

Abstract
In eukaryotic cells, autophagy is a process associated with programmed cell death. During this process, cytoplasmic proteins and organelles are engulfed by double‐membrane autophagosomes, which then fuse with lysosomes to form autolysosomes. These autolysosomes then degrade their contents to recycle the cellular components. Autophagy has been implicated in a wide variety of physiological and pathological processes that are closely related to tumorigenesis. In recent years, an increasing number of studies have indicated that nonsteroidal anti‐inflammatory drugs, such as celecoxib, meloxicam, sulindac, aspirin, sildenafil, rofecoxib, and sodium salicylate, have diverse effects in cancer that are mediated by the autophagy pathway. These nonsteroidal anti‐inflammatory drugs can modulate tumor autophagy through the PI3K/Akt/mTOR, MAPK/ERK1/2, P53/DRAM, AMPK/mTOR, Bip/GRP78, CHOP/ GADD153, and HGF/MET signaling pathways and inhibit lysosome function, leading to p53‐dependent G1 cell‐cycle arrest. In this review, we summarize the research progress in autophagy induced by nonsteroidal anti‐inflammatory drugs and the molecular mechanisms of autophagy in cancer cells to provide a reference for the potential benefits of nonsteroidal anti‐inflammatory drugs in cancer chemotherapy.
Keywords: Autophagy, nonsteroidal anti‐inflammatory drugs, programmed cell death
Background
Over the past few decades, much of cancer research has been focused on the mechanisms of apoptosis. However, apoptotic resistance has become a major obstacle in cancer treatment. Thus, studying the other mechanisms associated with programmed cell death has become increasingly important. Based on their associated morphological features, programmed cell death can be divided into apoptosis, autophagy, necrosis, and mitotic catastrophe. Recent studies have shown that changes in the external or internal environments (i.e., amino acid deficiency, insufficient glucose supply, reduced oxygen supply, and mitochondrial damage) can induce autophagy 1. Based on the substrate for each enzyme, autophagy can be subdivided into different pathways, including microautophagy, great‐autophagy, and molecular chaperone‐mediated autophagy.
Autophagy is a conserved catabolic process, and over the past decade, multiple studies have reported genetic and functional links between impaired autophagy and cancer, suggesting that autophagy is a mechanism of tumor suppression. During autophagy, cellular contents are enclosed within autophagosomes, which fuse with lysosomes to degrade and recycle their contents 2. In tumorigenesis, the function of autophagy is complex, since it is not only a prodeath mechanism but also a survival strategy under cellular stress 3, 4. The prodeath or prosurvival nature of the response may be related to tumor type, stage, and the ability of the tumor cells to sustain themselves. Some studies have reported that inhibition of autophagy enhances cellular apoptosis 5, 6.
Nonsteroidal anti‐inflammatory drugs (NSAIDs) are a structurally diverse group of drugs that are widely used to treat pain, inflammation, and fever, including acetylsalicylic acid, celecoxib, and acetaminophen. In 2016, the US Preventive Services Task Force recommended low‐dose aspirin for preventing colorectal cancer (CRC) in patients without a bleeding tendency 7. More recently, several novel studies have examined NSAIDs and cancer 8, and some researchers found that NSAIDs are closely related to autophagy, especially in hepatocellular carcinoma (HCC) 9, glioblastoma 10, neuroblastoma 11, acute leukemia 12, lung adenocarcinoma 13, oral cancer 14, breast cancer 15, ovarian cancer, colon cancer 16, bladder cancer17, and gastric cancer 18. In this article, we will review the mechanisms underlying the actions of NSAIDs in autophagy (Fig. 1; Tables 1 and 2). The use of NSAIDs in combination with chemotherapeutic drugs appears to be a promising approach for the treatment of drug‐resistant tumors that deserves further investigation.
Figure 1.
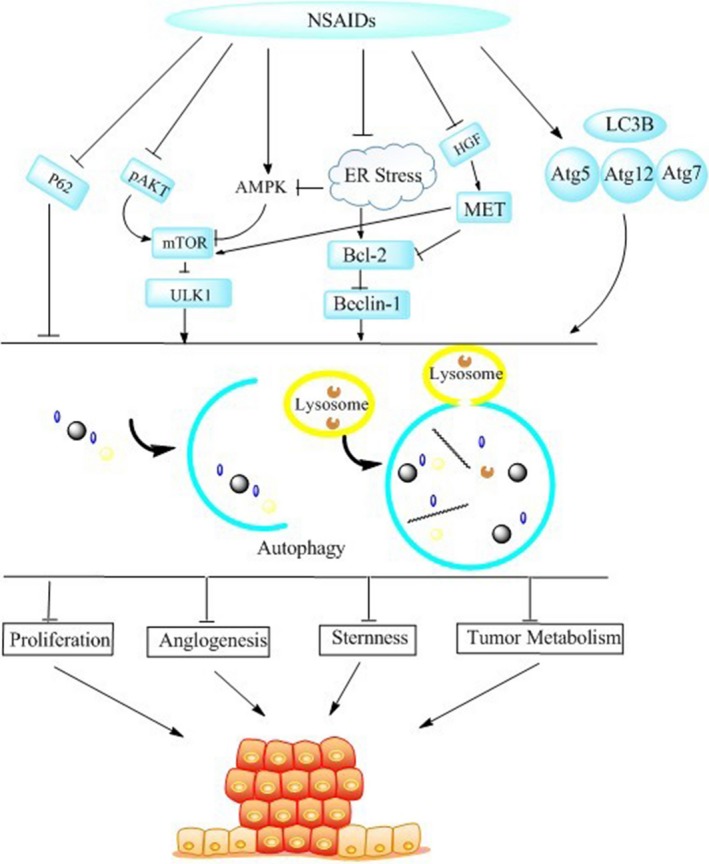
NSAIDs significantly induce autophagy. The most important pathway is mTOR signaling pathway, including PI3K/AKT and AMPK/mTOR. Similarly, NSAIDs also regulate directly the target genes, such as LC3B, P62, P53, Atg5, Atg12, and HIF‐1, the latest Discovered.
Table 1.
NSAIDs that induce tumor suppressing autophagy
| NSAIDs/chemotherapeutics | Mechanism of action | Cancer cell line | Animal models | Regulation of autophagy | Author (Refs.) |
|---|---|---|---|---|---|
| Celecoxib | Conversion of LC3 from LC3I to LC3II↑cleavage of caspases‐8,‐9, ‐3↑Bid↓ | HT‐29HCT116colorectal cancer cells | Enhance | Shengbing et al. 6 | |
| Celecoxib | ERS↑LC3‐II | MDA‐MB‐231MDA‐MB‐468Hs578t T47D JCBreast cancer cells | MCF‐7MDA‐MB‐468 | Enhance | Simmy et al. 15 |
| Celecoxib/γ‐irradiation | LC3‐II ↑GADD153/CHOP GRP78/BiP ↑P21Waf1 P27Kip1↑ P53↓ | U87MGU251MGGL261malignant glioma cells | Enhance | Kenshi et al. 36 | |
| Celecoxib/CPT‐11 | mTOR↑LC3‐II↑ | TNB9TS‐N‐2nu | Enhance | Setsuko et al. 11 | |
| Celecoxib/MDR | HGF/MET↓Bcl‐2 mTOR↓TGFb1,p16INK4b, P21Cip1 P27Kip1↑ | PLC/PRF/5P5 | Enhance | Roberto et al. 66 | |
| Celecoxib | p53 p21↑LC3‐II↑DNA synthesis↓ | U87MGU87MG‐E6LN229 | Enhance | Khong et al. 37 | |
| Celecoxib/Sildenafil | Beclin1↓Atg5↓ | GBM5/6/12/14 | Enhance | Laurence et al. 85 | |
| Celecoxib | P‐Akt↓ caspase‐8 ‐9↑procaspase‐8 ‐9↓ | SGC‐7901 | Enhance | MIN et al. 18 | |
| Celecoxib | LC3 II↑ LC3‐I↑P62↓ JNK↑ | PC3 | Enhance | Xin et al. 86 | |
| Sodium Salicylate | GD‐induced CuZnSOD↓HMGB↓ ROS production↓ | A549 | Enhance | Sung‐chul et al. 87 | |
| Aspirin | mTOR↓ AKT↓AMPK↑phosphorylation of S6K1,S6↓ | RKOSW480HCT116 | Enhance | Farhat et al. 62 | |
| Aspirin | p‐mTOR↓HIF‐1α↓VEGF‐A↓ULK1↑ LC3A↑ | H22S180 | Enhance | Qianqian et al. 65 | |
| Sulfasalazine | LC3‐II↑Atg5‐12↑p‐Akt↓ p‐ERK↑ | HSC‐4 | Enhance | Hye‐Yeon et al. 13 | |
| Sulfasalazine | NF‐κB↓P62↓ | U251 | Enhance | Jing et al. 88 | |
| Piroxicam/carboplatin | Vacuoles↑ | T245637 | Enhance | JéSSICA et al. 89 | |
| Indomethacin | Smad7↓ | RGM1 | Enhance | Ho‐Jae et al. 10 | |
| OSU‐03012 | ERK1/2↓Atg5↑ROS accumulation↑cleaved LC3↑ | Huh7Hep3BHepG2 | Huh7 | Enhance | Gao et al. 28 |
| OSU‐03012 | AKT↓ERK1/2↓ | GBM12 | Enhance | Laurence et al. 85 |
Table 2.
