

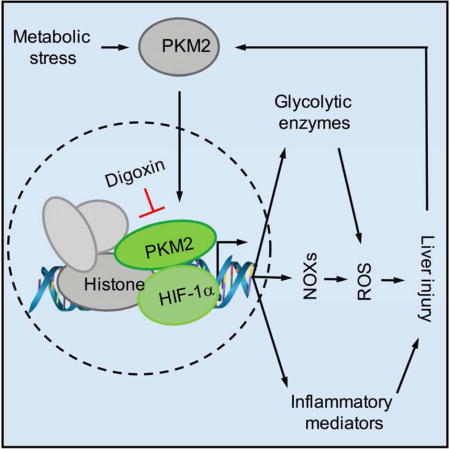
SUMMARY
Sterile inflammation after tissue damage is a ubiquitous response, and yet has the highest amplitude in the liver. This has major clinical consequences with alcoholic and non-alcoholic steatohepatitis (ASH and NASH) accounting for the majority of liver disease in industrialized countries, and both lacking therapy. Requirements for sustained sterile inflammation include increased oxidative stress, and activation of the HIF-1α signaling pathway. We demonstrate the ability of digoxin, a cardiac glycoside, to protect from liver inflammation and damage in ASH and NASH. Digoxin was effective in maintaining cellular redox homeostasis, and suppressing HIF-1α pathway activation. A proteomic screen revealed that digoxin binds pyruvate kinase M2 (PKM2), and independent of PKM2 kinase activity results in chromatin remodeling and down-regulation of HIF-1α transactivation. These data identify PKM2 as a mediator and therapeutic target for regulating liver sterile inflammation, and demonstrate a novel role for digoxin which can effectively protect the liver from ASH and NASH.
Keywords: Sterile inflammation, liver, Pyruvate Kinase M2, digoxin, HIF-1α, steatohepatitis
Graphical abstract
INTRODUCTION
The development of sterile inflammation after cell death is a ubiquitous response which occurs in all organs (Mehal, 2015). The amplitude of this response however varies widely, and the liver is notable for developing exceptionally strong sterile inflammation. This is seen in experimental models including acetaminophen toxicity, alcoholic steatohepatitis (ASH), and metabolic syndrome associated development of nonalcoholic steatohepatitis (NASH) (Kubes and Mehal, 2012; Marchesini et al., 2001). Such a high amplitude of sterile inflammation in the liver has major clinical consequences as NASH is by far the most common liver disease in industrialized countries, and is responsible for increasing amounts of liver damage and death (Fazel et al., 2016). It is also notable that diseases caused by sterile inflammation of the liver including ASH and NASH lack effective therapy.
The sterile inflammatory response in the liver incorporates several cell types and signaling pathways, and two processes among these are universally present. One is an increased tissue redox state, which plays a key role in the progression of inflammatory disorders (Mittal et al., 2014). Several cellular mechanisms contribute to the production of reactive oxygen species (ROS) in the liver, with increased leakage of protons across the inner mitochondrial membrane being an important one (Koliaki et al., 2015). Another is tissue hypoxia and activation of the HIF-1α pathway (Lefere et al., 2016). Acute and chronic alcohol exposure, and high fat diet (HFD) feeding result in liver hypoxia leading to HIF-1α pathway activation in ASH and NASH (Arteel et al., 1997; Arteel et al., 1996; Mantena et al., 2009). The importance of HIF-1α activation in NASH was shown by HIF-1α antisense nucleotide treatment resulting in protection against adverse metabolic effects induced by HFD (Nath et al., 2011; Shin et al., 2012). We have demonstrated that HIF-1α activation potentiates and sustains the amplitude of acute inflammatory responses, and is vital for the transition from acute self-limiting to a sustained chronic inflammation (Ouyang et al., 2013). There is also a close positive relationship between HIF-1α activation and increased ROS production due to up-regulation of NOX family of NADPH oxidases (Jiang and Torok, 2014). These mechanistic insights into the role of the HIF-1α pathway in sterile inflammation may have great clinical relevance due to the ability of cardiac glycosides (CGs) to inhibit HIF-1α activation (Zhang et al., 2008). CGs are well known in cardiology due to their effects of increasing ionotropy, and decreasing chronotropy, via partial inhibition of the Na+/K+ ATPase on the plasma membrane of myocytes (Smith, 1988). Of note the HIF-1α inhibitory effect of CGs is independent of its activity on the Na+/K+ ATPase, indicating CGs have additional unidentified molecular targets (Zhang et al., 2008).
In the present study, we describe a novel and potent role of digoxin in reducing the severity of steatosis, inflammation and hepatocellular damage in livers undergoing both acute and chronic sterile inflammatory injury. Digoxin improves oxidative stress during liver injury through maintaining cellular redox homeostasis, and the suppression of HIF-1α pathway activation and downstream signature genes in the liver. We have further identified pyruvate kinase isoform 2 (PKM2) as a digoxin-binding protein. PKM2 is the key metabolic regulator present predominantly in activated myeloid cells (Christofk et al., 2008; Palsson-McDermott et al., 2015; Tamada et al., 2012), and exists primarily as an enzymatically inactive monomer or dimer. The dimer of PKM2 can translocate to the nucleus, where it will interact with HIF-1α and regulate expression of numerous proglycolytic enzymes (Luo et al., 2011). Interestingly, digoxin does not alter PKM2 kinase activity but suppresses PKM2 dependent HIF-1α transcriptional activity. Protein immunoprecipitation combined with mass spectrometry demonstrate that active nuclear PKM2 directly interacts with multiple modified chromatin proteins, and digoxin reduces the binding of histones to PKM2 suggesting digoxin suppresses PKM2 promoted HIF-1α transactivation through chromatin modifications. These results identify PKM2 as a key molecule in liver sterile inflammation, and provide a molecular basis for the observed effects of digoxin in antagonizing liver injury from a wide range of toxic and metabolic insults.
RESULTS
Digoxin Reduces the Severity of Endotoxin-induced Hepatic Damage and Inflammation
To determine whether CGs limit acute hepatic damage and inflammation, we used an inflammatory stimuli-induced liver injury model by lipopolysaccharides (LPS) in D-galactosamine (D-GalN) sensitized mice. A critical involvement of HIF-1α pathway activation was determined in this type of tissue injury using myeloid-specific HIF-1α-null mice (Figure S1A), and myeloid cells were also identified as high expressers of PKM2 (Figure S1B). We then investigated the efficacy of digoxin on liver injury from LPS/D-GalN model in wild-type mice. Indicators of acute liver injury such as elevated serum transaminases, increased hepatic inflammatory infiltration, hemorrhage and hepatocyte death appeared 6 h after a single injection of LPS/D-GalN. A single intraperitoneal (i.p.) injection of digoxin 1 h prior to LPS/D-GalN treatment potently prevented hepatocellular damage and neutrophilic infiltrate across a wide dosage range (1.0-0.05 mg/kg) as determined by histological analysis (Figure 1A), and serum alanine transaminase (ALT) assay (Figure 1B). Inflammasome activation has recently emerged as a key feature in mediating the sterile inflammatory response to a wide range of liver pathologies (Kubes and Mehal, 2012; Mehal, 2014), and a maximal response is dependent on HIF-1α activation (Ouyang et al., 2013). Mice challenged with LPS/D-GalN showed a significant increase in mRNA abundance of pro-inflammatory cytokine IL-1β, NLRP3 as well as HIF-1α gene, and in protein levels of IL-1β in serum and active form of caspase-1 and IL-1β protein in whole liver (Figure 1C–G), demonstrating that the NLRP3 inflammasome was activated in LPS/D-GalN challenged liver tissue. Pretreatment of mice with digoxin dramatically prevented LPS/D-GalN induced inflammasome activation, which is consistent with reduced liver neutrophil infiltration and hepatocellular damage. Collectively, digoxin displayed dose-dependent protection against severe inflammatory liver injury induced by LPS. The administration of digoxin also resulted in significantly less hemorrhagic necrosis, neutrophil infiltrate and serum ALT levels in a toxin (thioacetamide, TAA) induced liver injury (Data not shown). This is a well-established animal model for assessing hepatic inflammation and damage response to injury (Zimmermann et al., 1989). Collectively these data show that digoxin represents parallel hepatoprotection in two different models of acute liver injury.
Figure 1. Digoxin reduces endotoxin-induced hepatic damage.
