

Abstract
Background
Breast cancer risk for BRCA1 and BRCA2 pathogenic mutation carriers is modified by risk factors that cluster in families, including genetic modifiers of risk. We considered genetic modifiers of risk for carriers of high-risk mutations in other breast cancer susceptibility genes.
Methods
In a family known to carry the high-risk mutation PALB2:c.3113G>A (p.Trp1038*), whole-exome sequencing was performed on germline DNA from four affected women, three of whom were mutation carriers.
Results
RNASEL:p.Glu265* was identified in one of the PALB2 carriers who had two primary invasive breast cancer diagnoses before 50 years. Gene-panel testing of BRCA1, BRCA2, PALB2 and RNASEL in the Australian Breast Cancer Family Registry identified five carriers of RNASEL:p.Glu265* in 591 early onset breast cancer cases. Three of the five women (60%) carrying RNASEL:p.Glu265* also carried a pathogenic mutation in a breast cancer susceptibility gene compared with 30 carriers of pathogenic mutations in the 586 non-carriers of RNASEL:p.Glu265* (5%) (p < 0.002). Taqman genotyping demonstrated that the allele frequency of RNASEL:p.Glu265* was similar in affected and unaffected Australian women, consistent with other populations.
Conclusion
Our study suggests that RNASEL:p.Glu265* may be a genetic modifier of risk for early-onset breast cancer predisposition in carriers of high-risk mutations. Much larger case-case and case-control studies are warranted to test the association observed in this report.
Keywords: RNASEL:P.Glu265*, Breast cancer, Modifier risk gene, Early-onset cancer
Background
There is marked variability in individual cancer risk between and within BRCA1 and BRCA2 mutation carrier families [1, 2]. Accumulating evidence reviewed in [3] indicates that breast cancer risk in mutation carriers is modified by several risk factors that cluster in families, including genetic modifiers of risk that influence mutation penetrance. Segregation analyses studies have demonstrated that risk prediction models that allow for genes to modify effect on breast cancer risk in BRCA1 and BRCA2 mutation carriers fit significantly better to familial data than models without a modifying component.
Genetic modifiers of risk for carriers of high-risk mutations in other breast cancer susceptibility genes, such as PALB2, are yet to be described. In this study, we examined the exomes of key members of a multiple-case family segregating the pathogenic PALB2:c.3113G>A (p.Trp1038*) mutation (Family A, Fig. 1) to explore the possibility that additional genetic factors could be responsible for modifying the breast cancer risk in this family.
Fig. 1.
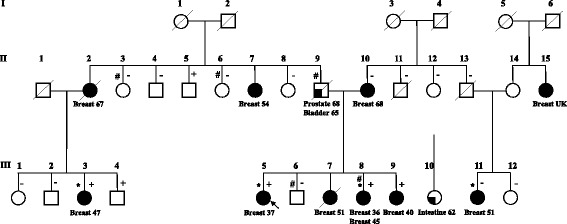
Pedigree of Family A (modified from Southey et al., [5]). + and –: carriers and non-carriers of PALB2:p.Trp1038*, respectively (data from [1]); #: carriers of RNASEL:p.Glu265*; *: individuals selected for whole-exome sequencing; arrow: proband. Breast cancer is indicated by black filled symbols, and other cancers are indicated by quarter-filled symbols
Methods
Subjects
The women in this study were ascertained via population-based sampling by the Australian site of the Breast Cancer Family Registry (ABCFR, [4]). Probands (defined as the first family members enrolled in the study, with or without a personal history of breast cancer) were identified from the Victoria and New South Wales cancer registries and invited to participate, regardless of family history.
All participants provided written informed consent for participation in this research program, which was approved by the ABCFR and the University of Melbourne Human Research Ethics Committee (Melbourne, VIC, Australia).
Whole-exome sequencing
Whole-exome sequencing (WES) was performed on the germline DNA of four affected women from Family A: the proband (III.5) and her sister (III.8), one maternal cousin (III.3) and one paternal cousin (III.11) (Fig. 1). Three of these women were carriers of PALB2:c.3113G>A (p.Trp1038*). Details of the family, the breast cancer diagnoses and histology are described in Southey et al. [5]. WES and bioinformatics analysis were described by Park et al. [6]. Briefly, genetic variants were assessed for relevance to cancer etiology. The highest priority group for further investigation included nonsense and frameshift variants and genetic variants predicted to affect consensus splice sites. Variants in genes that have been associated with cancer predisposition, encode proteins involved in cell cycle checkpoint control or DNA repair pathways and confirmed by Sanger sequencing were prioritised. By applying this approach, we identified the truncating variant RNASEL:p.Glu265*, which was previously reported to increase prostate cancer risk, as a candidate modifier variant.
Gene panel-testing
Gene-panel testing was performed on 591 probands participating to the ABCFR diagnosed under the age of 40 years who had biological sample available for testing.
