

Pathogens must metabolize host-derived compounds during infection and properly regulate the responsible pathways. Carnitine is a common eukaryotic-associated quaternary amine compound that can be catabolized by Pseudomonas aeruginosa. Here we expand on our understanding of how this metabolic pathway is regulated and provide details on how carnitine catabolism is intertwined with glycine betaine catabolism at the level of transcriptional control.
KEYWORDS: metabolism, osmoprotectant, quaternary amine, transcriptional regulation
ABSTRACT
The common environmental bacterium and opportunistic pathogen Pseudomonas aeruginosa encodes diverse metabolic pathways and associated regulatory networks allowing it to thrive in these different environments. In an effort to understand P. aeruginosa metabolism and detection of host-derived compounds, we previously identified CdhR and GbdR as members of the AraC transcription factor family that regulate catabolism of the quaternary amine compounds carnitine and glycine betaine, respectively. In this study, our goal was to further characterize regulation of carnitine catabolism by the transcription factor CdhR. CdhR binds in a concentration-dependent manner upstream of the carnitine catabolism operon promoter (PcaiXcdhCABhocS). We identified the CdhR binding site and determined that it overlaps with the GbdR binding site in the caiX-cdhR intergenic region. Carnitine catabolism is repressed by glucose and glycine betaine, and here we show this happens at the transcriptional level. Furthermore, we show that CdhR enhances its own expression and that GbdR contributes to cdhR expression by enhancing the level of basal expression. The intertwined regulation of caiX and cdhR transcription by GbdR and CdhR suggests that carnitine catabolism is under tight but tuneable control.
IMPORTANCE Pathogens must metabolize host-derived compounds during infection and properly regulate the responsible pathways. Carnitine is a common eukaryotic-associated quaternary amine compound that can be catabolized by Pseudomonas aeruginosa. Here we expand on our understanding of how this metabolic pathway is regulated and provide details on how carnitine catabolism is intertwined with glycine betaine catabolism at the level of transcriptional control.
INTRODUCTION
Pseudomonas aeruginosa is an opportunistic Gram-negative pathogen found in a wide variety of environments, often enriched in the drinking water distribution system, from which it can readily contaminate surfaces and medical devices in hospitals (1–4). The ability to transition from the preinfection niche to the host is likely a key to its success as a pathogen. P. aeruginosa encodes diverse pathways for metabolism of host-derived and host-independent carbon and nitrogen sources, allowing it to survive and thrive while undergoing these environmental transitions.
l-Carnitine (here referred to as “carnitine”) and O-acylcarnitines are quaternary amine compounds abundant in host tissues, one function of which is to shuttle fatty acids in and out of the mitochondria for β-oxidation in animals (5). There are no animal enzymes that can degrade carnitine (6): consequently any degradation in the host is due to bacteria, either by an aerobic pathway like P. aeruginosa (7) or an anaerobic pathway like many bacteria in the mammalian intestine (8). P. aeruginosa can acquire carnitine from the environment by import through the somewhat promiscuous ABC transporter CbcWV using the CaiX periplasmic substrate binding protein (9). The enzymes required for carnitine or short-chain acylcarnitine catabolism in P. aeruginosa are encoded in the carnitine catabolism operon, caiX-cdhCAB-hocS (Fig. 1A), enabling metabolism for osmoprotection, virulence factor induction, and nutrition (10–13). Medium- and long-chain acylcarnitines, with the exception of octanoylcarnitine, can be used as sole carbon sources as well (10), but the enzymes required for the hydrolysis of these compounds have not been identified.
FIG 1 .

Diagram of the P. aeruginosa PAO1 carnitine catabolism operon and the catabolic pathway. (A) Arrows represent the individual open reading frames of the carnitine catabolism operon and the regulator chdR. Below the arrow is the designated gene name. (B) Diagram of the cdhR-caiX intragenic region organized such that caiX transcription occurs left to right. The orange box denotes the cdhR 5′ UTR, the dark green box marks the position of the CdhR and GbdR binding sites (CdhR binding sequence listed below), and the light green box denotes the caiX 5′ UTR. (C) Diagram of the converging carnitine and choline catabolism pathways. Black arrows represent an enzymatic step in the catabolic pathway, and the gene names are italicized below. The blue arrows represent positive regulation by either CdhR or GbdR, and the T-bar represents repression by BetI.
Part of the metabolic flexibility of P. aeruginosa can be attributed to its large repertoire of regulatory proteins—more than 9% of the genome is dedicated to transcriptional regulation (14). One such regulator is CdhR, which is divergently transcribed from the carnitine catabolism operon and is required for growth on carnitine and induction of the carnitine operon (11). Aerobic carnitine degradation (Fig. 1B) leads to the formation of glycine betaine (GB), the catabolic genes for which are transcriptionally regulated by GbdR (15). GbdR and CdhR not only regulate catabolism of related quaternary amine compounds, but both are AraC family transcription factors that belong to the same glutamine amidotransferase-1-like (GATase-1) transcriptional regulator subfamily (GATRs) and are similar in sequence (62% positive and 44% identical) (16, 17).
In this study, we expand our understanding of the regulation of carnitine catabolism by identifying the CdhR binding site, determining essential binding site residues, and demonstrating that catabolite repression of carnitine catabolism by glucose and glycine betaine functions at the level of transcription of the carnitine operon. We show that GbdR can bind the intergenic region of caiX-cdhR in an orientation supporting regulation of cdhR. Finally, CdhR positively regulates its own expression in the presence of carnitine but represses basal expression in the absence of ligand, a repression that is alleviated when GbdR is present, suggesting a potential hierarchy of CdhR and GbdR binding at their overlapping binding sites.
RESULTS
Mapping the caiX promoter region.
The carnitine catabolism operon, caiX-cdhCAB-hocS (Fig. 1A), is driven from a promoter located between caiX and cdhR (Fig. 1B) and encodes proteins that are responsible for the hydrolysis of short-chain acylcarnitines (10) and degradation of carnitine to glycine betaine (GB) (11, 18) (Fig. 1C). cdhR is divergently transcribed from the carnitine catabolism operon (Fig. 1A) and was previously shown to be required for induction of caiX and growth on carnitine (11). Primer extension was used to define the transcriptional start site of caiX and cdhR. Plasmids containing the target regions were used to increase RNA copy number, particularly for cdhR, as native transcripts are at low abundance (19). The length of the caiX primer extension product placed the transcriptional start site 25 bases upstream of the translational start site at a thymine residue. The length of the cdhR primer extension product placed the transcriptional start site 63 bases upstream of the translational start site at a cytosine. The relative sizes of the two untranscribed regions (UTRs) and their spacing compared to the CdhR binding site are shown in Fig. 1B.
To expand upon our understanding of CdhR’s role in carnitine catabolism, we narrowed down the binding site of CdhR by promoter mapping of PcaiX using four sequentially shorter lacZYA transcriptional reporters. The constructs end at bp +3 from the caiX transcriptional start site and begin at bp −399 (pJAM22), bp −302 (pJAM23), bp −216 (pJAM24), and bp −112 (pJAM25) (Fig. 2A). In the presence of carnitine, all constructs except pJAM25 were induced, indicating that the binding site of CdhR is between bp −216 and −112 or overlaps −112 (Fig. 2A) in relation to the transcriptional start site.
FIG 2 .

Mapping the caiX promoter and its CdhR binding site. (A) Transcriptional fusions of lacZ to the upstream region of caiX start at bp +3 from the caiX transcriptional start site and end at base pairs marked in the figure. P. aeruginosa PA14 strains carrying each construct were grown in MOPS with 20 mM pyruvate and 20 µg·ml−1 gentamicin, with (black bars) or without (white bars) 1 mM carnitine. β-Galactosidase activities for these caiX transcriptional fusions are reported in Miller units. Error bars represent standard deviation from three biological replicates, and results are representative of three independent experiments. (B) EMSA with biotin-labeled caiX promoter DNA probe alone (lane 1) or with increasing concentrations of purified MBP-CdhR (lanes 2 and 3). An unlabeled (cold) caiX probe was used to compete for binding of MBP-CdhR from the labeled probe (lane 4). An unrelated dhc DNA probe was used to show specificity of MBP-CdhR binding to caiX (lanes 5 to 8).