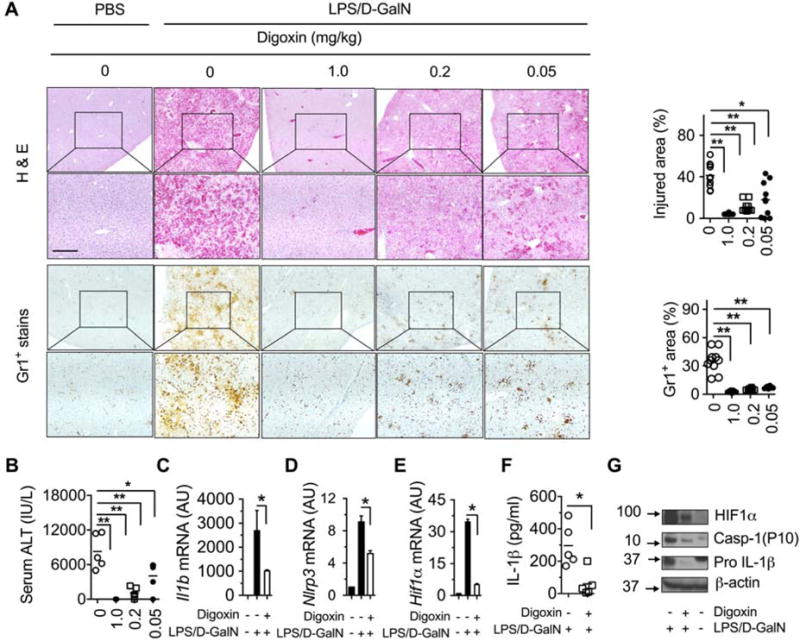
(A–G) Mice were injected with indicated dosages of digoxin or vehicle, followed by LPS/D-GalN 1h later and examination after a further 6 h.
(A–B) Dose-dependent reduction of hemorrhage, cellular necrosis, neutrophilic infiltrate and serum alanine transaminase in digoxin treated animals.
(C–E) Reduced mRNA levels of inflammatory genes in liver tissues by digoxin as measured by qRT-PCR.
(F) Reduced serum levels of IL-1β protein by digoxin as measured by ELISA.
(G) Reduced protein expression level of HIF-1α, caspase-1 (P10), and pro IL-1β genes by digoxin in liver as measured by western blot
Data represent mean ± SD (n=5). *p <0.05, **p<0.01. Scale bar, 100 μm.
Digoxin Reduces the Severity of Alcoholic and Non-alcoholic Steatohepatitis
Obesity and the metabolic syndrome have recently emerged as strong risk factors resulting in a series of changes in the liver that include steatosis, hepatocyte death, inflammation and fibrogenesis, and are collectively termed NASH (Marchesini and Marzocchi, 2007). Hypoxia is a principal causative factor and sensor for deleterious oxidative stress in both ASH and NASH, and HIF-1α functions as a master regulator of homeostatic responses to hypoxia and oxidative stresses by activating transcriptional events important for driving inflammation, metabolism and cell survival (Arteel et al., 1997; Ju et al., 2016; Lee et al., 2014; Majmundar et al., 2010; Nath et al., 2011; Palmer and Clegg, 2014). This is consistent with the development of liver hypoxia in diet induced obesity, and the inhibition of HIF-1α by anti-sense oligonucleotides resulting in a reduction of liver steatosis (Shin et al., 2012).
These features generate the possibility of HIF-1α pathway inhibition by digoxin as a therapeutic option for ASH and NASH. We examined whether digoxin is effective at improving chronic liver damage and inflammation in HFD derived NASH model. 8 weeks old mice were provided an ad lib 45 kcal% fat diet for 12 weeks, resulting in several features of NASH, including hepatic damage, steatosis, inflammatory cell infiltration, increased serum ALT level, and enhanced NAFLD activity scores (Figure 2A–E). Age-and gender- matched HFD mice were randomized into a control group, or experimental groups receiving a range of digoxin dosages. HFD fed mice were dosed twice a week by intraperitoneal (i.p.) injection of digoxin with 0, 1, 0.2 or 0.05 mg/kg. HFD fed mice treated with a dose range of digoxin showed dramatic reduction in hepatic damage, steatosis, and inflammation as determined by histological analysis (Figure 2A–B), serum ALT (Figure 2C) and leukocyte infiltration in the liver (Figure 2D–E). During the assay, mice remained fed ad libitum with HFD, and food intake did not change during the treatment period (Data not shown). Collectively, these data indicate that digoxin effectively prevents chronic hepatic damage, steatosis and inflammation in diet-induced obese mice. Due to the significant therapeutic implications we next determined whether digoxin could improve hepatic damage, steatosis and inflammation after the initiation of NASH. Mice were fed the HFD for 5 weeks, and then started with digoxin treatment. Histological analyses of NAFLD activity score (Figure 3A, B), liver leukocytes (Figure 3C, D), serum ALT level (Figure 3E), lipid quantification and triglyceride (TG) level (Figure 3F–H) showed that digoxin reduced the degree of hepatocellular damage, liver inflammation and lipid accumulation, even though the mice were maintained on the HFD.
Figure 2. Digoxin prevents chronic hepatic damage and inflammation in high fat diet model of non-alcoholic steatohepatitis.
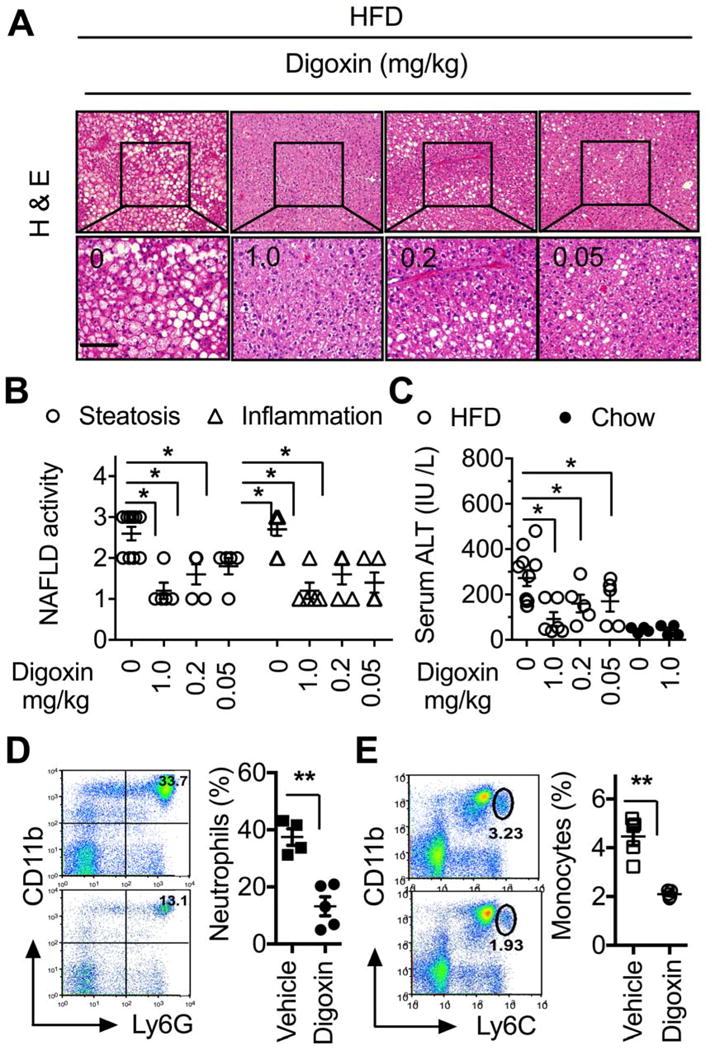
(A–E) Mice were injected Intraperitoneally with indicated dosages of digoxin or vehicle twice a week with concurrent HFD feeding for 12 weeks.
(A) Digoxin dose-dependently reduces HFD-induced steatosis by histological analysis of H & E stained sections. (B) Digoxin reduces HFD-induced hepatic steatosis and inflammation as scored quantitatively from H & E stained sections.
(C) Digoxin dose-dependently prevents HFD-induced hepatocellular damage as measured by the serum level of ALT.
Reduced liver neutrophil (D) and monocyte (E) infiltration by digoxin in HFD as measured by Ly6G/CD11b and Ly6C/CD11b positive cells in non-parenchymal cell populations using flow cytometry analysis.
Data represent mean ± SD (n=4–5). *p <0.05, **p<0.01. Scale bar, 100 μm.
Figure 3. Digoxin reduces hepatitis steatosis and inflammation when given therapeutically.
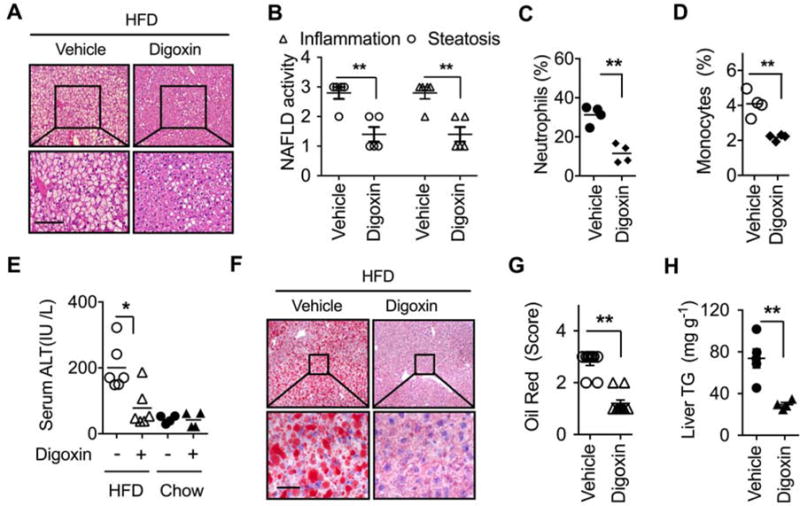
(A–H) Mice were fed with HFD for 5 weeks, and then given intraperitoneal injection of digoxin (1 mg/kg) or vehicle twice a week with continued HFD feeding for a total of 12 weeks.
