mtDNA locus is one-quarter the size of a diploid, nuclear locus (2N for a diploid locus; N/2 for a mtDNA locus).
, where N is the size of a Wright–Fisher population). Under random mating, deviations from Hardy–Weinberg equilibrium and mito-nuclear linkage disequilibrium are negligible. We assume throughout that the effective size of a mtDNA locus is one-quarter the size of a diploid, nuclear locus (2N for a diploid locus; N/2 for a mtDNA locus).
At the beginning of a coevolutionary cycle (arbitrarily labelled as generation t = 0), a male-harming m allele begins to spread within the population until it is eventually fixed. The frequency of m in the zygotes at generation t (t ≥ 0) is qmt. Following prior theory (e.g. [51,55,59–61]), we use deterministic models to approximate the expected evolutionary trajectory of the m allele, conditioned on its eventual fixation (see the electronic supplementary material). For the case of neutrally evolving mtDNA alleles (sf = 0; table 1), the expected frequency of m at generation t is
| 2.4a |
(see [55], p. 147; see also [61]), where the initial frequency of m is qm0 = 2/N. For the case of a sexually antagonistic m allele (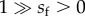 ), the expected frequency of m at generation t is
), the expected frequency of m at generation t is
| 2.4b |
(e.g. [62]), where 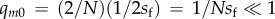 is the ‘effective’ initial frequency of the allele [51,59,60,63].
is the ‘effective’ initial frequency of the allele [51,59,60,63].
The frequency of the m allele mediates selection at the interacting nuclear locus. Consider an a allele that segregates at a frequency of qat in zygotes of generation t. Assuming Hardy–Weinberg and linkage equilibrium in the zygotes (as stated above), the expected frequencies of the three nuclear genotypes in adult males (after selection in generation t) are
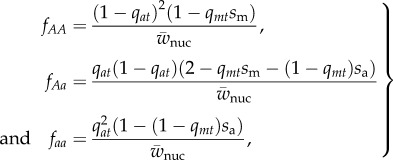 |
2.5 |
where 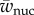 is the sum of the numerators of fAA, fAa and faa. The expected change in frequency of a due to selection in males is
is the sum of the numerators of fAA, fAa and faa. The expected change in frequency of a due to selection in males is
| 2.6 |
The total frequency change across a single generation, owing to selection, will be 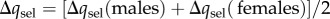 , where
, where 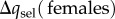 refers to the change in a over one generation as a consequence of selection in females (
refers to the change in a over one generation as a consequence of selection in females ( when the a is neutral for females). From equation (2.6), note that selection in males favours the a allele once m exceeds the critical frequency qmt = sa/(sm + sa), and otherwise allele A is favoured. Given the assumption of small selection coefficients and mutation rate of v (see above), the total expected change in the frequency of a will be
when the a is neutral for females). From equation (2.6), note that selection in males favours the a allele once m exceeds the critical frequency qmt = sa/(sm + sa), and otherwise allele A is favoured. Given the assumption of small selection coefficients and mutation rate of v (see above), the total expected change in the frequency of a will be 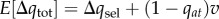 .
.
Mathematical analysis of the model focuses on two idealized scenarios. First, under sufficiently weak mutation at the compensatory locus, the time scale to resolve mother's curse is slow relative to the time to fixation of m. The load is then dominated by a lag until a compensatory mutation arises and invades the population. The male fitness component effectively ‘jumps’ between two states: a state of high relative fitness, and a state of low relative fitness (i.e. 1 – sm; figure 1). Assuming that the waiting time for a compensatory substitution is smaller than the interval between male-harming mtDNA substitutions (TC < TM, where TC is the mean time to compensation following the fixation of the male-harming allele; see above), the fraction of time spent in the low-fitness state will be flow = TC/TM, and male load, scaled per generation (equation (2.3)), is E[L | slow]/TM = smflow.
Figure 1.

Fluctuations in male fitness components resulting from coevolution between a mitochondrial and nuclear locus. The figure illustrates the case of sequential substitutions of male-harming mtDNA alleles, each followed by the substitution of a compensatory allele in the nuclear genome that restores male fitness. The mean time between substitutions at a mitochondrial locus is TM. The mean lag between a mitochondrial substitution and a nuclear compensatory substitution is TC. The time that males spend within the low-fitness state depends on the tempo of mitochondrial substitutions relative to the lag time of compensatory substitutions. (Online version in colour.)
