

Summary
Off-target effects (OTE) present a significant caveat of RNA interference (RNAi) caused by substantial complementarity between small interfering (si)RNAs and unintended mRNAs. We now discuss the existence of three types of seed dependent (s)OTEs: Type I involves unintended targeting through the guide strand seed of a siRNA. Type II is caused by the activity of the seed on the designated siRNA passenger strand when loaded into the RISC. Both type I and II sOTE will elicit unpredictable cellular responses. In contrast, during sOTE type III the guide strand seed preferentially targets essential survival genes resulting in Death Induced by Survival gene Elimination (DISE). In this review, we discuss DISE as a consequence of RNAi that may preferentially affect cancer cells.
Keywords: RNAi, Fas, cancer, cell death, DISE, OTE
Emerging New Concepts to Fight Cancer using Mechanisms Developed during Evolution
Cancer is a decimating malady that afflicts people across the globe. Based on WHO estimates, 8.8 million people died of cancer in 2015i. Most societies face a rising number of cancer incidences and a concurrent explosion in treatment costs. Moreover, the current trend of targeted therapy has failed to produce cures. Instead, most of these drugs merely extend patients' lifespans a few months. Today's reality starkly contrasts with the future imagined in 2003 by Andrew von Eschenbach, then head of the National Cancer Institute, when he set the ambitious goal to “eliminate suffering and death” due to cancer by the year 2015ii.
Radically novel approaches are therefore needed to effectively kill cancer cells and significantly extend patients' lives. One strategy is to identify and exploit evolutionary mechanisms developed by nature that have been repressing cancer in countless generations of multicellular organisms and thereby allowed our phylogenic tree to continue. The immune system represents one such important evolutionary mechanism that eliminates cancer in our bodies by recognizing abnormal/neoplastic cells expressing mutated peptides (tumor antigens), which result from DNA mutations. Indeed, recent successes in immune checkpoint therapy are expected to soon have an effect on the survival statistics of cancer. However, only a minority of patients, and only with certain cancers, currently benefit from immune checkpoint therapy [1]. Moreover, artificially de-repressing the immune system is not without side effects, which may include colitis, endocrinopathies, and liver toxicity [2].
The success of immune therapy in certain cancer patients exemplifies the power of utilizing natural, evolutionarily developed mechanisms, to specifically eliminate cancer cells in multicellular organisms. However, while the adaptive immune system developed about 500 million years ago [3], multicellular organisms may be as old as 2.1 billion years [4]. Recently, cancer was detected in hydra, a 600 million year old living fossil without an adaptive immune system [5]. Signaling pathways deregulated in human ovarian cancer cells are also deregulated in the cancer nodules found in this ancient organism, suggesting that cancer predates the immune system. A system much older than the immune system is the mechanism of RNA interference (RNAi). The components of the RNAi machinery are found in every known eukaryotic organism [6]. In this review we will discuss a main caveat of using RNAi, the phenomenon of off-target effect (OTE), in particular seed dependent OTEs (sOTE). As with any OTE, the biological response of cells would be expected to be unpredictable. We will review three different forms of sOTE and discuss how one of them causes a distinct biological response: simultaneous activation of multiple cell death pathways. Finally, we will discuss how this form of sOTE could be developed into a novel way to treat cancer.
RNA Interference
The RNAi machinery processes double-stranded RNAs into small RNAs that mediate the repression of partially complementary genes [7, 8]. In higher vertebrates, RNAi has been shown to have antitumor activity, mostly in the form of noncoding RNAs such as micro(mi)RNAs [9]. Mature miRNAs are small 19-22 nucleotide long noncoding RNAs generated in the nucleus from longer precursor primary miRNAs. These primary miRNAs are processed by the microprocessor complex Drosha/DGCR8 and exported into the cytosol by exportin 5 as smaller stem loop intermediates called pre-miRNAs. Dicer/TRBP then cleaves the pre-miRNAs, and the mature miRNAs are loaded into the RNA-induced silencing complex (RISC) [10, 11]. While in all cases, a double-stranded RNA duplex is required for RISC loading aided by Dicer, either one or both strands of a miRNA can be loaded into the RISC (either the 3p or the 5p arm of the pre-miR) and serve as the guide RNA that determines the specificity of the targeting. The strand that is not loaded into the RISC (the passenger strand) gets degraded [12]. Once in the RISC, the single-stranded miRNA guide strand then regulates expression of certain genes by targeting in most cases the 3′UTR of mRNAs, resulting in either degradation of the mRNA or translational silencing through various well-studied mechanisms [13, 14]. The extent of reverse complementarity determines the level of interaction between a miRNA and its mRNA target. Targeting specificity and mode of silencing is mostly determined by positions 2-7/8 (the seed sequence) at the 5′ end of the miRNA guide strand and auxiliary complementarity at the 3′ end [15].