(A) Digoxin reduces HFD-induced hepatic steatosis from histological analysis by H & E stained sections. (B) Digoxin reduces HFD-induced hepatic steatosis and inflammation as scored quantitatively from H & E stained sections. (C) Reduced liver neutrophil, and (D) monocyte infiltration by digoxin in HFD as measured by Ly6G/CD11b and Ly6C/CD11b positive cells in non-parenchymal cell populations using flow cytometry.
(E) Digoxin reduces HFD-induced hepatocellular damage as measured by the serum ALT.
(F–G) Digoxin attenuates hepatic steatosis as measured by oil red staining with quantification.
(H) Digoxin reduces liver TG content as measured by chemical analysis.
All data throughout the figure are shown as the mean ± SD from 4–5 mice in each group.
*p <0.05, **p<0.01.
Alcoholic steatohepatitis (ASH) shares the key features of increased ROS, HIF-1α activation, inflammation and steatosis with NASH (Mehal, 2013), and we therefore tested if digoxin could improve ASH in a well-accepted Lieber-Decarli ethanol liquid diet (5% ethanol) plus single ethanol binge mouse model during the chronic feeding (Bertola et al., 2013). Liver steatosis, damage and inflammation were characterized by histological analysis, quantification of neutrophil infiltration, and serum ALT and AST level (Figure S2). Digoxin (1.0-0.2 mg/kg) dose-dependently improved hepatic steatosis, neutrophil infiltration and hepatocellular damage in ASH (Figure S2A–D). The presence of the digoxin target pathway was confirmed in human liver tissues, which showed a greater degree of up-regulation of HIF-1α and HIF-1α dependent genes in severe, as compared to early ASH patients (Table S1, Figure S2E–I). To confirm that digoxin dosages used in the current study did not cause cardiac toxicity, EKG and echocardiography were performed on mice to monitor the cardiac function after digoxin treatment. The maximal dosage of digoxin did not result in a significant change in heart rate, PR interval, QRS width (by EKG monitoring), cardiac ejection fraction and fractional shortening (by cardiac echo measurement) over a 3-week period (Figure S3). Thus, we conclude that long-term treatment with digoxin reduces chronic liver damage, inflammation and steatosis in experimental models of NASH and ASH at doses that do not effect cardiac chronotropy and inotropy.
Digoxin Improves Abnormal Cellular Redox Status
Oxidative stress refers to elevated intracellular levels of ROS that participate in the major course of inflammatory, metabolic and proliferative liver diseases (Cichoz-Lach and Michalak, 2014). Mitochondria are a major source of ROS in hepatocytes, Kupffer cells, and neutrophils (Cichoz-Lach and Michalak, 2014), and the importance of ROS in liver injury is shown by the observation that reduction of ROS by a variety of manipulations protects the liver from a wide range of insults (Cederbaum et al., 2009; Li et al., 2015). We next determined whether digoxin suppresses ROS production both in vivo and in vitro from hepatocytes and immune cells. Consistent with previous reports both acute and chronic liver injury resulted in the production of high amounts of ROS from liver tissue, and peripheral neutrophils as evidenced in vivo using DHE (Dihydroethidium), and ex vivo using CM-H2DCFDA assays. Digoxin significantly reduced liver tissue (Figure 4 A–B) as well as peripheral neutrophil ROS levels (Figure 4C), indicating digoxin inhibits ROS production. Liver tissue can generate ROS through a process named oxidative burst mediated by NADPH oxidase 4 (NOX4) complex either within or outside the mitochondria in response to metabolic stress and inflammatory stimuli. To test if digoxin reduced hepatic oxidative stress due to mROS reduction in a cell intrinsic manner, human monocyte/macrophage like THP-1 cells were stimulated with either LPS or hydrogen peroxide, and the generation of superoxide mROS was quantified by mitoSOX dye using fluorescence activated cell sorting (FACS). LPS and hydrogen peroxide induced mROS production, which was significantly inhibited by digoxin (Figure 4D, E). We also detected an increased total ROS production in THP-1 cells treated with hydrogen peroxide, which digoxin significantly reduced (Figure 4F). Polymorphonuclear neutrophils (PMNs) generally maintain a high ability to produce ROS spontaneously in culture. Similar to liver tissue digoxin limited ROS production in human PMN cultures (Figure 4G, H). These data support a role for digoxin in improving hepatic cellular ROS induced by a wide range of stimuli.
Figure 4. Digoxin improves cellular redox status from a wide range of stimuli.
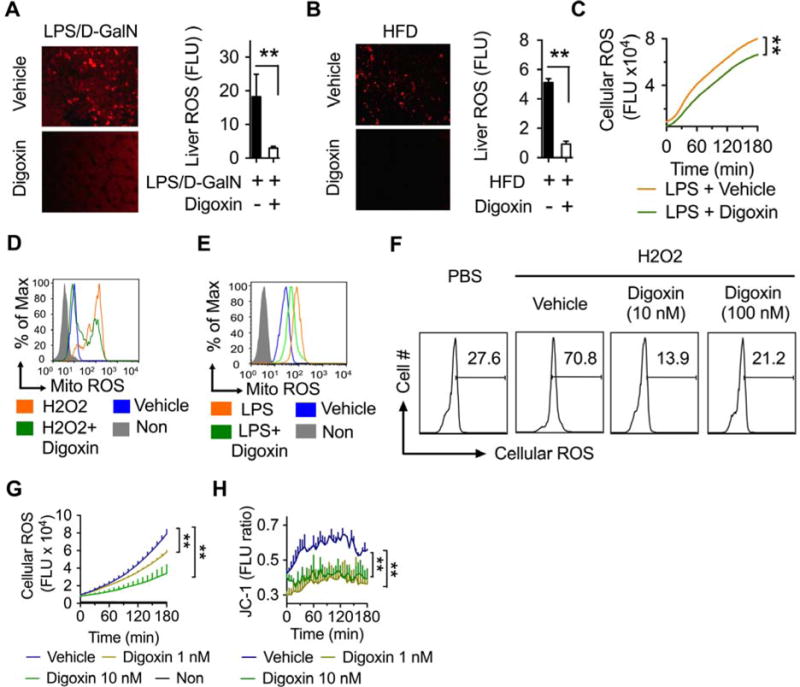
(A) Mice were injected with digoxin (1 mg/kg) or vehicle for 1 h, and then injected with LPS/D-GalN for 6 h. DHE was given i.v.15 min before sacrifice. Reduction in liver oxidative stress monitored by fluorescence microscopy is shown (left) and quantification in accounting multiple views of positive areas using Image J (right).
(B) Mice were intraperitoneally injected with digoxin (1 mg/kg) or vehicle twice a week with concurrent HFD feeding for 12 weeks. DHE was given i.v. 90 min before analysis. Reduction in liver oxidative stress was monitored by fluorescence microscopy (left) and quantified using Image J (right).
(C) Mice were treated as Figure 4A, except without DHE injection. Peritoneal neutrophils were harvested by lavage, and stained with CM-H2DCFDA followed by kinetic reading of ROS with fluorescent plate reader.
(D–E) THP-1 cells were treated with digoxin (10 nM) or vehicle for 3 h and then LPS (1μg/ml) (D), or H2O2 (10 mM) (E), for additional 3 h. MitoSOX ™ Red mitochondrial superoxide indicator was added 30 min before analysis to monitor mitochondrial ROS. The cells were collected and washed following flow cytometry analysis.
(F) THP-1 cells were treated with indicated dosages of digoxin or vehicle for 3 h and then H2O2 (10 mM) for additional 3 h. CM-H2DCFDA was added in last 30 min to monitor intracellular ROS. Cells were collected and washed following flow cytometry.
(G) Human primary neutrophils were isolated and treated with dosages of digoxin or vehicle for 3 h. The cells were then incubated with CM-H2DCFDA for 30 min, and washed following kinetic analysis of intracellular ROS with fluorescent plate reader.
(H) Human primary neutrophils were isolated and treated with dosages of digoxin, or vehicle for 3 h. Cells were then incubated with JC-1 for 30 min to monitor mitochondrial membrane potential. The kinetic analysis of mitochondrial membrane potential was performed using a fluorescent plate reader.
Digoxin Inhibits the Transcription of HIF-1α Signature Genes in Inflamed Liver
To identify pathways modified by digoxin we performed microarrays on liver tissues from the experimental NASH model with and without digoxin. The whole transcriptome data revealed that 23% of genes were detectable in the liver and digoxin up-regulated 12%, and down-regulated 11% (Figure 5A). Gene ontology (GO) annotations were performed using PANTHER classification system for the digoxin-modified genes. Analysis based on molecular functions revealed that digoxin-modified genes were mostly classed into the two major groups of gene products with binding and catalytic activity. The group of gene products with binding activity was further enriched for a number of nucleic acid binding and protein binding, while the group of genes with products of catalytic activity was selectively enriched for hydrolase activity and transferase activity.
