

Abstract
The integration of mechanophores, motifs that transduce mechanical forces into chemical reactions, allows creating materials with stress-dependent properties. Typical mechanophores are activated by cleaving weak covalent bonds, but these reactions can also be triggered by other stimuli, and this renders the behavior unspecific. Here we show that this problem can be overcome by extending the molecular-shuttle function of rotaxanes to mechanical activation. A mechanically interlocked mechanophore composed of a fluorophore-carrying macrocycle and a dumbbell-shaped molecule containing a matching quencher was integrated into a polyurethane elastomer. Deformation of this polymer causes a fluorescence turn-on, due to the spatial separation of fluorophore and quencher. This process is specific, efficient, instantly reversible, and elicits an easily detectable optical signal that correlates with the applied force.
The capability to visualize mechanical forces in polymers by an optical response is useful to investigate stress transfer and failure mechanisms and valuable for applications that range from tamper-proof packaging to structural integrity monitoring.1−4 Early attempts to create mechanochromic polymers involved the incorporation of conjugated segments, whose absorption changes due to conformational rearrangements,5 fluorophores with assembly dependent emission characteristics,6,7 and supramolecular mechanisms that allow translating mechanical forces into optical signals.8−10 The use of mechanophores that undergo mechanically induced chemical reactions is a newer approach toward polymers with mechanoresponsive behavior.11−15 Chromogenic responses can be achieved by coupling such mechanophores with chromophores, affording motifs in which force-induced bond cleavage leads to optical changes.1,16−24 Spiropyrans, which undergo electrocyclic ring-opening reactions to colored merocyanines,1,4,20,21 are the most-widely investigated mechanochromophores; other examples include diarylbibenzofuranones22,23 and hexaarylbiimidazoles,18 which dissociate into colored radicals. Though the integration of such motifs into macromolecules has afforded many mechanochromic materials, the fundamental operating principle has two important limitations. As the mechanism involves the force-induced reduction of chemical activation barriers, the same reactions generally occur upon exposure to heat or light, which renders the responses unspecific. For example, spiropyrans are well-known photochromic dyes25 and not only change color upon application of force but also exposure to light.1 Another problem is that most mechanically triggered chain scission reactions are irreversible, either because the reactants are spatially separated, the products react further, or the activation barrier for the reverse reaction is too high.13,17,19,20 Thus, even if the reverse reaction is a priori feasible, it is not necessarily enabled upon releasing the stress, but may require additional activation. Herein we show that these problems can be overcome by a new mechanophore type that exploits the molecular-shuttle function of rotaxanes26−28 and allows mechanically accessing and trapping an entropically unfavorable state, which cannot be stabilized by other stimuli. As a first example, we studied a mechanoluminophore formed by the interlocked assembly of a fluorophore-carrying cycle and a dumbbell-shaped molecule containing a matching quencher (Figure 1). The activation barrier for shuttling the cycle along the shaft is of the order of 10–20 kcal·mol–1 and can readily be overcome by thermal activation.26 Due to charge-transfer interactions, the cycle is preferably located around the quencher and the fluorophore is switched off. The force-induced displacement of the two elements, concomitant with a fluorescence turn-on, is possible by incorporating the rotaxane into a polymer, and macroscopic deformation of this material.
Figure 1.