Small interfering RNAs (siRNAs) act through a similar mechanism as miRNAs (Fig. 1). Endogenous noncoding small interfering RNAs (endo-siRNAs) are abundantly expressed in invertebrates and have been extensively studied in C. elegans and Drosophila [16, 17]. Mammalian cells lack an RNA dependent RNA polymerase believed to be required to amplify endo-siRNAs to a copy number high enough to mount a significant RNAi response. Nevertheless, endo-siRNAs in mammals have been convincingly described in embryonic stem cells, oocytes, and male germ cells [18-21]. In these cells, many of the endogenous siRNAs are derived from transposable elements like short interspersed elements (SINEs). More recent data point toward the existence of endo-siRNAs at low concentrations in human cancer cells [22]. mRNA-derived endo-siRNAs are unlikely to play a major role in somatic cells. However, while non-canonical sources of small RNAs such as fragments derived from highly abundant cellular RNAs (i.e. snoRNAs, tRNAs, rRNAs, mRNAs, or introns) have been described to bind Argonaute proteins [23-25], their potential RNAi activities have not been sufficiently explored. While most somatic mammalian cells are not believed to use endogenous siRNAs for gene regulation, all cells express the components needed for efficient RNAi believed to be mainly required for miRNA processing and function. This allowed for the technological development of siRNAs or small hairpin (sh)RNAs to induce efficient gene silencing.
Figure 1. Seed dependent RNAi off-target effects (sOTE).
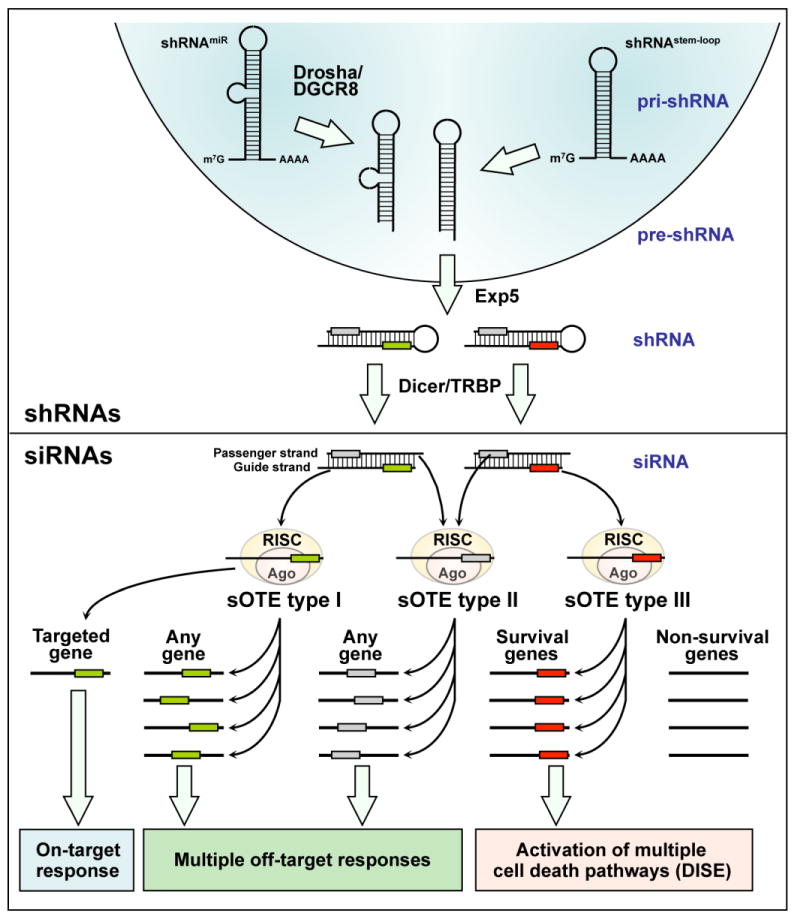
shRNAs, introduced most often in the form of a lentiviral vector and inserted into the genome, are transcribed by either RNA Pol II or III. They can either be simple stem loop shRNAs (shRNAstem-loop) or miRNA-based next generation shRNAs (shRNAmiR), which require Drosha/DGCR8 processing before they are exported into the cytosol by exportin 5 (Exp5). Each shRNA contains a sense/passenger strand and a complimentary antisense/guide strand. Dicer/TRBP trims these duplexes, thereby removing the loop region. Both trimmed shRNAs and exogenous and endogenous siRNAs are loaded into the RNA induced silencing complex (RISC) with their Argonaute proteins (Ago). Similar to miRNAs, positions 2-7 (the seed sequence) of the loaded guide strand greatly determines what target mRNA is silenced through complementarity of the seed sequence, mostly to the 3′UTR of the targeted mRNAs. In addition to the silencing of the mRNA that the siRNA was designed to target, siRNAs show three types of seed dependent off target effects (sOTE). sOTE type I: The guide strand gets properly loaded into the RISC but its seed sequence (green box) targets other genes expressed in treated cells that have complementarity to the siRNA seed. Because this type of sOTE can affect any type of gene, the resulting cellular responses are multiple. sOTE type II: the passenger strand of the siRNA, rather than the intended guide strand, is loaded into the RISC. Position 2-7/8 at the 5′ end of the passenger (now guide) strand (grey box) becomes the seed sequence, and targeting of mRNAs that show complementarity to the passenger strand seed sequence again results in multiple cellular responses. sOTE type III: Certain seed sequences in the guide strand of specialized RNAs (red box) preferentially target seed matches present in the 3′UTR of survival genes and to a lesser extent non-survival genes. This sOTE results in the cell death of the treated cells called DISE.