Figure 5. Digoxin inhibits the transcription of HIF-1α signature genes in inflamed liver.
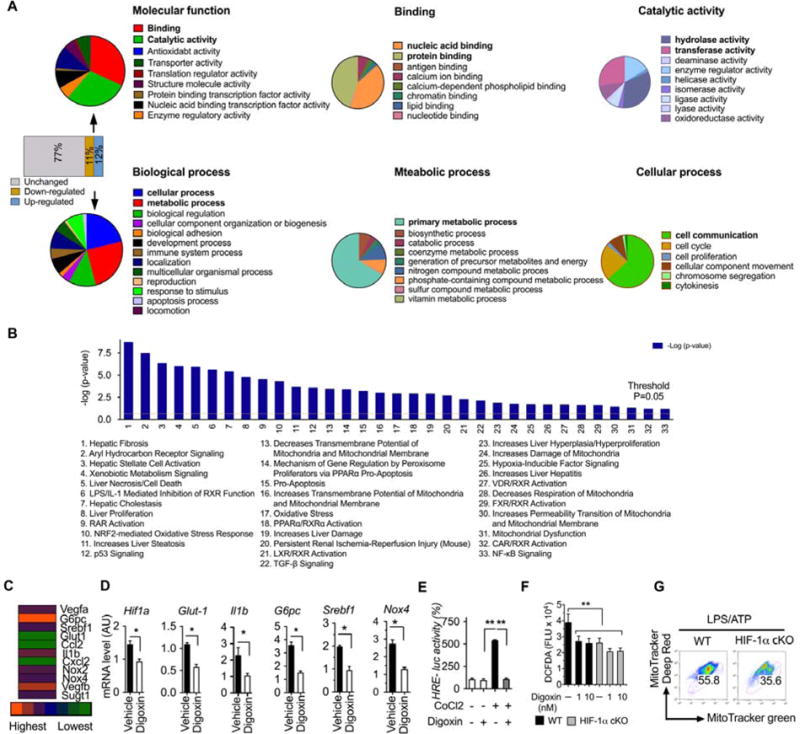
(A–D) Mice were intraperitoneally injected with digoxin (1 mg/kg) or vehicle twice a week with concurrent HFD feeding for 12 weeks.
(A) Microarray analysis of liver tissue was performed and digoxin regulated genes further narrowed down into multiple categories based on PANTHER analysis. (B) Significance of biological functions refers to the –log (p-value) obtained by the Ingenuity Pathway Analysis (IPA). Biological functions with a greater than 2-fold reduction are shown.
(C) Heat map showing the fold changes of digoxin down-regulated genes based microarray data (digoxin treated group versus vehicle group in HFD model). (D) Validation of gene expression for HIF-1α signatures from liver tissue was performed by RT-PCR.
(E) HEK293 T cells were transfected with HRE-luciferase construct for 12 h, and then cobalt chloride treatment in the presence of digoxin (10 nM) for additional 12 h. HRE luciferase activity was measured by dual-luciferase reporter assay. The relevant luciferase activity was based on renilla control.
(F) Primary neutrophils were collected from HIF-1αf/f-LysM (HIF-1α-cKO), and wild type controls. The cells were treated with digoxin as indicated dosages for 3 h, and then CM-H2DCFDA staining. The fluorescence density was measured kinetically by plate reader and the data showed at 6 h point.
(G) Bone marrow derived macrophages were collected from HIF-1α-cKO and wild type controls. The cells were stimulated with LPS for 6 h and challenged with ATP for 30 min. The FACS analysis was performed by Mito-Tracker Green and Mito-Tracker Deep Red. All data throughout the figure are shown as the mean ± SD from 5 mice in each group.
*p <0.05, **p<0.01.
Analysis based on GO biological function determined that the digoxin-modified genes were mainly enriched for cellular and metabolic process that were dominated by primary metabolic process, cell communication and cell cycle (Figure 5A). To further interpret the data in a biological context, the list of digoxin regulated genes with greater than two-fold decrease was analyzed using the Ingenuity Pathway Analysis (IPA) which identifies networks and biological functions which are most significant to the data set. Thirty-three biological functions had a greater than two-fold decrease in the livers of digoxin treated mice compared to controls (Figure 5B), with hepatic fibrosis, aryl hydrocarbon receptor signaling, hepatic stellate cell activation, xenobiotic metabolism signaling, liver necrosis, cell death showing the greatest difference. Further genome wide transcriptome analysis revealed that a unique set of HIF-1α signature genes were down-regulated by digoxin in HFD induced NASH liver tissue, including Vegf, G6pc, Glut-1 Srebf1 and Il1b (Figure 5C). The changes in HIF-1α signature genes were confirmed by RT-PCR (Figure 5D), indicating digoxin reprograms pathways downstream of HIF-1α in experimental NASH liver tissue. To confirm digoxin inhibition on HIF-1α transcriptional activity we tested the ability of digoxin to inhibit HRE (hypoxia response element) reporter activity in HRE-luciferase plasmid transfected cells in response to cobalt chloride, which is widely used to mimic hypoxia. Consistently, significant inhibition of digoxin on HRE promoter activation was observed, demonstrating digoxin has critical effects on the suppression of HIF-1α transcription (Figure 5E). In addition, inhibition of neutrophil ROS required the presence of HIF-1α, as digoxin did not reduce ROS production in HIF-1α deficient neutrophils (Figure 5F). Consistently, HIF-1α deficient macrophages showed ROS dependent mitochondria oxidative dysfunction in response to typical inflammasome activation (Figure 5G). These results collectively demonstrate that digoxin has critical effects in improving abnormal intracellular redox status and reducing oxidative stress due to its ability to modulate ROS scavenging antioxidant defense machinery in a HIF-1α dependent manner.
Digoxin Directly Binds to PKM2 and Inhibits PKM2-triggered Transactivation of the HIF-1α Gene
We next tried to understand the direct molecular mechanisms responsible for the digoxin effect on HIF-1α transcription. To identify digoxin interaction protein partners, we developed a new approach immobilizing digoxin onto epoxy-activated agarose beads, and incubating these beads with protein lysates from LPS stimulated macrophages. Digoxin bound proteins were eluted and separated by gel SDS-PAGE electrophoresis following protein visualization by silver staining (Figure 6A top, Figure S4). Interestingly, a protein band around 60 KDa was found identical to that seen on whole gel protein stain (Figure S4 right). We applied this band to LC-MS/MS analysis which identified pyruvate kinase M2 (PKM2) as the major digoxin binding protein (Figure 6A bottom). We next repeated this finding from digoxin bound protein elution by western blot using specific anti PKM2 antibody (Figure 6B). The specificity of this interaction was further determined by competitive protein binding assay in which macrophages protein lysate was pre-incubated with digoxin, and competitively resulted in loss of binding of PKM2 to the digoxin coated beads (Figure 6C). In addition to its well-known enzymatic activity of converting phosphoenolpyruvate (PEP) to pyruvate, PKM2 has non-canonical functions of regulating gene transcription and cell cycle progress via protein-protein interactions, and protein kinase activity (Lincet and Icard, 2015; Luo and Semenza, 2012). We first determined whether digoxin affects PKM2 kinase activity. Recombinant PKM2 caused significant increase in kinase activity, and this was not significantly alerted by digoxin (Figure 6D). Digoxin also did not affect active PKM2 protein translocation to nucleus where it is known to regulate transcription (Figure S5A). These data indicate that interventions that reduce PKM2 transcriptional activity will have a protective effect analogous to digoxin. The small molecule TEPP-46 which stabilizes the tetrameric form of PKM2, and reduces PKM2 nuclear translocation and transcription (Anastasiou et al., 2012; Palsson-McDermott et al., 2015). TEPP-46 treatment prior to LPS significantly reduced liver hemorrhage, hepatocyte necrosis and serum ALT level in D-GalN sensitized mice (Figure S5B). The results are consistent with reduction in PKM2 transcription as an important pathway in limiting liver inflammation and damage.
Figure 6. Digoxin directly binds to PKM2 and inhibits PKM2-HIF-1α axis activation.