Based upon CdhR-dependent transcriptional induction of caiX (11) (Fig. 2A), we assayed the capability of purified maltose binding protein (MBP)-CdhR to bind to the upstream activation sequence (UAS) of caiX. Using biotin-labeled caiX UAS as the DNA probe and purified MBP-CdhR, electrophoretic mobility shift assays (EMSAs) revealed that MBP-CdhR binds the caiX UAS in a concentration-dependent manner (Fig. 2B, lanes 1 to 3). The binding interaction between MBP-CdhR and caiX probe can be competed with unlabeled caiX probe (lane 4), but not an unlabeled dhcA probe (lane 5), and MBP-CdhR does not shift the nonspecific UAS, dhcA (lanes 6 to 8). These data demonstrate that MBP-CdhR binds specifically to the caiX UAS.
Identification of the CdhR binding site sequence and bases required for induction of caiX.
Promoter mapping and EMSAs determined the region of DNA necessary for carnitine-dependent caiX transcriptional induction and CdhR binding (Fig. 2). To further characterize the CdhR DNA contact site within the caiX UAS, we employed DNase I footprinting. There was a characteristic and site-specific protection of caiX UAS DNA as the MBP-CdhR concentration increased (Fig. 3A). Comparing the zone of protection to the A+G ladder, we were able to identify the CdhR contact region, stretching 34 bp (Fig. 3A). The CdhR binding site reveals that each half-site contains the sequence GGTCGC with a 15-bp spacer, which is very similar to the binding sites of three other related P. aeruginosa transcription factors, GbdR (17), SouR (20), and ArgR (21). To determine the importance of the half-site residues, we made mutations in the distal half-site by systematically changing partially overlapping dinucleotides to adenines (pJAM122 to pJAM127). Induction from these mutated cdhR binding sites demonstrated that the second guanine residue in the half-site is important for caiX expression, but complete abolishment of carnitine-dependent induction requires an additional mutation of the adjacent thymine residue (Fig. 3B).
FIG 3 .

The CdhR binding site and key residues for CdhR-dependent induction. (A) A DNase I footprinting assay was performed by taking the caiX UAS end labeled with 32P and adding increasing concentrations of MBP-CdhR, followed by DNase I treatment and nondenaturing 5% polyacrylamide TBE gel. The first lane of the gel is the A+G sequencing ladder, and the nanomolar concentration of MBP-CdhR is marked. (B) The caiX enhancer site was mutated by changing two bases at a time (in red and underlined) in the caiX distal binding site to adenosines and fused to lacZ. The P. aeruginosa PA14 wild type carrying each of the plasmids was grown in MOPS with 20 mM pyruvate at 20 µg·ml−1, with or without 1 mM carnitine for 4 h, and then β-galactosidase activity was reported as Miller units. Error bars represent standard deviations from three biological replicates, and results are representative of three independent experiments. Data were analyzed using a two-way analysis of variance (ANOVA) with a Sidak’s multiple-comparison posttest comparing each mutant’s pyruvate to carnitine. Abbreviations: P, pyruvate; C, carnitine; n.s., not significant; *, P < 0.05; ***, P < 0.001.
Glucose and glycine betaine repress the transcription of caiX.
In P. aeruginosa, the enzymatic activity of carnitine dehydrogenase (CDH) declines until no longer detectable if the cells are switched from carnitine to glucose as the sole carbon source (22). CDH activity oscillates when carnitine is the sole carbon source, predicted to occur when the catabolic product glycine betaine is produced, resulting in initiation of a negative-feedback oscillation loop (22). We used the caiX-lacZYA transcriptional reporter (pJAM22) to determine if catabolite repression is transcriptionally regulated. Maximal catabolite repression is seen at 4 mM glucose, while GB represses transcription to a lesser extent and at a higher concentration (Fig. 4).
FIG 4 .
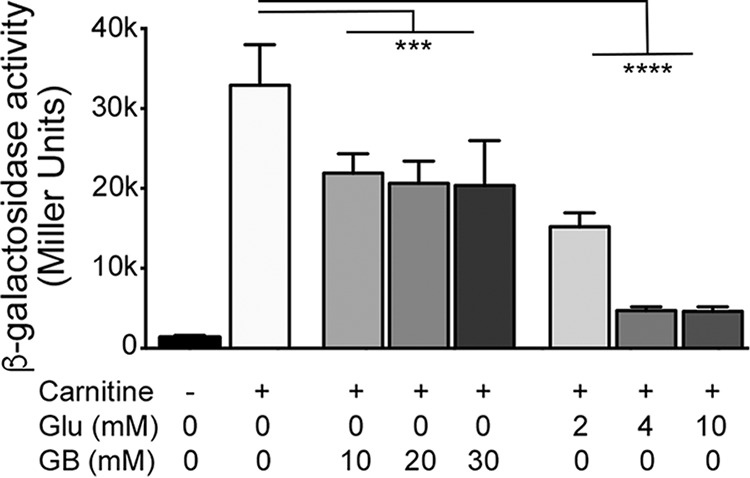
caiX transcription is repressed by glucose and glycine betaine. P. aeruginosa PA14 with PcaiX-lacZ (pJAM22) was grown in MOPS medium with 20 mM pyruvate and 20 µg·ml−1 gentamicin, with or without 1 mM carnitine. Glucose or glycine betaine was added at the millimolar concentrations noted. Cultures were induced for 4 h prior to measurement of β-galactosidase activity. Error bars represent standard deviations from three biological replicates, and results are representative of three independent experiments. Data were analyzed using a one-way ANOVA with a Dunnett’s multiple-comparison posttest comparing each condition to the condition with carnitine alone. Bars denote that all data underneath are different from carnitine alone with the same statistical certainty. Abbreviations: Glu, glucose; GB, glycine betaine; ***, P < 0.001; ****, P < 0.0001.
CdhR can bind, but does not regulate, the ABC transporter cbcXWV.
After establishing the CdhR binding sequence, GGTCGC-[N15]-GGTCGC, we searched for this sequence in the P. aeruginosa PAO1 genome using the DNA motif search tool from the Pseudomonas Genome Database website (23). Only two identical sites were identified within intergenic regions: caiX-cdhR and cbcX-sdaB, both of which are involved in carnitine metabolism (9, 11). CbcXWV is an ABC transporter, and the core transporter proteins CbcWV are required for growth on carnitine along with the substrate binding component CaiX (9). An MBP-CdhR EMSA with the cbcXWV UAS probe showed MBP-CdhR binding in a concentration-dependent manner (Fig. 5A), but quantitative reverse transcription-PCR (qRT-PCR) revealed that carnitine cannot support induction of cbcX (Fig. 5B). Deletion of cdhCA eliminates production of GB from carnitine (Fig. 1B) (11); therefore, for carnitine to lead to cbcXWV induction, GB must be produced to enable cbcXWV induction via GbdR (17).
FIG 5 .