Second, with a sufficiently high compensatory mutation rate, a alleles will spread rapidly once they are favoured. For the neutral model of mtDNA substitution, the evolutionary dynamics of the m allele are slow relative to the rate of spread of a, and the latter is therefore likely to fix before the former. In such cases, we can use a separation-of-timescales approximation (e.g. [62]), and model the substitution process at the nuclear locus as if a fixes instantaneously once selection favours it. The cumulative load becomes
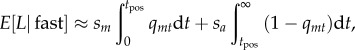 |
2.7a |
where qmt is given by equation (2.4a), and tpos is the time until positive selection favours the compensatory allele (the value of t for which qmt > sa/(sm + sa), based on equation (2.4a)).
When mtDNA mutations are positively selected in females, a compensatory allele is unlikely to invade until the m cytotype approaches fixation, but nevertheless, the load may be dominated by the time to fixation of the compensatory allele rather than the time until its initial appearance. In this case, the total load will be
| 2.7b |
where qat tracks the frequency of a compensatory allele that eventually becomes fixed.
(d). Simulations
Analytical results were verified using forward simulations that incorporate selection, mutation and multinomial sampling in a Wright–Fisher population of effective size N. The simulations track all six possible genotypes—AAM, AAm, AaM, Aam, aaM, aam—in each sex, allowing for the build-up of linkage disequilibrium between mitochondrial and nuclear loci. Each simulation run was initiated with the population fixed for the A allele, and initial frequencies of 2/N and 1–2/N for M and m alleles, respectively. Each run completes with the fixation of both a and m. For a given parameter set of sf, sm, sa, N and v, the average over simulation runs provides an estimate the exact cumulative contribution of mito-nuclear coevolution to the male load (equation (2.2)). All simulations were run in R v. 3.3.0 [64]. Additional details can be found in the electronic supplementary material.
3. Results
(a). Male-harming mtDNA substitutions with neutral effects in females
When male-harming mtDNA substitutions are neutral for females (sf = 0), the evolutionary dynamics of mitochondrial and nuclear substitutions depend on the magnitude of the compound parameter, N2vsm. When 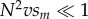 , each male-harming mtDNA allele fixes rapidly relative to the waiting time to invasion and fixation of its corresponding compensatory allele. Intuitively, this is because the mean time to fixation of a neutral mtDNA mutation is N generations, whereas the time to invasion of a favoured compensatory allele is roughly TC = 1/(Nvsm) generations. Thus, when
, each male-harming mtDNA allele fixes rapidly relative to the waiting time to invasion and fixation of its corresponding compensatory allele. Intuitively, this is because the mean time to fixation of a neutral mtDNA mutation is N generations, whereas the time to invasion of a favoured compensatory allele is roughly TC = 1/(Nvsm) generations. Thus, when 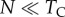 (or
(or 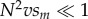 ), the contribution to the male load will be
), the contribution to the male load will be
| 3.1a |
Note that equation (3.1a) is inversely proportional to population size, reflecting adaptation limited by the availability of compensatory mutations. At the opposite extreme, when compensatory evolution is fast relative to the time scale of fixation for a mtDNA allele (N2vsm → ∞, justifying equation (2.7a)), the contribution to the male load is
| 3.1b |
(see the electronic supplementary material). From equation (3.1b), we see that in the limit of fast compensatory change, the male load now increases with population size. This positive relation between N and load reflects the impact of the fixation time of male-harming mtDNA alleles on the male load. Coevolution is no longer limited by the availability of genetic variation at compensatory loci; rather, the resolution of the male load is limited by the time until substitution of the mtDNA allele, which is proportional to N.
Forward simulations across a spectrum of N2vsm show that equations (3.1a) and (3.1b) work well within their relevant regions of parameter space (figure 2a; electronic supplementary material); their sum provides a general approximation for the male load:
| 3.2 |
Figure 2.

The male mitochondrial load due to male-harming/female-neutral mtDNA substitutions. (a) Results are shown for N = 10 000 and sm = sa = 0.02, with a variable mutation rate at the compensatory locus (v). Open circles show the average load from 1000 Wright–Fisher simulations of pairs of mito-nuclear substitutions. Mitochondrial load results are contrasted against the classical substitution load model (Haldane's [49] ‘cost of selection’, represented by C = ln(Ns)), which shows the load arising from the substitution of an unconditionally beneficial mtDNA mutation with fitness benefit of s = 0.02, and ‘effective’ initial frequency of 1/Ns (i.e. following [51,59]; see the electronic supplementary material). (b) Male mitochondrial load as a function of population size (N). Theoretical curves, based on equation (3.2), illustrate the impacts of the compensatory mutation rate (v) and male selection parameters (sm, sa) on the magnitude of the load. The minimum for each curve corresponds to Ncrit, the population size that minimizes the male load (see equation (3.3)).