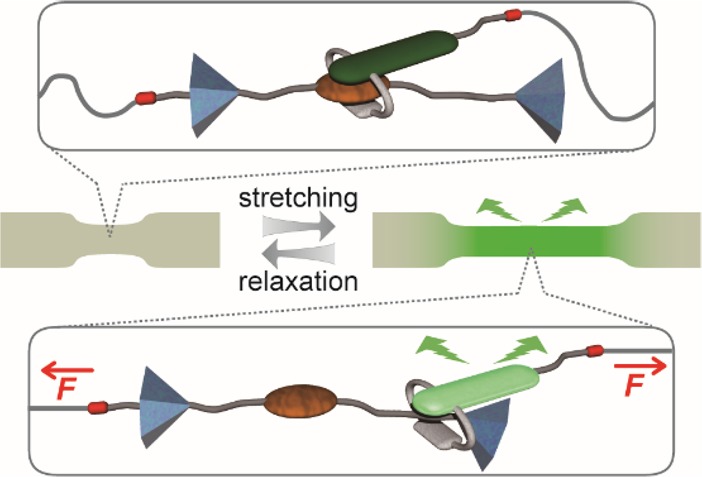
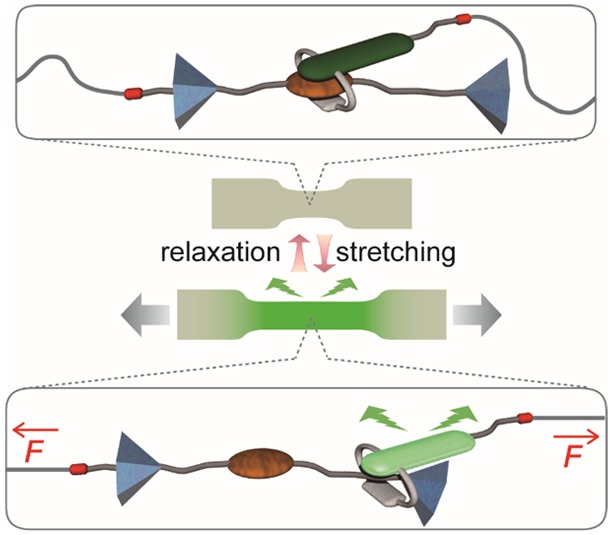
Operating principle of rotaxane-based mechanoluminophores. The interlocked assembly contains a fluorophore-carrying cycle (gray/green), a dumbbell-shaped molecule with a matching quencher (brown), and two stoppers (blue). Reactive groups (red) permit the integration into a polymer chain (gray). In the idle state, the cycle is preferably located around the quencher (top) and the fluorescence is quenched. The force-induced displacement of the two elements (bottom) causes a fluorescence turn-on.
The rotaxane-based mechanoluminophore 1 (Figure 2a) was inspired by works of Sanders and Stoddart, who pioneered neutral donor–acceptor catenanes29 and rotaxanes30 based on electron-rich 1,5-disubstituted naphthalene crown ethers and the electron-poor 1,4,5,8-naphthalenetetracarboxylic diimide (NpI) motif. The latter was expected to quench the emission of 4,7-bis(phenylethynyl)-2,1,3-benzothiadiazole (BTH). Thus, the widely used 1,5-dinaphtho[38]crown-10 cycle was modified to include this emitter. The [2]rotaxane 1 was prepared in a template-directed synthesis that relied on the self-assembly of this motif and NpI30 via a modified Huisgen 1,3-dipolar cycloaddition between an azide and an alkyne.31 Tris(p-tert-butylphenyl)phenylmethane stoppers32 were used to lock-in the structure and two hydroxyl groups were strategically placed on the cycle and the dumbbell to enable the covalent integration into polymer chains. The nonreactive [2]rotaxane 2, the free luminophore 3, and the free dumbbell 4 were prepared as reference materials (Figure 2a).
Figure 2.
