

Abstract
Objective
Attention-deficit/hyperactivity disorder (ADHD) is a common and highly heritable psychiatric condition. By exploiting the reported relationship between ADHD and educational attainment (EA), we here aimed to improve discovery of ADHD-associated genetic variants and investigate genetic overlap between these phenotypes.
Method
A conditional/conjunctional false discovery rate (condFDR/conjFDR) method was applied to genome-wide association study (GWAS) data on ADHD (2,064 trios, 896 cases, and 2,455 controls) and EA (n = 328,917) to identify ADHD-associated loci and loci overlapping between ADHD and EA. Identified single nucleotide polymorphisms (SNPs) were tested for association in an independent population-based study of ADHD symptoms (n = 17,666). Genetic correlation between ADHD and EA was estimated using LD score regression and Pearson correlation.
Results
At levels of condFDR < 0.01 and conjFDR < 0.05, we identified five ADHD-associated loci, three of these being shared between ADHD and EA. None of these loci had been identified in the primary ADHD GWAS, demonstrating the increased power provided by the condFDR/conjFDR analysis. Leading SNPs for 4 of 5 identified regions are in introns of protein coding genes: KDM4A, MEF2C, PINK1, RUNX1T1, while the remaining one is an intergenic SNP on chromosome 2 at 2p24. Consistent direction of effects in the independent study of ADHD symptoms was shown for 4 of 5 identified loci. A polygenic overlap between ADHD and EA was supported by significant genetic correlation (rg = −0.403, p = 7.90 × 10−8) and >10-fold mutual enrichment of SNPs associated with both traits.
Conclusion
We identified five novel loci associated with ADHD and provided evidence for a shared genetic basis between ADHD and EA. These findings could aid understanding of the genetic risk architecture of ADHD and its relation to EA.
Keywords: Attention-deficit/hyperactivity disorder, Educational attainment, conditional/conjunctional false discovery rate, genetic overlap
INTRODUCTION
Attention-deficit/hyperactivity disorder (ADHD) is a common neurodevelopmental condition, caused by interplay of genetic and environmental risk factors. Its prevalence is estimated to be 5% in school-aged children and 2.50% in adults.1 The heritability of ADHD is one of the highest reported among psychiatric disorders in epidemiological studies, estimated at 0.70–0.80.1 However, it has been difficult to identify genetic risk variants that account for the high heritability of ADHD, resulting in a relatively modest single nucleotide polymorphism (SNP)-based heritability, currently estimated at 0.28.2 This may be in part explained by its complex phenotypic structure (heterogeneous clinical features, developmental course and outcome, high rate of comorbid symptoms and disorders3) and genetic architecture with a highly polygenic etiology, with both common and rare variants contributing small effects.4 Moreover, large sample sizes are needed for reliable detection of such effects. The relatively small samples of existing ADHD genetic studies, as compared to those available for other psychiatric disorders,5,6 present an additional challenge. Up to now, no published genome-wide association studies (GWASs) have been able to detect genome-wide significant association (p < 5.00 × 10−8) for ADHD.
It is well-established that complex traits often have a polygenic structure with shared genetic background.7,8 Recently, a conditional/conjunctional false discovery rate (condFDR/conjFDR; see Table 1 for explanation of terminology) method was developed9 to exploit overlapping association across GWASs and thereby boost association signals in GWAS of one phenotype by combining it with genome-wide association data of another phenotype (condFDR) or enable detection of specific genetic loci shared between two phenotypes (conjFDR). If genetic overlap between two phenotypes exists, the method offers increased statistical power compared to conventional multiple hypotheses testing approaches.10,11 This method was successfully applied to discover novel associations and to detect shared genetic variants in various complex disorders, including neurological12,13 and psychiatric9 diseases.
Table 1.
Terminology
| • GWAS (genome-wide association study): a study that performs genome-wide scan of common genetic variants aiming to identify variants associated with the trait. |
| • LD (linkage disequilibrium): the statistical correlation between alleles at two loci. |
| • FDR (false discovery rate): a posterior probability that identified association is false. |
| • condFDR (conditional FDR): the method that uses association p-values of two traits (primary and conditional) to estimate a posterior probability that a variant has no association in a primary trait, given that p-values of the variant in both the primary and conditional traits are lower than their observed p-values. |
| • conjFDR (conjunctional FDR): an extension of condFDR method that estimates a posterior probability that a variant has no association for either phenotype or both at the same time, given that the p-values for both phenotypes are lower than the observed p-values. |
| • QQ (Quantile-Quantile) plot: a visual tool used to compare two probability distributions (e.g. observed vs expected) by plotting their quantiles against each other. |
| • Conditional QQ plot: a QQ plot comparing probability distributions of association p-values in primary trait for different strata of variants (e.g. variants with different levels of significance in conditional trait). |
| • eQTL (expression quantitative trait loci): genetic variants that affect gene expression levels. |
ADHD is consistently associated with lower levels of EA1,14: the percentage of US adolescents not completing high school is 5%, whereas it is approximately 35% for adolescents diagnosed with ADHD15. There are several ways in which ADHD may relate to lower EA, which are not mutually exclusive. First, the clinical and cognitive symptoms of ADHD (e.g. attention deficits) may directly perturb EA. Secondly, ADHD has a number of common comorbidities, including learning disabilities,16 mood disorders,16 and disruptive behavior,16 associated with lower EA. Another possibility is that ADHD and EA share causative factors. Recent findings demonstrate negative genetic correlation between ADHD and EA (rg = −0.305, se = 0.141, p = 3.00 × 10−2)17, suggesting that genetic variants conferring risk to ADHD may contribute to lower EA in the general population. Thus, we can hypothesize that ADHD and EA may have a shared genetic basis and may amplify association signal by combining these phenotypes in condFDR/conjFDR method.
In contrast to ADHD, where the currently published largest GWASs contain less than 4000 cases18,19, the latest GWAS on EA contains more than 300,000 individuals, uncovering multiple genome-wide significantly associated loci20. Combining this EA GWAS with moderately-powered GWAS of ADHD18 in the condFDR/conjFDR approach, we aimed here at identifying novel loci associated with ADHD as well as loci shared between ADHD and EA. The latter may provide insights into the molecular genetic mechanisms jointly influencing ADHD and EA and inform their biological underpinnings. Applying novel statistical methods, we also tested whether the observed phenotypic correlation between ADHD and EA implies a genetic correlation between these traits. Additionally, for the identified ADHD-associated variants, we assessed consistency of effect directions in an independent population-based study of ADHD symptoms and performed in silico analyses of their functional effects (eQTL, expression quantitative loci).
METHOD
Participant Samples
We used ADHD data from the Psychiatric Genomics Consortium (PGC)18. The data set contains information from 2,064 trios, 896 individuals with ADHD, and 2,455 controls. EA data were obtained from the Social Science Genetic Association Consortium (SSGAC)20, where EA was measured as the number of years of schooling completed that was harmonized between different educational systems. For our analyses, we used summary statistics generated by the meta-analysis of all discovery and replication cohorts, except the 23andMe sample (64 datasets with total n = 328,917).
Top association signals identified in our analyses were examined in the summary statistics from an independent GWAS of ADHD symptoms performed by EArly Genetics and Lifecourse Epidemiology (EAGLE) consortium21. Unlike the PGC case-control ADHD GWAS, EAGLE GWAS represents a meta-analysis of 9 population-based pediatric cohorts containing information on 17,666 children under the age of 13 years with measures of ADHD symptom scores.