RNAi Off-Target Effects
It is widely accepted that miRNAs target networks of genes, regulating complex cellular processes including development, cell differentiation, cell migration, and cell death [26]. Although promiscuous targeting is essential for endogenous miRNA function, the use of RNAi technology to study single gene functions hinges on selective gene silencing and repression of off-target effects (OTE) [27]. Additionally, RNAi technology suffers from other nonspecific features including general toxicity due to the presence of toxic sequence motifs [27, 28], poisoning/saturating of the RISC, or evocation of an interferon response [29]. There are a number of rules aimed at designing si/shRNAs that repress these nonspecific effects that may, otherwise, confound the study of single gene function [30]. Great progress has been made to modify siRNAs in a way that increases their uptake, stability, and at the same time increases targeting specificity. Both siRNAs and shRNAs have successfully been used in genome-wide studies to identify genes with specific properties [31-35].
While the goal is always to reduce OTE (both sequence dependent and independent) when designing reagents for RNAi, there are multiple forms of OTE that are not well differentiated, and often cannot be avoided. Certain forms of OTE are caused by a near perfect reverse complementarity between the siRNA and unintended targets. The chance of this happening is usually somewhat minimized by aligning the guide and/or the passenger strand of the targeting RNA to the human transcriptome (e.g. using BLAST) to predict if other complementary sequences may also be targeted. However, similar to miRNAs, a large part of the activity of a siRNA is determined by its seed sequence which, with 6-7 nts, is too short to predict potential crossreactivity with unintended targets. In the following, we will discuss three forms of seed dependent (s)OTE (Fig. 1):
-
Type I sOTE: Cross-reactivity of the si/shRNA with unintended targets in the genome due to sequences complementarity to the guide strand seed.
For a si/shRNA to target the mRNA it is designed to, the antisense/guide strand of the si/shRNA with its 5′ seed sequence, rather than the sense/passenger strand, must be predominantly loaded into the RISC. However, even when the antisense/guide strand is preferentially loaded, OTEs may still be observed. For this to happen, a minimal amount of sequence similarity, particularly in the seed sequence between the siRNAs and mRNAs in the genome is sufficient. Such complementarity can result in any level of knockdown correlating with the degree and location of complementarity between the si/shRNA and the mRNA sequence [36]. When this type of sOTE occurs, various unintended phenotypes may result. Thus, multiple non-overlapping si/shRNAs against the same mRNA are employed to confirm the specificity of the phenotypic effect of knockdown [37].
-
Type II sOTE: Cross-reactivity of the si/shRNA with unintended targets in the genome due to sequence complementarity to the passenger strand seed.
Loading of the designated passenger strand into the RISC can also result in the targeting of unintended genes, mediated by the differing seed sequence (positions 2-7/8) in the passenger strand. A number of rules have been established to design siRNAs that selectively silence the mRNA through loading of the guide strand. Several nucleotide sequence properties have been identified that can influence the preferential loading of the guide strand over the passenger strand into the RISC: (1) an A or U at the first position of the guide strand, (2) a C or G at the first position of the passenger strand [38] (to introduce a thermodynamic asymmetry), and (3) a low GC content in positions 2-7 of the seed sequence [39]. This positional composition biases the opening of the RNA duplex from the end with the preferred seed sequence. Such unwinding is important in strand selection. Phosphorylation of the 5′ end of the guide strand [40] and a balanced GC content in positions 8-16 [41] also biases loading of the desired guide strand. First generation siRNAs all had 2 nt 3′ overhangs mimicking Dicer cleavage products [42]. However, more recent siRNA structures allow Dicer to process the siRNA, allowing a more specific cleavage of the double-stranded RNA, facilitating loading to the RISC, in turn resulting in higher potency. Examples are the 27nt Dicer substrate short interfering RNAs (DsiRNA, [43]) or the 25 nt stealth siRNAs with no overhangs [44]. Virtually all commercially available artificial siRNAs are chemically modified to increase their stability and specificity [45].
In the case of shRNAs both sOTE type I and type II can be further intensified by imprecise cleavage of the stem loop precursor resulting in shifted potential seed sequences in both the passenger and guide strand. Many widely-used shRNAs are based on the first-generation stem loop shRNA platform (i.e. the TRC library). These shRNAs have been found to be prone to OTEs due to imprecise Dicer cleavage [46]. Second and third generation shRNAs that are based on miR-30a or miR-16-2, largely reduced the imprecision of the processing machinery [47] and have been shown to significantly reduce OTE activity [48].
-
Type III sOTE - DISE - A specific form of a seed dependent OTE affecting hundreds of survival genes.