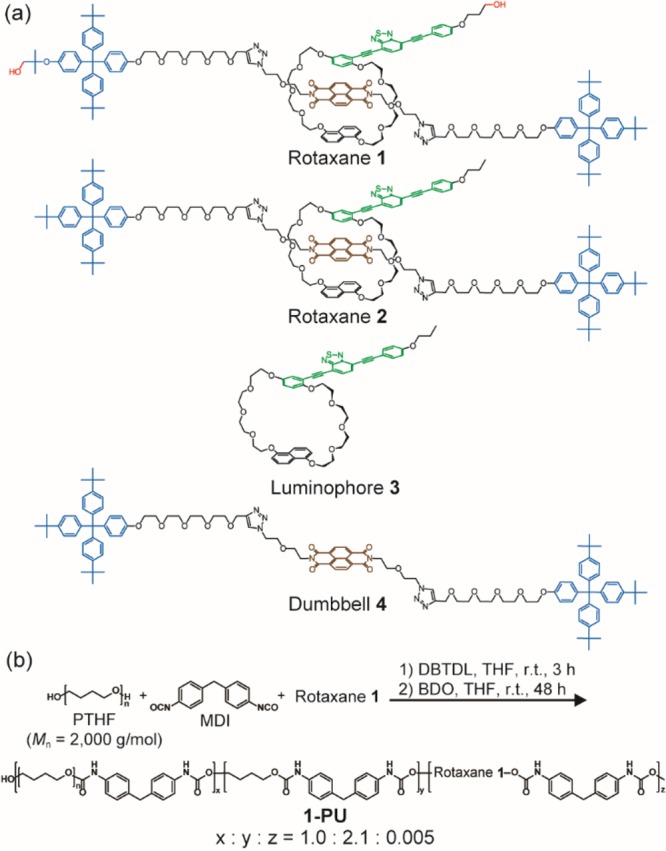
(a) Molecular structures of rotaxanes 1 and 2, luminophore 3, and dumbbell 4. (b) Synthesis and molecular structure of the mechanophore-containing polyurethane 1-PU. This polymer was prepared from poly(tetrahydrofuran) (PTHF), 4,4′-methylenebis(phenylisocyanate) (MDI), rotaxane 1, and 1,4-butanediol (BDO) in a polymerization catalyzed by dibutyltin dilaurate (DBTDL). The x:y and x:z ratios were determined by 1H NMR spectroscopy and the fraction of 1 in the monomer feed.
Upon excitation at 365 nm, solutions (c = 1 × 10–5 M) of the free luminophore 3 fluorescence brightly. The luminophore exhibits solvatochromic behavior (Figures S1 and S2) with emission maxima between 515 (hexane) and 601 nm (acetonitrile). Adding the free dumbbell 4 does not impact the fluorescence of a solution of 3 (Figure S3). Solutions of rotaxanes 1 and 2 show no emission, and the fluorophore’s absorption is slightly red-shifted (absorption maximum = 441 nm) relative to that of 3 (438 nm, Figures S4 and S5). Along with the characteristic shifts30 of the 1H NMR signals of the aromatic protons caused by the interaction of the NpI and the cyclic part in 1 and 2 (Figure S6), these data indicate the formation of [2]rotaxanes and demonstrate that in their idle state the BTH fluorescence is indeed quenched.
Adapting an established strategy,33−35 we incorporated rotaxane 1 in a statistical manner into a linear, segmented polyurethane elastomer through a low-temperature polyaddition reaction (1-PU, Figure 2b). To avoid interactions among multiple mechanophores, the rotaxane content was limited to ca. 0.4 wt % via the content of 1 in the monomer feed. The incorporation of 1 in the polymer was confirmed by UV–vis spectroscopy (Figure S7), but not detectable by NMR spectroscopy (Figure S8). We also synthesized a control polyurethane without mechanophore (PU) and formulated physical blends of this polymer with rotaxane 2 (2inPU) or a mixture of luminophore 3 and dumbbell 4 (3,4inPU). All compositions were processed into 60–130 μm thin films by solvent casting. Differential scanning calorimetry, thermogravimetric, and dynamic mechanical analyses show that the thermomechanical properties of 1-PU and PU are identical and match those of similar polyurethanes33,34 (Figures S9–S11). Uniaxial strain–stress measurements of 1-PU, PU, 2inPU, and 3,4inPU films reveal the expected elastic deformation behavior and show that the mechanical characteristics of the different compositions are within experimental error identical (Figure S12 and Table S1).
As intended, as-prepared 1-PU films do not exhibit any appreciable emission upon UV excitation, but upon deformation, the characteristic fluorescence of the BTH motif instantly appears (Figure 3a, Supporting Movie S1, Figure S13a). This behavior is reversible; as soon as the stress is released, the sample relaxes and ceases to emit light. By contrast, neither of the reference materials shows any mechanically induced fluorescence changes (Figure S13b,c, Supporting Movies S2 and S3). Films of 2inPU do not fluoresce in either unstretched or stretched state (Figure S14). On the other hand, 3,4inPU films fluoresce brightly, regardless of the strain applied (Figure S15). Thus, these results show that the mechanoluminophore 1 functions as intended and depicted in Figure 1; macroscopic forces are transferred to the rotaxanes and cause a displacement of the emitter-carrying cycle to the periphery of the dumbbell, which persists until the stress is released. The reference experiments confirm that the force transduction from the macroscopic to the molecular level requires a covalent integration of the mechanophore in the polymer.