NSAIDs that induce cytoprotective autophagy
| NSAIDs/chemotherapeutics | Mechanism of action | Cancer cell line | Autophagy inhibitor | Regulation of autophagy | Author (Refs.) |
|---|---|---|---|---|---|
| Celecoxib/ imatinib | Lysosome function↓ p62↑ | KBM5KBM5‐T315I | Inhibit | Ying et al. 7 | |
| Celecoxib | Lysosome function↓LC3‐II↓ p62↑ | HL‐60 | Inhibit | Ying et al. 12 | |
| Celecoxib | Atg12‐Atg5 conjugate↑LC3B↑ | NTUB1T24 | 3‐methyladenine Bafilomycin A1ATG7 siRNA | Enhance | Kuo‐How et al. 5 |
| Celecoxib | LC3‐II↑P62↓ | HT‐29 SW480HCT116 | 3‐methyladenineAtg8/LC3BsiRNA Vps34 siRNA | Enhance | Shengbing et al. 6 |
| Sulindac Sulfide | Cytochrome c↑ | HT‐29 | 3‐methyladenine | Enhance | Bauvy et al. 91 |
| Aspirin | Mcl‐1↓LC3II/I ratios early↑‐later↓p38↑ | HO‐8910H1299 A549HCT‐116 HT‐29 | 3‐methyladenineBafilomycin A1 | Enhance (short‐term)inhibit(long‐term) | Zhang et al. 16 |
| Meloxicam | Beclin 1, LC 3‐II↑p‐AKT↓ | HepG2Bel‐7402 Huh‐7 | 3‐methyladeninechloroquine | Enhance | Xiaofeng et al. 85 |
| Meloxicam | GRP78↑Beclin‐1, Atg5,Atg7 LC3↑P62↓ | HepG2Bel‐7402 cells | 3‐methyladenineAtg5 siRNA | Enhance | Jingtao et al. 85 |
Bcl‐2 family: regulation of autophagy
Bcl‐2 family proteins contain Bcl‐2 homology (BH) domains, and are divided into three types: antiapoptotic proteins (e.g., Bcl‐2, Mcl‐1, and Bcl‐w), proapoptotic proteins (e.g., Bax and Bak), and BH3‐only proteins (e.g., Beclin‐l and Bid). BH3‐only proteins play a role in promoting apoptosis by directly activating or antagonizing other BH‐containing proteins. The Bcl‐2 family of proteins regulates the mitochondrial membrane via dimerization, and their effects on autophagy can be regulated bidirectionally. Bcl‐2 family protein regulate autophagy by blocking calcium release from the endoplasmic reticulum, thereby inactivating calmodulin‐dependent kinase and adenosine monophosphate‐activated protein kinase (AMPK) and inhibiting mammalian target of rapamycin (mTOR) activity, which promotes autophagy 19. Beclin‐1 and Bcl‐2/Bcl‐xL interact via the Bcl‐2 homology (BH3) domain in Beclin‐1, and dissociation of Beclin‐1 from Bcl‐2/Bcl‐xL leads to increased autophagy 20. Beclin‐1 has been shown to be involved in the formation of autophagic vacuoles (Fig. 2), and other proapoptotic signals, such as Bad and Bax proteins, also activate autophagy 21. Beclin‐1, phosphoinositide 3‐kinase (PI3K), and Atg14 form a heterotrimer and bind the autophagy‐related proteins. In the presence of Atg4, the microtubule‐associated protein 1A/1B‐light chain 3 (LC3) precursor is cleaved into the soluble LC3‐I, which associates with phosphatidyl ethanolamine (PE) to form a LC3‐II‐PE conjugate. This LC3‐II‐PE conjugate is recruited to the autolysosomal membrane.
Figure 2.
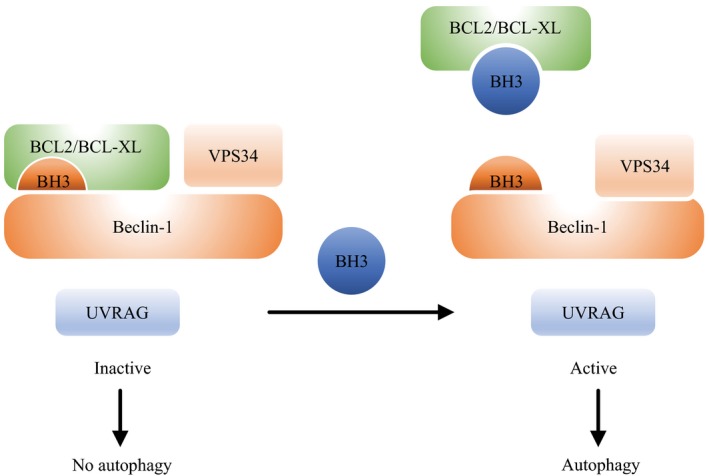
Among the initial steps of vesicle nucleation is the activation of mammalian Vps34, a class III phosphatidylinositol 3‐kinase (PI3K), to generate phosphatidylinositol‐3‐phosphate. Vps34 activation depends on the formation of a multiprotein complex of beclin‐1, UVRAG, and BH393.
Signaling pathways that control autophagy
Growth factor signaling: mitogen‐activated protein kinase/extracellular signal–activated protein kinases 1 and 2, phosphatidylinositol‐3‐kinase/Akt, and the mammalian target of rapamycin signaling pathway
The conserved mTOR protein complex includes mTORC1 and mTORC2. mTORC1 acts as a nutrient sensor and thus plays an important role in the regulation of autophagy 22. When mTORC1 is activated, autophagy is inhibited. The upstream regulators of mTORC1 are PI3K and Akt, and Akt kinase modulates the stability of the TSC1‐TSC2 protein complex. The TSC1‐TSC2 complex can also receive input from other signaling pathways, such as extracellular signal‐activated protein kinases 1 and 2, and some studies have shown that phosphorylation of TSC2 by Akt or ERK1/2 can dissociate the TSC1‐TSC2 complex, thereby activating mTORC1 23. Under hunger or hypoxia, the PI3K/Akt/mTOR signaling pathway and the Bcl‐2 family of proteins suppress autophagy 24.
Energy sensing pathway: AMPK positively regulates autophagy
Adenosine 5′‐monophosphate (AMP)‐activated protein kinase (AMPK) is a cellular energy sensor that is activated by hepatic protein kinase B1 (LKB1) when nutrient or energy expenditure [i.e., the adenosine monophosphate (AMP)/adenosine triphosphate (ATP) ratio] is reduced. In contrast to the Akt and ERK1/2 pathways, which activate mTOR 1, activated AMPK can phosphorylate and activate the TSC1‐TSC2 complex, which in turn inhibits formation of the mTORC1 complex 25. AMPK can act as a positive regulator of autophagy by directly activating ULK1, a serine/threonine‐protein kinase, through phosphorylation of ULK1 at Ser317 and Ser777. AMPK also inhibits the activity of mTORC1 by directly phosphorylating its receptor, a regulatory‐associated protein of mTOR 26.
