

Introduction
The atopic march refers to the natural history of allergic diseases as they develop over the course of infancy and childhood. Introduced by the American allergists A.F. Coca and R.A. Cooke in 1923, the term “atopy” became closely associated with the immunoglobulin (Ig) E molecule after its identification as the carrier of hypersensitivity.1 However, in the context of the atopic march, it is important to note that IgE is a pathophysiologic mediator of some, but not all, “atopic” diseases. As such, it is better to consider the atopic march as a progression of allergic conditions that have common genetic and environmental predisposing factors, share the immunologic feature of one or more allergen-specific T helper type 2 (TH2) responses, and are characterized by a “type 2” effector phase that can include generation of specific IgE, activation of granulocytes, and other innate features, such as mucous production and edema. Importantly, the presence of one allergic condition increases the risk for development of others, resulting in the additive feature of the atopic march.
Classically, the atopic march begins with atopic dermatitis (AD), and progresses to IgE-mediated food allergy (FA), asthma, and allergic rhinitis (AR) (Figure 1).2 Each of these are conditions carry a complex pathophysiology involving multiple facets of the immune system. For example, AD was once considered a manifestation of atopy itself, but is now thought to result from a combination of primary skin defect(s) and underlying genetic or environmental propensity to develop type 2 inflammation. While non-type 2 inflammation likely contributes to AD pathophysiology, for the purposes of this review we will focus on the role of type 2 inflammation, as it is the central tenet of the atopic march. Due to the type 2 inflammation associated with AD lesions, AD also represents an important route of allergen exposure by which systemic TH2 responses are initiated. Once an individual has commenced on the atopic march, it is difficult to halt the progression. Here, we start with a clinical vignette and then review the epidemiologic and translational evidence supporting the concept of the atopic march. We briefly review the immunologic mechanisms that are thought to underlie the atopic march, and discuss some of the clinical interventions aimed at preventing or intervening upon the atopic march.
Figure 1.
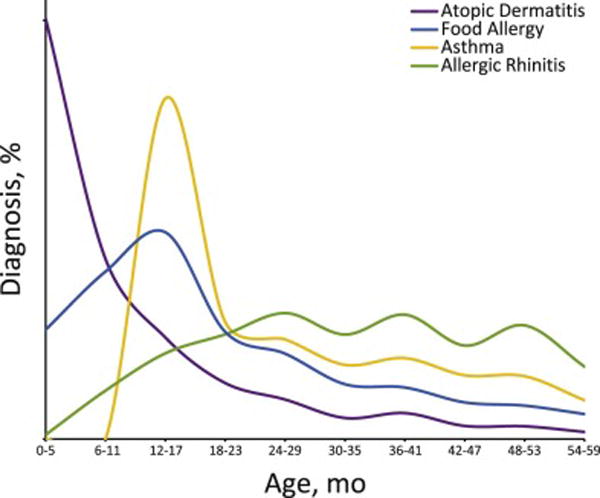
Age at diagnosis of common allergic conditions.
Clinical vignette
A 1-year-old Caucasian boy with a history of moderate AD was referred to an allergist due to concern for food allergy. The patient was exclusively breast fed until one year of age when he developed acute hives, vomiting, and respiratory distress after his first consumption of cow milk. His medical history was otherwise notable for infantile-onset AD that was poorly controlled with intermittent topical corticosteroid use. His family history included AD and asthma in his father, AD in his mother, and AD, FA, and asthma in his older sister. Skin testing confirmed the diagnosis of FA to milk, and the patient was instructed to avoid this food. An alternative AD therapeutic regimen was initiated which included more frequent and higher-dose corticosteroid use, and his skin improved over the following weeks.
The child was followed in allergy clinic and his AD significantly improved by the age of 2 years. That winter, he was admitted to the hospital for respiratory distress in the setting of a viral URI. The following spring, he developed AR and had positive skin testing to trees, grasses, and dust mite. By three years of age, he carried the diagnoses of FA, AR, and moderate persistent asthma requiring combination inhaled corticosteroid and long-acting beta agonist therapy. At four years of age, skin testing to cow milk revealed a wheel and flare of 1 and 2 mm respectively, and serum IgE to milk was 3.45 kU/L. The child subsequently passed a food challenge and introduced cow milk into his diet.