The above equation implies that for a given strength of selection (sm, sa) and compensatory mutation rate (v), there is a critical effective population size that minimizes the male mitochondrial load. This critical population size corresponds to
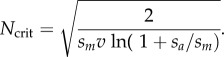 |
3.3 |
Below the threshold, there is a negative relation between N and the male load; above the threshold, the relation is positive (figure 2b). Intuitively, the critical population size increases as the compensatory mutation rate decreases, reflecting the greater influx of compensatory alleles in larger populations. A similar result applies if we rescale E[L] relative to the tempo at which male-harming/female-neutral mutations become fixed (i.e. equation (2.3)). Following standard population genetics theory (e.g. [65]), the average interval between neutrally evolving mtDNA substitutions is simply TM = 1/UN, where UN is the mitochondrial genomic mutation rate to male-harming alleles that are neutral to females. Consequently, the scaled load is E[L]/TM = UNE[L], which is minimized at the Ncrit in equation (3.3).
(b). Sexually antagonistic mtDNA substitutions
When male-harming mtDNA substitutions are sexually antagonistic (i.e. they benefit females), mito-nuclear substitutions are much more likely to fix sequentially because positive selection on each mtDNA mutation speeds the time to its fixation. When the strength of selection in females is of similar order to selection in males (sf/sm is not too small), compensatory alleles rarely begin to spread before the male-harming mtDNA allele is fixed (or nearly fixed) in the population. From equation (2.7b), the contribution to the male load becomes
| 3.4 |
(see the electronic supplementary material). Equation (3.4) closely approximates the male load when sm and sf have a similar order of magnitude (figure 3), and it underestimates the true load when compensatory mutation is strong and selection in females is weak (Nv is large; 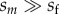 ; electronic supplementary material).
; electronic supplementary material).
Figure 3.

The male mitochondrial load due to sexually antagonistic mtDNA substitutions. Results are shown for N = 100 000, sm = sa = 0.02, and variable compensatory mutation rate, v. The solid line is the approximation from equation (3.4). Open circles show the average load from 1000 Wright–Fisher simulations of pairs of mito-nuclear substitutions. Mitochondrial load results are contrasted against the classical substitution load model (Haldane's [49] ‘cost of selection’, represented by C = ln(Ns)), which shows the load arising from the substitution of an unconditionally beneficial mtDNA mutation with fitness benefit of s = 0.02, and ‘effective’ initial frequency of 1/Ns (i.e. following [51,59]; see the electronic supplementary material).
From equation (3.4), the impact of individual bouts of mito-nuclear coevolution on the male load is minimized at an intermediate population size: Ncrit = 1/4v. The overall relation between population size and the male load also depends on the rate of male-harming mtDNA substitutions (K), which may also be dependent on N [66–68]. Most models of adaptive substitution predict a positive relation between population size and the rate of evolution. Some models predict a positive, linear association between K and N (e.g. [67,69]), while others predict that K is, at most, a diminishing-returns function of N [51,66]. We can generalize between these extremes by supposing that K ∝ Nb, where b is a constant in 0 < b < 1 that defines the strength of the diminishing return (e.g. K saturates at modest N when b is small). The net load is therefore proportional to NbE[L], and the critical population size falls within the range
| 3.5 |
where e ∼ 2.718 (note that b = 1 sets the lower bound of Ncrit, whereas b = 0 sets the upper bound). As before, the male load is minimized at an intermediate population size, with Ncrit decreasing as b increases.
4. Discussion
Strict maternal inheritance of mitochondrial genes facilitates accumulation of male-harming mtDNA mutations, which poses a problem for the long-term viability and fertility of males [3,6]. This is the dilemma of mother's curse [4]. Although compensatory evolution in the nuclear genome may ultimately resolve mother's curse, our theoretical analysis suggests that transient reductions in male fitness components are far from trivial—even in cases where the genetic architecture of compensatory adaptation is evolutionarily permissive.
Our results build upon a recent theoretical argument by Wade [32], who reasoned that compensatory evolution at nuclear genes provides an evolutionarily inefficient means for resolving mother's curse. By quantifying this inefficiency, we show that costs of mother's curse—though ultimately resolved—may nevertheless be substantial, particularly when benchmarked against other forms of genetic load. Haldane's [49] ‘cost of selection’ (or substitution load [50])—the transient load that arises during the substitution of a beneficial mutation—provides the most straightforward point of contrast with our results. Following standard theory ([49]; [70], p. 247; [56], p. 169; see also the electronic supplementary material), the load contributed by a mitochondrial substitution that equally benefits both sexes is C = –ln(p0), where p0 is the initial frequency of the beneficial allele. When adaptation uses new, beneficial mutations, then p0 ∼ 1/Ns for a mitochondrial mutant that eventually fixes [51,59] (see above), and the substitution load becomes C = ln(Ns). The male mitochondrial load easily exceeds the substitution load (C) over a wide range of parameter conditions (figures 2 and 3). For example, sexually antagonistic mtDNA mutations contribute to a male load of, at minimum, 4ln(Nsm) (see equation (3.4)), which is fourfold higher than Haldane's substitution load, given a similar order of selection (s ∼ sm). The effect is amplified for male-harming mtDNA mutations that are fixed by drift: while C increases logarithmically with Ns, the baseline load due to male-limited mtDNA substitutions increases linearly with Nsm (equation (3.2)), so that the latter may be orders of magnitude greater than the former.