Treatment of cancer cells with siRNAs, DsiRNAs or shRNAs derived from either the death receptor CD95 or its ligand CD95L induced a form of cell death: DISE (for death induced by survival gene elimination) [49]. DISE is a unique form of OTE we call sOTE type III (Fig. 1). DISE-inducing si/shRNAs kill cancer cells by preferentially targeting the 3′UTRs of a set of survival genes through a mechanism similar to miRNAs. It involves the simultaneous activation of multiple death pathways thus it cannot be blocked by any drug or by knockdown of any single gene [50]. Expression of either a CD95L-derived shRNA (shL3) or a CD95-derived shRNA (shR6) induced DISE in immortalized normal ovarian fibroblasts more efficiently than in matching non-immortalized cells [50], suggesting this form of cell death preferentially affects transformed cells. In addition, both of these shRNAs preferentially killed cancer cells with increased stemness [51]. A screen testing more than 4600 shRNAs identified numerous toxic RNAi active sequences present in the mRNAs of CD95 and CD95L [49]. Interestingly, deletion of the si/shRNA target sites by using CRISPR/Cas9 did not reduce death, and >80% of tested commercially available si- or shRNAs killed cancer cells even in the absence of the mRNA target gene expression, establishing DISE as the result of a sOTE. A number of previously described properties of OTE apply to DISE:
-
Some of the off-target-mediated silencing only requires a seed sequence of complementarity [52-54] similar to what has been described for miRNAs recognizing their targets.
In fact, most of the toxic activity of DISE-inducing siRNAs comes from the seed sequence of the guide strand [49]. As little as 6 nucleotides can determine whether a si/shRNA kills cancer cells through DISE or not.
-
OTE activity is mediated through the 3′UTR of target genes and not the 5′UTRs or ORFs [53].
This was established for a CD95L-derived and a CD95-derived DISE-inducing shRNA for which survival gene targeting occurred exclusively in the 3′UTR and not in the ORF [49].
-
Affected off-target transcripts tend to contain longer than average 3′UTRs [54].
While the 3′UTRs of the non-survival genes in general were longer than the 3′UTRs of survival genes (2440 versus 1799 nts, respectively, p=0.00001), it was preferentially the survival genes with longer 3′UTRs that were most downregulated in cells treated with DISE-inducing si/shRNAs (unpublished data).
-
Like miRNA targets, siRNA off-target transcripts tend to be highly expressed relative to a background set [54].
The survival genes targeted by DISE were more highly expressed than genes in a control group not required for survival (unpublished observation). Fisher's exact test showed that the more highly expressed survival genes were downregulated, whereas the non-survival control set genes were not despite the presence of seed matches in both sets [49].
In a study that included the 3′UTR of all human genes, no predictions could be made about which genes would be affected by sOTE [53]. By just considering the seed sequence false positive and false negative rates were at >99% and ∼93%, respectively. This would suggest that the cellular responses were unpredictable. However, most of these analyses involved the use of artificial si/shRNA libraries that were screened for their activity to silence reporter constructs. Effects on endogenous mRNAs and the responses by the cells were often not considered.
-
In summary, type I and type II sOTE are expected to elicit a plethora of unpredictable cellular responses depending on what mRNA or sets of mRNAs are affected (Fig. 1). In contrast sOTE type III elicits a more selected spectrum of responses: activation of multiple cell death pathways (Fig. 1). What, therefore, sets sOTE type III apart from sOTE type I and type II is that all DISE-inducing si/shRNAs kill cancer cells through a process inducing similar morphological changes and biochemical responses. In fact, we recently demonstrated that it was possible to identify DISE-inducing shRNAs from genes other than CD95 and CD95L solely by monitoring changes in cell morphology and biochemical responses [55].
While DISE certainly manifests somewhat differently in different cancer cells likely due to different transcriptomes, cancer cells tend to respond to various DISE-inducing si/shRNAs in similar ways. This suggests that these si/shRNAs trigger a common pathway that is inconsistent with a random OTE response. A previous report described an OTE as a non-random occurrence by demonstrating that seemingly unrelated siRNAs causing OTE could elicit very similar responses in cells. To identify a target that would resensitize small cell lung cell carcinoma to the Bcl-2 inhibitor ABT-737, a screen targeting 4000 genes using sets of SmartPools was performed [56]. This screen identified the Bcl-2 family member, MCL-1, as the major resistance factor. However, the majority of the siRNAs that were part of the top three scoring SmartPools in this screen— none of which were designed to target MCL-1—all targeted the 3′UTR of MCL-1 through 7mer seed pairing. This study provided the rationale to focus on MCL-1 as a new target for cancer cells that are resistant to Bcl-2 inhibitors. Interestingly, MCL-1 was identified in non-RNAi based genome-wide lethality screens as a critical survival gene [57, 58]. The identification of MCL1 as a factor rendering cancer cells resistant to Bcl-2 inhibitors may have been the first description of the DISE mechanism.