Atopic dermatitis and IgE-mediated Food Allergy: Early members of the atopic march
One of the most common pediatric conditions, AD is thought to affect between 7 and 12% of US children. AD is most commonly diagnosed in the first six months of life—prior to the development of FA, AR, and asthma.2, 3 AD likely results from disruption of the skin barrier due to intrinsic defects of epithelial cells in an individual with a genetic and/or environmental predisposition for type 2 inflammation. Defects in the epidermal barrier protein filaggrin, which are associated with both atopic dermatitis and allergic sensitization, are often cited as a quintessential example of such a barrier defect. However, other examples have now been identified including loss-of-function mutations in SPINK5 and the gene encoding corneodesmosin.4 Importantly, loss-of-function mutations in the filaggrin gene do not increases risk of food or aeroallergen sensitivity independently of AD status.5, 6 Therefore, while intrinsic epithelial barrier dysfunction is central to the pathophysiology of AD, there are other important genetic and/or environmental modifiers that are required for the development of allergic skin inflammation and progression of the atopic march. For example, polymorphisms in the gene encoding thymic stromal lymphopoietin (TSLP) and it’s receptor influence AD risk, FA, and asthma,7–9 while polymorphisms in the genes encoding IL-33 and its receptor are associated with increased risk of AD and asthma.10, 11 Together, these studies and others support the existence of a shared set of polymorphisms which predispose to AD and promote the subsequent development of other allergic conditions.8, 12, 13
Most proximally on the atopic march, the presence and severity of AD positively correlates with risk of developing FA. One recent review estimated that children with AD are as much as six times more likely to develop an FA compared to their healthy peers.14 Food-specific IgE responses can be detected in the first months of life, and peak at around 10% prevalence at 1 year of age.15 The fact that sensitization occurs prior to food ingestion in most cases suggests that sensitization to foods occurs via exposure through inflamed skin, as opposed to the gastrointestinal tract. Additionally, positive correlations have been found between the use of wheat or peanut-containing skin products and the development of wheat or peanut allergy, respectively,16, 17 and exposure to peanut dust in a child’s home positively correlates with likelihood of developing peanut allergy.18 Finally, allergen-specific T cells isolated from peanut-allergic patients express skin-related homing molecules,19, 20 providing additional evidence for the skin being the site of allergen sensitization in FA. Together, these findings support the theory that there is trans-cutaneous sensitization to food allergens in susceptible individuals.
Asthma and allergic rhinitis: Late members of the atopic march
AD is also strongly associated with the development of asthma and AR.21 In contrast to food-specific IgE responses, IgE responses to inhalant allergens develop later in childhood providing a possible explanation for the delayed age of onset for these conditions.15 The relationship between AD and respiratory allergy is influenced by AD severity—while around 20% of children with mild atopic dermatitis develop asthma, over 60% with severe atopic dermatitis develop asthma.21, 22 The presence of AD is also associated with increased asthma severity, and greater asthma persistence into adulthood.23, 24 Similar to FA, loss-of-function mutations in the filaggrin gene correlate with asthma susceptibility and severity in patients with AD, but not in those without AD—indicating that skin inflammation is required for allergic sensitization.5
Despite these data, it is important to note that not every patient with AD develops asthma, and not every patient with asthma has preceding AD. A recent retrospective analysis of two birth cohorts found eight separate patterns of atopic disease progression.25 Though limited by cohort size and parental survey as the reporting method, this study found that 10.5% of respondents followed the traditional pattern of the atopic march, while 15.5% had persistent AD, 5.7% had wheeze without AD, and 9.6% had rhinitis without AD. Together, these findings indicate that the atopic march is not present in all atopic individuals, and in particular those with adult-onset disease.
The presence of FA is also an independent risk factor for the development of AR and asthma. In a retrospective birth cohort study of almost 30,000 children, we found that the presence of FA was associated with development of asthma (odds ratio (OR) 2.16, 95% CI 1.94–2.40), and rhinitis (OR 2.72, 95% CI 2.45–3.03).2 Of the major food allergens, FA to peanut, milk, and egg were significantly associated with the subsequent development of asthma (OR 1.74, 1.38, and 1.60, respectively), and rhinitis (OR 2.59, 1.46, and 1.80, respectively). Additionally, there was an additive effect as patients with multiple food allergies were at increased risk of developing respiratory allergy as compared to patients with a single food allergy. A large meta-analysis of birth cohort studies recently confirmed that early life food sensitization increases risk of wheeze/asthma, eczema, and AR.26 It is important to note that it is difficult to control for confounding effects in these analyses, such as personal history of AD or family history of atopy. In subsequent analysis of our study, we found that FA predisposed to AR and asthma independently of AD status—suggesting additive risk of developing new allergic conditions as one progresses on the atopic march. Additionally, the ability to more accurately predict a patient’s risk of developing respiratory allergy by considering the presence of FA is of considerable clinical utility to the practicing clinician.