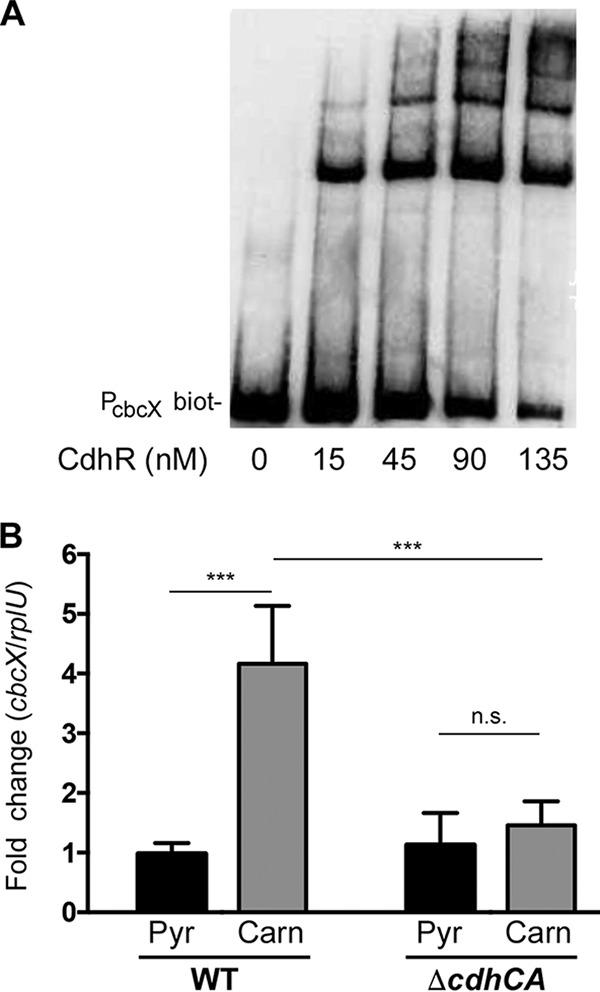
CdhR binds but does not regulate cbcXWV expression. (A) EMSA with a biotin-labeled (biot-) cbcX upstream region and purified MBP-CdhR in increasing concentrations. (B) Relative expression of cbcX was calculated based on the expression in WT pyruvate normalized to the rplU transcript using qRT-PCR. Three biological samples were run in triplicate, and the graph represents the mean values and standard deviation. Data were analyzed using a two-way ANOVA with a Tukey’s multiple comparison posttest comparing all strains and conditions to each other. Ends of the bars denote the comparison groups shown. Abbreviations: Pyr, pyruvate; Carn, carnitine; n.s., not significant; ***, P < 0.001.
Roles of GbdR and CdhR at the caiX-cdhR intergenic region.
The ability of CdhR to bind a known member of the GbdR regulon (cbcX) (17, 24), the detection of the GbdR binding consensus in the caiX-cdhR intergenic region (17), and the overlapping positions of the CdhR and GbdR consensus sites in the caiX UAS led us to investigate the role of GbdR in carnitine regulation. We predicted that GbdR would be able to bind the cdhR UAS, and an EMSA with purified MBP-GbdR demonstrated that MBP-GbdR binds the caiX-cdhR intergenic probe in a concentration-dependent manner (Fig. 6A). When the conserved CG residues for the GbdR-binding distal half-site were mutated to AA (GCCGC to GCAAC), binding was lost (Fig. 6A), as previously seen for similar mutations in the plcH and choE distal half-sites (17). Since GbdR binds the caiX-cdhR intergenic region, induction of caiX (pJAM22) was assessed in the wild type (WT) and a gbdR deletion mutant in both PA14 and PAO1 backgrounds. The gbdR deletion mutant has less induction of PcaiX-lacZ (Fig. 6C), but this defect is likely due to a defect in carnitine import (9) (Fig. 4).
FIG 6 .
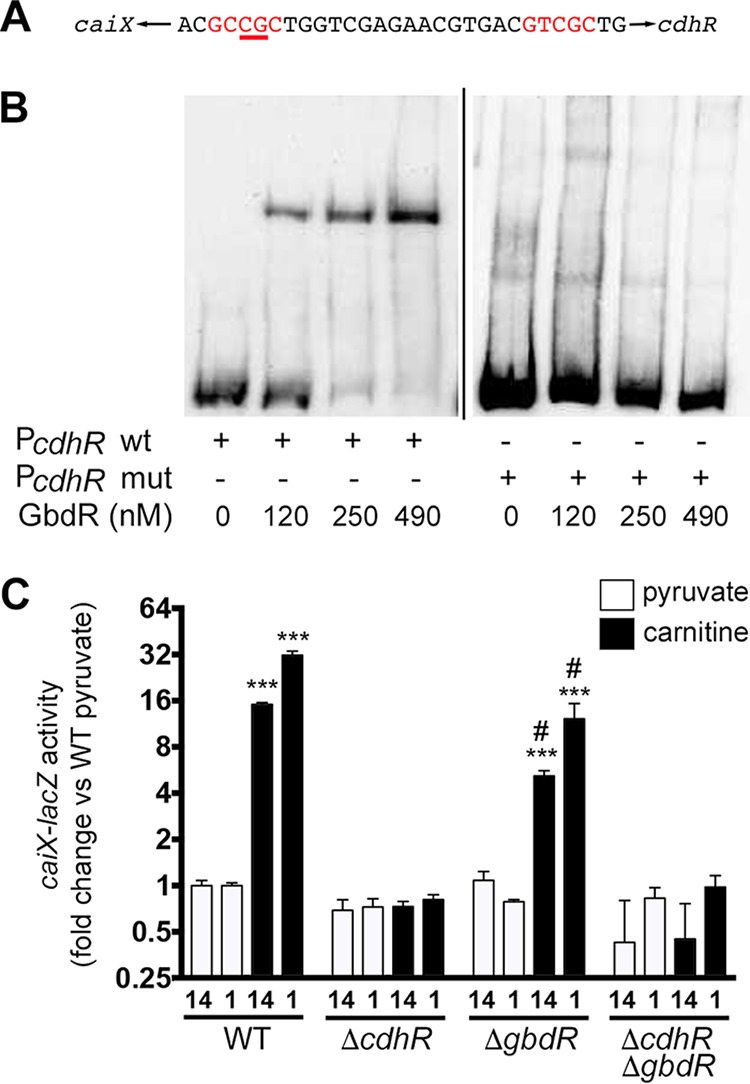
GbdR binds the caiX-cdhR intergenic region but does not induce transcription of caiX. (A) EMSAs were performed with increasing concentrations of purified MBP-GbdR with either the biotin-labeled cdhR wild-type probe or a mutant binding site probe. The mutated probe has the distal half-site CG residues (in relation to cdhR) changed to AA. (B) β-Galactosidase assay with a caiX-lacZ reporter plasmid (pJAM22) in wild-type, ΔcdhR, ΔgbdR, or ΔcdhR ΔgbdR strains of both PA14 (14) and PAO1 (1) grown in MOPS, 20 mM pyruvate, and 20 µg·ml−1 gentamicin. Induced cultures have an additional 1 mM carnitine. Error bars represent standard deviations from three biological replicates, and results are representative of three independent experiments. Data were analyzed with two-way ANOVA with a Tukey’s multiple-comparison test comparing all mutants and conditions within a given strain. (PA14 was not compared to PAO1.) Abbreviations: ***, P < 0.001 compared to WT with pyruvate; #, P < 0.01 compared to WT with carnitine.
After establishing that CdhR and GbdR bind the intergenic region of caiX-cdhR (Fig. 2B, 3A, and 6), we wanted to determine how these two transcription factors (TFs) impacted cdhR expression. Using two different translational reporter fusions—one carried on a plasmid in both PA14 and PAO1 backgrounds and one integrated into the chromosome at the attTn7 site—it became apparent that CdhR has a role in its own expression. In the wild type, carnitine increased expression of cdhR compared to the basal expression level (pyruvate) (Fig. 7A). In the absence of gbdR, carnitine still induces cdhR, but basal expression of cdhR is decreased compared to that of the wild type and a cdhR deletion mutant, suggesting that GbdR functions to relieve repression at this locus (Fig. 7A). The similarity of the activity in the double deletion mutant with the gbdR single deletion in the presence of carnitine suggests that carnitine detection by CdhR allows relief of the baseline CdhR-dependent repression. The effects of CdhR and GbdR on cdhR expression are also seen with single-cell expression levels (Fig. 7B), showing both the general effects seen with the population assessment (Fig. 7A), as well as the heterogeneity in individual cell expression. These findings are summarized in a genetic model (Fig. 8).
FIG 7 .