Detailed description of data used for analysis and data preprocessing steps is given in Supplement 1, available online.
Statistical Analyses
To assess genetic overlap between ADHD and EA and thus warrant subsequent condFDR/conjFDR analysis, we generated conditional QQ plots and fold-enrichment plots in both directions: conditioning ADHD on EA and vice versa9. To explore the nature of the polygenic overlap and test the hypothesis that the investigated phenotypes correlate genetically, we calculated Pearson correlations between association z-scores of ADHD and EA SNPs within nested subset (strata) of SNPs with increasing significance of p-values in either ADHD or EA (formal definition of SNP stratum is given in Supplement 1, available online). To further support this hypothesis, we estimated genetic correlation between ADHD and EA using LD score regression8. Details of these analyses are described in Supplement 1, available online.
To identify specific loci associated with ADHD, we applied the condFDR method described previously9. The condFDR method takes summary statistics that reflect genetic association of a phenotype of interest (primary) together with those of an auxiliary (conditional) phenotype and estimates a posterior probability that a SNP is null (has no association) in the primary phenotype, given that p-values of the SNP in both the primary and conditional phenotypes are lower than observed p-values. Thus, the condFDR method increases the power to discover loci associated with a primary phenotype by leveraging associations with a secondary phenotype. It does so by re-ranking SNPs compared to nominal p-value-based ranking9. In contrast, ranking SNPs based on unconditional FDR (e.g. using Benjamini–Hochberg or Benjamini–Yekutieli procedure) does not change their order (compared to nominal p-values).
Although both conditional QQ plots and genetic correlation based on the LD score regression can be useful to get a general idea of whether two traits have a significant genetic overlap, they are unable to find specific susceptibility loci shared by the traits. The conjFDR approach is an extension of condFDR allowing the identification of specific loci associated with both traits9. The conjFDR is defined as the maximum of the two condFDR values (taking one phenotype as primary and another as conditional and vice versa) for a specific SNP. Thus, the conjFDR approach estimates a posterior probability that an SNP is null for either phenotype or both at the same time, given that the p-values for both phenotypes are lower than the observed p-values. The method, therefore, uncovers loci associated with both phenotypes simultaneously.
To avoid inflation of the results due to LD-dependency in fold-enrichment and QQ plots as well as in condFDR/conjFDR analyses, we randomly pruned all SNPs across 500 iterations. For each iteration, all but one random SNP in each LD-independent region (clump of SNPs in strong LD, r2 > 0.2) were removed, and finally the results were averaged across all iterations. LD (r2 values) was estimated based on the 1000 Genomes Project phase 3 European sub-population data using PLINK.22
As for meta-analyses based on multiple data sources, the quality of our condFDR/conjFDR analysis will depend on the robustness of the primary data. More details about condFDR and conjFDR methods can be found in Supplement 1, available online, and in the original publication.9
Evaluation of the Detected ADHD Loci in an Independent Study of ADHD Symptoms
We used genetic data on association of ADHD symptoms obtained from the EAGLE consortium to test whether our results can be supported by data from the independent sample. For this purpose, we checked whether effects of the most significant SNPs in the loci identified by condFDR/conjFDR analyses are consistent between PGC ADHD and EAGLE data sets.
In Silico Identification of Allele-Specific Effects of Significant SNPs on Transcription
Identifying and investigating genetic variants that might affect gene expression (expression quantitative trait loci or eQTLs) may shed light on how associated variants may contribute to biological mechanisms underlying a phenotype. eQTLs vary significantly both between different tissues and over time.23 Existing GWASs on ADHD and EA clearly demonstrate remarkable enrichment of association signals in genomic regions implicated in regulation of gene expression in the brain.18,20 Hence, we focused on eQTL analysis of genes expressed in brain tissues. Significant associations identified with condFDR and conjFDR analyses were queried for known eQTLs using the GTEx portal (http://gtexportal.org) and the Braineac database (http://www.braineac.org). The latter database contains information on cis-eQTLs for 10 brain regions: cerebellar cortex, frontal cortex, hippocampus, medulla (specifically inferior olivary nucleus), occipital cortex (specifically primary visual cortex), putamen, substantia nigra, thalamus, temporal cortex, and intralobular white matter. Additionally, we checked age-dependent variations of expression in genes containing identified significant SNPs using the Human Brain Transcriptome database (http://hbatlas.org).24
RESULTS
Evaluation of Genetic Overlap and Correlation
In the absence of genetic overlap between two traits, it is expected that p-values for association with one trait are independent from the p-values for association with the other. However, conditional QQ plots in Figure 1 clearly demonstrate an increasing degree of leftward deflection for strata of more significant SNPs. This is observed both when conditioning ADHD on EA (Figure 1A) and vice versa (Figure 1B), suggesting substantial cross-trait polygenic enrichment. Enrichment of association signals for one trait among those of another is also clearly visible in the fold-enrichment plots, with more than 10-fold enrichment of SNPs from the strictest stratum (pconditonal trait < 1.00 × 10−3) for both traits (Figure S1, available online). Additionally, association z-scores of ADHD and EA demonstrate increasing negative correlation in more strictly defined strata of SNPs, both when strata are defined based on ADHD p-values (Figure 1C) and on EA p-values (Figure 1D). Moreover, LD score regression analysis also showed significant negative genetic correlation (rg = −0.403, se = 0.075, p = 7.90 × 10−8) between these phenotypes.
Figure 1.
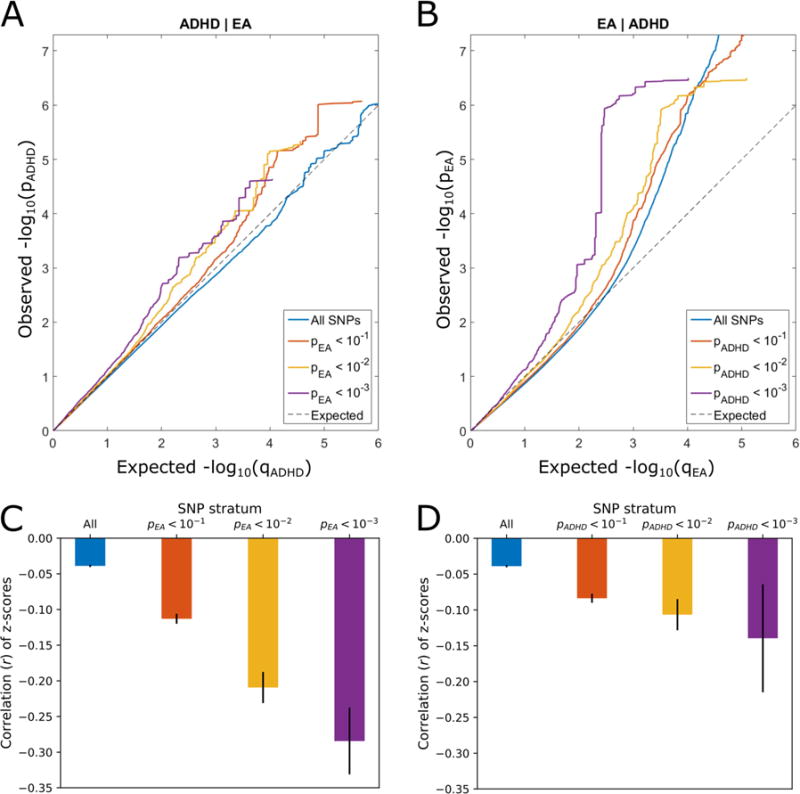
Conditional QQ plots and correlation plots. Note: Conditional QQ plots (A, B) demonstrate relation between expected (x axis) and observed (y axis) significance of markers in the primary trait when markers are stratified by their p-values in the conditional trait. A sequence of four nested strata is presented: all single nucleotide polymorphisms (SNPs) (i.e. p-values of the conditional trait ≤ 1.00), pconditional trait < .1, pconditional trait < .01, and pconditional trait < .001. A: attention-deficit/hyperactivity disorder (ADHD) conditioned on educational attainment (EA). B: EA conditioned on ADHD. Correlation plots (C, D) show Pearson’s correlation coefficients between association z-scores of ADHD and EA for the nested strata of SNPs (as introduced in the conditional QQ plots) averaged over 500 iterations of random pruning. Solid black lines indicate standard deviations. C: SNP strata are defined by the p-values of markers in educational attainment (ADHD|EA). D: SNP strata are defined by the p-values of markers in ADHD (EA|ADHD).