Finally, the clinical relationship between asthma and AR is well established, with up to three quarters of asthmatics reporting rhinitis symptoms.27 This association holds after controlling for total IgE, parental history of asthma, and allergen sensitization, suggesting that the coexistence of AR and asthma is not solely due to atopic predisposition. AR is also positively correlated with asthma severity, and AR treatment improves asthma control.28 Together, these observations indicate that the upper and lower airways behave as a physiological and pathophysiological unit, and have led to comprehensive recommendations from the Allergic Rhinitis and its Impact on Asthma (ARIA) guideline panel which emphasize the importance of appropriate treatment of AR in asthmatics.29
Immunologic mechanisms underlying the atopic march
As described previously, allergen exposure through inflamed skin is thought to be the primary route by which individuals initiate on the atopic march (Figure 2). This hypothesis is supported by data from animal models which indicate that trans-cutaneous allergen exposure promotes the development of specific T and B cell responses, and subsequent allergic disease. These models can take the form of genetic disruptions (as is the case with mice deficient in filaggrin), or skin irritation via mechanical or chemical means. It is worth noting that protease allergens (such as those contained in peanut, papaya, mites, insects, fungi, and some pollens) are unique in that they can induce sensitization when exposed to healthy skin, and act as an adjuvant for other allergens. For example, peanut exposure on the skin of mice increases the development of specific IgE responses to milk.30 Once an allergen has entered the skin, it has the opportunity to interact with the immune system. Skin is divided into two immune compartments: the epidermis, which contains predominantly Langerhans cells and CD8+ cytotoxic T lymphocytes, and the dermis, which contains dermal and plasmacytoid dendritic cells (DC), macrophages, mast cells, and innate and adaptive lymphocyte subsets.31
Figure 2.
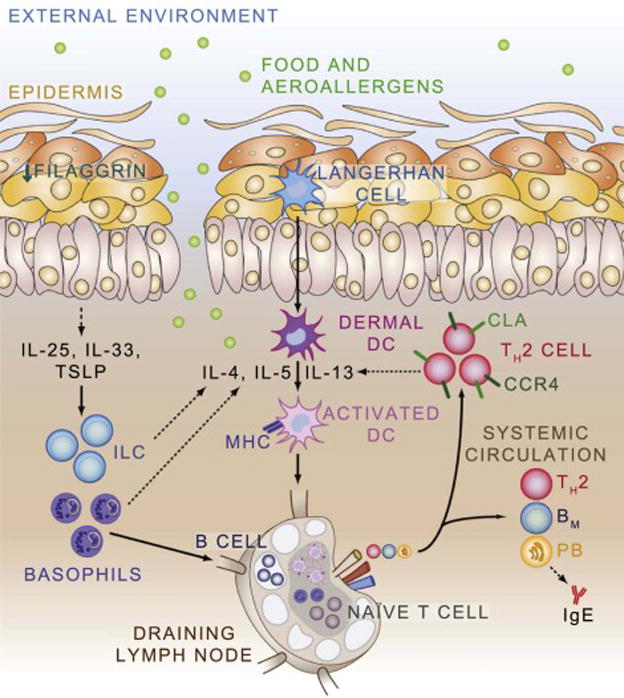
Model of the Atopic March. Deficiencies in epithelial proteins such as filaggrin, in conjunction with atopic predisposition, result in skin inflammation. Epithelial-derived cytokines such as Il-25, Il-33, and TSLP recruit and activate innate cell types including innate lymphoid cells (ILC) and basophils which produce cytokines that promote the activation of dendritic cells. Activated dendritic cells process allergen, upregulate MHC, and circulate to draining lymph nodes where they can interact with naïve T and B cells to promote the development of allergen-specific T and B cell responses. Allergen-specific T helper type 2 (TH2) cells home back to the skin through expression of C-C chemokine receptor type 4 (CCR4), cutaneous lymphocyte antigen (CLA), and other molecules. TH2 cells also enter the systemic circulation where they can exert effector response at distant tissue sites. Memory B cells (BM) recirculate in the blood and lymph, while plasmablasts (PB) home to the bone marrow where they differentiate in to plasma cells and produce allergen-specific IgE.
The inflammation observed in AD is associated with increased production of IL-4, IL-25, IL-33, and TSLP, which recruit IL-5 and IL-13 producing type-2 innate lymphoid cells and contribute to the development of type 2 inflammation.32 DCs and other immune cells migrate from the skin to draining lymph nodes, where they stimulate naive T cells to differentiate into allergen-specific TH2 cells.33 Once allergen-specific TH2 responses are present, they can exert effects systemically. For example, in mice it is established that epicutaneous sensitization can induce allergen-specific IgE and anaphylaxis,34 and allergic inflammation of the lung,35 esophagus,36 and GI tract.37 In the case of esophageal inflammation, the mechanism is IgE-independent, implying that pathogenic TH2 cells migrate from the skin to other tissue sites.