Figure 3.
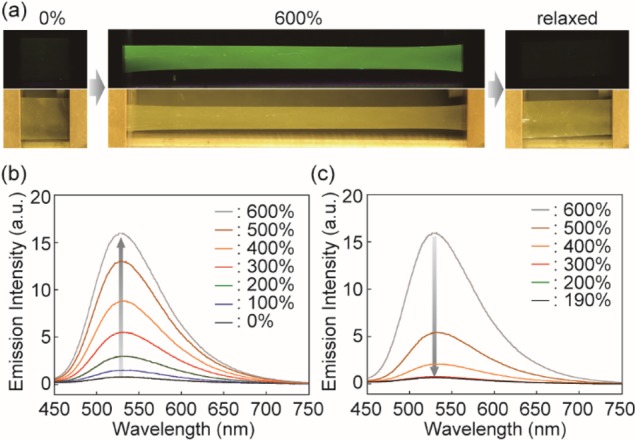
(a) Images of a 1-PU film show that the fluorescence is turned on upon deformation and switched off when the stress is removed. Top and bottom images were taken under irradiation with 365 nm light and ambient illumination, respectively. (b) Emission spectra of a pristine 1-PU film in the initial state and upon uniaxial deformation to the indicated strains. (c) Emission spectra of the sample used in (b) recorded upon gradually releasing the stress and thereby reducing the strain; the stress-free state had a residual elongation of 190%, due to hysteresis. All spectra were recorded with λex = 385 nm.
Several experiments were conducted to investigate the mechanochromic transduction in 1-PU in greater detail and relate optical response to macroscopic deformation. Figure 3b shows that the emission intensity increases with the applied strain λ = (L – L0)/L0 and the behavior is reversible (Figure 3c), even though 1-PU displays the typical hysteresis and cyclic softening (Figure 4a) associated with the evolution of the domain structure of segmented polyurethanes upon deformation.36 The stress and the emission intensity are clearly correlated (Figures 3b,c and Figure S16), as apparent from the overlay of the stress–strain curves and the emission intensities measured in a separate experiment (Figure 4a). A comparison of the emission intensities of fully stretched films (λ = 600%) of 1-PU and 3,4inPU suggests that deformation activates at least 29% of the mechanophores in 1-PU (Figure S17). The fluorescence intensities recorded over 20 loading–unloading cycles with strains of 190 and 600% (Figure 4b) reveal reversible and rapid switching between constant on/off states. A gradual decrease of the emission intensity is observed for a 1-PU film that was kept in the stretched state (Figure 4c, and Figure S18), mirroring the dynamic stress relaxation function.37 This suggests that the molecular relaxation processes of the polyurethane are intimately related to the stresses exerted on the covalently incorporated rotaxane. No optical changes were observed in a similar stress relaxation experiment with a 3,4inPU reference film (Figure S18).
Figure 4.