Identification of ADHD-Associated Loci and Loci Shared Between ADHD and EA
Using the condFDR/conjFDR method, we identified 5 LD-independent regions significantly associated with ADHD (condFDR < 0.01, conjFDR < 0.05), 3 of which were also identified as shared between ADHD and EA. From each of these regions, a single SNP with the lowest condFDR/conjFDR value (strongest association signal) was selected to represent their loci. These SNPs are presented in Table 2. Manhattan plots resulting from condFDR and conjFDR analyses are presented in Figures 2 and 3, respectively. Four out of five identified most significant SNPs revealed the opposite directions of effect in ADHD and EA.
Table 2.
Most Significant Single Nucleotide Polymorphisms (SNPs) for Each Linkage Disequilibrium (LD)-Independent Region Identified Either With Conditional False Discovery Rate (condFDR; condFDR <0. 01) or With Conjunctional False Discovery Rate (conjFDR; condFDR <0. 05) Analysis
| SNP | Chr Region | Position | condFDR | conjFDR | Location relative to gene | Genes in the region | p-value | Effect size | ||
|---|---|---|---|---|---|---|---|---|---|---|
| ADHD | EA | ADHD | EA | |||||||
| rs17414302 | 1p36.12 | 20976535 | 4.45×10−2 | 4.48×10−2 | intronic | PINK1 | 1.97×10−4 | 8.17×10−7 | −0.090 | 0.022 |
| rs618678 | 1p34.2 | 44133299 | 3.77×10−3 | 3.82×10−3 | intronic |
KDM4A PTPRF ST3GAL3 |
1.05×10−5 | 1.05×10−10 | −0.053 | 0.017 |
| rs4324303 | 2p24 | 13817678 | 2.17×10−3 | 5.75×10−1 | intergenic | − | 2.05×10−7 | 6.69×10−3 | −0.079 | 0.009 |
| rs412458 | 5q14.3 | 88029627 | 7.34×10−3 | 2.11×10−2 | intronic | MEF2C | 2.15×10−5 | 3.73×10−6 | 0.061 | −0.014 |
| rs4477079 | 8q21.3 | 93059038 | 4.37×10−3 | 3.65×10−1 | intronic | RUNX1T1 | 1.44×10−6 | 1.62×10−3 | −0.071 | −0.009 |
Note: CondFDR/conjFDR values that are below the predefined significance threshold of 0.01/0.05 are boldface. Chromosome and position are indicated according to GRCh37. For both attention-deficit/hyperactivity disorder (ADHD) and educational attainment (EA), p-values without genomic inflation correction are shown. The effect size is given as log10(OR) for ADHD and as Beta regression coefficient for EA. Genes in the region are defined as genes containing SNPs at either condFDR <0. 01 or condFDR <0. 05 and in LD (r2 > 0. 20) with the most significant SNP of the locus. Genes containing the leading SNP are marked in bold. Annotation was generated with Biomart Variant Effect Predictor (http://www.ensembl.org/Homo_sapiens/Tools/VEP).
Figure 2.
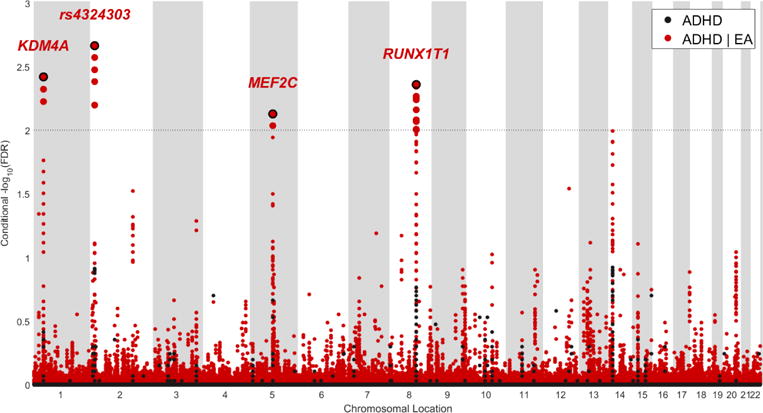
Manhattan of –log10(false discovery rate [FDR]B for attention-deficit/hyperactivity disorder (ADHD) conditional on educational attainment (EA). Note: The data are unpruned. The small points are non-significant single nucleotide polymorphisms (SNPs), the bold points represent significant SNPs (conditional false discovery rate [condFDR] < 0.01). Points corresponding to significant SNPs with lowest conditional FDR in each linkage disequilibrium (LD)-independent region (r2 > 0.20) have a black border and either the name of corresponding gene (for SNPs within the gene) or the rs-number (for an intergenic SNP) written above it. The horizontal grey dotted line shows the significance threshold of condFDR (0.01). Black dots stand for unconditional FDR values.
Figure 3.
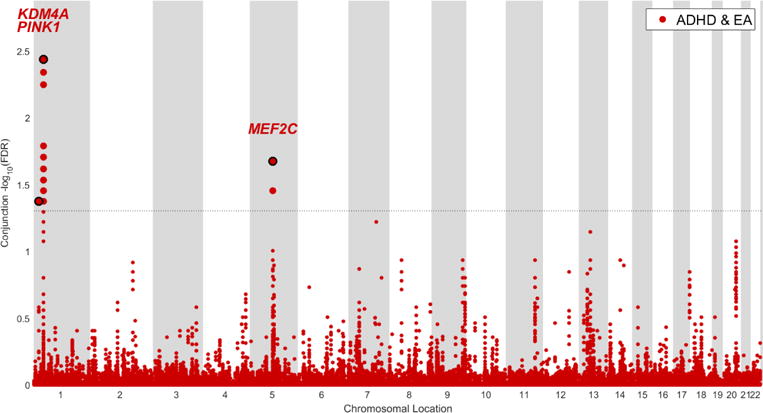
Manhattan plot of conjunctional –log10(false discovery rate [FDR]B for attention-deficit/hyperactivity disorder (ADHD) and educational attainment (EA). Note: The data are unpruned. The small points stand for non-significant single nucleotide polymorphisms (SNPs); the bold points represent significant SNPs (conjunctional false discovery rate [conjFDR] < 0.05). Points corresponding to significant SNPs with lowest conjunctional FDR in each linkage disequilibrium (LD)-independent region (r2 > 0.20) have a black border and the name of the corresponding gene written above it. The horizontal grey dotted line shows the significance threshold of conjFDR (0.05).