An additional mechanistic question related to the atopic march is whether the presence of one allergen-specific TH2 response potentiates to the development of additional TH2 responses. One mechanism by which this may occur is via a “bystander effect”—where existing inflammation acts as an adjuvant for the development of other TH2 responses. Basophils are one potential contributor to the bystander effect, as they are potent sources of IL-4, are recruited to draining LNs early in the response to infectious or allergic stimuli, and can cooperate with DCs to promote TH2 cell responses.38 Critically, blood basophil numbers in the steady-state, and basophil recruitment to lymph nodes after allergen exposure, are both directly related to serum IgE levels in an antigen-independent manner.39, 40 Thus, elevated IgE levels may potentiate basophil-facilitated TH2 responses both in the skin and at distant tissue sites.41–43 Together, these observations offer one immunologic mechanism by which the presence of a TH2 response to one allergen could potentiate the development of additional TH2 responses.
Clinical strategies aimed at prevention and intervention upon the atopic march
The epidemic increases in prevalence and severity of allergic conditions observed in recent decades are generally thought to have occurred too quickly to be attributed to genetic drift alone. As a result, considerable effort has been devoted to understanding the role of diet, hygiene, infections, allergens, air pollution, and other environmental factors in susceptibility to allergic disease.44 Particular attention has been given to the influence of pathologic or commensal microbial stimuli (and in particular commensal bacteria) on the mammalian immune system.45 Epidemiologic studies were the first to establish an increased risk for asthma and other allergic conditions with childhood antibiotic exposure.46 These associations held after adjusting for infections, and other potential confounders, suggesting detrimental effects of antibiotic exposure on non-pathologic microbes. Subsequent studies in animal models indicated that antibiotic exposure dramatically restructures the intestinal microbiome, and skews the immune system towards type 2 inflammation.39, 47
Clinical associations have also between made with childhood exposure to household pets, livestock, unpasteurized milk, and endotoxins, all of which are generally protective against allergic manifestations. Some of the most compelling recent data supporting a role for microbial exposure in atopic risk come from studies comparing geographically distinct but genetically related pediatric populations. In one study of children in Finland and Russia, a significant, dose-dependent reduction in the risk of atopy was associated with microbial exposure and prevalence of enteroviruses.48 In another study of Amish and Hutterite farm children, prevalence of asthma and allergic sensitization was 4 and 6 times lower in the Amish (who follow more traditional farming practices). This correlated with dust in Amish homes that had 6.8 times more endotoxin, which induced MyD88 and Trif-dependent inhibition of airway hyperreactivity and eosinophilia in a mouse asthma model.49 Given our improved understanding of the influence of microbial factors on allergic sensitization, it is not surprising that multiple studies have now attempted to prevent progression along the atopic march through various diet or probiotic-based interventions aimed at modifying the microbiome of various mucosal sites. However, the data to date are mixed, with some positive and some negative studies.50, 51
Studies have also examined the effects of optimal AD therapy, and immunotherapy, on the atopic march. One recent 3-year double-blind study examined the benefits of pimecrolimus therapy in 3 to 18-month-old patients with recent onset AD.52 No significant differences in the percentage of patients who developed FA, AR, or asthma were detected between the between the treatment and control groups, though baseline AD severity was positively correlated with development atopic comorbidities. Other small trials have shown that routine use of emollients reduces the incidence of AD by approximately half during the therapy period, though it remains to be seen if prophylactic emollients influence risk of allergic sensitization.53 There is also some evidence to suggest that subcutaneous and sublingual immunotherapy may prevent the progression to asthma in high-risk atopic patients.54 For example, one study examined the effect of receiving oral house dust mite (HDM) extract on asthma outcomes in 111 infants at high risk of atopy.55 Oral HDM therapy was well tolerated in the treatment group, but there was no significant preventive effect observed on HDM sensitization or the development of AD, FA, or wheeze. More in-depth investigation of the role of immunotherapy in the prevention of atopic disease is warranted.
Other efforts have attempted to modify the atopic march using prophylactic antihistamines. A large trial of 817 infants with AD aged 1 to 2 years showed no significant difference between treatment with high dose cetirizine or placebo with regard to development of asthma overall.56 However, a subgroup analysis indicated that infants sensitized to dust mite, grass, or both who were treated with cetirizine were significantly less likely to be diagnosed with asthma during the treatment period. However, a follow-up trial undertaken in children with atopic dermatitis and sensitivity to grass pollen and/or house dust mite allergens showed no benefit to levocetirizine therapy in delaying asthma diagnosis.57
Finally, a very promising avenue for disrupting the atopic march stems from the decades of work investigating the immunologic mechanisms and clinical relevance of oral tolerance.58 This work culminated in a randomized trial of peanut consumption in infants at risk for peanut allergy in 2015 that showed significant benefit to early peanut introduction.59 These findings led to new recommendations from the NIAID on how to prevent FA that emphasize facilitating oral peanut exposure prior to trans-cutaneous sensitization in individuals that are at risk for developing FA.60 However, it remains to be seen whether early introduction of peanut or other food allergens influences development of distal atopic conditions such as asthma or allergic rhinitis.