CdhR promotes cdhR expression, and GbdR dampens basal repression. (A) WT, ΔcdhR ΔgbdR, and ΔcdhR ΔgbdR strains in both PA14 (14) and PAO1 (1) backgrounds carrying a cdhR-lacZ translational plasmid reporter (pJAM135) were grown in MOPS with 20 mM pyruvate and 20 µg·ml−1 gentamicin, with or without 1 mM carnitine, and β-galactosidase activity was reported as fold change over WT pyruvate. Data were analyzed using a two-way ANOVA with a Sidak’s multiple-comparison posttest comparing to the WT pyruvate condition within each strain. (PA14 was not compared to PAO1.) *, P < 0.05. (B) PAO1 WT, ΔcdhR, and ΔgbdR strains, all with the translational fusion cdhR-yfp integrated at the attTn7 site, were grown on MOPS agar pads with 20 mM pyruvate and with or without 1 mM carnitine. Cells were imaged under phase-contrast and YFP fluorescence every 10 min at 32°C. Data were analyzed using a one-way ANOVA with a Dunnett’s multiple-comparison posttest comparing each time point within a strain to pyruvate at time zero (t = 0). ***, P < 0.001.
FIG 8 .
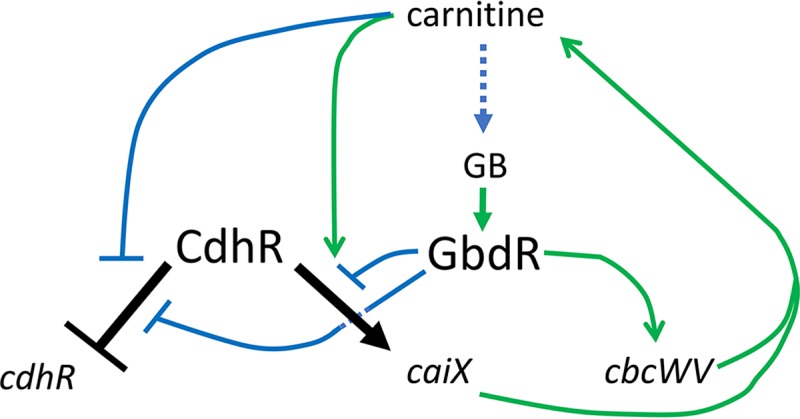
Regulation of caiX and cdhR by CdhR and GbdR. Shown is a genetic network diagram of the interactions known and proposed in this study. Arrows indicate positive interactions (induction or activation), and T-bars indicate negative interactions (repression or other inhibition). The regulatory steps are black for CdhR and green or blue for interactions between other members in the network. The dotted blue line from carnitine to glycine betaine (GB) marks the metabolic conversion noted in Fig. 1C.
DISCUSSION
The metabolic diversity of P. aeruginosa is controlled by a vast set of regulators, including one-component transcription factors that are often critical for regulating catabolism of alternate nutrient sources (25). One such transcription factor family that can control carbon metabolism and virulence is the AraC family (26, 27). In this study, we expand on our understanding of carnitine catabolism and show how two AraC family transcription factors, CdhR and GbdR, whose metabolic pathways converge at glycine betaine (GB) (Fig. 1C), are intertwined (Fig. 8). We identified the CdhR binding site in the caiX-cdhR intergenic region and through reporter and DNA binding assays reveal this site as an additional GbdR binding site. Through reporter fusions, we were able to show that CdhR regulates its own expression and is responsive to carnitine. We also show that GbdR binding to the caiX-cdhR region assists in regulating carnitine catabolism by inhibiting repression of cdhR transcription, thus maintaining the basal cdhR expression level.
The first question we addressed was how CdhR binds and regulates the carnitine operon. CaiF in Escherichia coli is the only other carnitine regulator that has been characterized to date, and it is a degenerate AraC TF that lacks the traditional N-terminal domain but maintains the helix-turn-helix (HTH) DNA binding domains (28). The CaiF amino acid sequence is 13% identical and 23% similar with 44% gaps compared to CdhR, with most of the similar regions within the HTH domains, therefore likely functioning in a manner much different than CdhR. caiF is transcribed in the opposite direction from the E. coli carnitine metabolism operon (caiTABCDE), which codes for utilization of carnitine as a terminal electron acceptor generating γ-butyrobetaine and binds to inverted repeats in the caiF UAS (28, 29). P. aeruginosa and other Gram-negative bacteria, capable of utilizing carnitine as a carbon and/or nitrogen source, contain a carnitine catabolism operon capable of generating glycine betaine and are organized in similar fashion to one another with an AraC family transcription factor divergently transcribed from the catabolic operon (Fig. 1A) (30). CdhR binds to direct repeats, and the binding site is upstream of the caiX promoter (Fig. 1B and 3A), categorizing CdhR as a class I activator (binds upstream and recruits RNA polymerase via the C-terminal domain of the alpha subunit [31]) that requires both half-sites for induction of caiX (Fig. 3B).
Kleber and Aurich analyzed the activity of carnitine dehydrogenase (CDH) with respect to glucose and glycine betaine (GB) and showed that glucose, as a preferred carbon source, is catabolite repressive (22), whereas glycine betaine leads to repression of CDH activity, resulting in oscillations of activity as carnitine is catabolized to glycine betaine (22). Our data demonstrate that repression of carnitine catabolism by glucose and glycine betaine can be controlled at the level of transcription (Fig. 4). Negative feedback by glycine betaine is likely GbdR dependent based on GbdR’s capability to bind the caiX-cdhR intergenic region (Fig. 6B) and GbdR’s responsiveness to GB (32). P. aeruginosa maintains intracellular glycine betaine pools, and GbdR fine-tuning of carnitine catabolism may be directly related to sustaining the homeostatic levels of glycine betaine, as the GB pool has a physiological impact on nutrients, osmoprotection, and virulence (33).
We performed an alignment using the Pseudomonas.com DNA motif search tool of the newly identified CdhR binding sequence to the PAO1 genome and identified two intergenic regions: caiX-cdhR and cbcX-sdaB (23). This led us to investigate if CdhR contributes to the regulation of carnitine import by the ABC transporter CbcWV in association with CaiX, which is required for growth on carnitine (9). Even though CdhR binds to the cbcXWV UAS in vitro, it does not contribute to cbcXWV expression (Fig. 5). We propose that an unknown transporter imports carnitine, which is metabolized to GB, and this GB drives expression of cbcXWV in a GbdR-dependent manner, which coupled with CdhR-dependent expression of caiX allows for a larger flux of carnitine needed to support growth (9). This is similar to the mechanism of choline import for cbcXWV induction, which is termed priming (24).
We previously characterized the GbdR regulon and identified the intergenic region of caiX-cdhR to have a GbdR binding site (17). Here we report that GbdR binds the caiX-cdhR intergenic region in vitro and the conserved CG residues are necessary for GbdR binding, as mutation of these residues to AA results in loss of binding (Fig. 6A). Based upon the conserved residues being located in the GbdR distal half-site (17), the orientation of GbdR binding is likely toward cdhR activation and not caiX. Interestingly, the GbdR and CdhR binding sites overlap in the caiX-cdhR intergenic region, which led to the hypothesis that GbdR has a role in regulating carnitine catabolism and, in particular, cdhR.
We propose a model (Fig. 8) showing the genetic network containing CdhR and GbdR. In the absence of carnitine, CdhR binds to its caiX-cdhR intergenic target sequence site in a manner that inhibits cdhR expression. GbdR competes for this binding site and limits CdhR-dependent inhibition of cdhR. Analysis of the DNA sequence up- and downstream of the CdhR binding site reveals multiple CdhR half-sites. These half-sites could participate in inhibition of CdhR basal expression by looping. CdhR inhibition of its own expression may be similar to the AraC “light-switch” mechanism, in which the regulator binds upstream sites to loop DNA and restrict polymerase access to the promoter (34). Another possibility is that CdhR oligomerizes along the DNA, nucleated at these half-sites, to dampen cdhR basal expression. In this model, GbdR would compete for binding, relieving CdhR-dependent cdhR repression. Upon CdhR detection of carnitine, a change occurs allowing increased expression of cdhR and caiX (Fig. 7). As the catabolic product GB builds up, the cell controls the flux of carnitine catabolism by GB-dependent transcriptional repression (Fig. 4), which is likely regulated by GbdR (Fig. 8).