(a). Scope of the model and additional costs of mtDNA inheritance
Our model focuses on an idealized scenario of mito-nuclear coadaptation in which (i) compensatory loci are male-limited with additive expression in males (table 1), (ii) nuclear loci are diploid and (iii) individuals are homoplasmic. We also focused on the fitness costs of mtDNA mutations that eventually fix, ignoring fitness costs of segregating mutations that are eventually lost due to purifying selection or genetic drift (but see electronic supplementary material, appendix V). As we discuss below, these assumptions are generally conservative, and should—if anything—underestimate the true magnitude of the load arising from mother's curse.
(i). Effects of pleiotropy at compensatory loci
Our model provides baseline predictions for the male load when compensatory loci are completely unconstrained by pleiotropic fitness effects in females. When compensatory alleles are expressed in both sexes, their effects are likely to be sexually antagonistic (reducing female fitness). This sexual antagonism in the nuclear genome should dampen the evolutionary capacity of nuclear loci to quickly resolve mother's curse, by reducing the proportion of compensatory alleles that are favoured by selection (effectively reducing v), and dampening the rate at which favoured alleles spread to fixation (e.g. [71,72]).
(ii). Effects of dominance at nuclear genes
Although additivity is assumed in our models, genetic incompatibilities may instead exhibit dominance, as they often do in contexts of interspecific genetic incompatibilities [73]. Non-additive effects of incompatibilities should have two consequences for mito-nuclear coadaptation and male fitness. First, to the extent that compensatory mutations are partially or completely recessive, their fixation probabilities will decline relative to the additive model (e.g. [52], p. 201). This should increase the waiting time to compensatory adaptation and elevate the male load. Deviations from additivity may also bias the relative contributions of sex-linked and autosomal (diploid) genes to the resolution of mother's curse. When compensatory alleles are recessive and evolution is mutation-limited, the waiting time to compensatory substitution should be lower under X-linked compared with autosomal inheritance; the converse is true for dominant compensatory alleles [74,75]. Such considerations may impact the chromosomal distribution of nuclear-encoded genes that are expressed in mitochondria (see [76] and references therein).
(iii). Effects of heteroplasmy
For simplicity, we modelled selection and evolution of mitochondrial genes under the assumption of homoplasmy (i.e. each individual inherits a single mitochondrial cytotype). In reality, each individual inherits many mitochondria from their mother, leaving some potential for heteroplasmy [77,78]. In the absence of selection on mtDNA mutations (i.e. under neutrality in females), transient heteroplasmy should have no effect on the evolutionary trajectories of male-harming substitutions (e.g. [77,78]). Likewise, when mtDNA alleles are sexually antagonistic, approximations based on homoplasmy are robust to heteroplasmy when mitochondrial genotypes additively affect fitness (as shown in [30]). Deviations from our results could occur in models that allow for heteroplasmy and strong dominance interactions among mitochondrial genotypes within an individual [30].
(iv). Further contributions to the male genetic load
Our models ignore three additional factors that are likely to further elevate the male genetic load. First, we have ignored fitness costs of mtDNA alleles that segregate in the population before they are ultimately eliminated by purifying selection in females, or genetic drift. As pointed out elsewhere [3,79] (electronic supplementary material, appendix V), transient polymorphisms can also contribute substantially to fitness variance among males, and deleterious mutation load in the mitochondrial genome. Second, we have ignored mtDNA substitutions with mildly deleterious effects on female fitness, which may fix under genetic drift and elevate the load of both sexes. Third, we have assumed that the genetic architecture of compensatory evolution is simple: that a single genetic substitution in the nuclear genome is sufficient to completely resolve mother's curse. If individual bouts of compensation require fixation of multiple alleles in the nuclear genome, then costs of mtDNA substitution may linger beyond our theoretical expectations.
5. Conclusion
Mitochondrial genetic effects on the organismal phenotype can be sexually asymmetric, with mutations that are primarily male-harming representing a component of the mutational landscape of the mitochondrial genome. Our study provides a theoretical framework for the potential role of mito-nuclear coevolution in sex-specific adaptation and disease. In particular, by demonstrating that the male mitochondrial load can be much larger than genetic load predictions from classical population genetics, we provide a theoretical rationale to expect persistent mother's curse effects in gonochoristic species—even in cases where the resolution of mother's curse is evolutionarily permissive. Finally, by showing that the male mitochondrial load is likely to exhibit a threshold-dependent relationship with population size, our model predicts that the fitness consequences of mother's curse may vary widely among species with different demographic histories. These predictions provide a strong rationale for systematically testing mother's curse predictions in species exhibiting pronounced differences in their effective population size.