Conclusion
The pronounced global increase in the prevalence and severity of atopic diseases over recent decades is of critical relevance to our population health. The concept of the atopic march has greatly improved our understanding of the pathophysiology of allergic conditions, and led to the development of exciting new therapeutic strategies for the prevention of atopy. Future research should be directed at better understanding environmental and genetic factors that predispose children to atopic conditions (including the role of commensal and pathologic microbes), as well as the fundamental immunologic mechanisms that lead to development of a TH2 response upon initial allergen exposure. Finally, it is important to consider the addition of new diseases to the atopic march, as supported by rigorous epidemiologic and mechanistic evidence. In sum, the atopic march remains a fundamental and well tested concept in the field of allergy that has practical relevance to the practicing Allergist, and has led to exciting new research questions and therapeutic opportunities.
Acknowledgments
DAH is supported by the CHOP NRSA Institutional Training in Pediatric Research Grant (T32 HD043021). JMS is supported by the Stuart Starr Endowed Chair of Pediatrics, The Children’s Hospital of Philadelphia Eosinophilic Esophagitis Fund, a Food Allergy Research & Education, Inc. Clinical Network grant, and the Consortium of Eosinophilic Gastrointestinal Disease Researchers (U54 AI117804).
Abbreviations
- Ig
immunoglobulin
- TH2
T helper type 2
- AD
atopic dermatitis
- FA
food allergy
- AR
allergic rhinitis
- EoE
eosinophilic esophagitis
- TSLP
thymic stromal lymphopoietin
- DC
dendritic cell
- HDM
house dust mite
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure: The authors have no financial relationships or other conflicts of interest relevant to this article to disclose
References
- 1.Cohen S, Dworetzky M, Frick OL. Coca and Cooke on the classification of hypersensitiveness. J Allergy Clin Immunol. 2003;111:205–210. doi: 10.1067/mai.2003.106. [DOI] [PubMed] [Google Scholar]
- 2.Hill DA, Grundmeier RW, Ram G, Spergel JM. The epidemiologic characteristics of healthcare provider-diagnosed eczema, asthma, allergic rhinitis, and food allergy in children: a retrospective cohort study. BMC Pediatr. 2016;16 doi: 10.1186/s12887-016-0673-z. 133-016-5540673-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bloom B, Jones LI, Freeman G. Summary health statistics for U.S. children: National Health Interview Survey, 2012. Vital Health Stat. 2013;10(258):1–81. [PubMed] [Google Scholar]
- 4.Marenholz I, Esparza-Gordillo J, Lee YA. The genetics of the skin barrier in eczema and other allergic disorders. Curr Opin Allergy Clin Immunol. 2015;15:426–434. doi: 10.1097/ACI.0000000000000194. [DOI] [PubMed] [Google Scholar]
- 5.Rogers AJ, Celedon JC, Lasky-Su JA, Weiss ST, Raby BA. Filaggrin mutations confer susceptibility to atopic dermatitis but not to asthma. J Allergy Clin Immunol. 2007;120:1332–1337. doi: 10.1016/j.jaci.2007.09.037. [DOI] [PubMed] [Google Scholar]
- 6.Thyssen JP, Tang L, Husemoen LL, et al. Filaggrin gene mutations are not associated with food and aeroallergen sensitization without concomitant atopic dermatitis in adults. J Allergy Clin Immunol. 2015;135:1375–8.e1. doi: 10.1016/j.jaci.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 7.Harada M, Hirota T, Jodo AI, et al. Thymic stromal lymphopoietin gene promoter polymorphisms are associated with susceptibility to bronchial asthma. Am J Respir Cell Mol Biol. 2011;44:787–793. doi: 10.1165/rcmb.2009-0418OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirota T, Nakayama T, Sato S, et al. Association study of childhood food allergy with GWAS-discovered loci of atopic dermatitis and eosinophilic esophagitis. J Allergy Clin Immunol. 2017 doi: 10.1016/j.jaci.2017.05.034. [DOI] [PubMed] [Google Scholar]
- 9.Margolis DJ, Kim B, Apter AJ, et al. Thymic stromal lymphopoietin variation, filaggrin loss of function, and the persistence of atopic dermatitis. JAMA Dermatol. 2014;150:254–259. doi: 10.1001/jamadermatol.2013.7954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shimizu M, Matsuda A, Yanagisawa K, et al. Functional SNPs in the distal promoter of the ST2 gene are associated with atopic dermatitis. Hum Mol Genet. 2005;14:2919–2927. doi: 10.1093/hmg/ddi323. [DOI] [PubMed] [Google Scholar]