Autophagy, a double‐edged sword in tumor progression
Autophagy has dual functions; in normal tissues, autophagy is a physiological process that can clear away damaged organelles to maintain cell metabolism at steady‐state. However, rising autophagy levels can cause “autophagic cell death.”27 Autophagic cell death, also known as type II programmed cell death, occurs in response to various anticancer therapies 28. Inducing autophagy has also been shown to improve the outcome of tumor treatments 29, 30, 31, 32, 33. However, when autophagy is cytoprotective, inhibition of autophagy has been shown to enhance the efficacy of antitumor treatment 34, 35, 36. Certain pathways of programmed cell death have been shown to be inhibited either in the presence of autophagic inhibitors or when the expression of the autophagy‐specific genes (ATGs) that regulate autophagy is reduced 37, 38. Recently Lu et al. 39 suggested that the balance between proautophagic and antiautophagic activity in the cellular microenvironment determines whether cancer cells die an autophagic death, remain dormant, or exit dormancy.
Autophagy: novel applications of NSAIDs
Anticancer effects and autophagy regulation of NSAIDs
A group of drugs that share similar mechanisms of action and are clearly distinct from other groups of drugs used for the treatment of inflammation, are collectively named “NSAIDs.”38 These drugs are inhibitors of the enzyme cyclooxygenase (COX) and affect the production of prostaglandin (PG) signaling molecules. There are two isoforms of COX; COX‐1, which is involved in the production of PGs in the gastrointestinal tract under basal conditions, and COX‐2, which is activated by growth factors, mitogens, and tumor promoters 28. COX‐2 is the rate‐limiting enzyme in prostaglandin synthesis. High expression of COX‐2 can promote the synthesis of prostaglandin E2 (PGE2), which induces cell proliferation and stimulates the expression of Bcl‐2 40. COX‐2 overexpression is closely related to the occurrence and development of tumors 41. Compelling data from large studies indicate that aspirin and other NSAIDs are associated with a decreased risk of colorectal, lung, and other carcinomas 27, 39, 40. It was reported that prostaglandin E2 (PGE2) may modulate various immune responses by inhibiting apoptosis and autophagy, and stimulate cancer cell proliferation 37, 42. However, the underlying mechanism is unclear. One study investigated the relationship between prostaglandin E2 (PGE2) production and autophagic cell death in human patellar tendon fibroblasts (HPTFs) in vitro 41. Since the 1970s, numerous large epidemiologic studies and meta‐analyses have strongly supported a protective association between the ingestion of aspirin/other NSAIDs and adenocarcinomas 38, 43, 44. A meta‐analysis of 11 randomized clinical trials by Chen et al. 45 reported that celecoxib was beneficial for the treatment of various types of advanced cancer. Specifically, NSAIDs have various antitumor effects related to apoptosis, autophagy, and tumor immunity. However, numerous questions remain unanswered. For example, which patients can be effectively treated with NSAIDs? How can efficacy be improved? What roles could NSAIDs play in chemotherapy in combination therapy or as an adjunct? Some studies have shown that increased inflammation is associated with an increased risk of cancer, and that obesity further increases the inflammatory response. For obese people, oral aspirin is more effective for preventing tumors 46. Some studies have shown that the effect of aspirin on tumor prevention is dose‐dependent 47. However, another study found that 100 mg/day is the best dose of aspirin for reducing the incidence of adverse reactions, such as bleeding 48. Accumulating evidence suggests that the use of low‐dose aspirin can prevent and reduce the risk of cancer 49. The combination of chemotherapy drugs with NSAIDs, which reduce the side effects of chemotherapy, has been shown to be very effective. Some research has reported that combination treatment with imatinib and celecoxib had an additive synergistic cytotoxicity effect in imatinib‐resistant chronic myeloid leukemia cells (Q > 0.85) 9. Celecoxib, acting as an alternative autophagy inhibitor, might serve as a new tool to enhance the antitumor activity of therapeutic agents, especially those that induce cytoprotective autophagy. However, these issues require further study.
NSAID‐induced autophagy has diverse anticancer effects by regulating Beclin‐1, LC3‐II, p62, and Atg5‐12
The autophagy‐related gene BECN1 encodes Beclin‐1, which is an important regulator of autophagy. BECN1 is located on 17q21 and it encodes a 450‐amino acid polypeptide 8. In many tumors, research has shown that Beclin‐1 could be used as a marker of tumor suppressor loss during tumor development 29, 30. The autophagosome‐associated protein light chain 3 (LC3), also called autophagy‐related gene 8 (Atg8), is a marker of autophagy that exists in two forms, LC3‐I and LC3‐II, that are produced posttranslationally. At the onset of the autophagy cascade, cytosolic LC3‐I is first converted to membrane‐bound LC3‐II. This conversion is triggered by celecoxib, and LC3‐II is thought to be present on autophagosome membranes. Zhong et al. 50 reported that meloxicam induced cell cytoprotective autophagy by upregulating Beclin‐1 and LC3‐II in HCC cells. Specific inhibition of autophagy by 3‐methyladenine and chloroquine (CQ) enhanced the proapoptotic effects of meloxicam by upregulating the expression of Bax 50. However, Suzuki et al. 51 showed that LC3‐II expression is enhanced by celecoxib treatment in hypoxic glioblastoma cells when compared to the expression levels in normoxic glioblastoma cells. Autophagic cell death also occurred. The same results were obtained in various tumor cell lines, such as TNUB1 urothelial carcinoma cells 56, U87MG glioblastoma cells 10 and MCF‐7 breast cancer cells 15 in vivo and in vitro. Not only did meloxicam and celecoxib notably increase the levels of Beclin‐1 and LC3‐II/LC3‐I but they also significantly reduced p62 levels. P62 is degraded during autophagy and can be used as a marker of autophagic flux 52. P62 also accumulates in autophagy‐deficient cells 52. Huang and Sinicrope 67 hypothesized that cotreatment of HCT116 cells with celecoxib and a small‐molecule Bcl‐2/Bcl‐xL antagonist (ABT‐737) reduced the level of p62 protein compared to reduction induced by either drug alone, and increased the conversion levels of autophagosome‐associated protein LC3. The adaptor protein p62 is a well‐known marker of autophagy and participates in crosstalk with important signaling pathways, including the nuclear factor‐κB (NF‐κB) signaling pathway 26, 53. Substantial research has confirmed that the adaptor protein p62 participates in the regulation of NF‐κB signaling, although the mechanism is not fully understood 54. However, Yu et al. 55 showed that p62 is involved in cisplatin‐resistance in ovarian cancer cells through autophagy.
It is interesting to note that celecoxib had the opposite effect on autophagy in chronic myelogenous leukemia (CML) than in solid tumors, as previously reported 9. Ying et al. published that celecoxib prevents autophagic flux by inhibiting lysosome function at its late stages and enhances the cytotoxicity of imatinib in imatinib‐resistant KBM5‐T315I cells. The expression levels of p62 protein were increased when cells were treated with celecoxib. The same results were obtained using cells from patients with chronic myelogenous leukemia (CML). The main finding of this study, that celecoxib acts as an autophagy suppresser in HL‐60 cells, will help to expand the applications of celecoxib to different forms of leukemia. Another group reported that celecoxib suppresses autophagy in HL‐60 cells by altering the acidic environment of the lysosomes, and that celecoxib induces cell apoptosis and necrosis in HL‐60 cells 12. Since celecoxib can act as an inhibitor of autophagy, it could be used as a new tool for enhancing the antitumor activity of therapeutic agents, especially agents that induce cytoprotective autophagy in acute myeloid leukemia (AML) cells (Fig. 1).
There are more than 20 proteins involved in the autophagy pathway downstream of mTOR. Research has shown that the levels of the conjugated Atg5‐Atg12 protein increase significantly after treatment with celecoxib, meloxicam, or OSU‐03012 5, 41, 56. OSU‐03012 is a derivative of celecoxib, a COX‐2 inhibitor, with anticancer activity that has been shown to induce the death of various types of cancer cells 57. The overexpression of autophagy‐related gene5 (Atg5) not only promotes autophagy but also promotes apoptosis 58. The proapoptotic effect of autophagy‐related gene5 (Atg5) is related to its regulation of Bcl‐xL (i.e., through the release of cytochrome C and the activation of calpain and caspases) 59.