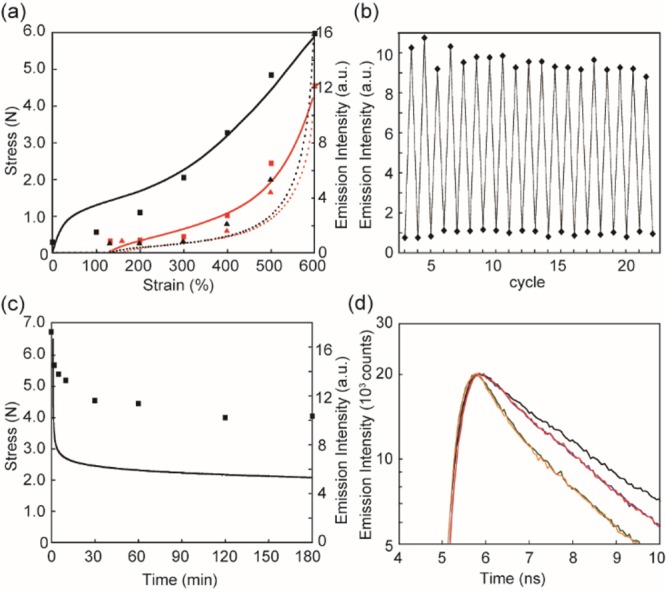
(a) Overlay of strain (lines) and emission intensity (symbols) measured separately upon uniaxially deforming pristine 1-PU films (black solid line and squares), releasing the stress (black dotted line and triangles), deforming the sample again (red solid line and squares), and releasing the stress again (red dotted line and triangles). (b) Emission intensities recorded upon deforming a 1-PU film for 20 cycles (first and second cycles omitted). The emission intensities were determined at strains of 190 (relaxed state) and 600%. (c) Nominal stress (line) and emission intensity (symbols) measured as a function of time after deforming a pristine 1-PU film to a strain of 600% and keeping the strain constant. (d) Emission decay profiles of a pristine 1-PU film recorded upon uniaxial deformation to nominal strains of 400 and 600% (green and blue lines) and after releasing the stress and stretching the sample again to 400 and 600% (orange and red lines). The emission decay profile of a 3,4inPU reference film (black line) is also shown. All emission intensities were measured at 530 nm, with λex = 385 (a–c) or 405 nm (d). The intensities in panels a–c were extracted from uncorrected emission spectra.
Emission lifetime measurements provide further support of the molecular mechanism at play (Figure 4d, Figures S19 and S20). In a moderately deformed 1-PU film (λ = 400%), the luminophores are closer to the quencher groups and the emission lifetime is shorter than in a maximally deformed film (λ = 600%), whose emission lifetime approaches that of the 3,4inPU reference sample, which does not change upon deformation (Figure S20). To demonstrate that the mechanical switching in 1-PU is indeed a highly specific process, we monitored the fluorescence of this material at elevated temperature. Gratifyingly, no obvious changes of the emission color and intensity were observed (Figure S21). Even at 100 °C, the luminescence of an unstretched film remains switched off, whereas stretched samples fluoresce brightly. However, annealing 1-PU at 150 °C caused an increase of the emission intensity at 530 nm over time, which suggests dethreading and/or decomposition of the rotaxane at this temperature (Figure S22). Chain cleavage could also be achieved by ultrasonication of a THF solution of 1-PU, which led to a gradual and significant decrease of the molecular weight and an increase of the emission intensity (Figure S23). The fact that no emission turn-on but a similar molecular-weight decrease was observed when a corresponding solution of PU and rotaxane 2 was similarly treated (Figure S24) suggests that at least some of the mechanophores are irreversibly cleaved upon sonication of 1-PU.
In summary, a new mechanophore type was accessed by incorporating a weakly interacting fluorophore/quencher pair that displays conformation-dependent optical characteristics in a mechanically interlocked rotaxane. The deformation of a rubbery polyurethane containing the first mechanoluminophore based on this design leads to rapid and reversible fluorescence switching, and the optical response correlates with the macroscopic deformation. The concept should be applicable to other interlocked molecules, including catenanes and knots, various chromophore systems, notably charge-transfer pairs that change their absorption color upon dissociation, and different polymer types. In addition to the attractive features already mentioned, it appears straightforward to tailor the optical properties via the choice of the chromophores, and the forces required to dissociate the fluorophores and quenchers are very small. Thus, such supramolecular mechanophores may also be useful to visualize ultrasmall mechanical forces, such as those generated by living cells.
Acknowledgments
We thank Ms. Miho Yamada for HRMS measurements. Y.S. acknowledges support from JSPS Overseas Research Fellowships, JSPS KAKENHI (JP16H00818), the Asahi Glass Foundation, the Iketani Science and Technology Foundation, and the Ogasawara Foundation for the Promotion of Science and Engineering. This work was supported by the Swiss National Center of Competence in Research Bio-Inspired Materials. The research leading to these results has received funding from the European Research Council under the European Union’s Seventh Framework Programme (FP7/2007-2013)/ERC grant agreement no. 291490-MERESPO.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b12405.