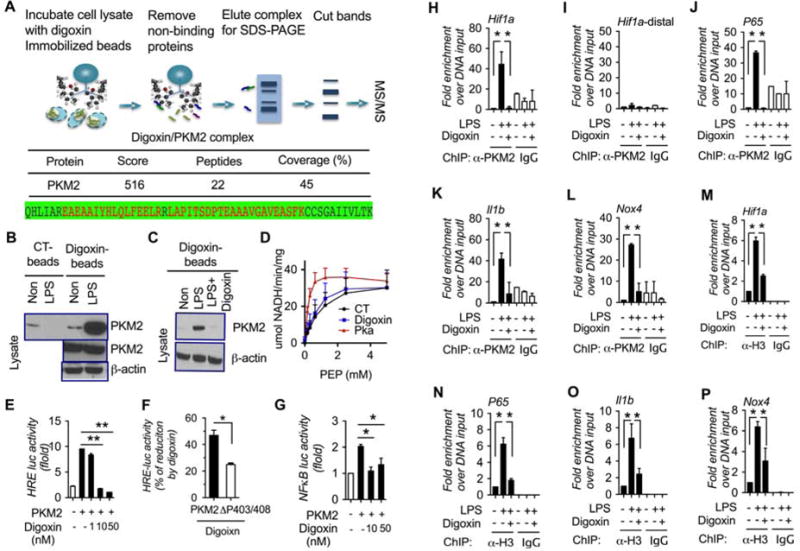
(A) Whole cell lysates from LPS stimulated Raw264.7 cells were applied on digoxin-immobilized affinity resin (upper). LC-Mass spectrum (LC-MS/MS) of digoxin bound proteins identified PKM2 (middle), and the detected peptide sequence of PKM2 was identical and shown as red (bottom).
(B) Cell lysates from LPS stimulated Raw264.7 cells were applied on digoxin immobilized affinity resin and eluted proteins examined by western blot using anti-PKM2 antibody.
(C) Cell lysates from LPS stimulated Raw264.7 cells were incubated with or without digoxin (10 mM) for overnight, and then applied on digoxin immobilized affinity resin and eluted proteins examined by western blot using anti-PKM2 antibody.
(D) PKM2 enzyme activity was measured in the presence of digoxin (50 nM) and PKM2 activator (Pka 550602) in pancreatic β cells (INS).
(E–G) HEK293T cells were co-transfected with HRE-luciferase (E, F), or NFκB-luciferase (G) construct for 12 h, treated with digoxin for additional 12 h. The luciferase activity was measured by dual-luciferase reporter assay and normalized to renilla control.
(H–P) Raw264.7 cells were treated with digoxin (50 nM) for 3 h, and the stimulated with LPS for 6 h, and whole cell lysates were assayed by ChIP-PCR using anti-PKM2 antibody (H–L), and anti-histone H3 (M–P), and the DNA binding was determined by qPCR.
We subsequently determined whether digoxin affects PKM2-triggered transcription events. PKM2 ectopic expression resulted in a significant increase of HRE as well as NFκB promoter activity in a reporter luciferase assay, which was dose-dependently inhibited by digoxin (Figure 6E and G). PKM2 hydroxylation at sites proline-403 and proline-408 increases the interaction of PKM2 with HIF-1α, and potentiates PKM2-mediated HIF-1α transcription (Luo et al., 2011). To test if the ability of digoxin to reduce PKM2 activation of HRE was also dependent on these known PKM2 regulatory sites, a mutant PKM2 with double mutation of the pro-403/408 sites to alanine (ΔP403/408A) was tested. The ΔP403/408A-PKM2 was significantly less sensitive to digoxin mediated reduction of PKM2 mediated HRE promoter activity (Figure 6F). This demonstrates overlap between the known PKM2 regulatory sites and the potential site of action of digoxin. Further, the two CG analogs digitonin and proscillaridin A, but not the controls with similar structures also displayed the inhibitory efficacy on PKM2 trigged HRE promoter activity (Figure S6A). In contrast digoxin did not significantly affect FXR, LXR and PPARα promoter luciferase activities (Figure S6B–D), demonstrating the selectivity of the inhibition of HIF-1α and NFκB. These findings are consistent with the reports that active PKM2 can trans-locate into the nucleus, where it is recruited with HIF-1α to HREs, and enhances HIF-1α occupancy, p300 recruitment, and H3K9 acetylation (Luo et al., 2011), and PKM2 regulates HIF-1α gene expression via NFκB activation (Azoitei et al., 2016). To further determine the effects of digoxin on the extent of PKM2 regulatory pathways, we performed chromatin immune-precipitation PCR (ChIP-PCR) analysis using mouse macrophages treated with LPS in the presence or absence of digoxin. Interestingly, LPS treatment resulted in a significant increase of PKM2 binding in the HIF-1α, P65 subunit of NFκB, IL-1β, and NOX4 promoter region, but not the distal enhancer region of HIF-1α, while digoxin broadly inhibited these binding activities (Figure 6H–L). ChIP-PCR analysis further confirmed that LPS enhanced the histone H3 binding activities to promoter regions of the same genes, while digoxin broadly inhibited these binding activities (Figure 6M–P). Collectively, these results demonstrate that digoxin inhibits the transcription of PKM2 dependent genes that are relevant to metabolic process at a chromatin level.
Digoxin Displays the Ability to Suppress the Protein Interaction of PKM2 to Chromatin
To further identify the possibility of digoxin to alter the interaction of PKM2 with nuclear proteins, PKM2 immunoprecipitation was performed from the nuclear lysate of LPS treated mouse macrophage with or without digoxin treatment. Nuclear PKM2 bound protein complexes were subjected to LC-MS/MS proteomic screen to specifically identify PKM2 binding proteins in the nucleus. The binding of PKM2 to numerous histone proteins was found to be regulated by digoxin (Figure 7A). To confirm these proteomic findings, PKM2 co-immunoprecipitation assays were repeated using the same cell lysate from above. Endogenous histone H3 and acetylated histone H3 were specifically precipitated by anti-PKM2 antibody, but not by control IgG, and the amount precipitated was significantly less in macrophages exposed to digoxin (Figure 7B). These data demonstrate that PKM2 physically interacts with histone proteins known to be important in up-regulating HIF-1α and NFκB response elements, and digoxin reduces such interactions. Consistent with our findings, broad gene-network analysis of the association of PKM2 with HIF-1α, and PKM2 with histone proteins based on STRING program algorithms with the results of automatic literature-mining searches revealed that PKM2 has related interactions with HIF-1α including EP300, EPAS1 and VHL, and with histones including his2A and his3A (Figures S7A–B). To directly confirm the requirement for PKM2 and HIF-1α in the ability of digoxin to inhibit inflammatory responses, we generated PKM2 and HIF-1α knockout Raw264.7 macrophages using lentiviral CRISPR technology. LPS induced mRNA expression levels of Il1b and Nox4 were dramatically decreased in both Pkm2 and Hif1a stable knockout conditions. Hif1a mRNA expression also decreased in PKM2 knockout cells, indicating HIF-1α acts downstream of PKM2. Digoxin did not result in further reduction of the already low levels of Il1b and Nox4 in the absence of Pkm2 or Hif1a (Figure 7C). Collectively, our results indicate that digoxin reduces liver injury in NASH/ASH via a direct interaction with PKM2, and reducing PKM2-triggered transactivation of HIF-1α and PKM2- histone interactions which control the gene transcription that are closely relevant to cellular oxidative stress and liver inflammation (Figure 7D).
Figure 7. Digoxin disrupts the protein interaction of PKM2 to histones in the nucleus.
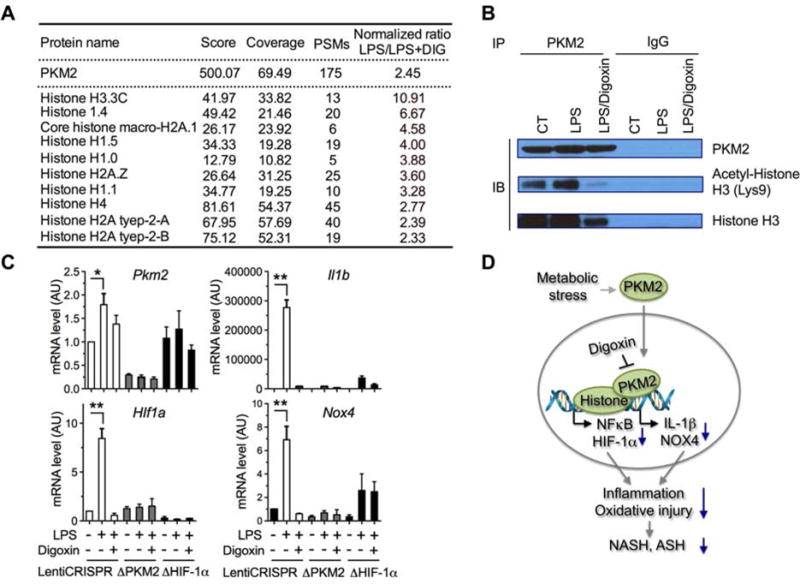
(A) Nuclear protein lysates from LPS stimulated Raw264.7 cells were immune-precipitated using anti-PKM2 antibody. PKM2 bound proteins were identified by LC-MS/MS. The PKM2 bound histones were extracted as A.
(B) The nuclear cell lysates from (A) were applied for PKM2 immune-precipitation and blotted with indicated antibodies.
(C) PKM2, HIF-1α stable knockout Raw264.7 macrophages were stimulated with LPS in the presence or absence of digoxin (50 nM) for 6 h. Reduced mRNA levels of inflammatory genes as indicated by digoxin were measured using qRT-PCR. The data are representative as the mean ± SD from quadruplicate wells of three independent experiments in one colony and two more independent experiments in additional colony of stable cells.
(D) Proposed mechanism of digoxin on PKM2 triggered transactivation.