Identified Loci and Related Genes
Two loci (represented in Table 2 by variants rs618678 and rs412458) were identified both in condFDR and conjFDR analyses. rs618678 represents the strongest signal in the conjFDR analysis (conjFDR = 3.82 × 10−3) and the second strongest in the condFDR analysis (condFDR = 3.77 × 10−3). This SNP is an intronic variant within KDM4A on chromosome 1p34.2 (Figure 4B). Figure 4B and Figure S2B (available online) show the genetic context of rs618678, indicating, respectively, the conjFDR and condFDR values of adjacent SNPs. It is worth noting that in our analysis, rs618678 tags a broad region of association. As can be seen in Figure 4B, multiple significant SNPs in strong LD (r2 > 0.60) with rs618678 were detected in this region, spanning over more than 200000 basepairs (bp). Besides KDM4A, the region also contains PTPRF (located in 1p34.2, upstream of KDM4A) and ST3GAL3 (1p34.1, directly downstream KDM4A) genes. The latter was also identified in the eQTL analysis (discussed below). Another significant signal identified in both condFDR (condFDR = 7.34 × 10−3) and conjFDR (conjFDR = 2.11 × 10−2) analyses is represented by rs412458, an intronic variant within MEF2C on chromosome 5q14.3 (Figure S2A, D, available online).
Figure 4.
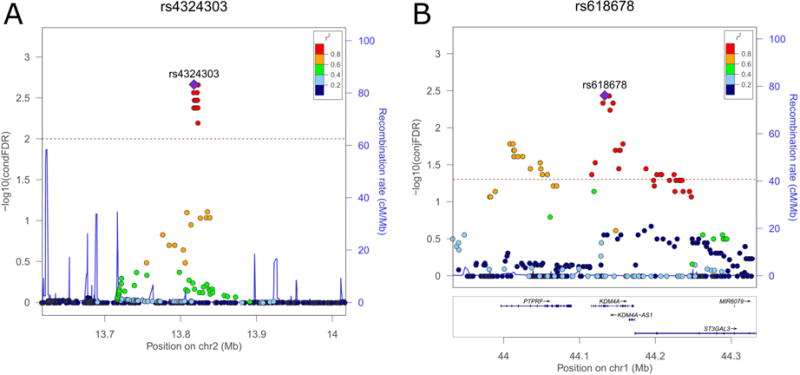
Genetic context of the strongest associations identified in conditional false discovery rate (condFDR) (A) and conjunctional false discovery rate (conjFDR) (B) analyses. Note: Values for both genotyped and imputed variants are shown on the left y-axis as –log10(condFDR) and −log10(conjFDR), respectively. In each subplot, a single nucleotide polymorphism (SNP) with the strongest association is shown in the large purple square. The color of the remaining markers reflects the degree of linkage disequilibrium (LD) with the strongest-associated SNP measured as r2 coefficient (described in the legend). The recombination rate is plotted as a solid blue line; its value in centimorgan/megabase (cM/Mb) is indicated on the right y-axis. The red dotted lines indicate the FDR thresholds (0.01 for condFDR and 0.05 for conjFDR). A: surrounding of the strongest association in condFDR analysis: rs4324303 (condFDR = 2.17 × 10−3). B: surrounding of the strongest association in conjFDR analysis: rs618678 (conjFDR = 3.82 × 10−3). Figures are generated with LocusZoom.52
Two loci were identified by condFDR, but not conjFDR. The strongest signal was detected at rs4324303 (condFDR = 2.17 × 10−3), that is in the intergenic region on chromosome 2p24 (Figure 4A). Multiple significant variants tagged by rs4477079 (condFDR = 4.37 × 10−3) were also identified on chromosome 8 within RUNX1T1 (Figure S2C, available online).
Finally, conjFDR analysis identified a shared variant (conjFDR = 4.48 × 10−2) at PINK1 (rs17414302, intronic, 1p36.12) (Figure S2E, available online). There were no LD-linked SNPs in the direct vicinity and only 25 SNPs in LD (r2 > 0.20) with this variant, residing upstream of PINK1, at about 100000 bp.
None of the SNPs identified either in condFDR or conjFDR reached genome-wide significance in previously published GWAS of ADHD.18 Rs618678 reached genome-wide significance in EA (p = 1.05 × 10−10)20. Rs412458, which was identified by both condFDR and conjFDR, was not reported as genome-wide significant by the published EA GWAS (p = 3.73 × 10−6), but it is in LD (r2 = 0.35) with rs588282 that did reach genome-wide significance in that study (previously reported p = 1.69 × 10−10). Other loci identified in our analyses were below genome-wide significance threshold in EA. It is also worth noting that the unconditional FDR values for all identified SNPs were above 0.01 and 0.05 in condFDR and conjFDR analysis, respectively.
Evaluation of the Detected ADHD Loci in an Independent Study of ADHD Symptoms
To assess the robustness of our results, we examined the loci identified in either the condFDR or conjFDR analyses (Table 2) in the association summary statistics from the independent GWAS of ADHD symptoms conducted by EAGLE consortium.21 Four out of five loci (represented by SNPs rs17414302, rs412458, rs618678, rs4324303) have the same direction of effect in the PGC and EAGLE GWASs while the last locus (represented by rs4477079 SNP) has an opposite direction of effect in these GWASs. These results are presented in Table S1 (available online).
In Silico Identification of Allele-Specific Effects on Transcription
According to Human Brain Transcriptome data,24 all six implicated genes (Table 2, genes in the region) have a pronounced expression in different brain regions during the whole life cycle (Figure S3, available online). Therefore, alterations in the expression level of these genes (where the detected SNPs are located) may affect a broad variety of processes over an extended period. We scanned the Braineac database to check whether SNPs identified in either the condFDR or conjFDR analyses are associated with gene expression in brain tissues. We found that four of five SNPs from Table 2 may operate as eQTLs, significantly (p < .001) associated with the expression of 13 different genes in several brain regions (Table S2, available online). Among those 13 genes, the most significant eQTL was observed between rs618678 and ST3GAL3. Further, significant eQTL effects of rs618678 on ST3GAL3 were identified in muscle-skeletal tissue (p = 3.40 × 10−5) in the GTEx database (https://gtexportal.org/), but not in the brain tissue.
DISCUSSION
The present study sought to investigate the genetic overlap between ADHD and EA, to leverage their potentially common genetics in order to improve the discovery of ADHD-associated loci and help our understanding of the correlation between EA and ADHD observed in epidemiological studies. It is, however, worth emphasizing the broad potential of the applied methodology, which can be used to leverage the great variety of existing GWAS data for dissecting the molecular genetic basis underlying complex human traits and disorders and their shared genetic etiology.
We identified significant genetic overlap between ADHD and EA supported by a pronounced genetic correlation (rg = −0.403, se = 0.075, p = 7.90 × 10−8), consistent enrichment of shared variants in conditional QQ plots (Figure 1A, B), more than 10-fold mutual enrichment of SNPs associated with both traits (Figure S1, available online) and growing negative correlation of association z-scores for the nested SNP strata with increasing significance in both traits (Figure 1C, D). These findings encourage the hypothesis that there is a shared genetic basis underlying ADHD and EA where in general ADHD risk alleles are associated with lower EA.