In conclusion, we show that CdhR regulates the carnitine catabolic operon by directly binding the caiX UAS, and this is likely the only site in the genome where its potential binding is effective in altering gene transcription. We also show that catabolic repression of carnitine catabolism can function through direct repression of carnitine catabolic operon transcription. Finally, we show the role of GbdR in regulation at this site. These data suggest a system for fine-tuning carnitine catabolism in relation to other carbon sources and that both GbdR and CdhR alter transcription from the caiX-cdhR intergenic regulatory region.
MATERIALS AND METHODS
Strains and growth conditions.
P. aeruginosa wild-type strains PAO1 and PA14 and their derivatives (Table 1) were maintained on Pseudomonas isolation agar (PIA [Difco]) plates or Lennox broth (LB) liquid, and when necessary 50 or 40 µg·ml−1 gentamicin was added to the media, respectively. Escherichia coli NEB5α or T7 Express E. coli (NEB C3016) cells were maintained on LB plates with 10 µg·ml−1 gentamicin, LB liquid with 7 µg·ml−1 gentamicin, LB plates or liquid with 125 µg·ml−1 carbenicillin, or LB plates or liquid with 100 µg·ml−1 kanamycin.
TABLE 1 .
Strains and plasmids used in this study
| Strain or plasmid | Genotype or description | Reference or source |
|---|---|---|
| Strains | ||
| P. aeruginosa PAO1 | ||
| MJ79 | Wild type | 14 |
| MJ80 | ΔgbdR | 15 |
| JM236 | ΔcdhR | This study |
| JM253 | Wild type attTn7::88-89intYFPCFP-2 | This study |
| JM339 | ΔcdhR attTn7::88-89intYFPCFP-2 | This study |
| JM340 | ΔgbdR attTn7::88-89intYFPCFP-2 | This study |
| MJ784 | ΔgbdR ΔcdhR | This study |
| P. aeruginosa PA14 | ||
| MJ101 | Wild type | 42 |
| MJ11 | ΔcdhR | 11 |
| MJ26 | ΔgbdR | 15 |
| MJ262 | ΔcdhCA | 11 |
| JM179 | ΔcdhR ΔgbdR | This study |
| E. coli | ||
| MJ340 | Wild-type S17λpir | |
| DH5α | NEB C2987 | NEB |
| T7Express | NEB C3016 | NEB |
| Plasmids | ||
| pMQ30 | Suicide vector, Gmr | 36 |
| pMQ80 | High-copy-no. Pseudomonas vector, Gmr | 36 |
| pMal-C2X | T7-expressing vector, MBP N-terminal tag, Ampr | NEB |
| pTNS2 | Plasmid carrying attTn7 transposase | 43 |
| pUC18-mini-Tn7T-Gm | Gmr on mini-Tn7T | 35 |
| pUC18-mini-Tn7T-Gm-eyfp | Gmr on mini-Tn7T with YFP | 35 |
| pUCP22 | High-copy-no. Pseudomonas stabilization vector, Gmr | 44 |
| pMW5 | lacZYA in pUCP22 | 32 |
| pMW79 | PA14 genomic clone of PA5380-PA5389 in pMQ80 | 11 |
| pPA5380KO | gbdR deletion construct in pEX18-Gm | 15 |
| pJAM22 | Promoter caiX-lacZYA transcriptional fusion A | This study |
| pJAM23 | Promoter caiX-lacZYA transcriptional fusion B | This study |
| pJAM24 | Promoter caiX-lacZYA transcriptional fusion C | This study |
| pJAM25 | Promoter caiX-lacZYA transcriptional fusion D | This study |
| pJAM50 | PA5389 in pMal-C2X | This study |
| pJAM76 | YFP-CFP in pMQ80 | This study |
| pJAM86 | CFP PA5388-PA5389 intergenic region YFP in pUC18mini, DR2 | This study |
| pJAM90 | PA5389 deletion construct in pMQ30 | This study |
| pJAM122 | Promoter caiX-lacZYA transcriptional fusion | This study |
| pJAM123 | Promoter caiX-lacZYA mut 1 transcriptional fusion | This study |
| pJAM124 | Promoter caiX-lacZYA mut 2 transcriptional fusion | This study |
| pJAM125 | Promoter caiX-lacZYA mut 3 transcriptional fusion | This study |
| pJAM126 | Promoter caiX-lacZYA mut 4 transcriptional fusion | This study |
| pJAM127 | Promoter caiX-lacZYA mut 5 transcriptional fusion | This study |
| pJAM130 | Promoter caiX-lacZYA mut 6 transcriptional fusion | This study |
| pJAM131 | C terminus of lacZ in pUCP22 | This study |
| pJAM135 | PcdhRlacZYA in pUCP22 | This study |
Deletion constructs.
A deletion of PA5389 (cdhR) in PAO1 was made in the wild-type background (MJ79). The upstream and downstream regions of PA5389 were PCR amplified from PAO1 genomic DNA with the primers 5389GOIF1KpnI, 5389SOEGOIR1, 5389SOEGOIF1, and 5389GOIR1BamHI (Table 2). The splice overlap extension PCR product was ligated into the Zero Blunt plasmid pCR-Blunt (Invitrogen), excised with EcoRI, and ligated into similarly cut pMQ30 to generate pJAM90. Conjugation of donor E. coli S17λpir carrying pJAM90 with PAO1 and subsequent screening were done as previously described (10), generating strain JM236. The deletion strain, JM179, was verified by lack of growth on carnitine and by PCR.
TABLE 2 .
Primers used in this study
| Primer | Sequence (5′ to 3′)a |
|---|---|
| Deletion constructs | |
| 5389GOIF1KpnI | ATAGGGTACCGAAGAACACCACCCACTGCT |
| 5389SOEGOIR1 | AAGTACGAAGGCGACTCGACCATGGAGAAGCCCATTACCGAGAAGC |
| 5389SOEGOIF1 | GCTTCTCGGTAATGGGCTTCTCCATGGTCGAGTCGCCTTCGTACTT |
| 5389GOIR1BamHI | ATCGTCTTCGCTGTTTTTCC |
| Protein expression construct | |
| 5389Mal-c2xF | GCATCAGAATTCTCCCAGGACTTCTGGTTTCT |
| 5389Mal-c2xR | GCATCAAAGCTTTCAGCCTCGCTCAGCTCGA |
| Primer extension | |
| 5388primerextension | 5′-Fluorescein 6-FAM-ACTGGCCAGGATCAGCAGG |
| 5389primerextension | 5′-Fluorescein 6-FAM-AGACAGTATCGGCCTCAGGAA |
| EMSA probes | |
| PA5388promF3 | AAGCTTGTGCCAGCGGTAGAGGTC |
| PA5388promR | TGAGGTACCTTGATTGTTTTTCTGCGAGGT |
| PA5388promRbiot | Biotin-TTGATTGTTTTTCTGCGAGGT |
| 5389EMSA-F | ATGAAAGCTTGCAGCAGGAGAAACCAGAAG |
| 5389EMSA-R-biot | Biotin-TTGATTGTTTTTCTGCGAGGT |
| 5389EMSA-Mut3F | GGACGGCGGCGAAGCGCACTGCGAAGACC |
| cbcXprom-F | CCGGCAAAGACCACTATGAT |
| cbcXprom-R-biot | Biotin-GAACTCCTCTGCAGGGTAAGG |
| dhcprom-F-biot | Biotin-GAGGCTTTCCTCCAGGCTCT |
| dhcprom-R | GGATGGTACCCTCTTCCGGCTCTTGTGATT |
| dhcprom-F | GAACTCCTCTGCAGGGTAAGG |
| Transcriptional reporters | |
| PA5388promR | TGAGGTACCTTGATTGTTTTTCTGCGAGGT |
| PA5388promF1 | ATGAAAGCTTACAGCAGGTCGCCTTTCTT |
| PA5388promF3 | AAGCTTGTGCCAGCGGTAGAGGTC |
| PA5388promF2 | ATGAAAGCTTGCAGCAGGAGAAACCAGAAG |
| PA5388promF4 | AAGCTTCTGCAGTGCAAGAGCTGGT |
| P5388pos | ATGAAAGCTTCGCTTGGCAATGGCCAGGTCGCT |
| P5388mut1 | ATGAAAGCTTCGCTTGGCAATGGCCAAATCGCT |
| P5388mut2 | ATGAAAGCTTCGCTTGGCAATGGCCAGAACGCT |
| P5388mut3 | ATGAAAGCTTCGCTTGGCAATGGCCAGGAAGCT |
| P5388mut4 | ATGAAAGCTTCGCTTGGCAATGGCCAGGTAACT |
| P5388mut5 | ATGAAAGCTTCGCTTGGCAATGGCCAGGTCAAT |
| P5388mut6 | ATGAAAGCTTCGCTTGGCAATGGCCAGATCGCT |
| Translational reporters | |
| 2-lacZCtermFhindcla | GCAAGCTTATTATCGATGAGCGTGGTGGTTATGC |
| 2-lacZCtermRsmakpn | CGGTACCCGGGGATCCTTATTTTTGACACCAGACC |
| YFP R HindIII | GCATCAAAGCTTATTACTTGTACAGCTCGTCCA |
| YFP F Kpn Sal | GACAGCGGTACCAATCGTCGACCATATGCTGAGCAAGGGCGAGG |
| 88-89YC-DR#2ycF | GGGCACCACCCCGGTGAACAGCTCCTCGCCCTTGCTCAGCATGGGGCGCTCCGGGGTTGA |
| 88-89YC-DR#2ycR | CGGCACCACGCCGGTGAACAGCTCCTCGCCCTTGCTCAGCATCGGTCTCCCCTCGTGCGG |
6-FAM, 6-carboxyfluorescein.