Regulation of NSAID toxicity by endoplasmic reticulum stress proteins
The endoplasmic reticulum (ER) is a cellular stress response system 60, 61 that has two main functions: (1) under moderate stress, this system defends against and neutralizes the insult to restore homeostasis. A critical component of this protective response is glucose‐regulated protein 78 (GRP78), a chaperone and calcium‐binding protein whose expression is increased in response to stress 62. (2) Under excessive stress, the ER system switches to its proapoptotic mode and initiates apoptosis. A central player in this process is CHOP (also known as GADD153), a transcription factor that alters the transcriptional profile of cells and triggers the proapoptotic pathway 63. ER stress has been previously reported to induce autophagy 64, 65. When misfolded proteins accumulate in the ER under stress, the stress activates the unfolded protein response, which augments the expression of proteins involved in the recovery process. As previously reported, celecoxib is a known inducer of ER stress. Induction of autophagy after celecoxib treatment may be associated with the cellular response to ER stress 66. Autophagy is known to play an important role in coping with multiple forms of cellular stress, including nutrient or growth factor deprivation, hypoxia, or damaged organelles 65, 67, 68, 69. Celecoxib can induce the activation of stress‐related molecules, such as phospho‐eIF2a, activating transcription factor 4 (ATF‐4), phosphor‐stress‐activated protein kinase/c‐Jun NH(2)‐terminal kinase (SAPK/JNK), and phospho‐c‐Jun. The induction of autophagy after celecoxib treatment may be associated with these stress responses.
Some researchers reported that the Bip/GRP78 signaling pathway activates NSAID‐induced autophagy 64, 65. Celecoxib is a calcium pump inhibitor that has potential therapeutic effects. However, Johnson et al. 65 reported that celecoxib could increase intracellular calcium ion levels, and induce the ER‐mediated effects of GRP78 and CHOP. Under normoxic conditions, the levels of the ER stress marker GRP78 increased as the concentration of celecoxib increased in all tested cell lines over the concentration range examined 65. In addition, GRP78, CHOP, and LC3‐II expression levels in the U87MG and U251MG cell lines are comparable after treatment with celecoxib with or without radiation, particularly under hypoxic conditions 65.
NSAIDs activate autophagy by blocking mTOR signaling
Multiple signal transduction mechanisms are known to regulate autophagy; however, the mTOR pathway is the most important pathway in the onset of autophagy 29. mTOR is mainly activated by the PI3K/Akt/mTOR signaling pathway, and it is a central checkpoint that negatively regulates autophagy. In contrast, anticancer drugs that inhibit the PI3K/Akt/mTOR axis augment autophagy progression. Additionally, adenosine monophosphate‐activated protein kinase (AMPK) is a pivotal cellular energy sensor that recognizes cellular ATP starvation; its downstream targets include the negative regulator of mTOR 67. AMPK‐mediated inhibition of mTOR phosphorylation and activity induces autophagic responses in a variety of different cell types 68. For example, Din et al. 72 found that aspirin activates AMPK and reduces mTOR signaling in CRC cells by inhibiting the mTOR effectors S6K1 and 4E‐BP1. Aspirin and metformin (an activator of AMPK) promote the inhibition of mTOR and Akt, as well as autophagy in colorectal cancer cells 69, 70. As described previously, Zhao et al. 71 proposed that aspirin may inhibit angiogenesis and induce autophagy by inhibiting the mTOR signaling pathways in murine hepatocarcinoma and sarcoma models; the expression levels of p‐mTOR, hypoxia‐inducible factor 1‐alpha (HIF‐1α), and vascular endothelial growth factor‐A (VEGF‐A) were decreased, while the expression levels of Unc‐51‐like kinase (ULK1) and LC3A were increased following treatment of the H22 and S180 cell lines with aspirin and everolimus (an inhibitor of mTOR). Other studies have shown that there is no significant change in the messenger RNA (mRNA) levels of Akt following treatment with celecoxib 18. However, phospho‐Akt levels decreased in a time‐ and dose‐dependent manner 18. Celecoxib regulates autophagy via the PI3K/Akt signaling pathway in SGC‐7901 gastric cancer cells. Researchers have shown that different NSAIDs have the same effect as autophagy‐inducing drugs, such as meloxicam 50, sodium salicylate 71, and sulfasalazine 14. Mazzanti et al. 72 reported similar results; exposure of cells to a low concentration of celecoxib downregulates mTOR expression and causes G1 cell‐cycle arrest with autophagy in the human multidrug‐resistant overexpressing HCC cell line PLC/PRF/5, whereas higher concentrations of celecoxib trigger apoptosis. Furthermore, Setsuko et al. 62 found that mTOR gene expression was downregulated in drug‐resistant tumors when treated with a low dose of irinotecan (CPT‐11) in combination with celecoxib for 20 consecutive days. However, in chemosensitive neuroblastoma xenografts, mTOR gene expression was significantly upregulated compared to that in untreated controls. Hence, we can speculate that the effect of NSAIDs in tumor cells switches between autophagy and apoptosis, depending on the concentration and the time of administration. Low concentrations of NSAIDs can induce autophagy at early stages, whereas higher concentrations of NSAIDs can inhibit autophagy and induce apoptosis at later stages (Fig. 3). More research is required to determine whether drug resistance has an effect on the mTOR pathway, which can affect the progression of autophagy.
Figure 3.
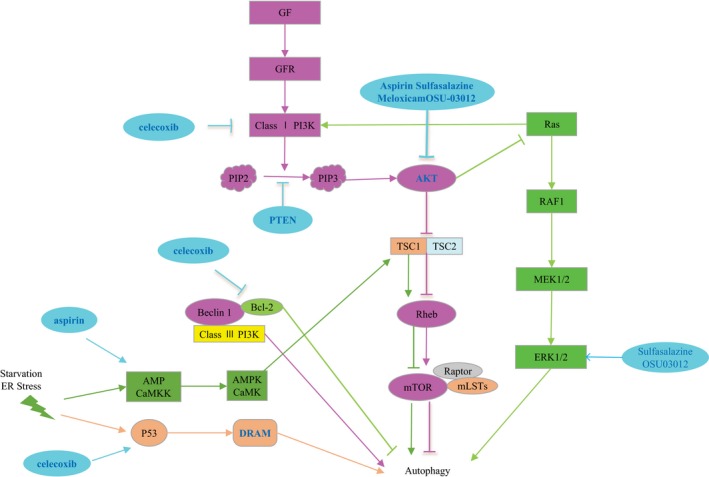
Upstream cell signaling pathway of autophagy. Fuchsia: growth factor activates PI3K/AKT/mTOR signaling pathway; dark green: starvation and endoplasmic reticulum stress promote autophagy through AMPK/CaMKK signaling pathway; dark yellow: P53/DRAM signaling pathway; pale green: Ras signaling pathway can not only promote autophagy but also inhibit autophagy; and light blue: various NSAIDs modulate different signaling pathways to promote autophagy.
NSAIDs inhibit tumor cell viability through p53‐dependent autophagy
Mutational inactivation of the tumor suppressor gene p53 is frequently detected in human tumors, and p53 mutation/inactivation is reported in 63–65% of high‐grade gliomas 74. The presence of DNA damage initiates a signaling cascade, p53 activation (by phosphorylation at Ser15 and Ser20), and subsequent transcriptional activation by p53 75. Genotoxic stress caused by DNA‐damaging agents also induces p53‐dependent autophagy 76, 77, 78. Some reports have suggested that celecoxib inhibits glioma proliferation through p53‐dependent induction of autophagy, but not apoptosis 10. Celecoxib did not induce significant levels of autophagy in U87MGPFT, U87MG‐E6, or U373MG human glioblastoma cell lines, which lack functional p53. The mechanisms of p53‐dependent autophagy are not fully understood, but are thought to involve both transcription‐independent functions (e.g., activation of the nutrient energy sensor AMPK) and transcription‐dependent functions (e.g., upregulation of the mTOR inhibitors phosphatase and tensin homolog (PTEN) and TSC1 or the p53‐regulated autophagy and cell death gene DRAM) 78 (Fig. 3). The antiproliferative mechanism of COX‐2 inhibitors is unclear, and how they induce autophagy in tumors is unknown. Liu et al. 18, 79 hypothesized that crosstalk between p53 80, PI3K/Akt 81, and Bcl‐2‐beclin‐1 pathways was involved in the autophagy/apoptosis switch 82. Thus, the role of p53 in celecoxib‐induced autophagy must be clarified.