- 11.Savenije OE, Mahachie John JM, Granell R, et al. Association of IL33-IL-1 receptor-like 1 (IL1RL1) pathway polymorphisms with wheezing phenotypes and asthma in childhood. J Allergy Clin Immunol. 2014;134:170–177. doi: 10.1016/j.jaci.2013.12.1080. [DOI] [PubMed] [Google Scholar]
- 12.Weidinger S, Willis-Owen SA, Kamatani Y, et al. A genome-wide association study of atopic dermatitis identifies loci with overlapping effects on asthma and psoriasis. Hum Mol Genet. 2013;22:4841–4856. doi: 10.1093/hmg/ddt317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marenholz I, Esparza-Gordillo J, Ruschendorf F, et al. Meta-analysis identifies seven susceptibility loci involved in the atopic march. Nat Commun. 2015;6:8804. doi: 10.1038/ncomms9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsakok T, Marrs T, Mohsin M, et al. Does atopic dermatitis cause food allergy? A systematic review. J Allergy Clin Immunol. 2016;137:1071–1078. doi: 10.1016/j.jaci.2015.10.049. [DOI] [PubMed] [Google Scholar]
- 15.Kulig M, Bergmann R, Klettke U, Wahn V, Tacke U, Wahn U. Natural course of sensitization to food and inhalant allergens during the first 6 years of life. J Allergy Clin Immunol. 1999;103:1173–1179. doi: 10.1016/s0091-6749(99)70195-8. [DOI] [PubMed] [Google Scholar]
- 16.Lack G, Fox D, Northstone K, Golding J, Avon Longitudinal Study of Parents and Children Study Team Factors associated with the development of peanut allergy in childhood. N Engl J Med. 2003;348:977–985. doi: 10.1056/NEJMoa013536. [DOI] [PubMed] [Google Scholar]
- 17.Fukutomi Y, Taniguchi M, Nakamura H, Akiyama K. Epidemiological link between wheat allergy and exposure to hydrolyzed wheat protein in facial soap. Allergy. 2014;69:1405–1411. doi: 10.1111/all.12481. [DOI] [PubMed] [Google Scholar]
- 18.Brough HA, Liu AH, Sicherer S, et al. Atopic dermatitis increases the effect of exposure to peanut antigen in dust on peanut sensitization and likely peanut allergy. J Allergy Clin Immunol. 2015;135:164–170. doi: 10.1016/j.jaci.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DeLong JH, Simpson KH, Wambre E, James EA, Robinson D, Kwok WW. Ara h 1-reactive T cells in individuals with peanut allergy. J Allergy Clin Immunol. 2011;127:1211–8.e3. doi: 10.1016/j.jaci.2011.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan SM, Turcanu V, Stephens AC, Fox AT, Grieve AP, Lack G. Cutaneous lymphocyte antigen and alpha4beta7 T-lymphocyte responses are associated with peanut allergy and tolerance in children. Allergy. 2012;67:336–342. doi: 10.1111/j.1398-9995.2011.02765.x. [DOI] [PubMed] [Google Scholar]
- 21.Gustafsson D, Sjoberg O, Foucard T. Development of allergies and asthma in infants and young children with atopic dermatitis–a prospective follow-up to 7 years of age. Allergy. 2000;55:240–245. doi: 10.1034/j.1398-9995.2000.00391.x. [DOI] [PubMed] [Google Scholar]
- 22.Pearce N, Ait-Khaled N, Beasley R, et al. Worldwide trends in the prevalence of asthma symptoms: phase III of the International Study of Asthma and Allergies in Childhood (ISAAC) Thorax. 2007;62:758–766. doi: 10.1136/thx.2006.070169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Strachan DP, Butland BK, Anderson HR. Incidence and prognosis of asthma and wheezing illness from early childhood to age 33 in a national British cohort. BMJ. 1996;312:1195–1199. doi: 10.1136/bmj.312.7040.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinez FD, Wright AL, Taussig LM, Holberg CJ, Halonen M, Morgan WJ. Asthma and wheezing in the first six years of life. The Group Health Medical Associates. N Engl J Med. 1995;332:133–138. doi: 10.1056/NEJM199501193320301. [DOI] [PubMed] [Google Scholar]
- 25.Belgrave DC, Granell R, Simpson A, et al. Developmental profiles of eczema, wheeze, and rhinitis: two population-based birth cohort studies. PLoS Med. 2014;11:e1001748. doi: 10.1371/journal.pmed.1001748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alduraywish SA, Lodge CJ, Campbell B, et al. The march from early life food sensitization to allergic disease: a systematic review and meta-analyses of birth cohort studies. Allergy. 2016;71:77–89. doi: 10.1111/all.12784. [DOI] [PubMed] [Google Scholar]