A double mutant of cdhR and gbdR in PA14 was made using the PA14 ΔgbdR strain MJ26 as the recipient strain for mating with E. coli S17λpir carrying pJAM90 as described above. The double mutant strain was verified by PCR. The PAO1 cdhR gbdR double mutant was made using the PAO1 ΔcdhR strain JM236 as the recipient strain for mating with E. coli S17λpir carrying pPA5280KO (15). The double mutant was verified by PCR and its inability to grow on glycine betaine.
Construction of transcriptional reporter constructs.
Promoter mapping of PA5388 (caiX) was done using four different truncations of the caiX upstream region cloned as transcriptional reporter constructs. pMW79 was used as the PCR template for amplification of the upstream region of caiX, using PA5388promR as the reverse primer for all products with the forward primers PA5388promF1, PA5388promF2, PA5388promF3, and PA5388promF4. These four PCR products were ligated into the pCR-Blunt, digested with KpnI and HindIII, and ligated into similarly cut pMW5 to yield four different P5388lacZYA transcriptional fusions (pJAM22 to pJAM25). Each was transformed by electroporation into PA14 wild type and selected on PIA-gentamicin plates.
To assess which residues were essential for induction of caiX, seven different transcriptional reporters were constructed by amplifying the caiX binding site from pMW79 using the PA5388promR primer and seven different forward primers: P5388pos, P5388mut1, P5388mut2, P5388mut3, P5388mut4, P5388mut5, and P5388mut6. PCR products were digested with HindIII and KpnI and ligated into similarly cut pMW5. The resulting plasmids, pJAM122 to pJAM127 and pJAM130, were transformed into PA14 wild type by electroporation and selected on PIA-gentamicin.
Construction of cdhR translational reporter constructs.
To determine control of cdhR, a divergent fluorescent translational reporter with yellow fluorescent protein (YFP) and cyan fluorescent protein (CFP) on either side of the intergenic region of PA5388 and PA5389 was constructed. YFP was amplified using primers YFP F Kpn Sal and YFP R HindIII from the template pUC18miniTn7T-Gm-eyfp (35). The YFP fragment was ligated into pCR Zero Blunt plasmid, digested with HindIII and KpnI, and subsequently ligated into similarly cut pMQ80. The resulting plasmid was digested with EcoRI and SacI and ligated with an EcoRI- and SacI-cut P. aeruginosa-codon-biased CFP, to generate pJAM76. The PA5388-PA5389 intergenic region was amplified using primers 88-89YC-DR#2ycF and 88-89YCDR#2ycR. pJAM76 was digested with SalI to linearize the plasmid for recombination of the PA5388-PA5389 intergenic regions using yeast cloning in Saccharomyces cerevisiae via the method of Shanks et al. (36). The resultant plasmid was digested with HindIII, and the insert was ligated into similarly cut pUC18-mini-Tn7T-Gm, yielding pJAM86, which was coelectroporated with pTNS2 to insert CFP-PA5388-PA5389-YFP onto the chromosome at the attTn7 site (35) of the PAO1 wild-type, PAO1 ΔcdhR, and PAO1 ΔgbdR strains.
A PA5389 translational lacZ fusion was made by inserting a gBlock (IDT) that has 320 bp of the upstream region of PA5389 into the translational start site of PA5389 and then from the translational start site of lacZ to +850 bases in the lacZ gene into pJAM131. pJAM131 was built by PCR amplification of the C-terminal end of lacZ with primers 2-lacZCtermFhindcla and 2-lacZCtermRsmakpn, digested with HindIII and KpnI, and ligated into a similarly cut pUCP22. The gBlock was digested with HindIII and ClaI and ligated into similarly cut pJAM131 to make pJAM135.
Cloning and expression of MBP-CdhR.
The plasmid pJAM50, expressing a maltose binding protein (MBP) fusion to the amino terminus of CdhR, was made by amplifying cdhR with primers 5389Mal-C2XF and 5389Mal-C2XR and ligating the product into pCR-Blunt. The cdhR coding segment was excised with EcoRI and HindIII and ligated into the similarly cut pMal-C2X (NEB), transformed by electroporation into T7 Express E. coli (NEB C2566), and selected on LB-carbenicillin to generate strain JM153.
To express MBP-CdhR, 1 liter of JM153 was grown at 37°C in LB with 75 µg·ml−1 carbenicillin to an optical density at 600 nm (OD600) of 0.4. The culture was induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and grown for an additional 3 h. Cells were collected by centrifugation and resuspended in 20 mM Tris HCl–150 mM NaCl (pH 7.4) with 1× Halt protease inhibitor cocktail (Thermo) and lysed by French press. DNase I was added to the lysate and incubate room temperature for 15 min. Samples were centrifuged at 13,000 rpm for 30 min at 4°C to separate the soluble and insoluble fractions. The soluble fraction was filtered through a 0.22-µm-pore filter and applied to an Affi-Gel heparin gel (Bio Rad) column. The column was washed with column buffer (10 mM KH2PO4, 150 mM NaCl [pH 7.4] in 1× phosphate-buffered saline [PBS]), and MBP-CdhR was eluted from the column with column buffer plus 1.5 M NaCl. Fractions containing MBP-CdhR were run on a 12% SDS denaturing gel and stained with Coomassie brilliant blue (Thermo) to verify purity and determine which fractions to use. Elutions containing MBP-CdhR were dialyzed in a 20,000 molecular weight cutoff (MWCO) Slidealyzer (Thermo) overnight at 4°C in buffer (20 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA [pH 7.4]).
Primer extension.
The transcriptional start sites of caiX and cdhR were determined by growing PA14 the wild type carrying pMW79 or PA14 wild type carrying pJAM135, respectively, in morpholinepropanesulfonic acid (MOPS) medium with 20 mM pyruvate, 5 mM glucose, and 20 µg·ml−1 gentamicin overnight at 37°C. Cells were washed, resuspended to an OD600 of 0.3 in MOPS supplemented with 20 mM l-carnitine or pyruvate and 20 µg·ml−1 gentamicin in a 12-well plate, and grown for 7 h at 37°C while shaking. Cells were collected by centrifugation, and RNA was purified using the RNeasy minikit (Qiagen) as per the manufacturer’s instructions. Primer extension was completed using the purified RNA as the template for cDNA generation using Superscript II reverse transcriptase (Invitrogen) and the 5′-fluorescein-labeled primers 5388primerextension and 5389primerextension. The cDNA products were analyzed by capillary electrophoresis to determine the size of the DNA fragment of the induced carnitine sample versus the pyruvate control sample by comparison to DNA size standards.