In our recent analysis, we found a remarkable correlation between the experimentally determined toxicity of CD95L-derived shRNAs and an in silico predicted toxicity index (TI) [49]. We defined the TI as the ratio of the number of seed matches in the 3′UTRs of ∼1800 survival genes [57] divided by the number of seed matches in the 3′UTR of ∼400 control genes not required for survival (both normalized for the number of genes in each group).
While survival genes are generally more highly expressed than genes not critical for cell survival, the TI was still a useful tool to predict toxic seeds when we based it on a group of 850 survival genes (a subgroup to the ∼1800 survival genes) compared to a group of expression matched 850 non-survival genes [49]. This suggests that survival genes may contain different types of seed matches (based on base composition or sequence) when compared to non-survival genes.
What could be so special about DISE-inducing seeds? It was previously shown in a study in Drosophila that genes that regulate development and differentiation (the “targets”) are enriched in seed matches of miRNAs, whereas genes regulating cell survival (the “antitargets”) have an underrepresentation of such seed matches [59]. For instance, the expression of an abundant miRNA such as miR-200c in an epithelial cell would not co-occur together with the expression of a critical survival gene containing miR-200c seed matches in its 3′UTR. This would kill the cell. Evolution must therefore have selected against the presence of seed matches of highly expressed miRNAs in the 3′UTR of survival genes. Differences in the classes of GO terms enriched in genes carrying few targets versus many targets were also reported for mammalian cells [60]. It is likely DISE-inducing si/shRNAs carry seed sequences that preferentially target seed matches present in the 3′UTRs of the “anti-targets”. DISE-inducing si/shRNAs may therefore target naturally existing sequences that evolved during evolution.
A major question is whether DISE occurs endogenously. Are small toxic RNAs ever generated from coding or noncoding genes that kill cells through this mechanism? While there is no conclusive evidence of this at this point, one set of preliminary experiments points at such a mechanism at least being possible. When we overexpressed CD95L, we found that it killed cancer cells independent of CD95 activation or full-length CD95L protein expression (unpublished data). We reason that multiple genes in the genome (the donor or D genes) could simultaneously give rise to a significant pool of such toxic small sequences with RNAi-activity that kill cells by targeting the 3′UTR of survival genes (the S genes) (Fig. 2). This model is now testable.
Figure 2. Model for DISE.
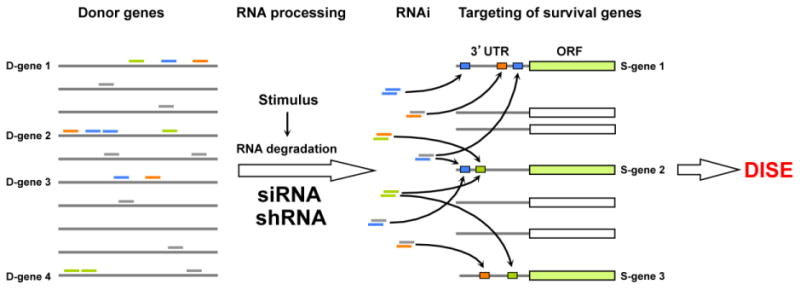
A number of genes (donor (D) genes) in the human genome contain sequences that when transcribed into RNA are degraded/processed to generate small RNAs, by forming heteroduplexes within the same mRNA (in stem regions) or with other small RNA fragments, and are then loaded onto Ago proteins and the RISC. CD95L could be one of the D-genes. Such endogenous siRNAs and many si- and shRNAs designed to selectively silence genes target seed matches present in the 3′UTRs of survival genes (S genes) instead, resulting in the induction of DISE.
Due to its fundamental targeting of survival genes it first seemed unlikely that DISE could be developed into a novel form of cancer therapy. However, we recently demonstrated that DISE can be triggered in vivo by delivering CD95L-derived siRNAs via HDL mimetic bioactive nanoparticles to treat ovarian cancer in mouse xenografts [61]. The CD95L derived siRNA used in our study could also kill a mouse cancer cell line, yet the treatment had no toxic effect on the mice, suggesting that DISE preferentially affects cancer cells. Remarkably, while cells in vitro and in vivo became resistant to treatment, they did not become resistant to the DISE mechanism itself. We recently extended these findings by using siRNAs that are 10-100 times more toxic to both human and mouse cancer cells than the sequences tested before, and again, treatment with the siRNA loaded nanoparticles while reducing tumor growth did not show toxicity in the treated mice (unpublished data).