In comparison to the previous study, exploring the topic of genetic overlap between ADHD and EA,17 our analysis employs a much larger data set of EA, allowing for a more reliable detection of genetic overlap (Figure 1; rg = −0.403, se = 0.075, p = 7.90 × 10−8). It is also worth noting that we report a genetic correlation that is stronger than previously observed using the same ADHD data and a smaller (N = 101,069) EA dataset (rg = −0.305, se = 0.141, p = 3.00 × 10−2)17. Moreover, our study provides further insights into the shared genetic basis of ADHD and EA by identifying specific genetic loci jointly influencing these phenotypes. Further studies are warranted to determine in what way these genetic variants influence ADHD and EA. It is feasible that the shared genetic effects may influence EA through an intermediary phenotype such as reading disability, which is comorbid to ADHD,25 or through more basic neurobiological systems.
By combining GWAS summary statistics data on ADHD and EA18,20 in the condFDR/conjFDR analyses, we enhanced discovery in the moderately powered ADHD GWAS and found five novel LD-independent loci associated with ADHD (Table 2). None of the loci identified in our analyses reached genome-wide significance in the ADHD GWAS,18 while rs618678 and rs412458 reached genome-wide significance in the GWAS of EA.20 Four of five loci have opposite directions of effect in the PGC case-control ADHD study18 and EA study20 (Table 2) and consistent directions of effect in the independent population-based study of ADHD symptoms from the EAGLE consortium21 (Table S1, available online). The only SNP (rs4477079) having the same direction of effect in the PGC ADHD data set and EA also has inconsistent effect directions in the PGC ADHD and EAGLE ADHD datasets. Despite the relatively small GWAS sample sizes on ADHD by the PGC18 and EAGLE21 consortia, and their differences in definitions of phenotype, observed consistency of effect directions of the identified variants supports the credibility of the findings and the statistical approach. The fact that the majority of identified SNPs had opposite directions of effect in ADHD and EA is in line with the observed negative genetic correlation and corresponds to the expectations that can be drawn from existing clinical studies demonstrating poor academic performance and decreased rates of high school graduation and postsecondary education in individuals with diagnosed ADHD.14 Altogether, these findings provide new insights into the genetic architecture of ADHD, suggesting shared molecular genetic mechanisms with EA. Furthermore, the findings may suggest that individuals with a high load of ADHD genetic risk factors, but not necessarily with the disorder itself, may be at higher risk for lower EA.
The most significant locus shared between ADHD and EA (rs618678) is located on chromosome 1 and represents a broad region of association spanning over more than 200,000 bp in 1p34.2 and 1p34.1 (Figure 4B; Figure S2B, available online). This region contains three protein coding genes: PTPRF, KDM4A and ST3GAL3. rs618678 is an intronic variant within KDM4A, a member of the Jumonji domain 2 family, which encodes a protein that demethylates histone residues, and acts as an epigenetic transcriptional regulator.26 Genome-wide significant variants within KDM4A were reported in a recent GWAS of schizophrenia,5 a disorder that may share a genetic background with ADHD. The protein encoded by PTPRF is a member of the protein tyrosine phosphatase (PTP) family, which regulates a variety of cellular processes, including cell growth, differentiation, mitotic cycle, and oncogenic transformation. Mouse studies showed that PTPRF promotes neurogenesis in the hippocampus,27 a brain region linked to memory. ST3GAL3 encodes a sialyltransferase responsible for the terminal sialylation of brain gangliosides and glycoproteins, which constitute a major part of the surface glycan coat of neurons and glia and act as an interface for cellular interactions.28 Interestingly, mutations of ST3GAL3 may impair the development of higher cognitive functions29 and are associated with severe infantile epilepsy.30 Our eQTL analysis with Braineac database revealed strong associations of rs618678 with altered expression of ST3GAL3 (Table S2, available online), suggesting that this may be a potential mechanism whereby this locus affects ADHD and EA. However, this association was not detected using the GTEx database. The discrepancy between the results from the different eQTL datasets could be attributed to differences in methodological techniques or sample configuration between the eQTL databases, or reflect the relatively small sample sizes. The eQTL results should be re-assessed when larger brain-eQTL databases are available.
The second locus shared between ADHD and EA (rs412458) is an intronic variant within MEF2C (Figure S2A, D, available online), which has multiple LD-linked variants with low condFDR/conjFDR values. MEF2C encodes one of four transcription factors constituting the myocyte enhancer factor 2 (MEF2) family.31 MEF2 is involved in neuronal survival and may regulate the growth and pruning of neurons as well as the number of synapses in the hippocampus, with potential relevance for memory and learning.32 Mutations of MEF2C cause severe mental retardation with stereotypic movements, seizures, and/or cerebral malformations.33 Further, genome-wide significant SNPs within MEF2C have been reported to be associated with schizophrenia,5 which shares polygenic risk with ADHD.34 In addition, mutations in MEF2 genes have been found in patients with different neurological disorders, including Rett-like disorder and Parkinson’s diseases.32 MEF2C expression is particularly enriched in the cerebral cortex35 (Figure S3, available online).
The third locus identified as susceptible for both ADHD and EA by conjFDR is an intronic variant within PINK1 on chromosome 1 (rs17414302). PINK1 encodes a serine/threonine protein kinase that primarily localizes to mitochondria and protects against progressive mitochondrial damage and dysfunction.36 This protein is thought to be involved in regulating neurite morphogenesis, enhancing anterograde mitochondrial transport and density of mitochondria in dendrites and upregulating expression of neuronal differentiation proteins.37 PINK1 is important for the maintenance of mitochondria in part by selective degradation of compromised mitochondria (mitophagy).38 Mutations in this gene are a common cause of autosomal recessive Parkinson’s disease.39 However, rs17414302 represents an isolated signal with rather poor LD support (Figure S2E, available online), and it should thus be examined in more detail.
The strongest SNP association with ADHD revealed by the condFDR analysis was rs4324303. This SNP was not significant in the conjFDR analysis, but showed consistent direction of effect with ADHD symptoms in the EAGLE sample, possibly suggesting a putative role specific to ADHD. Rs4324303 is an intergenic variant located approximately 1 mega base upstream of the nearest protein coding gene (TRIB2). It is therefore difficult to speculate about the potential role of this variant in different cellular processes.
Another variant identified by the condFDR analysis is rs4477079, an intronic variant within RUNX1T1 on chromosome 2. RUNX1T1 acts as a co-repressor of Notch40 and Wnt41 pathways. RUNX1T1 was reported to have high expression levels in adult and fetal brains42 and may influence the axon guidance process.43 RUNX1T1 was previously identified among the top associations (although not reaching genome-wide significance) in the context of oppositional defiant disorder (ODD), which is a frequent psychiatric disorder seen in individuals with ADHD.44 Notably, unlike the other loci identified in our analyses, this locus shows an inconsistent direction of effect between PGC ADHD risk and quantitative measures of ADHD symptoms in pediatric populations (Table S1, available online) and a co-directional effect between PGC ADHD risk and EA (Table 2). The latter is contrary to expectations based on previous findings. The role of RUNX1T thus remains puzzling, and further studies are needed to clarify it.