NSAID‐induced autophagy enhances radiosensitivity and the cytotoxicity of chemotherapy drugs
Many studies have shown that antitumor therapy, including radiotherapy and chemotherapy, can induce apoptosis, but it can also induce autophagy. Under these conditions, autophagy is often a protective mechanism against various treatments to inhibit apoptosis of tumor cells, which are often drug‐ and radiation‐resistant. Therefore, if autophagy can be inhibited in tumor cells during antitumor therapy, the apoptosis signaling pathway will need to compensate by initiating the programmed cell death of tumor cells, which could improve the efficacy of antitumor treatments.
The role of autophagy in tumor resistance has also become the focus of recent research 83. In a variety of tumor models, autophagy can remove abnormal proteins, organelles, and reactive oxygen species from within the cell, which plays a role in its drug resistance. When apoptosis in tumor cells was induced by chemotherapeutic drugs, autophagic activity was increased, which led to drug resistance; however, the combination of chemotherapeutic drugs with an autophagy inhibitor could reduce this drug resistance 73. The following is a review of autophagy inhibitors, which are frequently used for protective autophagy: 3‐methyladenine (3‐MA), which inhibits class III PI3K activity; chloroquine (CQ) and hydroxychloroquine (HCQ), which inhibit the fusion of autophagic bodies and lysosomes by altering lysosomal pH values; bafilomycin A1, which inhibits the activity of vacuolar‐ATPase; and small interfering RNA (siRNA) which inhibits the expression of autophagy‐related gene (Atg) (Table 2). However, under certain conditions, autophagy can also be involved in apoptosis induction. In fact, apoptosis may become the major mode of cell death. High mobility group protein B1 (HMGB1) is a protein released by tumor cells in response to the cytotoxic effects of chemotherapeutic agents. HMGB1 activates autophagy through the PI3K/Akt signaling pathway to enhance drug resistance. The ability of salicylate to prevent necrotic death may contribute to its anti‐inflammatory action and may suppress tumor development by switching the cell death pathway from tumor‐promoting necrotic cell death to tumor‐suppressive autophagic cell death. Switching the method of cell death may provide a new strategy for the development of antitumor therapies 74. Other studies suggest that P‐glycoprotein (P‐gp) expression and the activity of the hepatocyte growth factor (HGF)/MET autocrine loop are correlated in HCC cells. HGF/MET autocrine loop activity can lead to the overexpression of Bcl‐2 and mTOR, which inhibit the translation initiation factor eIF2 alpha, making the tumor cells resistant to both autophagy and apoptosis 73. Celecoxib inhibits the expression of the P‐gp protein in the loop, thereby reversing resistance. Kaneko et al. found that celecoxib could alleviate the negative effect of imatinib on autophagy by upregulating p62 expression 9. Sensitivity to chemotherapy in patients significantly improved, and combination treatment with imatinib and celecoxib had synergistic cytotoxicity effects in imatinib‐resistant cells 7. Inhibition of autophagy has been shown to enhance antitumor drug efficacy. Concurrently, some researchers proposed that celecoxib enhances the radiosensitivity of hypoxic glioblastoma cells by inducing strong ER stress signals 51. Celecoxib is a promising radio‐sensitizing drug for clinical use in patients with glioblastoma. However, some researchers have expressed different views. Researchers have found that celecoxib enhances the cytotoxicity of imatinib in imatinib‐resistant KBM5‐T315I CML cell lines 9. The underlying mechanism may be related to imatinib‐induced autophagy, which could be reversed by celecoxib. Zhang et al. 16, 35 found that long‐term combination treatment with aspirin and a small‐molecule Bcl‐2/Bcl‐xL antagonist (ABT‐737) could synergistically induce apoptosis both in A549 and H1299 cell lines. Meanwhile, combination treatment with short‐term aspirin and the small‐molecule Bcl‐2/Bcl‐xL antagonist (ABT‐737) caused a greater autophagic response than that to either drug alone, and autophagy switched from a cytoprotective signal to a death‐promoting signal 16. P38 acts as the cellular switch between these two different cell death pathways (i.e., autophagy and apoptosis) induced by cotreatment with aspirin and ABT‐737 16. Hence, the dose and duration of administration can also be two factors that alter the sensitization effects of NSAIDs (Fig. 4). The sensitivity of tumor cells to celecoxib‐induced cellular autophagy is likely to be concentration‐ and/or tumor‐type dependent. These different effects, in a large part, depend on the type of tumor, disease stage, and method of treatment 30, 84.
Figure 4.
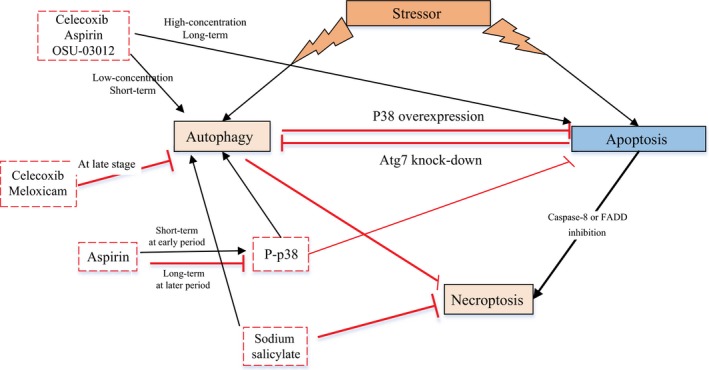
Interaction between different types of programmed cell death and NSAIDs. At late stage and longer term, NSAIDs (e.g., celecoxib, meloxicam, and aspirin) switch from autophagy to apoptosis. At early period and shorter term, they switch from apoptosis to autophagy.
Side effects of NSAIDs and possible mechanisms
Conventional nonselective NSAIDs inhibit both COX enzymes. Inhibition of COX‐1 can lead to acute gastric mucosal lesions 85. Celecoxib is the first selective COX‐2 inhibitor approved for clinical use in the treatment of osteoarthritis and acute pain. It can significantly reduce the incidence of adverse gastrointestinal effects. However, recent studies have shown that long‐term use of selective COX‐2 inhibitors increases the risk of adverse cardiovascular events, especially myocardial infarction 85, 86. This is may be due to the mechanism of NSAIDs by which cardiovascular events are induced, mainly by inhibiting COX‐2 in endothelial cells, thereby inhibiting the production of prostacyclin I (prostacyclin, PGI2). PGI2 can dilate blood vessels and inhibit platelet aggregation. A reduction in PGI2 production increases the risk of vascular injury and tendency for thrombus. To balance the benefits of aspirin and the risk of bleeding, patients should be evaluated, by analyzing them for PIK3CA mutations. To use NSAIDs to effectively prevent tumor formation and progression, we can evaluate COX‐2 expression and gene polymorphisms. The proposed anticancer mechanism for NSAIDs is as follows. NSAIDS exert anticancer effects by reducing the frequency of gene mutations, inhibiting prostaglandin peroxidase synthase‐2 activity, inhibiting inflammation, and regulating autophagy. Although both observational and randomized studies provided convincing evidence for aspirin as an effective chemopreventive agent for CRC 87 it is not recommended for primary prevention of CRC in people at average risk of chronic venous diseases (CVD) because of the potential risks. Aspirin is used for secondary prevention of CRC, particularly in certain subgroups, such as those with wild‐type KRAS tumors 88. Future studies are needed to find the optimal timing, dose, and duration of NSAID use and to identify subgroups of patients with cancer for whom the benefits of aspirin outweigh its risks.
Summary and future perspectives
NSAIDs have been used as anti‐inflammatory, analgesic, and antithrombotic drugs 69. Hence, the safety and side effect profiles of these drugs are well understood. These drugs are good clinical candidates for further exploration of their underlying mechanisms of chemoprevention in cancer 21. The antitumor effects of NSAIDs are related to their autophagy‐modulating effects: either activation or inhibition of autophagy. Thus, it is important to determine the fate of tumor cells treated with NSAIDs. The net effect of autophagy in cancer may depend on the type of tumor, stage of tumorigenesis, tumor microenvironment, as well as genetic and epigenetic factors. One of the most effective treatment methods is to combine lower dose and longer term NSAID administration with chemotherapeutic drugs. NSAIDs are commonly used antipyretic and analgesic drug. With further research, NSAIDs could play an important role in combating tumor progression, especially with the discovery of new mechanisms and the development of molecular biology techniques to study autophagy. Understanding the effects of NSAIDs and their antitumor effects at the molecular and cellular levels will improve our understanding of tumor pathogenesis and uncover potential anticancer targets.
Conflict of Interest
All the authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants performed by any of the authors.