DISCUSSION
The emergence of sterile inflammatory diseases within the umbrella of metabolic syndrome is one of the most pressing current medical challenges. The liver is notably affected with the development of the related conditions of alcoholic hepatitis, and nonalcoholic steatohepatitis (ASH and NASH). There are no approved therapies for ASH or NASH, and therapeutic development is limited by the relatively poor understanding of the initiating steps in sterile inflammation, and the dysregulation of a wide range of pathways making it difficult to know which ones to target. We took a two-step approach to identifying new therapeutic approaches by first selecting a core pathway, activation of which is required for the development of sterile inflammation in a wide range of settings. We wished to identify an approved drug which can inhibit this pathway, allowing for the possibility of rapid transition to the clinic. The requirement of HIF-1α pathway activation in macrophages for the development of an inflammatory response made this a candidate for a core pathway, and the ability of cardiac glycosides, to inhibit HIF-1α fulfilled the second criteria. The fact that the mechanism by which digoxin inhibited HIF-1α was unknown provided an opportunity for discovery of a new pathway regulating NASH.
Our data shows that digoxin reduces oxidative stress during liver injury through maintaining cellular redox homeostasis and protects the liver from a wide variety of insults. Digoxin also suppresses HIF-1α pathway activation and downstream signature genes by binding the known HIF-1α activator PKM2 and suppressing PKM2 dependent HIF-1α transcriptional activity. Further digoxin reduced the binding of PKM2 to histones, suggesting digoxin suppresses PKM2 promoted HIF-1α transactivation through chromatin modifications.
An imbalance in redox homeostasis, with the development of ROS is common to the development of a wide range of liver injury including ASH and NASH. Cellular ROS can increase via a wide range of pathways including mitochondrial damage, reduction in anti-oxidants, and subsequent to HIF-1α upregulation (Cederbaum, 2003; Feldstein and Bailey, 2011; Paik et al., 2014). Cellular death due to increased ROS resulting in a sterile inflammatory response is a key feature in ASH and NASH, and we have shown the ability of digoxin to improve mitochondrial and cellular ROS in a wide range of in vitro and in vivo systems (Figure 4A–C). This is consistent with the broad protective effects of digoxin in liver injury.
Transcription analysis of HFD livers with and without digoxin confirmed that digoxin affects a very wide range of genes, with enrichment of gene products with binding activity to nucleic acids and proteins, and hydrolase and transferase activity. Pathway analysis showed that genes involved in primary metabolic processes and cell communication were dominantly effected. Direct investigation of HIF-1α dependent genes in HFD induced NASH revealed a significant reduction in HIF-1α, and a set of HIF-1α dependent genes by digoxin (Figure 5C–D). The importance of the HIF-1α pathway in human alcoholic hepatitis was corroborated by the greater signal of HIF-1α, and HIF-1α dependent genes, in severe alcoholic hepatitis compared to early alcoholic hepatitis (Figure S2 E–I). We confirmed that digoxin is able to reduce HIF-1α transcriptional activity by using a HRE reporter luciferase assay, and also confirmed that the antioxidant effect of digoxin was dependent on HIF-1α (Figure 5 E–G). Collectively these data demonstrate that digoxin induces significant changes in gene transcripts related to HIF-1α, primary metabolic processes and nucleic acid binding among others, and that the antioxidant effect of ROS is dependent on HIF-1α.
The ability of digoxin to bind to PKM2 is an unexpected finding and provides novel insights into PKM2 biology, and the role of PKM2 in sterile inflammatory liver diseases. PKM2 is best known as the rate-limiting glycolytic enzyme that catalyzes the conversion of phosphoenopyruvate (PEP) and ADP to pyruvate and ATP (Luo and Semenza, 2012). In addition to its pyruvate kinase function PKM2 interacts with HIF-1α in the nucleus and functions as a transcriptional co-activator for HIF-1α, resulting in stimulation of HIF-1α responsive genes (Luo et al., 2011). Binding of digoxin to PKM2 does not alter its pyruvate kinase ability or reduce its nuclear translocation (Figure 6D and S5). It does however reduce the binding of PKM2 to HRE and NFκB promoter containing genes and to histone proteins. Functionally this reduces the ability of PKM2 to upregulate transcription of HRE and NFκB and the downstream inflammatory and ROS cascade (Figure 6E–G).
Digoxin is a cardiac glycoside consisting of three sugars and the aglycone digxigenin. It is isolated from the foxglove plant (Digitalis lanata), and along with other cardiac glycosides is present in many plants at lower concentrations. There is extensive clinical experience with digoxin, and its only clinical use to date has been to reduce inotropy and increase chronotropy. Digoxin is notable for producing cardiotoxicity at concentrations that are close to its effective concentration. Remarkably however at lower doses digoxin does not have any cardiac or other toxicity. The current data have identified an entirely novel application of this old drug and, at doses that are significantly below the dose required for the cardiac effect.
These data identify PKM2 as novel target for manipulation of sterile inflammation in the liver, and demonstrates that it is possible to pharmacologically regulate the nuclear transcription functions of PKM2, independently from the enzyme activity. Digoxin is the first PKM2 binding drug to be identified, and has the ability to do so at doses below required for a cardiac effect.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PKM2 (D78A4) | Cell Signaling Technology | Cat#:4053; BRID: AB_1904096 |
| Histone H3 | Cell Signaling Technology | Cat #:2650; BRID: AB_2115124 |
| IL-1beta | R & D | Cat#:AF401-NA; BRID:AB_416684 |
| Caspase-1p10 (M20) | Santa Cruz Biotechnology | Cat#: SC-514; BRID: AB_2068895 |
| NLRP3 (Cyro-2) | AdipoGen Life Sciences | Cat#: AG-20B-0014; BRID: AB 2490202 |
| Ly6C (CL21)-FITC | BD Biosciences | Cat#: 553104; BRID: AB_394628 |
| Ly6G (1A8)-PE | BD Biosciences | Cat#:551461; BRID: AB_394208 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Digoxin | Sigma | D6003 |
| 2-mercaptoethanol | Thermo Fisher Scientific | 21985-023 |
| diisocyanato hexane | Sigma | 52649 |
| M-CSF | PeproTech | 315-02 |
| methylamine | Sigma | 426466 |
| Lipofectamine™ 2000 | Thermo Fisher Scientific | 11668027 |
| D-galactosamine | Calbiochem | CAS-1772-03-08 |
| Critical Commercial Assays | ||
| PolymorphPrep | AXIS-SHIELD DENSITY GRADIENT MEDIA | AXS-1114683 |
| TentaGel NH2 Resin | Advanced Chem Tech | SJ6011 |
| Nuclear Extraction Kit | EMD Millipore Corporation | 2900 |
| 2x SYBR Green I Master | Roche | Cat#: 04707516001 |
| Deposited Data | ||
| Gene microarray of HFD liver tissue | GSE108839 | |
| Experimental Models: Cell Lines | ||
| Human: THP-1 cells | ATCC | TIB-202 |
| Human: HEK293 T cells | ATCC | CRL-11268 |
| Mouse: Raw264.7 | ATCC | TIB-71 |
| Experimental Models: Organisms/Strains | ||
| Lysozyme-Cre mouse | Jackson Laboratories | #004781 |
| C57BI/6 | National Cancer Institute | C57BI/6 |
| Software and Algorithms | ||
| FlowJo, Version 10 | FlowJo | https://www.flowjo.com |
| GraphPad Prism version 7 | Prism | https://www.graphpad.com |
| Other | ||
| Regular chow diet | Harlan Teklad | TD.2018 |
| High fat diet | Research Diets Inc | D12451 |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Wajahat Zafar Mehal (wajahat.mehal@yale.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
C57BL/6J mice were from the National Cancer Institute (Frederick, MD) and were group housed (5 per cage) while they were fed either a high fat diet (Research Diets Inc, #D12451, New Brunswick, NJ; 45% calories from fat, 35% from carbohydrate, 20% from protein) or a chow diet (Harlan Teklad #TD.2916, Madison, WI; 12% calories from fat, 48.5% from carbohydrate, 16.4% from protein), ad lib for 12 weeks. HIF-1αflox/flox mice were obtained from from Dr. R. Medzhitov (Yale University, New Haven, CT). The lysozyme-Cre (LysMcre) mice were obtained from Jackson Laboratories (Stock # 004781, Bar Harbor, ME). We then generated LysMcre+/− HIF-1αflox/flox (abbreviated HIF1–1α cKO) mice by intercrossing HIF-1αflox/flox and LysMcre mice. HIF-1αflox/flox mice without LysMcre gene were used as controls for all experiments. Animals were maintained in group-housing on a 12-h light/12-h dark cycle with free access to water and standard rodent chow. Housing rooms were maintained at 22–23 degrees Celsius. Male wild-type littermate controls were used in all experiments. Mice were randomly assigned to experimental groups. At the conclusion of each study, mice were euthanized by general isoflurane anesthesia with additional cervical dislocation. All animal studies were approved by the Yale University Institutional Animal Care and Use Committee and were performed in accordance with all regulatory standards.