The authors declare no competing financial interest.
Supplementary Material
References
- Davis D. A.; Hamilton A.; Yang J.; Cremar L. D.; Van Gough D.; Potisek S. L.; Ong M. T.; Braun P. V.; Martinez T. J.; White S. R.; Moore J. S.; Sottos N. R. Nature 2009, 459, 68–72. 10.1038/nature07970. [DOI] [PubMed] [Google Scholar]
- Calvino C.; Neumann L.; Weder C.; Schrettl S. J. Polym. Sci., Part A: Polym. Chem. 2017, 55, 640–652. 10.1002/pola.28445. [DOI] [Google Scholar]
- Caruso M. M.; Davis D. A.; Shen Q.; Odom S. A.; Sottos N. R.; White S. R.; Moore J. S. Chem. Rev. 2009, 109, 5755–5798. 10.1021/cr9001353. [DOI] [PubMed] [Google Scholar]
- Li J.; Nagamani C.; Moore J. S. Acc. Chem. Res. 2015, 48, 2181–2190. 10.1021/acs.accounts.5b00184. [DOI] [PubMed] [Google Scholar]
- Rubner M. F. Macromolecules 1986, 19, 2129–2138. 10.1021/ma00162a005. [DOI] [Google Scholar]
- Löwe C.; Weder C. Adv. Mater. 2002, 14, 1625–1629. . [DOI] [Google Scholar]
- Crenshaw B. R.; Weder C. Chem. Mater. 2003, 15, 4717–4724. 10.1021/cm034447t. [DOI] [Google Scholar]
- Lavrenova A.; Balkenende D. W. R.; Sagara Y.; Schrettl S.; Simon Y. C.; Weder C. J. Am. Chem. Soc. 2017, 139, 4302–4305. 10.1021/jacs.7b00342. [DOI] [PubMed] [Google Scholar]
- Balkenende D. W.; Coulibaly S.; Balog S.; Simon Y. C.; Fiore G. L.; Weder C. J. Am. Chem. Soc. 2014, 136, 10493–10498. 10.1021/ja5051633. [DOI] [PubMed] [Google Scholar]
- Sagara Y.; Yamane S.; Mitani M.; Weder C.; Kato T. Adv. Mater. 2016, 28, 1073–1095. 10.1002/adma.201502589. [DOI] [PubMed] [Google Scholar]
- Hickenboth C. R.; Moore J. S.; White S. R.; Sottos N. R.; Baudry J.; Wilson S. R. Nature 2007, 446, 423–427. 10.1038/nature05681. [DOI] [PubMed] [Google Scholar]
- Piermattei A.; Karthikeyan S.; Sijbesma R. P. Nat. Chem. 2009, 1, 133–137. 10.1038/nchem.167. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Spiering A. J. H.; Karthikeyan S.; Peters G. W. M.; Meijer E. W.; Sijbesma R. P. Nat. Chem. 2012, 4, 559–562. 10.1038/nchem.1358. [DOI] [PubMed] [Google Scholar]
- Ducrot E.; Chen Y.; Bulters M.; Sijbesma R. P.; Creton C. Science 2014, 344, 186–189. 10.1126/science.1248494. [DOI] [PubMed] [Google Scholar]
- Ramirez A. L. B.; Kean Z. S.; Orlicki J. A.; Champhekar M.; Elsakr S. M.; Krause W. E.; Craig S. L. Nat. Chem. 2013, 5, 757–761. 10.1038/nchem.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z.; Mercer J. A. M.; Zhu X.; Romaniuk J. A. H.; Pfattner R.; Cegelski L.; Martinez T. J.; Burns N. Z.; Xia Y. Science 2017, 357, 475–479. 10.1126/science.aan2797. [DOI] [PubMed] [Google Scholar]
- Robb M. J.; Kim T. A.; Halmes A. J.; White S. R.; Sottos N. R.; Moore J. S. J. Am. Chem. Soc. 2016, 138, 12328–12331. 10.1021/jacs.6b07610. [DOI] [PubMed] [Google Scholar]