To further evaluate the ADHD-associated variants identified in this study utilizing the data from PGC ADHD case-control and EA GWASs, we examined our top hits in light of the ADHD symptoms’ GWAS. Four of five loci identified here revealed consistent direction of effect in the independent GWAS of ADHD symptoms (Table S1, available online). Of note, twin studies provide strong evidence that the diagnosis of ADHD can be considered the extreme of a continuous trait,45 and several studies show that the polygenic risk score computed from an association study of ADHD diagnosis predicts the variability of ADHD symptoms in population samples.21,46 Additionally, it has been shown that the continuous measure of ADHD (such as symptom score) and the ADHD diagnosis share over 90% of their genetic background.47 Thus, the results of the performed exploration may be viewed as confirmatory of our findings.
It is also worth mentioning that two loci identified in our analyses (corresponding to rs618678 and rs412458 in Table 2) were reported to reach genome-wide significance in the largest GWAS on ADHD performed to date, with a total number of 20,183 individuals with ADHD and 35,191 controls. In this GWAS, ADHD diagnosis was based on either the International Classification of Diseases-10th Revision (ICD-10) or DSM-IV. The study is yet unpublished, but preprint is available in bioRxiv.47
As children with ADHD have been reported to be at high risk for academic failure, school dropout, grade repetition, and placement in special education,48,49 it is likely that the prevalence of ADHD cases among individuals with lower EA would be increased compared to the prevalence among individuals with higher EA. Moreover, ADHD is known to have a complex pattern of comorbid conditions1 (including dyslexia,25 ODD,50 and others), many of them also associated with lower EA. This potential overlap of phenotypes prevents us from translating the genetic correlation into actual pleiotropy, which is defined as the same gene variant affecting independent diseases or traits. Furthermore, it is challenging to evaluate small effect sizes and speculate about molecular mechanisms behind the effective variants when examining such potentially overlapping phenotypes. Another general problem is that the effects of the associated variants are small, and their functional roles have not been directly investigated. Associated genetic loci contain several genes, and it is difficult to establish an arrow of causality when studying association between traits. Thus, the question of whether ADHD is diagnosed because of observed educational problems or because ADHD is the cause of subsequent educational problems—or there is another common underlying factor—needs further exploration.
Also of possible relevance is the sample overlap between PGC ADHD and EA datasets (both GWASs include the WTCCC58C cohort51), which may inflate the results of our FDR analyses. However, the results of LD score regression, which are in line with those of our FDR analyses, are not affected by the sample overlap.8
We identified five loci associated with ADHD and provided evidence for a shared genetic basis between ADHD and EA, implicating three genetic loci in this overlap. Four of five identified loci showed consistent effects in the independent data set of ADHD symptoms, and inverse correlation with EA, in line with prior epidemiological and genetic studies. Altogether, the findings provide new insights into the relationship between ADHD and EA, suggesting shared molecular genetic mechanisms. On a cautious note, the identified risk variants are not informative clinically due to their small effect sizes. Further research is required to clarify the biological effects of the identified genetic variants and how these may influence EA and ADHD pathogenesis.
Supplementary Material
Lay summary.
Attention-deficit/hyperactivity disorder (ADHD) is a common and heritable condition, but its genetic underpinnings remain largely unknown. We show evidence for shared genetic basis between ADHD and educational attainment and identify five novel ADHD-associated gene loci. The findings may provide an insight into the genetic risk architecture of ADHD and its relation to educational attainment.
Clinical guidance.
There are multiple identifiable genetic risk loci in ADHD with small effect sizes.
The currently identified risk loci cannot be used for clinical diagnostics or treatment guidance.
There is a genetic overlap between ADHD and educational attainment.
Genes shared between ADHD and educational attainment may provide a conceptual framework for understanding why children with ADHD tend to have academic underachievement.
The findings emphasize the need for identifying and helping children with ADHD in the school setting.
Acknowledgments
This work was supported by the Research Council of Norway (248778, 223273, 213694, 248980), the KG Jebsen Stiftelsen (SKGJ MED 008), the National Institutes of Health (R01GM104400), and the European Union’s Horizon 2020 research and innovation programme under grant agreement no. 667302. Dr. Wang was also supported by The Research Council of Norway through a FRIPRO Mobility Grant (contract no 251134). The FRIPRO Mobility grant scheme (FRICON) is co-funded by the European Union’s Seventh Framework Programme for research, technological development and demonstration under Marie Curie grant agreement no 608695. Dr. Faraone has received grant and research support from the K.G. Jebsen Centre for Research on Neuropsychiatric Disorders, the University of Bergen, Bergen, Norway, the European Union’s Seventh Framework Programme for research, technological development, and demonstration, the European Union’s Horizon 2020 research and innovation programme, and the National Institute of Mental Health.
The authors thank the Psychiatric Genetics Consortium (PGC) and the Social Science Genetic Association Consortium (SSGAC) for access to GWAS data. The authors thank Thomas Bjella, PhD, of the Oslo University Hospital & Institute of Clinical Medicine, for support with the database.
Disclosure: Dr. Faraone has received income, potential income, travel expenses, continuing education support, research support from, and/or has served on the advisory boards of/as a consultant to Lundbeck, Rhodes, Arbor, KenPharm, Ironshore, Neurovance, Impact, Takeda, Shire, Akili Interactive Labs, CogCubed, Alcobra, VAYA Pharma, Sunovion, Genomind, and NeuroLifeSciences. In previous years, he has received income or research support from Shire, Neurovance, Alcobra, Otsuka, McNeil, Janssen, Novartis, Pfizer, and Eli Lilly and Co. He has served as editor of the American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. His institution (SUNY) has US patent US20130217707 A1 for the use of sodium hydrogen exchange inhibitors in the treatment of ADHD. He has received royalties from books published by Guilford Press (Straight Talk about Your Child’s Mental Health), Oxford University Press (Schizophrenia: The Facts), and Elsevier (ADHD: Non-Pharmacologic Interventions). He has held stock in CogCubed and Ironshore. He is the principal investigator of http://adhdinadults.com/.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Drs. Shadrin, Smeland, Zayats, Schork, Frei, Bettella, Witoelar, Li, Eriksen, Krull, Djurovic, Reichborn-Kjennerud, Thompson, Johansson, Haavik, Dale, Wang, and Andreassen report no biomedical financial interests or potential conflicts of interest.
This article is discussed in an editorial by Dr. Lauren McGrath on page xx.
Clinical guidance is available at the end of this article.
Supplemental material cited in this article is available online.
Contributor Information
Dr. Alexey A. Shadrin, NORMENT, KG Jebsen Centre for Psychosis Research, Institute of Clinical Medicine, University of Oslo, Oslo, Norway; Division of Mental Health and Addiction, Oslo University Hospital, Oslo, Norway.
Dr. Olav B. Smeland, NORMENT, KG Jebsen Centre for Psychosis Research, Institute of Clinical Medicine, University of Oslo, Oslo, Norway; Division of Mental Health and Addiction, Oslo University Hospital, Oslo, Norway.
Dr. Tetyana Zayats, K.G. Jebsen Centre for Neuropsychiatric Disorders, University of Bergen, Bergen, Norway.
Dr. Andrew J. Schork, University of California, San Diego; Institute of Biological Psychiatry, Medical Health Center, Sct. Hans Hospital and University of Copenhagen, Copenhagen, Denmark.
Dr. Oleksandr Frei, NORMENT, KG Jebsen Centre for Psychosis Research, Institute of Clinical Medicine, University of Oslo, Oslo, Norway; Division of Mental Health and Addiction, Oslo University Hospital, Oslo, Norway.
Dr. Francesco Bettella, NORMENT, KG Jebsen Centre for Psychosis Research, Institute of Clinical Medicine, University of Oslo, Oslo, Norway; Division of Mental Health and Addiction, Oslo University Hospital, Oslo, Norway.