Supplementary Material
Acknowledgements
We thank two anonymous reviewers, whose suggestions led to substantial improvements to the paper.
Data accessibility
Details of the model, and R code for the simulations, are included in the electronic supplementary material.
Authors' contributions
T.C., M.F.C., E.H.M. and D.K.D. conceived the project, drafted and edited the manuscript. T.C. developed and analysed the models.
Competing interests
We have no competing interests.
Funding
This research was supported by funds from the Australian Research Council, the School of Biological Sciences at Monash University, a Marie Skłodowska-Curie Fellowship (to M.F.C.), and a Royal Society University Research Fellowship (to E.H.M.)
References
- 1.Birky CW. 1995. Uniparental inheritance of mitochondrial and chloroplast genes: mechanisms and evolution. Proc. Natl Acad. Sci. USA 92, 11 331–11 338. ( 10.1073/pnas.92.25.11331) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frank SA. 1989. The evolutionary dynamics of cytoplasmic male sterility. Am. Nat. 133, 345–376. ( 10.1086/284923) [DOI] [Google Scholar]
- 3.Frank SA, Hurst LD. 1996. Mitochondria and male disease. Nature 383, 224 ( 10.1038/383224a0) [DOI] [PubMed] [Google Scholar]
- 4.Gemmell NJ, Metcalf VJ, Allendorf FW. 2004. Mother's curse: the effect of mtDNA on individual fitness and population viability. Trends Ecol. Evol. 19, 238–244. ( 10.1016/j.tree.2004.02.002) [DOI] [PubMed] [Google Scholar]
- 5.Dowling DK. 2014. Evolutionary perspectives on the links between mitochondrial genotype and disease phenotype. Biochim. Biophys. Acta 1840, 1393–1403. ( 10.1016/j.bbagen.2013.11.013) [DOI] [PubMed] [Google Scholar]
- 6.Beekman M, Dowling DK, Aanen DK. 2014. The costs of being male: are there sex-specific effects of uniparental mitochondrial inheritance? Phil. Trans. R. Soc. B 369, 20130440 ( 10.1098/rstb.2013.0440) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schnable PS, Wise RP. 1998. The molecular basis of cytoplasmic male sterility and fertility restoration. Trends Plant Sci. 3, 175–180. ( 10.1016/S1360-1385(98)01235-7) [DOI] [Google Scholar]
- 8.Chase CD. 2007. Cytoplasmic male sterility: a window to the world of plant mitochondrial–nuclear interactions. Trends Genet. 23, 81–90. ( 10.1016/j.tig.2006.12.004) [DOI] [PubMed] [Google Scholar]
- 9.Nakada K, Sato A, Yoshida K, Morita T, Tanaka H, Inoue SI, Yonekawa H, Hayashi JI. 2006. Mitochondria-related male infertility. Proc. Natl Acad. Sci. USA 103, 15 148–15 153. ( 10.1073/pnas.0604641103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith S, Turbill C, Suchentrunk F. 2010. Introducing mother's curse: low male fertility associated with an imported mtDNA haplotype in a captive colony of brown hares. Mol. Ecol. 19, 36–43. ( 10.1111/j.1365-294X.2009.04444.x) [DOI] [PubMed] [Google Scholar]
- 11.Patel MR, et al. 2016. A mitochondrial DNA hypomorph of cytochrome oxidase specifically impairs male fertility in Drosophila melanogaster. eLife 5, e16923 ( 10.7554/eLife.16923) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martikainen MK, Grady JP, Ng YS, Alston CL, Gorman GS, Taylor RW, McFarland R, Turnbull DM. 2017. Decreased male reproductive success in association with mitochondrial disfunction. Eur. J. Hum. Genet. 25, 1162–1164. ( 10.1038/ejhg.2017.114) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wallace D, Singh G, Lott M, Hodge J, Schurr T, Lezza A, Elsas L, Nikoskelainen E. 1988. Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science 242, 1427–1430. ( 10.1126/science.3201231) [DOI] [PubMed] [Google Scholar]
- 14.Milot E, Moreau C, Gagnon A, Cohen AA, Brais B, Labuda D. 2017. Mother's curse neutralizes natural selection against a human genetic disease over three centuries. Nat. Ecol. Evol. 1, 1400–1406. ( 10.1038/s41559-017-0276-6) [DOI] [PubMed] [Google Scholar]
- 15.Innocenti P, Morrow EH, Dowling DK. 2011. Experimental evidence supports a sex-specific selective sieve in mitochondrial genome evolution. Science 332, 845–848. ( 10.1126/science.1201157) [DOI] [PubMed] [Google Scholar]