- 27.Leynaert B, Neukirch C, Kony S, et al. Association between asthma and rhinitis according to atopic sensitization in a population-based study. J Allergy Clin Immunol. 2004;113:86–93. doi: 10.1016/j.jaci.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 28.Ponte EV, Franco R, Nascimento HF, et al. Lack of control of severe asthma is associated with co-existence of moderate-to-severe rhinitis. Allergy. 2008;63:564–569. doi: 10.1111/j.1398-9995.2007.01624.x. [DOI] [PubMed] [Google Scholar]
- 29.Camargos P, Ibiapina C, Lasmar L, Cruz AA. Obtaining concomitant control of allergic rhinitis and asthma with a nasally inhaled corticosteroid. Allergy. 2007;62:310–316. doi: 10.1111/j.1398-9995.2007.01241.x. [DOI] [PubMed] [Google Scholar]
- 30.Brozek JL, Bousquet J, Agache I, et al. Allergic Rhinitis and its Impact on Asthma (ARIA) guidelines-2016 revision. J Allergy Clin Immunol. 2017 doi: 10.1016/j.jaci.2017.03.050. [DOI] [PubMed] [Google Scholar]
- 31.Tordesillas L, Goswami R, Benede S, et al. Skin exposure promotes a Th2-dependent sensitization to peanut allergens. J Clin Invest. 2014;124:4965–4975. doi: 10.1172/JCI75660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kashem SW, Haniffa M, Kaplan DH. Antigen-Presenting Cells in the Skin. Annu Rev Immunol. 2017;35:469–499. doi: 10.1146/annurev-immunol-051116-052215. [DOI] [PubMed] [Google Scholar]
- 33.Tait Wojno ED, Artis D. Emerging concepts and future challenges in innate lymphoid cell biology. J Exp Med. 2016;213:2229–2248. doi: 10.1084/jem.20160525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Worbs T, Hammerschmidt SI, Forster R. Dendritic cell migration in health and disease. Nat Rev Immunol. 2017;17:30–48. doi: 10.1038/nri.2016.116. [DOI] [PubMed] [Google Scholar]
- 35.Galand C, Leyva-Castillo JM, Yoon J, et al. IL-33 promotes food anaphylaxis in epicutaneously sensitized mice by targeting mast cells. J Allergy Clin Immunol. 2016;138:1356–1366. doi: 10.1016/j.jaci.2016.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spergel JM, Mizoguchi E, Brewer JP, Martin TR, Bhan AK, Geha RS. Epicutaneous sensitization with protein antigen induces localized allergic dermatitis and hyperresponsiveness to methacholine after single exposure to aerosolized antigen in mice. J Clin Invest. 1998;101:1614–1622. doi: 10.1172/JCI1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Noti M, Wojno ED, Kim BS, et al. Thymic stromal lymphopoietin-elicited basophil responses promote eosinophilic esophagitis. Nat Med. 2013;19:1005–1013. doi: 10.1038/nm.3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Han H, Roan F, Johnston LK, Smith DE, Bryce PJ, Ziegler SF. IL-33 promotes gastrointestianl allergy in a TSLP-independent manner. Mucosal Immunol. 2017 doi: 10.1038/mi.2017.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Siracusa MC, Kim BS, Spergel JM, Artis D. Basophils and allergic inflammation. J Allergy Clin Immunol. 2013;132:789–801. doi: 10.1016/j.jaci.2013.07.046. quiz 788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hill DA, Siracusa MC, Abt MC, et al. Commensal bacteria-derived signals regulate basophil hematopoiesis and allergic inflammation. Nat Med. 2012;18:538–546. doi: 10.1038/nm.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hill DA, Siracusa MC, Ruymann KR, Tait Wojno ED, Artis D, Spergel JM. Omalizumab therapy is associated with reduced circulating basophil populations in asthmatic children. Allergy. 2014;69:674–677. doi: 10.1111/all.12375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim BS, Wang K, Siracusa MC, et al. Basophils promote innate lymphoid cell responses in inflamed skin. J Immunol. 2014;193:3717–3725. doi: 10.4049/jimmunol.1401307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hussain M, Borcard L, Walsh KP, et al. Basophil-derived IL-4 promotes epicutaneous antigen sensitization concomitant with the development of food allergy. J Allergy Clin Immunol. 2017 doi: 10.1016/j.jaci.2017.02.035. [DOI] [PubMed] [Google Scholar]