Dr. Aree Witoelar, NORMENT, KG Jebsen Centre for Psychosis Research, Institute of Clinical Medicine, University of Oslo, Oslo, Norway; Division of Mental Health and Addiction, Oslo University Hospital, Oslo, Norway.
Dr. Wen Li, NORMENT, KG Jebsen Centre for Psychosis Research, Institute of Clinical Medicine, University of Oslo, Oslo, Norway; Division of Mental Health and Addiction, Oslo University Hospital, Oslo, Norway.
Dr. Jon A. Eriksen, NORMENT, KG Jebsen Centre for Psychosis Research, Institute of Clinical Medicine, University of Oslo, Oslo, Norway; Division of Mental Health and Addiction, Oslo University Hospital, Oslo, Norway.
Dr. Florian Krull, NORMENT, KG Jebsen Centre for Psychosis Research, Institute of Clinical Medicine, University of Oslo, Oslo, Norway; Division of Mental Health and Addiction, Oslo University Hospital, Oslo, Norway.
Dr. Srdjan Djurovic, Oslo University Hospital, OsloNORMENT, KG Jebsen Centre for Psychosis Research, University of Bergen.
Dr. Stephen V. Faraone, KG Jebsen Centre for Neuropsychiatric Disorders, University of Bergen, SUNY Upstate Medical University, Syracuse, New York.
Dr. Ted Reichborn-Kjennerud, Division of Mental Health, Norwegian Institute of Public Health, Oslo, and Institute of Clinical Medicine, University of Oslo.
Dr. Wesley K. Thompson, University of California, San Diego.
Dr. Stefan Johansson, K.G. Jebsen Centre for Neuropsychiatric Disorders, University of Bergen, Bergen, Norway; Center for Medical Genetics and Molecular Medicine, Haukeland University Hospital, Bergen, Norway.
Dr. Jan Haavik, K.G. Jebsen Centre for Neuropsychiatric Disorders, University of Bergen, Bergen, Norway; Division of Psychiatry, Haukeland University Hospital.
Dr. Anders M. Dale, NORMENT, KG Jebsen Centre for Psychosis Research, Institute of Clinical Medicine, University of Oslo, and University of California, San Diego.
Dr. Yunpeng Wang, NORMENT, KG Jebsen Centre for Psychosis Research, Institute of Clinical Medicine, University of Oslo, Oslo, Norway; Division of Mental Health and Addiction, Oslo University Hospital, Oslo, Norway; University of California, San Diego, La Jolla, CA.
Dr. Ole A. Andreassen, NORMENT, KG Jebsen Centre for Psychosis Research, Institute of Clinical Medicine, University of Oslo, Oslo, Norway; Division of Mental Health and Addiction, Oslo University Hospital, Oslo, Norway.
References
- 1.Faraone SV, Asherson P, Banaschewski T, et al. Attention-deficit/hyperactivity disorder. Nat Rev Dis Primers. 2015;1:15020. doi: 10.1038/nrdp.2015.20. [DOI] [PubMed] [Google Scholar]
- 2.Cross-Disorder Group of the Psychiatric Genomics C. Lee SH, Ripke S, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet. 2013;45:984–994. doi: 10.1038/ng.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Freitag CM, Rohde LA, Lempp T, Romanos M. Phenotypic and measurement influences on heritability estimates in childhood ADHD. Eur Child Adolesc Psychiatry. 2010;19:311–323. doi: 10.1007/s00787-010-0097-5. [DOI] [PubMed] [Google Scholar]
- 4.Martin J, O’Donovan MC, Thapar A, Langley K, Williams N. The relative contribution of common and rare genetic variants to ADHD. Transl Psychiatry. 2015;5:e506. doi: 10.1038/tp.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schizophrenia Working Group of the Psychiatric Genomics C. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bipolar Disorder Working Group PGC. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet. 2011;43:977–983. doi: 10.1038/ng.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Visscher PM, Brown MA, McCarthy MI, Yang J. Five years of GWAS discovery. Am J Hum Genet. 2012;90:7–24. doi: 10.1016/j.ajhg.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bulik-Sullivan B, Finucane HK, Anttila V, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47:1236–1241. doi: 10.1038/ng.3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andreassen OA, Djurovic S, Thompson WK, et al. Improved detection of common variants associated with schizophrenia by leveraging pleiotropy with cardiovascular-disease risk factors. Am J Hum Genet. 2013;92:197–209. doi: 10.1016/j.ajhg.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Desikan RS, Schork AJ, Wang Y, et al. Polygenic Overlap Between C-Reactive Protein, Plasma Lipids, and Alzheimer Disease. Circulation. 2015;131:2061–2069. doi: 10.1161/CIRCULATIONAHA.115.015489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andreassen OA, Thompson WK, Dale AM. Boosting the power of schizophrenia genetics by leveraging new statistical tools. Schizophr Bull. 2014;40:13–17. doi: 10.1093/schbul/sbt168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferrari R, Wang Y, Vandrovcova J, et al. Genetic architecture of sporadic frontotemporal dementia and overlap with Alzheimer’s and Parkinson’s diseases. J Neurol Neurosurg Psychiatry. 2017;88:152–164. doi: 10.1136/jnnp-2016-314411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yokoyama JS, Wang Y, Schork AJ, et al. Association Between Genetic Traits for Immune-Mediated Diseases and Alzheimer Disease. JAMA Neurol. 2016;73:691–697. doi: 10.1001/jamaneurol.2016.0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Voigt RG, Katusic SK, Colligan RC, Killian JM, Weaver AL, Barbaresi WJ. Academic Achievement in Adults with a History of Childhood Attention-Deficit/Hyperactivity Disorder: A Population-Based Prospective Study. J Dev Behav Pediatr. 2017;38:1–11. doi: 10.1097/DBP.0000000000000358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barkley RA. Major life activity and health outcomes associated with attention-deficit/hyperactivity disorder. J Clin Psychiatry. 2002;63(Suppl 12):10–15. [PubMed] [Google Scholar]
- 16.Biederman J, Newcorn J, Sprich S. Comorbidity of attention deficit hyperactivity disorder with conduct, depressive, anxiety, and other disorders. Am J Psychiatry. 1991;148(5):564–577. doi: 10.1176/ajp.148.5.564. [DOI] [PubMed] [Google Scholar]
- 17.Hagenaars SP, Harris SE, Davies G, et al. Shared genetic aetiology between cognitive functions and physical and mental health in UK Biobank (N=112 151) and 24 GWAS consortia. Mol Psychiatry. 2016;21:1624–1632. doi: 10.1038/mp.2015.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neale BM, Medland SE, Ripke S, et al. Meta-analysis of genome-wide association studies of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2010;49:884–897. doi: 10.1016/j.jaac.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zayats T, Jacobsen KK, Kleppe R, et al. Exome chip analyses in adult attention deficit hyperactivity disorder. Transl Psychiatry. 2016;6:e923. doi: 10.1038/tp.2016.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okbay A, Beauchamp JP, Fontana MA, et al. Genome-wide association study identifies 74 loci associated with educational attainment. Nature. 2016;533:539–542. doi: 10.1038/nature17671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Middeldorp CM, Hammerschlag AR, Ouwens KG, et al. A Genome-Wide Association Meta-Analysis of Attention-Deficit/Hyperactivity Disorder Symptoms in Population-Based Pediatric Cohorts. J Am Acad Child Adolesc Psychiatry. 2016;55(10):896–905. e896. doi: 10.1016/j.jaac.2016.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Francesconi M, Lehner B. The effects of genetic variation on gene expression dynamics during development. Nature. 2014;505(7482):208–211. doi: 10.1038/nature12772. [DOI] [PubMed] [Google Scholar]