Cancer Medicine 2018; 7(2):471–484
This study was funded by grants from Jiangsu Cadre Health Care Research Project (BJ15028) and the National Natural Science Foundation of China (81503528).
Contributor Information
Jian‐wei Lu, Email: lujw@medmail.com.cn.
Ji‐feng Feng, Email: jifeng_feng@163.com.
References
- 1. Chiarugi, P. , and Giannoni E.. 2008. Anoikis: a necessary death program for anchorage‐dependent cells. Biochem. Pharmacol. 76:1352–1364. [DOI] [PubMed] [Google Scholar]
- 2. Sato, K. , Tsuchihara K., Fujii S., Sugiyama M., Goya T., Atomi Y., et al. 2007. Autophagy is activated in colorectal cancer cells and contributes to the tolerance to nutrient deprivation. Cancer Res. 67:9677–9684. [DOI] [PubMed] [Google Scholar]
- 3. Hour, T. C. , Chen J., Huang C. Y., Guan J. Y., Lu S. H., Hsieh C. Y., et al. 2000. Characterization of chemoresistance mechanisms in a series of cisplatin‐resistant transitional carcinoma cell lines. Anticancer Res. 20:3221–3225. [PubMed] [Google Scholar]
- 4. Yu, H. J. , Tsai T. C., Hsieh T. S., Chiu T. Y.. 1992. Characterization of a newly established human bladder carcinoma cell line, NTUB1. J. Formos. Med. Assoc. 91:608–613. [PubMed] [Google Scholar]
- 5. Huang, K.‐H. , Kuo K.‐L., Ho I.‐L., Chang H. C., Chuang Y. T., Lin W. C., et al. 2013. Celecoxib‐induced cytotoxic effect is potentiated by inhibition of autophagy in human urothelial carcinoma cells. PLoS ONE 8:e82034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huang, S. , and Sinicrope F. A.. 2010. Celecoxib‐induced apoptosis is enhanced by ABT‐737 and by inhibition of autophagy in human colorectal cancer cells. Autophagy 6:256–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bibbins‐Domingo, K. , and U.S. Preventive Services Task Force . 2016. Aspirin use for the primary prevention of cardiovascular disease and colorectal cancer: U.S. Preventive Services Task Force recommendation statement. Ann. Intern. Med. 164:836–845. [DOI] [PubMed] [Google Scholar]
- 8. Sahni, S. , Merlot A. M., Krishan S., Jansson P. J., Richardson D. R., et al. 2014. Gene of the month: BECN1. J. Clin. Pathol. 67:656–660. [DOI] [PubMed] [Google Scholar]
- 9. Lu, Y. , Liu L.‐L., Liu S.‐S., Fang Z. G., Zou Y., Deng X. B., et al. 2016. Celecoxib suppresses autophagy and enhances cytotoxicity of imatinib in imatinib‐resistant chronic myeloid leukemia cells. J. Transl. Med. 14:270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kang, K. B. , Zhu C., Yong S. K., Gao Q., and Wong M. C.. 2009. Enhanced sensitivity of celecoxib in human glioblastoma cells: induction of DNA damage leading to p53‐dependent G1 cell cycle arrest and autophagy. Mol. Cancer. 8:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kaneko, S. , Kaneko M., and Fukushima T.. 2013. Enhanced antitumor effect of lower‐dose and longer‐term CPT‐11 treatment in combination with low‐dose celecoxib against neuroblastoma xenografts. Int. J. Clin. Oncol. 18:116–125. [DOI] [PubMed] [Google Scholar]
- 12. Lu, Y. , Liu X. F., Liu T. R., Fan R. F., Xu Y. C., Zhang X. Z., et al. 2016. Celecoxib exerts antitumor effects in HL‐60 acute leukemia cells and inhibits autophagy by affecting lysosome function. Biomed. Pharmacother. 84:1551–1557. [DOI] [PubMed] [Google Scholar]
- 13. Lim, S. C. , Kim S. M., Choi J. E., Kim C. H., Duong H. Q., Han S. I., et al. 2008. Sodium salicylate switches glucose depletion‐induced necrosis to autophagy and inhibits high mobility group box protein 1 release in A549 lung adenocarcinoma cells. Oncol. Rep. 19:1165–1171. [PubMed] [Google Scholar]
- 14. Han, H. ‐Y. , Kim H., Jeong S. ‐H., Lim D. S., and Ryu M. H.. 2014. Sulfasalazine induces autophagic cell death in oral cancer cells via Akt and ERK pathways. Asian Pac. J. Cancer Prev. 15:6939–6944. [DOI] [PubMed] [Google Scholar]
- 15. Thomas, S. , Sharma N., Encouse B., Cho H., Agarwal P., Gaffney K. J., et al. 2012. Preferential killing of triple‐negative breast cancer cells in vitro and in vivo when pharmacological aggravators of endoplasmic reticulum stress are combined with autophagy inhibitors. Cancer Lett. 325:63–71. [DOI] [PubMed] [Google Scholar]
- 16. Zhang, C. , Shi J., Mao S. Y., Xu Y. S., Zhang D., Feng L. Y., et al. 2015. Role of p38 MAPK in enhanced human cancer cells killing by the combination of aspirin and ABT‐737. J. Cell Mol. Med. 19:408–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Silva, J. , Arantes‐Rodrigues R., Pinto‐Leite R., Faustino‐Rocha A. I., Fidalgo‐Gonçalves L. I., Santos L., et al. 2017. Synergistic effect of carboplatin and piroxicam on two bladder cancer cell lines. Anticancer Res. 37:1737–1745. [DOI] [PubMed] [Google Scholar]
- 18. Liu, M. , Li C. ‐M., Chen Z. ‐F., Ji R., Guo Q. H., Li Q., et al. 2014. Celecoxib regulates apoptosis and autophagy via the PI3K/Akt signaling pathway in SGC‐7901 gastric cancer cells. Int. J. Mol. Med. 33:1451–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hoyer‐Hansen, M. , Bastholm L., Szyniarowski P., Campanella M., Szabadkai G., Farkas T., et al. 2007. Control of macroautophagy by calcium, calmodul independent kinase kinase‐beta, and Bcl‐2. Mol Cell 2007;25 19:3–205. [DOI] [PubMed] [Google Scholar]
- 20. Maiuri, M. C. , Le Toumelin G., Criollo A., Rain J. C., Gautier F., Juin P., et al. 2007. Functional and physical interaction between Bcl‐x (l) and a BH3‐like domain in Beclin‐1. EMBO J. 26:2527–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ghavami, S. , Sharma P., Yeganeh B., Ojo O. O., Jha A., Mutawe M. M., et al. 2014. Airway mesenchymal cell death by mevalonate cascade inhibition: integration of autophagy, unfolded protein response and apoptosis focusing on Bcl2 family proteins. Biochim. Biophys. Acta 1843:1259–1271. [DOI] [PubMed] [Google Scholar]
- 22. Jung, C. H. , Ro S. H., Cao J., Otto N. M., and Kim D. H.. 2010. mTOR regulation of autophagy. FEBS Lett. 584:1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ma, L. , Chen Z., Erdjument‐Bromage H., Tempst P., and Pandolfi P. P.. 2005. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 121:179–193. [DOI] [PubMed] [Google Scholar]
- 24. Luo, S. , and Rubinsztein D. C.. 2007. Atg5 and Bcl‐2 provide novel insights into the interplay between apoptosis and autophagy. Cell Death Differ. 14:1247–1250. [DOI] [PubMed] [Google Scholar]
- 25. Schaaf, M. B. , Cojocari D., Keulers T. G., Jutten B., Starmans M. H., de Jong M. C., et al. 2013. The autophagy associated gene, ULK1, promotes tolerance to chronic and acute hypoxia. Radiother. Oncol. 108:529–534. [DOI] [PubMed] [Google Scholar]
- 26. Gwinn, D. M. , Shackelford D. B., Egan D. F., Mihaylova M. M., Mery A., Vasquez D. S., et al. 2008. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30:214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Scott, R. C. , Juhasz G., and Neufeld T. P.. 2007. Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr. Biol. 17:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sinha, S. , and Levine B.. 2009. The autophagy effector Beclin 1: a novel BH3‐only protein. Oncogene 27:137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang, P. M. , Liu Y. L., Lin Y. C., Shun C. T., Wu M. S., Chen C. C., et al. 2010. Inhibition of autophagy enhances anticancer effects of atorvastatin in digestive malignancies. Cancer Res. 70:7699–7709. [DOI] [PubMed] [Google Scholar]