- 44.Venturelli N, Lexmond WS, Ohsaki A, et al. Allergic skin sensitization promotes eosinophilic esophagitis through the IL-33-basophil axis in mice. J Allergy Clin Immunol. 2016;138:1367–1380.e5. doi: 10.1016/j.jaci.2016.02.034. [DOI] [PubMed] [Google Scholar]
- 45.Platts-Mills TA. The allergy epidemics: 1870–2010. J Allergy Clin Immunol. 2015;136:3–13. doi: 10.1016/j.jaci.2015.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hill DA, Artis D. Intestinal bacteria and the regulation of immune cell homeostasis. Annu Rev Immunol. 2010;28:623–667. doi: 10.1146/annurev-immunol-030409-101330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kummeling I, Stelma FF, Dagnelie PC, et al. Early life exposure to antibiotics and the subsequent development of eczema, wheeze, and allergic sensitization in the first 2 years of life: the KOALA Birth Cohort Study. Pediatrics. 2007;119:e225–31. doi: 10.1542/peds.2006-0896. [DOI] [PubMed] [Google Scholar]
- 48.Hill DA, Hoffmann C, Abt MC, et al. Metagenomic analyses reveal antibiotic-induced temporal and spatial changes in intestinal microbiota with associated alterations in immune cell homeostasis. Mucosal Immunol. 2010;3:148–158. doi: 10.1038/mi.2009.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seiskari T, Kondrashova A, Viskari H, et al. Allergic sensitization and microbial load–a comparison between Finland and Russian Karelia. Clin Exp Immunol. 2007;148:47–52. doi: 10.1111/j.1365-2249.2007.03333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stein MM, Hrusch CL, Gozdz J, et al. Innate Immunity and Asthma Risk in Amish and Hutterite Farm Children. N Engl J Med. 2016;375:411–421. doi: 10.1056/NEJMoa1508749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Friedman NJ, Zeiger RS. The role of breast-feeding in the development of allergies and asthma. J Allergy Clin Immunol. 2005;115:1238–1248. doi: 10.1016/j.jaci.2005.01.069. [DOI] [PubMed] [Google Scholar]
- 52.Simpson MR, Dotterud CK, Storro O, Johnsen R, Oien T. Perinatal probiotic supplementation in the prevention of allergy related disease: 6 year follow up of a randomised controlled trial. BMC Dermatol. 2015;15:13-015-0030-1. doi: 10.1186/s12895-015-0030-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schneider L, Hanifin J, Boguniewicz M, et al. Study of the Atopic March: Development of Atopic Comorbidities. Pediatr Dermatol. 2016;33:388–398. doi: 10.1111/pde.12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lowe AJ, Su JC, Allen KJ, et al. A randomised trial of a barrier lipid replacement strategy for the prevention of atopic dermatitis and allergic sensitisation: The PEBBLES Pilot Study. Br J Dermatol. 2017 doi: 10.1111/bjd.15747. [DOI] [PubMed] [Google Scholar]
- 55.Zolkipli Z, Roberts G, Cornelius V, et al. Randomized controlled trial of primary prevention of atopy using house dust mite allergen oral immunotherapy in early childhood. J Allergy Clin Immunol. 2015;136:1541–7. e1–11. doi: 10.1016/j.jaci.2015.04.045. [DOI] [PubMed] [Google Scholar]
- 56.Warner JO, ETAC Study Group Early Treatment of the Atopic Child. A double-blinded, randomized, placebo-controlled trial of cetirizine in preventing the onset of asthma in children with atopic dermatitis: 18 months’ treatment and 18 months’ posttreatment follow-up. J Allergy Clin Immunol. 2001;108:929–937. doi: 10.1067/mai.2001.120015. [DOI] [PubMed] [Google Scholar]
- 57.Simons FE, Early Prevention of Asthma in Atopic Children (EPAAC) Study Group Safety of levocetirizine treatment in young atopic children: An 18-month study. Pediatr Allergy Immunol. 2007;18:535–542. doi: 10.1111/j.1399-3038.2007.00558.x. [DOI] [PubMed] [Google Scholar]
- 58.Sicherer SH, Sampson HA. Food allergy: recent advances in pathophysiology and treatment. Annu Rev Med. 2009;60:261–277. doi: 10.1146/annurev.med.60.042407.205711. [DOI] [PubMed] [Google Scholar]
- 59.Du Toit G, Roberts G, Sayre PH, et al. Randomized trial of peanut consumption in infants at risk for peanut allergy. N Engl J Med. 2015;372:803–813. doi: 10.1056/NEJMoa1414850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fleischer DM, Sicherer S, Greenhawt M, et al. Consensus communication on early peanut introduction and the prevention of peanut allergy in high-risk infants. Ann Allergy Asthma Immunol. 2015;115:87–90. doi: 10.1016/j.anai.2015.06.001. [DOI] [PubMed] [Google Scholar]