β-Galactosidase assays.
Strains were grown overnight at 37°C in MOPS minimal medium supplemented with 20 mM pyruvate, 5 mM glucose, and 20 µg·ml−1 gentamicin. Cells were collected by centrifugation, washed, resuspended in MOPS, and inoculated into MOPS with 20 mM pyruvate and 20 µg·ml−1 gentamicin, with or without 1 mM carnitine as the inducing compound. Induction was carried out for 4 h at 37°C, and β-galactosidase assays were performed according to Miller (37).
Catabolite repression.
The PA14 wild type carrying pJAM22 was grown overnight at 37°C in MOPS with 20 mM pyruvate, 5 mM glucose, and 20 µg·ml−1 gentamicin. Cells were collected by centrifugation, washed, resuspended in MOPS, and then inoculated into MOPS with 20 mM pyruvate and 20 µg·ml−1 gentamicin at a final OD600 of 0.05. All samples had a 1 mM concentration of the inducting compound carnitine, except for the noninducing control (pyruvate alone). Catabolite repression samples had glucose at 2, 4, or 10 mM or had glycine betaine at 20 or 40 mM. Cultures were induced for 4 h at 37°C, and β-galactosidase assays were performed according to Miller (37).
EMSAs.
To determine binding of MBP-CdhR or MBP-GbdR to promoters, EMSAs were performed as previously described (17). Briefly, caiX, dhc, cbcX, cdhR, or cdhR mutant promoter DNA fragments were made by PCR amplification from PA14 genomic DNA template with primers listed in the EMSA primer section in Table 2 and verified by sequencing. The DNA probes were dialyzed in 1/4 Tris-EDTA (TE) on a 0.025-µm-pore filter for 20 min. EMSAs were conducted following the LightShift chemiluminescent EMSA kit instructions (Pierce) as modified by Hampel et al. (17) with labeled probes used at 1 fmol·µl−1 and unlabeled competitor at 600 fmol·µl−1. Samples were electrophoresed on 5% Tris-borate-EDTA (TBE) nondenaturing gels, transferred to BioDyne-B nylon membrane (Thermo), and detected using the Thermo chemiluminescent nucleic acid detection module per the manufacturer’s instructions.
DNase I footprinting.
DNase I footprinting was performed as described previously (17). Briefly, the target DNA was made by labeling the 5′ end of either PA5388promR or PA5388promF2 primer with 32P using T4 polynucleotide kinase and [γ-32P]ATP and amplifying the PA14 caiX upstream region. The radiolabeled PCR product was purified from a 5% polyacrylamide Tris-borate-EDTA gel. The MBP-CdhR footprinting assay was conducted as in Brenowitz et al. (38), as modified in the study by Hampel et al. (17).
Fluorescence microscopy.
P. aeruginosa PAO1 WT, ΔcdhR, and ΔgbdR strains with the divergent fluorescent reporter PA5388-PA5389 intergenic region CFPYFP-DR2 (pJAM86) at the attTn7 site, were grown overnight in MOPS with 20 mM pyruvate and 5 mM glucose at 37°C. Cells were collected by centrifugation, washed, and resuspended in MOPS to an OD600 of 1.0. A 1/20 dilution was made, and 1 µl was placed on the center of an agar pad (1.5% low-melting-point agarose, MOPS medium with 20 mM pyruvate, and with or without 1 mM carnitine). The agar pad was placed cell side down on a 50-mm glass bottom cell culture dish (Warner Instruments). Samples were imaged on a Nikon Ti-E every 10 min for 6 h at 32°C. Image stacks were imported into Fiji 2.0.0-rc-29/1.49 s (39) using the Bio-Formats importer 5.1.1 (40). A rolling variance filter on the contrast channel was used to define background areas, and after an additional threshold was applied, the “find maxima” macro was used to define cell areas. Using masks from the contrast channel, mean pixel intensity measurements were taken from the YFP channel. A framewise background intensity correction was performed for each cell using R 3.2.0 (41).
ACKNOWLEDGMENTS
We thank Kristin Schutz for technical assistance in strain generation.
J.A.M. was supported by a National Institutes of Health Institutional NRSA Fellowship (T32 AI055402). J.A.M. and M.J.W. were supported by grants from the National Institute of General Medical Sciences (P20 GM103496) and the National Institute of Allergy and Infectious Disease (R01 AI103003). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Green SK, Schroth MN, Cho JJ, Kominos SK, Vitanza-Jack VB. 1974. Agricultural plants and soil as a reservoir for Pseudomonas aeruginosa. Appl Microbiol 28:987–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lyczak JB, Cannon CL, Pier GB. 2000. Establishment of Pseudomonas aeruginosa infection: lessons from a versatile opportunist. Microbes Infect 2:1051–1060. doi: 10.1016/S1286-4579(00)01259-4. [DOI] [PubMed] [Google Scholar]
- 3.van der Kooij D, Oranje JP, Hijnen WA. 1982. Growth of Pseudomonas aeruginosa in tap water in relation to utilization of substrates at concentrations of a few micrograms per liter. Appl Environ Microbiol 44:1086–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rogues AM, Boulestreau H, Lashéras A, Boyer A, Gruson D, Merle C, Castaing Y, Bébear CM, Gachie JP. 2007. Contribution of tap water to patient colonisation with Pseudomonas aeruginosa in a medical intensive care unit. J Hosp Infect 67:72–78. doi: 10.1016/j.jhin.2007.06.019. [DOI] [PubMed] [Google Scholar]
- 5.Bremer J. 1983. Carnitine—metabolism and functions. Physiol Rev 63:1420–1480. doi: 10.1152/physrev.1983.63.4.1420. [DOI] [PubMed] [Google Scholar]
- 6.Rebouche CJ, Chenard CA. 1991. Metabolic fate of dietary carnitine in human adults: identification and quantification of urinary and fecal metabolites. J Nutr 121:539–546. doi: 10.1093/jn/121.4.539. [DOI] [PubMed] [Google Scholar]
- 7.Lindstedt G, Lindstedt S, Midtvedt T, Tofft M. 1967. The formation and degradation of carnitine in Pseudomonas. Biochemistry 6:1262–1270. doi: 10.1021/bi00857a006. [DOI] [Google Scholar]
- 8.Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, Smith JD, DiDonato JA, Chen J, Li H, Wu GD, Lewis JD, Warrier M, Brown JM, Krauss RM, Tang WH, Bushman FD, Lusis AJ, Hazen SL. 2013. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med 19:576–585. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen C, Malek AA, Wargo MJ, Hogan DA, Beattie GA. 2010. The ATP-binding cassette transporter Cbc (choline/betaine/carnitine) recruits multiple substrate-binding proteins with strong specificity for distinct quaternary ammonium compounds. Mol Microbiol 75:29–45. doi: 10.1111/j.1365-2958.2009.06962.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meadows JA, Wargo MJ. 2013. Characterization of Pseudomonas aeruginosa growth on O-acylcarnitines and identification of a short-chain acylcarnitine hydrolase. Appl Environ Microbiol 79:3355–3363. doi: 10.1128/AEM.03943-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wargo MJ, Hogan DA. 2009. Identification of genes required for Pseudomonas aeruginosa carnitine catabolism. Microbiology 155:2411–2419. doi: 10.1099/mic.0.028787-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lucchesi GI, Lisa TA, Casale CH, Domenech CE. 1995. Carnitine resembles choline in the induction of cholinesterase, acid phosphatase, and phospholipase C and in its action as an osmoprotectant in Pseudomonas aeruginosa. Curr Microbiol 30:55–60. doi: 10.1007/BF00294525. [DOI] [PubMed] [Google Scholar]
- 13.Kleber HP, Schöpp W, Sorger H, Tauchert H, Aurich H. 1967. Formation of 3-dehydrocarnitine from l-carnitine through the action of a Pseudomonas aeruginosa enzyme. Acta Biol Med Ger 19:659–667. [PubMed] [Google Scholar]