- 16.Mossman JA, Tross JG, Li N, Wu Z, Rand DM. 2016a. Mitochondrial-nuclear interactions mediate sex-specific transcriptional profiles in Drosophila. Genetics 204, 613–630. ( 10.1534/genetics.116.192328) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mossman JA, Tross JG, Jourjine NA, Li N, Wu Z, Rand DM. 2017. Mitonuclear interactions mediate transcriptional responses to hypoxia in Drosophila. Mol. Biol. Evol. 34, 447–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Camus MF, Clancy DJ, Dowling DK. 2012. Mitochondria, maternal inheritance, and male aging. Curr. Biol. 22, 1717–1721. ( 10.1016/j.cub.2012.07.018) [DOI] [PubMed] [Google Scholar]
- 19.Camus MF, Wolf JBW, Morrow EH, Dowling DK. 2015. Single nucleotides in the mtDNA sequence modify mitochondrial molecular function and are associated with sex-specific effects on fertility and aging. Curr. Biol. 25, 2717–2722. ( 10.1016/j.cub.2015.09.012) [DOI] [PubMed] [Google Scholar]
- 20.Clancy DJ, Hime GR, Shirras AD. 2011. Cytoplasmic sterility in Drosophila melanogaster associated with a mitochondrial CYTB variant. Heredity 107, 374–376. ( 10.1038/hdy.2011.12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mossman JA, Biancani LM, Zhu CT, Rand DM. 2016b. Mitonuclear epistasis for development time and its modification by diet in Drosophila. Genetics 203, 463–484. ( 10.1534/genetics.116.187286) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eyre-Walker A. 2017. Mitochondrial replacement therapy: are mito-nuclear interactions likely to be a problem? Genetics 205, 1365–1372. ( 10.1534/genetics.116.196436) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wolff JN, Tompkins DM, Gemmell NJ, Dowling DK. 2016. Mitonuclear interactions, mtDNA-mediated thermal plasticity, and implications for the Trojan female technique for pest control. Sci. Rep. 6, 30016 ( 10.1038/srep30016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yee WKW, Sutton KL, Dowling DK. 2013. In vivo male fertility is affected by naturally occurring mitochondrial haplotypes. Curr. Biol. 23, R55–R56. ( 10.1016/j.cub.2012.12.002) [DOI] [PubMed] [Google Scholar]
- 25.Reinhardt K, Dowling DK, Morrow EH. 2013. Mitochondrial replacement, evolution, and the clinic. Science 341, 1345–1346. ( 10.1126/science.1237146) [DOI] [PubMed] [Google Scholar]
- 26.Wade MJ, Brandvain Y. 2009. Reversing mother's curse: selection on male mitochondrial fitness effects. Evolution 63, 1084–1089. ( 10.1111/j.1558-5646.2009.00614.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Unckless RL, Herren JK. 2009. Population genetics of sexually antagonistic mitochondrial mutants under inbreeding. J. Theor. Biol. 260, 132–136. ( 10.1016/j.jtbi.2009.06.004) [DOI] [PubMed] [Google Scholar]
- 28.Hedrick P. 2012. Reversing mother's curse revisited. Evolution 66, 612–6116. ( 10.1111/j.1558-5646.2011.01465.x) [DOI] [PubMed] [Google Scholar]
- 29.Zhang H, Guillaume F, Engelstädter J. 2012. The dynamics of mitochondrial mutations causing male infertility in spatially structured populations. Evolution 66, 3179–3188. ( 10.1111/j.1558-5646.2012.01675.x) [DOI] [PubMed] [Google Scholar]
- 30.Kuijper B, Lane N, Pomiankowski A. 2015. Can paternal leakage maintain sexually antagonistic polymorphism in the cytoplasm. J. Evol. Biol. 28, 468–480. ( 10.1111/jeb.12582) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Engelstädter J, Charlat S. 2006. Outbreeding selects for spiteful cytoplasmic elements. Proc. R. Soc. B 273, 923–929. ( 10.1098/rspb.2005.3411) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wade MJ. 2014. Paradox of mother's curse and the maternally provisioned offspring microbiome. Cold Spring Harb. Perspect. Biol. 6, a017541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rand DM, Haney RA, Fry AJ. 2004. Cytonuclear coevolution: the genomics of cooperation. Trends Ecol. Evol. 19, 645–653. ( 10.1016/j.tree.2004.10.003) [DOI] [PubMed] [Google Scholar]
- 34.Burton RS, Barreto FS. 2012. A disproportionate role for mtDNA in Dobzhansky-Muller incompatibilities? Mol. Ecol. 21, 4942–4957. ( 10.1111/mec.12006) [DOI] [PubMed] [Google Scholar]