- 30. Kanzawa, T. , Zhang L., Xiao L., Germano I. M., Kondo Y., Kondo S., et al. 2005. Arsenic trioxide induces autophagic cell death in malignant glioma cells by upregulation of mitochondrial cell death protein BNIP3. Oncogene 24:980–991. [DOI] [PubMed] [Google Scholar]
- 31. Kim, K. W. , Mutter R. W., Cao C., Albert J. M., Freeman M., Hallahan D. E., et al. 2006. Autophagy for cancer therapy through inhibition of pro‐apoptotic proteins and mammalian target of rapamycin signaling. J. Biol. Chem. 281:36883–36890. [DOI] [PubMed] [Google Scholar]
- 32. Chresta, C. M. , Davies B. R., Hickson I., Harding T., Cosulich S., Critchlow S. E., et al. 2010. AZD8055 is a potent, selective, and orally bioavailable ATP‐competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 70:288–298. [DOI] [PubMed] [Google Scholar]
- 33. Kimple, R. J. , Grabowski S., Pepez M., Collichio F., Ewend M. G., and Morris D. E.. 2010. Concurrent temozolomide and radiation, a reasonable option for elderly patients with glioblastoma multiforme? Am. J. Clin. Oncol. 33:265–270. [DOI] [PubMed] [Google Scholar]
- 34. Lu, Z. , Luo R. Z., Lu Y., Zhang X., Yu Q., Khare S., et al. 2008. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J Clin Invest 118:3917–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kanzawa, T. , Germano I. M., Komata T., Ito H., Kondo Y., and Kondo S.. 2004. Role of autophagy in temozolomide‐induced cytotoxicity for malignant glioma cells. Cell Death Differ. 11:448–457. [DOI] [PubMed] [Google Scholar]
- 36. Ito, H. , Daido S., Kanzawa T., Kondo S., and Kondo Y.. 2005. Radiation‐induced autophagy is associated with LC3 and its inhibition sensitizes malignant glioma cells. Int. J. Oncol. 26:1401–1410. [PubMed] [Google Scholar]
- 37. Jorgensen, H. G. , Allan E. K., Jordanides N. E., Mountford J. C., and Holyoake T. L.. 2007. Nilotinib exerts equipotent anti‐proliferative effects to imatinib and does not induce apoptosis in CD34+ CML cells. Blood 109:4016–4019. [DOI] [PubMed] [Google Scholar]
- 38. Abedin, M. J. , Wang D., McDonnel M. A., Lehmann U., and Kelekar A.. 2007. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ. 14:500–510. [DOI] [PubMed] [Google Scholar]
- 39. Pattingre, S. , Bauvy C., and Codogno P.. 2003. Amino acids interfere with the ERK1/2‐dependent control of macroautophagy by controlling the activation of Raf‐1 in human colon cancer HT‐29 cells. J. Biol. Chem. 278:16667–16674. [DOI] [PubMed] [Google Scholar]
- 40. Fosslien, E. 2000. Molecular pathology of cyclooxygenase‐2 in neoplasia. Ann. Clin. Lab. Sci. 30:3–21. [PubMed] [Google Scholar]
- 41. Gao, M. , Yeh P. Y., Lu Y. S., Hsu C. H., Chen K. F., Lee W. C., et al. 2008. OSU‐03012, a novel celecoxib derivative, induces reactive oxygen speciesrelated autophagy in hepatocellular carcinoma. Cancer Res. 68:9348–9357. [DOI] [PubMed] [Google Scholar]
- 42. Konno, H. , Baba M., Shoji T., Ohta M., Suzuki S., and Nakamura S.. 2002. Cyclooxygenase‐2 expression correlates with uPAR levels and is responsible for poor prognosis of colorectal cancer. Clin. Exp. Metastasis 19:527–534. [DOI] [PubMed] [Google Scholar]
- 43. Jin, S. , and White E.. 2007. Role of autophagy in cancer: management of metabolic stress. Autophagy 3:28–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Amaravadi, R. K. , and Thompson C. B.. 2007. The roles of therapy induced autophagy and necrosis in cancer treatment. Clinical Cancer Res 13:7271–7279. [DOI] [PubMed] [Google Scholar]
- 45. Chen, J. , Shen P., Zhang X. C., Zhao M. D., Zhang X. G., and Yang L.. 2014. Efficacy and safety profile of celecoxib for treating advanced cancers: a meta‐analysis of 11 randomized clinical trials. Clin. Ther. 36:1253–1263. [DOI] [PubMed] [Google Scholar]
- 46. Movahedi, M. , Bishop D. T., Macrae F., Mecklin J. P., Moeslein G., Olschwang S., et al. 2015. Obesity, aspirin, and risk of colorectal cancer in carriers of hereditary colorectal cancer: a prospective investigation in the CAPP2 study. J. Clin. Oncol. 33:3591–3597. [DOI] [PubMed] [Google Scholar]
- 47. Cao, Y. , Nishihara R., Wu K., Wang M., Ogino S., Willett W. C., et al. 2016. Population‐wide impact of long‐term use of aspirin and the risk for cancer. J Am Med Assoc Oncol 2:762–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lotrionte, M. , Biasucci L. M., Peruzzi M., Frati G., Giordano A., and Biondi‐Zoccai G.. 2016. Which aspirin dose and preparation is best for the long‐term prevention of cardiovascular disease and cancer? Evidence from a systematic review and network meta‐analysis. Prog. Cardiovasc. Dis. 58:495–504. [DOI] [PubMed] [Google Scholar]
- 49. Dulai, P. S. , Singh S., Marquez E., Khera R., Prokop L. J., Limburg P. J., et al. 2016. Chemoprevention of colorectal cancer in individuals with previous colorectal neoplasia: systematic review and network meta‐analysis. Br. Med. J. 355:i6188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dong, X. F. , Li R., Xiu P., Dong X., Xu Z., Zhai B., et al. 2014. Meloxicam executes its antitumor effects against hepatocellular carcinoma in COX‐2‐dependent and ‐independent pathways. PLoS ONE 9:e92864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Suzuki, K. , Gerelchuluun A., Hong Z., Sun L., Zenkoh J., Moritake T., et al. 2013. Celecoxib enhances radiosensitivity of hypoxic glioblastoma cells through endoplasmic reticulum stress. Neuro Oncol 15:1186–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mathew, R. , Karp C. M., Beaudoin B., Vuong N., Chen G., Chen H. Y., et al. 2009. Autophagy suppresses tumorigenesis through elimination of p62. Cell 137:1062–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Moscat, J. , and Diaz‐Meco M. T.. 2009. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 137:1001–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Duran, A. , Linares J. F., Galvez A. S., Wikenheiser K., Flores J. M., Diaz‐Meco M. T., et al. 2008. The signaling adaptor p62 is an important NF‐kappaB mediator in tumorigenesis. Cancer Cell 13:343–354. [DOI] [PubMed] [Google Scholar]
- 55. Yu, H. , Su J., Xu Y., Kang J., Li H., Zhang L., et al. 2011. p62/SQSTM1 involved in cisplatin resistance in human ovarian cancer cells by clearing ubiquitinated proteins. Eur. J. Cancer 47:1585–1594. [DOI] [PubMed] [Google Scholar]
- 56. Zhu, J. , Huang J. W., Tseng P. H., Yang Y. T., Fowble J., Shiau C. W., et al. 2004. From the cyclooxygenase‐2 inhibitor celecoxib to a novel class of 3‐phosphoinositide‐dependent protein kinase‐1 inhibitors. Cancer Res. 64:4309–4318. [DOI] [PubMed] [Google Scholar]
- 57. Matsushita, M. , Suzuki N. N., Obara K., Fujioka Y., Ohsumi Y., Inagaki F., et al. 2007. Structure of Atg5.Atg16, a complex essential for autophagy. J. Biol. Chem. 282:6763–6772. [DOI] [PubMed] [Google Scholar]
- 58. Yousefi, S. , Perozzo R., Schmid I., et al. 2006. Calpain‐mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 8:1124–1132. [DOI] [PubMed] [Google Scholar]
- 59. Schontha, A. H. 2012. Targeting endoplasmic reticulum stress for cancer therapy. Front Biosci. 4:412–431. [DOI] [PubMed] [Google Scholar]
- 60. Tabas, I. , and Ron D.. 2011. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 13:184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lee, A. S. 2007. GRP78 induction in cancer: therapeutic and prognostic implications. Cancer Res. 67:3496–3499. [DOI] [PubMed] [Google Scholar]
- 62. Oyadomari, S. , and Mori M.. 2004. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 11:381–389. [DOI] [PubMed] [Google Scholar]