- 24.Kang HJ, Kawasawa YI, Cheng F, et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Germano E, Gagliano A, Curatolo P. Comorbidity of ADHD and dyslexia. Dev Neuropsychol. 2010;35:475–493. doi: 10.1080/87565641.2010.494748. [DOI] [PubMed] [Google Scholar]
- 26.Guerra-Calderas L, Gonzalez-Barrios R, Herrera LA, Cantu de Leon D, Soto-Reyes E. The role of the histone demethylase KDM4A in cancer. Cancer Genet. 2015;208:215–224. doi: 10.1016/j.cancergen.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 27.Bernabeu R, Yang T, Xie Y, Mehta B, Ma SY, Longo FM. Downregulation of the LAR protein tyrosine phosphatase receptor is associated with increased dentate gyrus neurogenesis and an increased number of granule cell layer neurons. Mol Cell Neurosci. 2006;31:723–738. doi: 10.1016/j.mcn.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 28.Yoo SW, Motari MG, Susuki K, et al. Sialylation regulates brain structure and function. FASEB J. 2015;29(7):3040–3053. doi: 10.1096/fj.15-270983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu H, Eggers K, Chen W, et al. ST3GAL3 mutations impair the development of higher cognitive functions. Am J Hum Genet. 2011;89(3):407–414. doi: 10.1016/j.ajhg.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Edvardson S, Baumann AM, Muhlenhoff M, et al. West syndrome caused by ST3Gal-III deficiency. Epilepsia. 2013;54(2):e24–27. doi: 10.1111/epi.12050. [DOI] [PubMed] [Google Scholar]
- 31.Potthoff MJ, Olson EN. MEF2: a central regulator of diverse developmental programs. Development. 2007;134:4131–4140. doi: 10.1242/dev.008367. [DOI] [PubMed] [Google Scholar]
- 32.Dietrich JB. The MEF2 family and the brain: from molecules to memory. Cell Tissue Res. 2013;352:179–190. doi: 10.1007/s00441-013-1565-2. [DOI] [PubMed] [Google Scholar]
- 33.Le Meur N, Holder-Espinasse M, Jaillard S, et al. MEF2C haploinsufficiency caused by either microdeletion of the 5q14.3 region or mutation is responsible for severe mental retardation with stereotypic movements, epilepsy and/or cerebral malformations. J Med Genet. 2010;47:22–29. doi: 10.1136/jmg.2009.069732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nivard MG, Gage SH, Hottenga JJ, et al. Genetic Overlap Between Schizophrenia and Developmental Psychopathology: Longitudinal and Multivariate Polygenic Risk Prediction of Common Psychiatric Traits During Development. [published online ahead of print Mar 2017] Schizophr Bull. doi: 10.1093/schbul/sbx031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin X, Shah S, Bulleit RF. The expression of MEF2 genes is implicated in CNS neuronal differentiation. Brain Res Mol Brain Res. 1996;42(2):307–316. doi: 10.1016/s0169-328x(96)00135-0. [DOI] [PubMed] [Google Scholar]
- 36.Narendra DP, Jin SM, Tanaka A, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8(1):e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dagda RK, Pien I, Wang R, et al. Beyond the mitochondrion: cytosolic PINK1 remodels dendrites through protein kinase A. J Neurochem. 2014;128(6):864–877. doi: 10.1111/jnc.12494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scarffe LA, Stevens DA, Dawson VL, Dawson TM. Parkin and PINK1: much more than mitophagy. Trends Neurosci. 2014;37(6):315–324. doi: 10.1016/j.tins.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304(5674):1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 40.Salat D, Liefke R, Wiedenmann J, Borggrefe T, Oswald F. ETO, but not leukemogenic fusion protein AML1/ETO, augments RBP-Jkappa/SHARP-mediated repression of notch target genes. Mol Cell Biol. 2008;28(10):3502–3512. doi: 10.1128/MCB.01966-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moore AC, Amann JM, Williams CS, et al. Myeloid translocation gene family members associate with T-cell factors (TCFs) and influence TCF-dependent transcription. Mol Cell Biol. 2008;28(3):977–987. doi: 10.1128/MCB.01242-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wolford JK, Prochazka M. Structure and expression of the human MTG8/ETO gene. Gene. 1998;212(1):103–109. doi: 10.1016/s0378-1119(98)00141-3. [DOI] [PubMed] [Google Scholar]
- 43.Terman JR, Kolodkin AL. Nervy links protein kinase a to plexin-mediated semaphorin repulsion. Science. 2004;303(5661):1204–1207. doi: 10.1126/science.1092121. [DOI] [PubMed] [Google Scholar]
- 44.Aebi M, van Donkelaar MM, Poelmans G, et al. Gene-set and multivariate genome-wide association analysis of oppositional defiant behavior subtypes in attention-deficit/hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet. 2016;171(5):573–588. doi: 10.1002/ajmg.b.32346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Levy F, Hay DA, McStephen M, Wood C, Waldman I. Attention-deficit hyperactivity disorder: a category or a continuum? Genetic analysis of a large-scale twin study. J Am Acad Child Adolesc Psychiatry. 1997;36(6):737–744. doi: 10.1097/00004583-199706000-00009. [DOI] [PubMed] [Google Scholar]
- 46.Groen-Blokhuis MM, Middeldorp CM, Kan KJ, et al. Attention-deficit/hyperactivity disorder polygenic risk scores predict attention problems in a population-based sample of children. J Am Acad Child Adolesc Psychiatry. 2014;53(10):1123–1129. e1126. doi: 10.1016/j.jaac.2014.06.014. [DOI] [PubMed] [Google Scholar]
- 47.Demontis D, Walters RK, Martin J, et al. Discovery Of The First Genome-Wide Significant Risk Loci For ADHD. bioRxiv. 2017 doi: 10.1038/s41588-018-0269-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heiligenstein E, Guenther G, Levy A, Savino F, Fulwiler J. Psychological and academic functioning in college students with attention deficit hyperactivity disorder. J Am Coll Health. 1999;47(4):181–185. doi: 10.1080/07448489909595644. [DOI] [PubMed] [Google Scholar]
- 49.Gray SA, Fettes P, Woltering S, Mawjee K, Tannock R. Symptom Manifestation and Impairments in College Students With ADHD. J Learn Disabil. 2016;49(6):616–630. doi: 10.1177/0022219415576523. [DOI] [PubMed] [Google Scholar]
- 50.Wilens TE, Biederman J, Brown S, et al. Psychiatric comorbidity and functioning in clinically referred preschool children and school-age youths with ADHD. J Am Acad Child Adolesc Psychiatry. 2002;41:262–268. doi: 10.1097/00004583-200203000-00005. [DOI] [PubMed] [Google Scholar]
- 51.Power C, Elliott J. Cohort profile: 1958 British birth cohort (National Child Development Study) Int J Epidemiol. 2006;35:34–41. doi: 10.1093/ije/dyi183. [DOI] [PubMed] [Google Scholar]
- 52.Pruim RJ, Welch RP, Sanna S, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–2337. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.