What could be the reasons for the reduced susceptibility of normal cells to DISE? We reported that both Drosha and Dicer k.o. HCT116 cells were hypersensitive to DISE induced by CD95/CD95L derived si- and/or shRNAs [49]. This could be explained by the fact that both these k.o. cells are virtually devoid of miRNAs. Since we also showed toxicity of a DISE-inducing siRNA can be alleviated by transfecting cells with a nontoxic variant of the same siRNA, it is possible that wild-type cells are protected from DISE by high levels of miRNAs occupying the RISC. In both Drosha and Dicer k.o. cells, the RISC could be more available to toxic small RNAi active RNAs. It has been reported that a fundamental difference between all cancer cells and their matching normal cells is a global downregulation of miRNAs in all cancers [62]. The higher miRNA expression of normal tissues may protect them from DISE. This model seems to be in conflict with previous reports that demonstrated that the stability of Argonaute proteins is regulated by bound miRNAs [63, 64]. Both Drosophila S2 cells and mouse embryonic fibroblasts lacking Dicer expression [63], or mouse embryonic stem cells with knocked out Dicer or DGCR8 [64] contained less Ago proteins. However, neither Dicer nor Drosha k.o. HCT116 cells showed reduced Ago2 expression [49]. Another reason for the difference in DISE sensitivity between normal and cancer cells could therefore lie in the different stability of Ago proteins between normal and cancer cells.
A major question that arises from these findings is why not all si/shRNAs with high on-target activity towards a survival gene are more toxic to cancer cells than normal cells. Our model of endogenous miRNA RISC occupancy may be most relevant to si/shRNAs with sOTE III activity because an siRNA that was designed and tested to target an oncotarget with high selectivity will still be toxic to cells with low RISC availability. In contrast, DISE-inducing si/shRNAs that target countless survival genes with low efficiency might be more active in cells with high RISC availability such as cancer cell and hence more toxic.
Concluding Remarks
Based on its fundamental and universal nature, it might be possible to develop DISE to treat cancer. Maybe the most important question on the path to a possible new cancer therapy is why do DISE-inducing siRNAs not target survival genes in normal cells and kill them? Early in vivo data suggest that there may exist a selective sensitivity of cancer cells compared to normal tissues but this requires more study. Another fundamental question that needs to be addressed is what are the sequence motifs or the rules that determine whether a seed sequence in an si- or even in a miRNA preferentially targets survival genes. While most miRNAs do not target survival genes, there might be a few that do. DISE might also mediate or contribute to massive cell death caused by other known insults. These insults could include radiation or chemotherapeutic drugs, lysis of eukaryotic cells by viruses, or attack by cytotoxic killer cells. Future studies will have to test whether any endogenous noncoding RNA loaded into the RISC acts by targeting cellular survival genes.
Box - Trends.
RNAi is widely used to perform genome-wide screens.
A number of rules are established to reduce the number of OTEs during RNAi.
Because RNAi employs the same mechanism used by miRNAs to target hundreds of genes, OTEs are inherent to the process. While it is desirable for functional studies to only target one gene, it is inevitable that multiple genes will be targeted.
Once loaded into the RISC, all single-stranded oligonucleotides will follow the same targeting rules, regardless of whether they are derived from an endogenous miRNA or from an endogenous or exogenous siRNA.
Box - Outstanding Questions.
Under what circumstances are endogenous RNAi-active sequences generated and kill cancer cells through DISE?
What types of RNAs, and how many of them, contain DISE-inducing sequences?
What determines whether an siRNA causes sOTE type I/II or sOTE type III (DISE)?
Why are cancer cells, and among them cancer stem cells, more sensitive to DISE than normal cells?
Can DISE be used for cancer therapy?
Is DISE the manifestation of an evolutionary conserved mechanism, and if it is, when did it evolve and is there any evidence of DISE induction in invertebrates or single cell organisms?
Are there disease situations in which DISE is accidentally triggered that contribute to human pathology?
Acknowledgments
This work was funded by training grants T32CA070085 (to M.P.) and T32CA009560 (to W.P.) and R35CA197450 (to M.E.P.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Topalian SL, et al. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27(4):450–61. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weber JS, et al. Toxicities of Immunotherapy for the Practitioner. J Clin Oncol. 2015;33(18):2092–9. doi: 10.1200/JCO.2014.60.0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flajnik MF, Kasahara M. Origin and evolution of the adaptive immune system: genetic events and selective pressures. Nat Rev Genet. 2010;11(1):47–59. doi: 10.1038/nrg2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.El Albani A, et al. Large colonial organisms with coordinated growth in oxygenated environments 2.1 Gyr ago. Nature. 2010;466(7302):100–4. doi: 10.1038/nature09166. [DOI] [PubMed] [Google Scholar]
- 5.Domazet-Loso T, et al. Naturally occurring tumours in the basal metazoan Hydra. Nat Commun. 2014;5:4222. doi: 10.1038/ncomms5222. [DOI] [PubMed] [Google Scholar]