- 35.Wolff JN, Ladoukakis ED, Enríquez JA, Dowling DK. 2014. Mitonuclear interactions: evolutionary consequences over multiple biological scales. Phil. Trans. R. Soc. B 369, 20130443 ( 10.1098/rstb.2013.0443) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rand DM, Clark AG, Kann LM. 2001. Sexually antagonistic cytonuclear fitness interactions in Drosophila melanogaster. Genetics 159, 173–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.James AC, Ballard JWO. 2003. Mitochondrial genotype affects fitness in Drosophila simulans. Genetics 164, 187–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roubertoux PL, et al. 2003. Mitochondrial DNA modifies cognition in interaction with the nuclear genome and age in mice. Nat. Genet. 35, 65–69. ( 10.1038/ng1230) [DOI] [PubMed] [Google Scholar]
- 39.Ellison CK, Burton RS. 2008. Interpopulation hybrid breakdown maps to the mitochondrial genome. Evolution 62, 631–638. ( 10.1111/j.1558-5646.2007.00305.x) [DOI] [PubMed] [Google Scholar]
- 40.Dowling DK, Meerupati T, Arnqvist G. 2010. Cytonuclear interactions and the economics of mating in seed beetles. Am. Nat. 176, 131–140. ( 10.1086/653671) [DOI] [PubMed] [Google Scholar]
- 41.Meiklejohn CD, Holmbeck MA, Siddiq MA, Abt DN, Rand DM, Montooth KL. 2013. An incompatibility between a mitochondrial tRNA and its nuclear-encoded tRNA synthetase compromises development and fitness in Drosophila. PLoS Genet. 9, e1003238 ( 10.1371/journal.pgen.1003238) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yee WKW, Rogell B, Lemos B, Dowling DK. 2015. Intergenomic interactions between mitochondrial and Y-linked genes shape male mating patterns and fertility in Drosophila melanogaster. Evolution 69, 2876–2890. ( 10.1111/evo.12788) [DOI] [PubMed] [Google Scholar]
- 43.Dobler R, Rogell B, Budar F, Dowling DK. 2014. A meta-analysis of the strength and nature of cytoplasmic genetic effects. J. Evol. Biol. 27, 2021–2034. ( 10.1111/jeb.12468) [DOI] [PubMed] [Google Scholar]
- 44.Immonen E, Collet M, Goenaga J, Arnqvist G. 2016. Direct and indirect genetic effects of sex-specific mitonuclear epistasis on reproductive ageing. Heredity 116, 338–347. ( 10.1038/hdy.2015.112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Charlesworth D. 1981. A further study of the problems of the maintenance of females in gynodioecious species. Heredity 46, 27–39. ( 10.1038/hdy.1981.3) [DOI] [Google Scholar]
- 46.Gregorius HR, Ross MD. 1984. Selection with gene-cytoplasm interactions. I. Maintenance of cytoplasm polymorphisms. Genetics 107, 165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Babcock CS, Asmussen MA. 1996. Effects of differential selection in the sexes on cytonuclear polymorphism and disequilibria. Genetics 144, 39–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jacobs MS, Wade MJ. 2003. A synthetic review of the theory of gynodioecy. Am. Nat. 161, 837–851. ( 10.1086/375174) [DOI] [PubMed] [Google Scholar]
- 49.Haldane JBS. 1957. The cost of selection. J. Genet. 55, 511–524. ( 10.1007/BF02984069) [DOI] [Google Scholar]
- 50.Crow JF. 1970. Genetic loads and the cost of natural selection. In Mathematical topics in population genetics (ed. Kojima K.), pp. 128–177. Berlin, Germany: Springer. [Google Scholar]
- 51.Maynard Smith J. 1976. What determines the rate of evolution? Am. Nat. 110, 331–338. ( 10.1086/283071) [DOI] [Google Scholar]
- 52.Haldane JBS. 1931. A mathematical theory of natural selection. VIII. Stable metapopulations. Proc. Camb. Phil. Soc. 27, 137–142. ( 10.1017/S0305004100009439) [DOI] [Google Scholar]
- 53.Kimura M. 1985. The role of compensatory neutral mutations in molecular evolution. J. Genet. 64, 7–19. ( 10.1007/BF02923549) [DOI] [Google Scholar]
- 54.Stephan W. 1996. The rate of compensatory evolution. Genetics 144, 419–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ewens WJ. 2004. Mathematical population genetics. I. Theoretical introduction, 2nd edn New York, NY: Springer. [Google Scholar]
- 56.Charlesworth B, Charlesworth D. 2010. Elements of evolutionary genetics. Greenwood Village, CO: Roberts and Company Publishers. [Google Scholar]
- 57.Lande R. 1994. Risk of population extinction from fixation of new deleterious mutations. Evolution 48, 1460–1469. ( 10.1111/j.1558-5646.1994.tb02188.x) [DOI] [PubMed] [Google Scholar]