- Verstraeten F.; Gostl R.; Sijbesma R. P. Chem. Commun. 2016, 52, 8608–8611. 10.1039/C6CC04312G. [DOI] [PubMed] [Google Scholar]
- Gostl R.; Sijbesma R. P. Chem. Sci. 2016, 7, 370–375. 10.1039/C5SC03297K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossweiler G. R.; Hewage G. B.; Soriano G.; Wang Q.; Welshofer G. W.; Zhao X.; Craig S. L. ACS Macro Lett. 2014, 3, 216–219. 10.1021/mz500031q. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Zhang H.; Fang X.; Lin Y.; Xu Y.; Weng W. ACS Macro Lett. 2014, 3, 141–145. 10.1021/mz400600r. [DOI] [PubMed] [Google Scholar]
- Imato K.; Kanehara T.; Ohishi T.; Nishihara M.; Yajima H.; Ito M.; Takahara A.; Otsuka H. ACS Macro Lett. 2015, 4, 1307–1311. 10.1021/acsmacrolett.5b00717. [DOI] [PubMed] [Google Scholar]
- Imato K.; Irie A.; Kosuge T.; Ohishi T.; Nishihara M.; Takahara A.; Otsuka H. Angew. Chem., Int. Ed. 2015, 54, 6168–6172. 10.1002/anie.201412413. [DOI] [PubMed] [Google Scholar]
- Wang T.; Zhang N.; Dai J.; Li Z.; Bai W.; Bai R. ACS Appl. Mater. Interfaces 2017, 9, 11874–11881. 10.1021/acsami.7b00176. [DOI] [PubMed] [Google Scholar]
- Berkovic G.; Krongauz V.; Weiss V. Chem. Rev. 2000, 100, 1741–1754. 10.1021/cr9800715. [DOI] [PubMed] [Google Scholar]
- Stoddart J. F. Chem. Soc. Rev. 2009, 38, 1802–1820. 10.1039/b819333a. [DOI] [PubMed] [Google Scholar]
- Bissell R. A.; Cordova E.; Kaifer A. E.; Stoddart J. F. Nature 1994, 369, 133–137. 10.1038/369133a0. [DOI] [Google Scholar]
- Erbas-Cakmak S.; Leigh D. A.; McTernan C. T.; Nussbaumer A. L. Chem. Rev. 2015, 115, 10081–10206. 10.1021/acs.chemrev.5b00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton D. G.; Davies J. E.; Prodi L.; Sanders J. K. M. Chem. - Eur. J. 1998, 4, 608–620. . [DOI] [Google Scholar]
- Jacquot de Rouville H.-P.; Iehl J.; Bruns C. J.; McGrier P. L.; Frasconi M.; Sarjeant A. A.; Stoddart J. F. Org. Lett. 2012, 14, 5188–5191. 10.1021/ol3022963. [DOI] [PubMed] [Google Scholar]
- Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. . [DOI] [PubMed] [Google Scholar]
- Gibson H. W.; Lee S. H.; Engen P. T.; Lecavalier P.; Sze J.; Shen Y. X.; Bheda M. J. Org. Chem. 1993, 58, 3748–3756. 10.1021/jo00066a030. [DOI] [Google Scholar]
- Crenshaw B. R.; Weder C. Macromolecules 2006, 39, 9581–9589. 10.1021/ma061685b. [DOI] [Google Scholar]
- Ayer M. A.; Simon Y. C.; Weder C. Macromolecules 2016, 49, 2917–2927. 10.1021/acs.macromol.6b00418. [DOI] [Google Scholar]
- Belaissaoui A.; Shimada S.; Ohishi A.; Tamaoki N. Tetrahedron Lett. 2003, 44, 2307–2310. 10.1016/S0040-4039(03)00242-9. [DOI] [Google Scholar]
- Yi J.; Boyce M. C.; Lee G. F.; Balizer E. Polymer 2006, 47, 319–329. 10.1016/j.polymer.2005.10.107. [DOI] [Google Scholar]
- Qi H. J.; Boyce M. C. Mech. Mater. 2005, 37, 817–839. 10.1016/j.mechmat.2004.08.001. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.