Primary Cell Cultures
Mouse peritoneal neutrophils (PMNs) and macrophages (PECs) were isolated by peritoneal lavage after intraperitoneal injection of 4% thioglycollate solution (B2551, Fluka). Cells were cultured in DMEM medium (10% FBS), penicillin/streptomycin and L-glutamine. Mouse bone marrow derived macrophages were isolated, and were differentiated for 7 days in complete RPMI-1640 medium supplemented with 2 mM L-glutamine, 100U/ml penicillin, 100 μg/ml streptomycin, 50 μM 2-mercaptoethanol, 10% FBS and 20 ng/ml M-CSF. All the cells were obtained from male origin.
Fresh peripheral Blood from healthy subjects was obtained with written informed consent in accordance with the Yale University ethics committee. PMN were isolated using PolymorphPrep according to previous report (Genestier et al., 2005). Purity was determined by FACS analysis with specific anti Gr-1 Ab and was determined to be > 95%. Spontaneous ROS of PMNs was measured by culturing cells (2 × 105 cells/200 μl) in RPMI-1640 medium containing 1% FBS. All the cells were obtained from male healthy subjects.
Cell Lines
Mouse Raw264.7 and human THP-1 cells were obtained from male origin; Human Hela cells were from female origin; Human HEK293 cells were suggested to be of male origin.
METHOD DETAILS
Preparation of Digoxin Immobilized Affinity Resin
TentaGel NH2 Resin (0.22 mmole/g loading, 0.5 g, 1 eq) was activated with diisocyanato hexane (1.1 mmol, 10eq) in DMF (5 mL) in a BioRad PolyPrep reaction vessel to provide isocyanate functionalized beads. Activated resin was washed 3x 10 mL DMF and was reacted with Digoxin (1.1 mmole, 10 eq) in 0.1% DMAP in DMF (5 mL). After digoxin coupling the beads were washed 3x DMF, 3x MeOH, 3x DCM and dried. Digoxin negative control beads were prepared by quenching the isocyanate functionalized beads with methylamine (0.22 mmole, 2 eq) in 0.1% DMAP in DMF (5 mL), and washed 3x with DMF, 3x with MeOH, 3x with DCM and dried under vacuum.
Immunoprecipitation and Western Blot
Whole-cell lysates were obtained through RIPA buffer lysis, and isolation of nuclear extract was performed using Nuclear Extraction Kit. Total PKM2 was immunoprecipitated in nuclear extract from Raw264.7 cells using anti-PKM2 (D78A4) antibody followed by SDS-PAGE, protein silver staining and western blot.
Quantitative Real-Time RT–PCR
Total RNA was extracted using TRIZOL (Invitrogen), and cDNA was generated with an oligo (dT) primer and the Superscript II system (Invitrogen, USA) followed by analysis using LightCycler 480 system (Roche). Real-time PCR was performed using SYBR Green real-time PCR analysis (Roche). Expression of GAPDH was used to standardize the samples, and the results were expressed as a ratio relative to control. Primer sequences used for quantitative PCR were previously reported (Ouyang, 2013).
Generation of PKM2, HIF-1α stable knockout cells by CRISPR/Cas9 system
Mouse Hif1α sgRNA, Pkm2 sgRNA and Scrambled sgRNA CRISPR/Cas9 All-in-One lentivirus set were purchased from Applied Biological Materials (Cat # K4459515, K011). Pkm2 sgRNA was custom designed to ensure Pkm2 variant 2 (NM_011099) is the target and Pkm2 variant 1 (NM_001253883.1) is not affected.
Mouse Raw264.7 macrophages were transduced overnight with the lentiviral particles at the multiplicity of infection (MOI) of 50 for two repeats. The transduced cells were then transferred into proper culture plate, and the cell colonies were isolated after puromycin (6 μg/ml) selection. The Pkm2 and Hif1α knockout cells used in the experiment were selected from multiple cell colonies, and knockout efficacy of targeting gene was measured by qRT-PCR using specific primers.
Transfection and luciferase reporter assay
Human HEK 293T cells were transiently transfected with NFκB, HRE, LXR, PPARα, RXR promoter luciferase reporter construct and induced plasmids together with Renilla luciferase (Rluc) control reporter vector by Lipofectamine™ 2000 reagent. All the luciferase activities were measured and normalized to Rluc or β-galactosidase activity and the normalized value with the percentage of control group was indicated.
LPS/D-galactosamine Model of Acute Liver Injury, TAA Model of Acute Liver Injury, and HFD model of Steatohepatitis and Chronic Liver Injury
Eight-week-old male C57BL/6J mice were IP injected with 1mg/kg body weight LPS (Sigma) and 500mg/kg body weight D-galactosamine for 5 h, 200mg/kg body weight TAA (Sigma) for 24 h.
Eight-week-old male C57BL/6J mice were Mice were on High Fat Diet (HFD, 45% fat, D12451 Research Diets) or chow. Animals were studied after 12 weeks of feeding.
At the end of the protocols, the whole livers and serum were collected for histological, cytological, biochemical, and molecular analysis.
Chromatin Immunoprecipitation (ChIP) Quantitative PCR (ChIP-qPCR)
ChIP was performed using ChIP assay kit (Upstate). Raw264.7 cell Lysates were incubated with primary antibodies; Anti-PKM2 antibody, Anti-Histone H3, negative control anti-IgG, and positive control Pol II Antibody. Q-PCR was carried out using SYBR green (Roche), and the specific primers were used as follows: mouse HIF-1α promoter region, 5′-ctgggcaaactgttaccg-3′ (forward) and 5′-cctgcaagaaacgctgaa-3′ (reverse); mouse Hif1α promoter distal region, 5′-ctgtttctgggctgtcaagt-3′ (forward) and 5′- agtgaccccatcttaccaca-3′ (reverse); mouse Nox4 promoter region, 5′-CTTGGGCTTCTGACTGTGA-3′ (forward), 5′- GAGCCGGAAGTAGCCTTT-3′ (reverse); mouse p65 promoter region, 5′- CAGGTGTCTGCCTAGTCCTC-3′ (forward), 5′-TTCCACACGTTTTCCTAAGC-3′ (reverse); mouse Il1b promoter region, 5′-CACAGAAGCACCATCCAGT-3′ (forward), 5′-AGATGCACACCCAGAAGTG-3′.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are expressed as the mean ± s.e.m. and were compared by one-way ANOVA and Student-Newman-Keuls test. Student’s t test was used for two-group analyses, except mentioned specifically in the text. Differences in values were considered significant at p < 0.05.
Supplementary Material
Highlights.
Digoxin protects the liver from a wide range of sterile injury
Digoxin down-regulates HIF-1α transcription initiated by sterile injury
Digoxin inhibits the transcription of PKM2 dependent genes
Digoxin binds PKM2 and attenuates the interaction between PKM2 and histones
In Brief.
XXX et al show that the cardiac glycoside, digoxin, protects against liver disease by modulating the HIF-1a/oxidative stress pathway in sterile inflammation. Digoxin binds PKM2, limiting its availability to regulate transcription of pro-inflammatory genes. This study identifies a novel protective role for digoxin in alcoholic and non-alcoholic steatohepatitis.
Acknowledgments
This study was funded by VA Merit award and NIH grants 2R56DK0 5U01AA021912-02 (to R. B. and W. Z. M); P30 pilot grant (to X. O.); K08DK092281 (to R. H.). The Yale Liver Center core facilities were funded by NIH by grant DK P30-034989. P.P. was supported by the Intramural Research Program of NIAAA/NIH, B.T.N by the Hungarian-American Enterprise Scholarship Fund. National Institute on Alcohol Abuse and Alcoholism (NIAAA) (1U01AA021908-01 and 1U01AA020821). Joaq.C was supported by Juan Rodes Scholarship by AEEH (Asociacion Española para el Estudio del Higado). Peter Gareiss from (P.G.) the Yale Center for Molecular Discovery prepared the digoxin immobilized affinity resin. Rolando Garcia-Milian (R.G.M) from Cushing/Whitney Medical Library of Yale University for the support of IPA program analysis.
W.Z.M and X.O. are listed as co-inventors on a preliminary patent application by Yale University for the use of digoxin in hepatoprotection.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTION
X.O., F.S.W, B.G., R.B., Joaq.C., P.S., Joan. C., N.J.T., R.G.K., P.P., W.Z.M.: Hypothesis generation, conceptual design, data analysis and manuscript preparation.
X.O., S.N.H., J.Y.Z., D.F., R.L.P., S.Y.C., R.H., Y. C., W. H.Y., I.G.M., B.T.N.: Conducting experiments and data analysis.