- 6.Swarts DC, et al. The evolutionary journey of Argonaute proteins. Nat Struct Mol Biol. 2014;21(9):743–53. doi: 10.1038/nsmb.2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 8.Hannon GJ. RNA interference. Nature. 2002;418(6894):244–51. doi: 10.1038/418244a. [DOI] [PubMed] [Google Scholar]
- 9.Hammond SM. MicroRNAs as tumor suppressors. Nat Genet. 2007;39(5):582–3. doi: 10.1038/ng0507-582. [DOI] [PubMed] [Google Scholar]
- 10.Krol J, et al. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11(9):597–610. doi: 10.1038/nrg2843. [DOI] [PubMed] [Google Scholar]
- 11.Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014;15(8):509–24. doi: 10.1038/nrm3838. [DOI] [PubMed] [Google Scholar]
- 12.Wilson RC, Doudna JA. Molecular mechanisms of RNA interference. Annu Rev Biophys. 2013;42:217–39. doi: 10.1146/annurev-biophys-083012-130404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meister G. Argonaute proteins: functional insights and emerging roles. Nat Rev Genet. 2013;14(7):447–59. doi: 10.1038/nrg3462. [DOI] [PubMed] [Google Scholar]
- 14.Ghildiyal M, Zamore PD. Small silencing RNAs: an expanding universe. Nat Rev Genet. 2009;10(2):94–108. doi: 10.1038/nrg2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lewis BP, et al. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 16.Okamura K, Lai EC. Endogenous small interfering RNAs in animals. Nat Rev Mol Cell Biol. 2008;9(9):673–8. doi: 10.1038/nrm2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tchernitsa O, et al. Systematic evaluation of the miRNA-ome and its downstream effects on mRNA expression identifies gastric cancer progression. J Pathol. 2010;222(3):310–9. doi: 10.1002/path.2759. [DOI] [PubMed] [Google Scholar]
- 18.Song R, et al. Male germ cells express abundant endogenous siRNAs. Proc Natl Acad Sci U S A. 2011;108(32):13159–64. doi: 10.1073/pnas.1108567108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tam OH, et al. Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature. 2008;453(7194):534–8. doi: 10.1038/nature06904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watanabe T, et al. Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature. 2008;453(7194):539–43. doi: 10.1038/nature06908. [DOI] [PubMed] [Google Scholar]
- 21.Babiarz JE, et al. Mouse ES cells express endogenous shRNAs, siRNAs, and other Microprocessor-independent, Dicer-dependent small RNAs. Genes Dev. 2008;22(20):2773–85. doi: 10.1101/gad.1705308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Werner A, et al. Contribution of natural antisense transcription to an endogenous siRNA signature in human cells. BMC Genomics. 2014;15:19. doi: 10.1186/1471-2164-15-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar P, et al. Meta-analysis of tRNA derived RNA fragments reveals that they are evolutionarily conserved and associate with AGO proteins to recognize specific RNA targets. BMC Biol. 2014;12:78. doi: 10.1186/s12915-014-0078-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ender C, et al. A human snoRNA with microRNA-like functions. Mol Cell. 2008;32(4):519–28. doi: 10.1016/j.molcel.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 25.Hansen TB, et al. Argonaute-associated short introns are a novel class of gene regulators. Nat Commun. 2016;7:11538. doi: 10.1038/ncomms11538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schickel R, et al. MicroRNAs: key players in the immune system, differentiation, tumorigenesis and cell death. Oncogene. 2008;27(45):5959–74. doi: 10.1038/onc.2008.274. [DOI] [PubMed] [Google Scholar]
- 27.Petri S, Meister G. siRNA design principles and off-target effects. Methods Mol Biol. 2013;986:59–71. doi: 10.1007/978-1-62703-311-4_4. [DOI] [PubMed] [Google Scholar]
- 28.Fedorov Y, et al. Off-target effects by siRNA can induce toxic phenotype. RNA. 2006;12(7):1188–96. doi: 10.1261/rna.28106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marques JT, Williams BR. Activation of the mammalian immune system by siRNAs. Nat Biotechnol. 2005;23(11):1399–405. doi: 10.1038/nbt1161. [DOI] [PubMed] [Google Scholar]
- 30.Bramsen JB, et al. A large-scale chemical modification screen identifies design rules to generate siRNAs with high activity, high stability and low toxicity. Nucleic Acids Res. 2009;37(9):2867–81. doi: 10.1093/nar/gkp106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mohr SE, et al. RNAi screening comes of age: improved techniques and complementary approaches. Nat Rev Mol Cell Biol. 2014;15(9):591–600. doi: 10.1038/nrm3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo J, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137(5):835–48. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schlabach MR, et al. Cancer proliferation gene discovery through functional genomics. Science. 2008;319(5863):620–4. doi: 10.1126/science.1149200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Silva JM, et al. Profiling essential genes in human mammary cells by multiplex RNAi screening. Science. 2008;319(5863):617–20. doi: 10.1126/science.1149185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cowley GS, et al. Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Sci Data. 2014;1:140035. doi: 10.1038/sdata.2014.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jackson AL, Linsley PS. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat Rev Drug Discov. 2010;9(1):57–67. doi: 10.1038/nrd3010. [DOI] [PubMed] [Google Scholar]