- 58.Lande R. 1998. Risk of population extinction from fixation of deleterious and reverse mutations. Genetica 102, 21–27. ( 10.1023/A:1017018405648) [DOI] [PubMed] [Google Scholar]
- 59.Maynard Smith J. 1971. What use is sex? J. Theor. Biol. 30, 319–335. ( 10.1016/0022-5193(71)90058-0) [DOI] [PubMed] [Google Scholar]
- 60.Betancourt AJ, Kim Y, Orr HA. 2004. A pseudohitchhiking model of X vs. autosomal diversity. Genetics 168, 2261–2269. ( 10.1534/genetics.104.030999) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Takahasi KR. 2009. Coalescent under the evolution of coadaptation. Mol. Ecol. 18, 5018–5029. ( 10.1111/j.1365-294X.2009.04424.x) [DOI] [PubMed] [Google Scholar]
- 62.Otto SP, Day T. 2007. A biologist’s guide to mathematical modeling in ecology and evolution. Princeton, NJ: Princeton University Press. [Google Scholar]
- 63.Orr HA, Unckless RL. 2014. The population genetics of evolutionary rescue. PLoS Genet. 10, e1004551 ( 10.1371/journal.pgen.1004551) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Core Team R. 2016. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- 65.Kimura M, Ohta T. 1971. Theoretical aspects of population genetics. Princeton, NJ: Princeton University Press. [PubMed] [Google Scholar]
- 66.Gillespie JH. 2004. Why k=4Nus is silly. In The evolution of population biology (eds Singh RS, Uyenoyama MK), pp. 178–192. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 67.McCandlish DM, Stoltzfus A. 2014. Modeling evolution using the probability of fixation: history and implications. Quart. Rev. Biol. 89, 225–252. ( 10.1086/677571) [DOI] [PubMed] [Google Scholar]
- 68.Lanfear R, Kokko H, Eyre-Walker A. 2014. Population size and the rate of evolution. Trends Ecol. Evol. 29, 33–41. [DOI] [PubMed] [Google Scholar]
- 69.Kimura M. 1979. Model of effectively neutral mutations in which selective constraint is incorporated. Proc. Natl Acad. Sci. USA 76, 3440–3444. ( 10.1073/pnas.76.7.3440) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Crow JF, Kimura M. 1970. An introduction to population genetics theory. New York, NY: Harper and Row. [Google Scholar]
- 71.Otto SP. 2004. Two steps forward, one steo back: the pleiotropic effects of favoured alleles. Proc. R. Soc. Lond. B 271, 705–714. ( 10.1098/rspb.2003.2635) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Connallon T, Clark AG. 2013. Antagonistic versus nonantagonistic models of balancing selection: characterizing the relative timescales and hitchhiking effects of partial selective sweeps. Evolution 67, 908–917. ( 10.1111/j.1558-5646.2012.01800.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Coyne JA, Orr HA. 2004. Speciation. Sunderland, MA: Sinauer Associates. [Google Scholar]
- 74.Charlesworth B, Coyne JA, Barton NH. 1987. The relative rates of evolution of sex chromosomes and autosomes. Am. Nat. 130, 113–146. ( 10.1086/284701) [DOI] [Google Scholar]
- 75.Meisel RP, Connallon T. 2013. The faster-X effect: integrating theory and data. Trends Genet. 29, 537–544. ( 10.1016/j.tig.2013.05.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dean R, Zimmer F, Mank JE. 2015. Deficit of mitonuclear genes on the human X chromosome predates sex chromosome formation. Genome Biol. Evol. 7, 636–641. ( 10.1093/gbe/evv017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bergstrom CT, Pritchard J. 1998. Germline bottlenecks and the evolutionary maintenance of mitochondrial genomes. Genetics 149, 2135–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Roze D, Rousset F, Michalakis Y. 2005. Germline bottlenecks, biparental inheritance and selection on mitochondrial variants: a two-level selection model. Genetics 170, 1385–1399. ( 10.1534/genetics.104.039495) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Smith SRT, Connallon T. 2017. The contribution of the mitochondrial genome to sex-specific fitness variance. Evolution 71, 1417–1424. ( 10.1111/evo.13238) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Details of the model, and R code for the simulations, are included in the electronic supplementary material.