References
- Anastasiou D, Yu Y, Israelsen WJ, Jiang JK, Boxer MB, Hong BS, Tempel W, Dimov S, Shen M, Jha A, et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nature chemical biology. 2012;8:839–847. doi: 10.1038/nchembio.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arteel GE, Iimuro Y, Yin M, Raleigh JA, Thurman RG. Chronic enteral ethanol treatment causes hypoxia in rat liver tissue in vivo. Hepatology. 1997;25:920–926. doi: 10.1002/hep.510250422. [DOI] [PubMed] [Google Scholar]
- Arteel GE, Raleigh JA, Bradford BU, Thurman RG. Acute alcohol produces hypoxia directly in rat liver tissue in vivo: role of Kupffer cells. Am J Physiol. 1996;271:G494–500. doi: 10.1152/ajpgi.1996.271.3.G494. [DOI] [PubMed] [Google Scholar]
- Azoitei N, Becher A, Steinestel K, Rouhi A, Diepold K, Genze F, Simmet T, Seufferlein T. PKM2 promotes tumor angiogenesis by regulating HIF-1alpha through NF-kappaB activation. Molecular cancer. 2016;15:3. doi: 10.1186/s12943-015-0490-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model) Nat Protoc. 2013;8:627–637. doi: 10.1038/nprot.2013.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cederbaum AI. Iron and CYP2E1-dependent oxidative stress and toxicity. Alcohol. 2003;30:115–120. doi: 10.1016/s0741-8329(03)00104-6. [DOI] [PubMed] [Google Scholar]
- Cederbaum AI, Lu Y, Wu D. Role of oxidative stress in alcohol-induced liver injury. Arch Toxicol. 2009;83:519–548. doi: 10.1007/s00204-009-0432-0. [DOI] [PubMed] [Google Scholar]
- Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- Cichoz-Lach H, Michalak A. Oxidative stress as a crucial factor in liver diseases. World J Gastroenterol. 2014;20:8082–8091. doi: 10.3748/wjg.v20.i25.8082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazel Y, Koenig AB, Sayiner M, Goodman ZD, Younossi ZM. Epidemiology and natural history of non-alcoholic fatty liver disease. Metabolism. 2016 doi: 10.1016/j.metabol.2016.01.012. [DOI] [PubMed] [Google Scholar]
- Feldstein AE, Bailey SM. Emerging role of redox dysregulation in alcoholic and nonalcoholic fatty liver disease. Antioxid Redox Signal. 2011;15:421–424. doi: 10.1089/ars.2011.3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genestier AL, Michallet MC, Prevost G, Bellot G, Chalabreysse L, Peyrol S, Thivolet F, Etienne J, Lina G, Vallette FM, et al. Staphylococcus aureus Panton-Valentine leukocidin directly targets mitochondria and induces Bax-independent apoptosis of human neutrophils. J Clin Invest. 2005;115:3117–3127. doi: 10.1172/JCI22684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang JX, Torok NJ. NADPH Oxidases in Chronic Liver Diseases. Adv Hepatol. 2014;2014 doi: 10.1155/2014/742931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju C, Colgan SP, Eltzschig HK. Hypoxia-inducible factors as molecular targets for liver diseases. J Mol Med (Berl) 2016;94:613–627. doi: 10.1007/s00109-016-1408-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, Herder C, Carstensen M, Krausch M, Knoefel WT, et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015;21:739–746. doi: 10.1016/j.cmet.2015.04.004. [DOI] [PubMed] [Google Scholar]
- Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology. 2012;143:1158–1172. doi: 10.1053/j.gastro.2012.09.008. [DOI] [PubMed] [Google Scholar]
- Lee YS, Kim JW, Osborne O, Oh da Y, Sasik R, Schenk S, Chen A, Chung H, Murphy A, Watkins SM, et al. Increased adipocyte O2 consumption triggers HIF-1alpha, causing inflammation and insulin resistance in obesity. Cell. 2014;157:1339–1352. doi: 10.1016/j.cell.2014.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefere S, Van Steenkiste C, Verhelst X, Van Vlierberghe H, Devisscher L, Geerts A. Hypoxia-regulated mechanisms in the pathogenesis of obesity and non-alcoholic fatty liver disease. Cell Mol Life Sci. 2016 doi: 10.1007/s00018-016-2222-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Tan HY, Wang N, Zhang ZJ, Lao L, Wong CW, Feng Y. The Role of Oxidative Stress and Antioxidants in Liver Diseases. Int J Mol Sci. 2015;16:26087–26124. doi: 10.3390/ijms161125942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincet H, Icard P. How do glycolytic enzymes favour cancer cell proliferation by nonmetabolic functions? Oncogene. 2015;34:3751–3759. doi: 10.1038/onc.2014.320. [DOI] [PubMed] [Google Scholar]
- Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, Cole RN, Pandey A, Semenza GL. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145:732–744. doi: 10.1016/j.cell.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Semenza GL. Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol Metab. 2012;23:560–566. doi: 10.1016/j.tem.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Molecular cell. 2010;40:294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantena SK, Vaughn DP, Andringa KK, Eccleston HB, King AL, Abrams GA, Doeller JE, Kraus DW, Darley-Usmar VM, Bailey SM. High fat diet induces dysregulation of hepatic oxygen gradients and mitochondrial function in vivo. Biochem J. 2009;417:183–193. doi: 10.1042/BJ20080868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesini G, Brizi M, Bianchi G, Tomassetti S, Bugianesi E, Lenzi M, McCullough AJ, Natale S, Forlani G, Melchionda N. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes. 2001;50:1844–1850. doi: 10.2337/diabetes.50.8.1844. [DOI] [PubMed] [Google Scholar]
- Marchesini G, Marzocchi R. Metabolic syndrome and NASH. Clinics in liver disease. 2007;11:105–117. ix. doi: 10.1016/j.cld.2007.02.013. [DOI] [PubMed] [Google Scholar]
- Mehal WZ. The Gordian Knot of dysbiosis, obesity and NAFLD. Nat Rev Gastroenterol Hepatol. 2013;10:637–644. doi: 10.1038/nrgastro.2013.146. [DOI] [PubMed] [Google Scholar]
- Mehal WZ. The inflammasome in liver injury and non-alcoholic fatty liver disease. Dig Dis. 2014;32:507–515. doi: 10.1159/000360495. [DOI] [PubMed] [Google Scholar]
- Mehal WZ. Cells on Fire. Sci Am. 2015;312:44–49. doi: 10.1038/scientificamerican0615-44. [DOI] [PubMed] [Google Scholar]
- Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxidants & redox signaling. 2014;20:1126–1167. doi: 10.1089/ars.2012.5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath B, Levin I, Csak T, Petrasek J, Mueller C, Kodys K, Catalano D, Mandrekar P, Szabo G. Hepatocyte-specific hypoxia-inducible factor-1alpha is a determinant of lipid accumulation and liver injury in alcohol-induced steatosis in mice. Hepatology. 2011;53:1526–1537. doi: 10.1002/hep.24256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang X, Ghani A, Malik A, Wilder T, Colegio OR, Flavell RA, Cronstein BN, Mehal WZ. Adenosine is required for sustained inflammasome activation via the A(2)A receptor and the HIF-1alpha pathway. Nat Commun. 2013;4:2909. doi: 10.1038/ncomms3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang XGA, Malik A, Wilder T, Flavell RA, Cronstein BN, Mehal WZ. Adenosine is required for inflammasome activation via the A2A receptor and a HIF-1α pathway Nat Commun. 2013 doi: 10.1038/ncomms3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik YH, Kim J, Aoyama T, De Minicis S, Bataller R, Brenner DA. Role of NADPH oxidases in liver fibrosis. Antioxid Redox Signal. 2014;20:2854–2872. doi: 10.1089/ars.2013.5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer BF, Clegg DJ. Ascent to altitude as a weight loss method: the good and bad of hypoxia inducible factor activation. Obesity. 2014;22:311–317. doi: 10.1002/oby.20499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, van den Bosch MW, Quinn SR, Domingo-Fernandez R, Johnston DG, et al. Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015;21:65–80. doi: 10.1016/j.cmet.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin MK, Drager LF, Yao Q, Bevans-Fonti S, Yoo DY, Jun JC, Aja S, Bhanot S, Polotsky VY. Metabolic consequences of high-fat diet are attenuated by suppression of HIF-1alpha. PloS one. 2012;7:e46562. doi: 10.1371/journal.pone.0046562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TW. Digitalis. Mechanisms of action and clinical use. N Engl J Med. 1988;318:358–365. doi: 10.1056/NEJM198802113180606. [DOI] [PubMed] [Google Scholar]
- Tamada M, Suematsu M, Saya H. Pyruvate kinase M2: multiple faces for conferring benefits on cancer cells. Clin Cancer Res. 2012;18:5554–5561. doi: 10.1158/1078-0432.CCR-12-0859. [DOI] [PubMed] [Google Scholar]
- Zhang H, Qian DZ, Tan YS, Lee K, Gao P, Ren YR, Rey S, Hammers H, Chang D, Pili R, et al. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc Natl Acad Sci U S A. 2008;105:19579–19586. doi: 10.1073/pnas.0809763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann C, Ferenci P, Pifl C, Yurdaydin C, Ebner J, Lassmann H, Roth E, Hortnagl H. Hepatic encephalopathy in thioacetamide-induced acute liver failure in rats: characterization of an improved model and study of amino acid-ergic neurotransmission. Hepatology. 1989;9:594–601. doi: 10.1002/hep.1840090414. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.