- 63. Pyrko, P. , Kardosh A., Liu Y. T., Soriano N., Xiong W., Chow R. H., et al. 2007. Calcium‐activated endoplasmic reticulum stress as a major component of tumor cell death induced by 2,5‐dimethyl‐celecoxib, a non‐coxib analogue of celecoxib. Mol. Cancer Ther. 6:1262–1275. [DOI] [PubMed] [Google Scholar]
- 64. Johnson, A. J. , Hsu A. L., Lin H. P., Xueqin S. O., Ching‐Shih C. H., et al. 2002. The cyclo‐oxygenase‐2 inhibitor celecoxib perturbs intracellular calcium by inhibiting endoplasmic reticulum Ca2+‐ATPases: a plausible link with its anti‐tumour effect and cardiovascular risks. Biochem J 366:831–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kroemer, G. , Marino G., and Levine B.. 2010. Autophagy and the integrated stress response. Mol. Cell 40:280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhu, X. , Zhou M., Liu G., Huang X., He W., Gou X., et al. 2017. Autophagy activated by the c‐Jun N‐terminal kinase‐mediated pathway protects human prostate cancer PC3 cells from celecoxib‐induced apoptosis. Exp Ther Med 13:2348–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kim, Y. C. , and Guan K. L.. 2015. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest 125:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chapuis, N. , Tamburini J., Green A. S., Willems L., Bardet V., Park S., et al. 2010. Perspectives on inhibiting mTOR as a future treatment strategy for hematological malignancies. Leukemia 24:1686–1699. [DOI] [PubMed] [Google Scholar]
- 69. Yang, C. S. , Kim J. J., Lee H. M., Jin H. S., Lee S. H., Park J. H., et al. 2014. The AMPK‐PPARGC1A pathway is required for antimicrobial host defense through activation of autophagy. Autophagy 10:785–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wang, L. T. , Chen B. L., Wu C. T., Huang K. H., Chiang C. K., and Liu S. H.. 2013. Protective role of AMP activated protein kinase‐evoked autophagy on an in vitro model of ischemia/reperfusion‐induced renal tubular cell injury. PLoS ONE 8:79814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zhao, Q. , Wang Z., Wang Z., Wu L., Zhang W., et al. 2016. Aspirin may inhibit angiogenesis and induce autophagy by inhibiting mTOR signaling pathway in murine hepatocarcinoma and sarcoma models. Oncol Lett 12:2804–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Din, F. V. , Valanciute A., Houde V. P., Zibrova D., Green K. A., Sakamoto K., et al. 2012. Aspirin inhibits mTOR signaling, activates AMP‐activated protein kinase, and induces autophagy in colorectal cancer cells. Geoenterology 142:1504–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mazzanti, R. , Platini F., Bottini C., Fantappiè O., Solazzo M., Tessitore L., et al. 2009. Down‐regulation of the HGF/MET autocrine loop induced by celecoxib and mediated by P‐gp in MDR‐positive human hepatocellular carcinoma cell line. Biochem. Pharmacol. 78:21–32. [DOI] [PubMed] [Google Scholar]
- 74. Newcomb, E. W. , Madonia W. J., Pisharody S., Lang F. F., Koslow M., Miller D. C., et al. 1993. A correlative study of p53 protein alteration and p53 gene mutation in glioblastoma multiforme. Brain Pathol. 3:229–235. [DOI] [PubMed] [Google Scholar]
- 75. Norbury, C. J. , and Zhivotovsky B.. 2004. DNA damage‐induced apoptosis. Oncogene 23:2797–2808. [DOI] [PubMed] [Google Scholar]
- 76. Zeng, X. , Yan T., Schupp J. E., Seo Y., and Kinsella T. J.. 2007. DNA mismatch repair initiates 6‐thioguanine–induced autophagy through p53 activation in human tumor cells. Clin. Cancer Res. 13:1315–1321. [DOI] [PubMed] [Google Scholar]
- 77. Feng, Z. , Zhang H., Levine A. J., and Jin S.. 2005. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl Acad. Sci. USA 102:8204–8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Crighton, D. , Wilkinson S., O'Prey J., Syed N., Smith P., Harrison P. R., et al. 2006. DRAM, a p53‐induced modulator of autophagy, is critical for apoptosis. Cell 126:121–134. [DOI] [PubMed] [Google Scholar]
- 79. Kang, R. , Zeh H. J., Lotze M. T., et al. 2011. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 18:571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Liu, J. , Lin Y., Yang H., Deng Q., Chen G., He J., et al. 2011. The expression of p33 (ING1), p53, and autophagy‐related gene Beclin1 in patients with non‐small cell lung cancer. Tumor Biol 32:1113–1121. [DOI] [PubMed] [Google Scholar]
- 81. Cheng, Y. , Ren X., Zhang Y., Patel R., Sharma A., Wu H., et al. 2011. eEF‐2 kinase dictates cross‐talk between autophagy and apoptosis induced by Akt inhibition, thereby modulating cytotoxicity of novel Akt inhibitor MK‐2206. Cancer Res. 71:2654–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Djavaheri‐Mergny, M. , Maiuri M. C., and Kroemer G.. 2010. Crosstalk between apoptosis and autophagy by caspase‐mediated cleavage of Beclin 1. Oncogene 29:1717–1719. [DOI] [PubMed] [Google Scholar]
- 83. Liang, J. , Shao S. H., Xu Z. X., Hennessy B., Ding Z., Larrea M., et al. 2007. The energy sensing LKB1‐AMPK pathway regulates p27 (kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nature Cell Biol 9:218–224. [DOI] [PubMed] [Google Scholar]
- 84. Booth, L. , Roberts J. L., Cruickshanks N., Tavallai S., Webb T., Samuel P., et al. 2015. PDE5 inhibitors enhance celecoxib killing in multiple tumor types. J. Cell. Physiol. 230:1115–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zhong, J. , Dong X., Xiu P., Wang F., Liu J., Wei H., et al. 2015. Blocking autophagy enhances meloxicam lethality to hepatocellular carcinoma by promotion of endoplasmic reticulum stress. Cell Prolif. 48:691–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Pellicano, R. 2014. Gastrointestinal damage by non‐steroidal anti‐inflammatory drugs: updated clinical considerations. Minerva Gastroenterol. Dietol. 60:255–261. [PubMed] [Google Scholar]
- 87. White, E. , and DiPaola R. S.. 2009. The double‐edged sword of autophagy modulation in cancer. Clin. Cancer Res. 15:5308–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Baron, J. A. 2009. Aspirin and NSAIDs for the prevention of colorectal cancer. Recent Results Cancer Res. 181:223–229. [DOI] [PubMed] [Google Scholar]
- 89. Hua, X. , Phipps A. I., Burnett‐Hartman A. N., Adams S. V., Hardikar S., Cohen S. A., et al. 2017. Timing of aspirin and other nonsteroidal anti‐inflammatory drug use among patients with colorectal cancer in relation to tumor markers and survival. J. Clin. Oncol. 15:JCO 2017723569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ho‐Jae, L. , Min Park J., and Ki Baik H.. 2017. Mitigated NSAID induced apoptotic and autophagic cell death with Smad7 overexpression. J Clin Biochem Nutr 60:55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Bauvy, C. , Gane P., Arico S., Codogno P., and Ogier‐Denis E.. 2001. Autophagy delays sulindac sulfide‐induced apoptosis in the human intestinal colon cancer cell line HT‐29. Exp. Cell Res. 268:139–149. [DOI] [PubMed] [Google Scholar]
- 92. Su, J. , Liu F., Xia M., Xu Y., Li X., Kang J., et al. 2015. p62 participates in the inhibition of NF‐κB signaling and apoptosis induced by sulfasalazine in human glioma U251 cells. Oncol. Rep. 34:235–243. [DOI] [PubMed] [Google Scholar]
- 93. Maiuri, M. C. , Zalckvar E., Kimchi A., and Kroemer G.. 2007. Self‐eating and self‐killing: crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 8:741–752. [DOI] [PubMed] [Google Scholar]