- 14.Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FS, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Smith K, Spencer D, Wong GK, Wu Z, Paulsen IT, Reizer J, Saier MH, Hancock RE, Lory S, Olson MV. 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- 15.Wargo MJ, Szwergold BS, Hogan DA. 2008. Identification of two gene clusters and a transcriptional regulator required for Pseudomonas aeruginosa glycine betaine catabolism. J Bacteriol 190:2690–2699. doi: 10.1128/JB.01393-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nock AM, Wargo MJ. 2016. Choline catabolism in Burkholderia thailandensis is regulated by multiple glutamine amidotransferase 1-containing AraC family transcriptional regulators. J Bacteriol 198:2503–2514. doi: 10.1128/JB.00372-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hampel KJ, Labauve AE, Meadows JA, Fitzsimmons LF, Nock AM, Wargo MJ. 2014. Characterization of the GbdR regulon in Pseudomonas aeruginosa. J Bacteriol 196:7–15. doi: 10.1128/JB.01055-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bastard K, Smith AA, Vergne-Vaxelaire C, Perret A, Zaparucha A, De Melo-Minardi R, Mariage A, Boutard M, Debard A, Lechaplais C, Pelle C, Pellouin V, Perchat N, Petit JL, Kreimeyer A, Medigue C, Weissenbach J, Artiguenave F, De Berardinis V, Vallenet D, Salanoubat M. 2014. Revealing the hidden functional diversity of an enzyme family. Nat Chem Biol 10:42–49. doi: 10.1038/nchembio.1387. [DOI] [PubMed] [Google Scholar]
- 19.Kolodrubetz D, Schleif R. 1981. Identification of araC protein and two-dimensional gels, its in vivo instability and normal level. J Mol Biol 149:133–139. doi: 10.1016/0022-2836(81)90265-5. [DOI] [PubMed] [Google Scholar]
- 20.Willsey GG, Wargo MJ. 2015. Sarcosine catabolism in Pseudomonas aeruginosa is transcriptionally regulated by SouR. J Bacteriol 198:301–310. doi: 10.1128/JB.00739-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park SM, Lu CD, Abdelal AT. 1997. Purification and characterization of an arginine regulatory protein, ArgR, from Pseudomonas aeruginosa and its interactions with the control regions for the car, argF, and aru operons. J Bacteriol 179:5309–5317. doi: 10.1128/jb.179.17.5309-5317.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kleber HP, Aurich H. 1967. Damped oscillations in the synthesis of carnitine dehydrogenase by Pseudomonas aeruginosa. Hoppe Seylers Z Physiol Chem 348:1727–1729. [PubMed] [Google Scholar]
- 23.Winsor GL, Lam DK, Fleming L, Lo R, Whiteside MD, Yu NY, Hancock RE, Brinkman FS. 2011. Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res 39:D596–D600. doi: 10.1093/nar/gkq869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malek AA, Chen C, Wargo MJ, Beattie GA, Hogan DA. 2011. Roles of three transporters, CbcXWV, BetT1, and BetT3, in Pseudomonas aeruginosa choline uptake for catabolism. J Bacteriol 193:3033–3041. doi: 10.1128/JB.00160-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yahr TL, Greenberg EP. 2004. The genetic basis for the commitment to chronic versus acute infection in Pseudomonas aeruginosa. Mol Cell 16:497–498. doi: 10.1016/j.molcel.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 26.Yang J, Tauschek M, Robins-Browne RM. 2011. Control of bacterial virulence by AraC-like regulators that respond to chemical signals. Trends Microbiol 19:128–135. doi: 10.1016/j.tim.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 27.Martin RG, Rosner JL. 2001. The AraC transcriptional activators. Curr Opin Microbiol 4:132–137. doi: 10.1016/S1369-5274(00)00178-8. [DOI] [PubMed] [Google Scholar]
- 28.Eichler K, Buchet A, Lemke R, Kleber HP, Mandrand-Berthelot MA. 1996. Identification and characterization of the caiF gene encoding a potential transcriptional activator of carnitine metabolism in Escherichia coli. J Bacteriol 178:1248–1257. doi: 10.1128/jb.178.5.1248-1257.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buchet A, Nasser W, Eichler K, Mandrand-Berthelot MA. 1999. Positive co-regulation of the Escherichia coli carnitine pathway cai and fix operons by CRP and the CaiF activator. Mol Microbiol 34:562–575. doi: 10.1046/j.1365-2958.1999.01622.x. [DOI] [PubMed] [Google Scholar]
- 30.Uanschou C, Frieht R, Pittner F. 2005. What to learn from a comparative genomic sequence analysis of l-carnitine dehydrogenase. Monatsh Chem 136:1365–1381. doi: 10.1007/s00706-005-0331-x. [DOI] [Google Scholar]
- 31.Browning DF, Busby SJ. 2016. Local and global regulation of transcription initiation in bacteria. Nat Rev Microbiol 14:638–650. doi: 10.1038/nrmicro.2016.103. [DOI] [PubMed] [Google Scholar]
- 32.Wargo MJ, Ho TC, Gross MJ, Whittaker LA, Hogan DA. 2009. GbdR regulates Pseudomonas aeruginosa plcH and pchP transcription in response to choline catabolites. Infect Immun 77:1103–1111. doi: 10.1128/IAI.01008-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fitzsimmons LF, Hampel KJ, Wargo MJ. 2012. Cellular choline and glycine betaine pools impact osmoprotection and phospholipase C production in Pseudomonas aeruginosa. J Bacteriol 194:4718–4726. doi: 10.1128/JB.00596-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schleif R. 2010. AraC protein, regulation of the l-arabinose operon in Escherichia coli, and the light switch mechanism of AraC action. FEMS Microbiol Rev 34:779–796. doi: 10.1111/j.1574-6976.2010.00226.x. [DOI] [PubMed] [Google Scholar]
- 35.Choi KH, Schweizer HP. 2006. Mini-Tn7 insertion in bacteria with single attTn7 sites: example Pseudomonas aeruginosa. Nat Protoc 1:153–161. doi: 10.1038/nprot.2006.24. [DOI] [PubMed] [Google Scholar]
- 36.Shanks RM, Caiazza NC, Hinsa SM, Toutain CM, O’Toole GA. 2006. Saccharomyces cerevisiae-based molecular tool kit for manipulation of genes from Gram-negative bacteria. Appl Environ Microbiol 72:5027–5036. doi: 10.1128/AEM.00682-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller J. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 38.Brenowitz M, Senear DF, Kingston RE. 2001. DNase I footprint analysis of protein-DNA binding. Curr Protoc Mol Biol Chapter 12:Unit 12.4. doi: 10.1002/0471142727.mb1204s07. [DOI] [PubMed] [Google Scholar]
- 39.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Linkert M, Rueden CT, Allan C, Burel JM, Moore W, Patterson A, Loranger B, Moore J, Neves C, Macdonald D, Tarkowska A, Sticco C, Hill E, Rossner M, Eliceiri KW, Swedlow JR. 2010. Metadata matters: access to image data in the real world. J Cell Biol 189:777–782. doi: 10.1083/jcb.201004104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.R Development Core Team 2015. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 42.Rahme LG, Stevens EJ, Wolfort SF, Shao J, Tompkins RG, Ausubel FM. 1995. Common virulence factors for bacterial pathogenicity in plants and animals. Science 268:1899–1902. doi: 10.1126/science.7604262. [DOI] [PubMed] [Google Scholar]
- 43.Choi KH, Gaynor JB, White KG, Lopez C, Bosio CM, Karkhoff-Schweizer RR, Schweizer HP. 2005. A Tn7-based broad-range bacterial cloning and expression system. Nat Methods 2:443–448. doi: 10.1038/nmeth765. [DOI] [PubMed] [Google Scholar]
- 44.Schweizer HP. 1991. Escherichia-Pseudomonas shuttle vectors derived from pUC18/19. Gene 97:109–121. doi: 10.1016/0378-1119(91)90016-5. [DOI] [PubMed] [Google Scholar]