- 37.Yuan B, et al. siRNA Selection Server: an automated siRNA oligonucleotide prediction server. Nucleic Acids Res. 2004;32(Web Server issue):W130–4. doi: 10.1093/nar/gkh366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fellmann C, et al. Functional identification of optimized RNAi triggers using a massively parallel sensor assay. Mol Cell. 2011;41(6):733–46. doi: 10.1016/j.molcel.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ui-Tei K, et al. Guidelines for the selection of highly effective siRNA sequences for mammalian and chick RNA interference. Nucleic Acids Res. 2004;32(3):936–48. doi: 10.1093/nar/gkh247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iwasaki S, et al. Defining fundamental steps in the assembly of the Drosophila RNAi enzyme complex. Nature. 2015;521(7553):533–6. doi: 10.1038/nature14254. [DOI] [PubMed] [Google Scholar]
- 41.Kamola PJ, et al. The siRNA Non-seed Region and Its Target Sequences Are Auxiliary Determinants of Off-Target Effects. PLoS Comput Biol. 2015;11(12):e1004656. doi: 10.1371/journal.pcbi.1004656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma JB, et al. Structural basis for overhang-specific small interfering RNA recognition by the PAZ domain. Nature. 2004;429(6989):318–22. doi: 10.1038/nature02519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rose SD, et al. Functional polarity is introduced by Dicer processing of short substrate RNAs. Nucleic Acids Res. 2005;33(13):4140–56. doi: 10.1093/nar/gki732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith C. Sharpening the tools of RNA interference. Nature Methods. 2006;3:475–486. [Google Scholar]
- 45.Chiu YL, Rana TM. siRNA function in RNAi: a chemical modification analysis. RNA. 2003;9(9):1034–48. doi: 10.1261/rna.5103703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gu S, et al. The loop position of shRNAs and pre-miRNAs is critical for the accuracy of dicer processing in vivo. Cell. 2012;151(4):900–11. doi: 10.1016/j.cell.2012.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Watanabe C, et al. Quantitative evaluation of first, second, and third generation hairpin systems reveals the limit of mammalian vector-based RNAi. RNA Biol. 2016;13(1):25–33. doi: 10.1080/15476286.2015.1128062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fellmann C, et al. An optimized microRNA backbone for effective single-copy RNAi. Cell Rep. 2013;5(6):1704–13. doi: 10.1016/j.celrep.2013.11.020. [DOI] [PubMed] [Google Scholar]
- 49.Putzbach W, et al. Many si/shRNAs can kill cancer cells by targeting multiple survival genes through an off-target mechanism. eLife. 2017;6:e29702. doi: 10.7554/eLife.29702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hadji A, et al. Death induced by CD95 or CD95 ligand elimination. Cell Reports. 2014;10:208–222. doi: 10.1016/j.celrep.2014.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ceppi P, et al. CD95 and CD95L promote and protect cancer stem cells. Nature Commun. 2014;5:5238. doi: 10.1038/ncomms6238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin X, et al. siRNA-mediated off-target gene silencing triggered by a 7 nt complementation. Nucleic Acids Res. 2005;33(14):4527–35. doi: 10.1093/nar/gki762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Birmingham A, et al. 3′ UTR seed matches, but not overall identity, are associated with RNAi off-targets. Nat Methods. 2006;3(3):199–204. doi: 10.1038/nmeth854. [DOI] [PubMed] [Google Scholar]
- 54.Jackson AL, et al. Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. RNA. 2006;12(7):1179–87. doi: 10.1261/rna.25706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Patel M, Peter ME. Identification of DISE-inducing shRNAs by monitoring cellular responses. Cell Cycle. 2017 doi: 10.1080/15384101.2017.1383576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lin X, et al. ‘Seed’ analysis of off-target siRNAs reveals an essential role of Mcl-1 in resistance to the small-molecule Bcl-2/Bcl-XL inhibitor ABT-737. Oncogene. 2007;26(27):3972–9. doi: 10.1038/sj.onc.1210166. [DOI] [PubMed] [Google Scholar]
- 57.Wang T, et al. Identification and characterization of essential genes in the human genome. Science. 2015;350(6264):1096–101. doi: 10.1126/science.aac7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Blomen VA, et al. Gene essentiality and synthetic lethality in haploid human cells. Science. 2015;350(6264):1092–6. doi: 10.1126/science.aac7557. [DOI] [PubMed] [Google Scholar]
- 59.Stark A, et al. Animal MicroRNAs confer robustness to gene expression and have a significant impact on 3′UTR evolution. Cell. 2005;123(6):1133–46. doi: 10.1016/j.cell.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 60.Zare H, et al. An evolutionarily biased distribution of miRNA sites toward regulatory genes with high promoter-driven intrinsic transcriptional noise. BMC Evol Biol. 2014;14:74. doi: 10.1186/1471-2148-14-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Murmann AE, et al. Induction of DISE in ovarian cancer cells in vivo. Oncotarget. 2017;8:84643–84658. doi: 10.18632/oncotarget.21471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lu J, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435(7043):834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 63.Smibert P, et al. Homeostatic control of Argonaute stability by microRNA availability. Nat Struct Mol Biol. 2013;20(7):789–95. doi: 10.1038/nsmb.2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martinez NJ, Gregory RI. Argonaute2 expression is post-transcriptionally coupled to microRNA abundance. RNA. 2013;19(5):605–12. doi: 10.1261/rna.036434.112. [DOI] [PMC free article] [PubMed] [Google Scholar]