Amplicon-based sequencing of the coding regions and proximal intron-exon junctions of RNASEL (NM_021133.3), BRCA1 (NM_007294.3), BRCA2 (NM_000059.3) and PALB2 (NM_024675.3) using the Hi-Plex protocol [7]. Massively parallel sequencing (150 bp paired-end) was performed on the MiSeq (Illumina, San Diego, CA, USA). Bioinformatics analysis and variant calling performed using ROVER as described in [8].
Classification of variants
Classification of genetic variants in BRCA1 and BRCA2 was performed in accordance with the Evidence-based Network for the Interpretation of Germline Mutant Alleles (ENIGMA) consortium’s recommendations (April 2016 update) [9].
Consistent with ENIGMA classification criteria, all loss-of-function genetic variants in PALB2 were considered “pathogenic”, unless there was evidence to the contrary.
Although ATM was not part of the panel-test, affected probands diagnosed under the age of 40 participating in the ABCFR have been genotyped for ATM:c.7271T>G by Chenevix-Trench et al. [10]. There is overwhelming data to support the association of this variant with breast cancer risk similar in magnitude to BRCA2 mutations (e.g. [11]).
Genotyping of RNASEL:P.Glu265*
RNASEL:p.E265* carrier frequency was determined by genotyping all probands participating in the ABCFR diagnosed with breast cancer regardless of age of onset (n = 1445, 591 of which were mutation-screened), and age-matched unaffected controls (n = 827). Clinical characteristics of the participants are presented in Table 1.
Table 1.
Clinical characteristics of the participants to the population-based Australian Breast Cancer Family Registry that were genotyped for RNASEL:p.Glu265*
| Cases (n = 1445) | Controls (n = 827) | |
|---|---|---|
| Age at diagnosis (years) | 42 [23–59] | n/a |
| Age at recruitment (years) | 43 [23–60] | 41 [20–60] |
| Ethnicity | ||
| White/Caucasian | 1345 (93%) | 710 (85.9%) |
| Asian and Pacific Islander | 85 (5.9%) | 32 (3.9%) |
| Other | 8 (0.6%) | 7 (0.8%) |
| Unknown | 7 (0.5%) | 78 (9.4%) |
| Laterality | ||
| Right breast | 713 (49.3%) | n/a |
| Left breast | 732 (50.7%) | n/a |
| Estrogen Receptor Status | ||
| Negative | 439 (30.4%) | n/a |
| Positive | 815 (56.4%) | n/a |
| Unknown | 191 (13.2%) | n/a |
| Grade | ||
| GI | 212 (14.6%) | n/a |
| GII | 526 (36.4%) | n/a |
| GIII | 555 (38.4%) | n/a |
| GIV | 3 (0.2%) | n/a |
| Unknown | 149 (10.4%) | n/a |
n/a: not applicable
GI, grade I; GII, grade II; GIII, Grade III; GIV, Grade IV
Genotyping was performed using a custom Taqman probe-based assay (ThermoFisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. Probe sequences are available upon request. The reactions were amplified and analysed on the LightCycler480 (Roche, Penzberg, Germany).
Statistical analysis
The difference in the prevalence of RNASEL:p.Glu265* in pathogenic mutation carriers and non-carriers (case only analysis) was tested using a two-sided Fisher’s Exact test.
Results
Whole-exome sequencing
The mutation PALB2:c.3113G>A (p.Trp1038*) was identified in the paternal lineage of Family A by Southey et al. [5]. There were ten diagnoses of breast cancer, five of which occurred under the age of 50 years, in nine women in the extended family (Fig. 1). WES was performed on four women and identified RNASEL:p.Glu265* in III.8, who is known to carry the PALB2 mutation and had two primary diagnoses of breast cancer at age 36 and 45 years. Further genotyping identified RNASEL:p.Glu265* in four additional relatives unaffected by cancer. II.9 (an obligate carrier) had diagnoses of prostate and bladder cancer.
Gene-panel testing
Panel-testing by targeted-sequencing of 591 early-onset affected probands from the ABCFR identified five additional carriers of RNASEL:p.Glu265*. No other pathogenic genetic variants (as per ENIGMA classification criteria) were observed in RNASEL.
Among the five carriers of RNASEL:p.Glu265*, there was one carrier of BRCA2:c.6275_6276delTT(p.Leu2092Profs), one carrier of BRCA1:c.4239del (p.Glu1413Aspfs) and one carrier of ATM:c.7271T>G (p.Val2424Glu) (previously identified by Chenevix-Trench [10]). In total, 3/5 early-onset affected probands carriers of RNASEL:p.Glu265* also harboured a pathogenic mutation in a known breast cancer susceptibility gene.
Their family pedigrees are presented in Fig. 2. The family of the BRCA1:p.Glu1413Aspfs carrier could not be further tested (Fig. 2a). Mutation screening in the family carrying BRCA2:p.Glu1413Aspfs revealed two additional carriers of RNASEL:p.Glu265*: the proband’s mother, who had been diagnosed with breast cancer and leukemia (age at diagnoses 65 and 83 years respectively) and one of the proband’s brothers (Fig. 2b). The affected sister of the proband was found to carry BRCA2:p.Glu1413Aspfs but not the RNASEL mutation. In the family carrying ATM:p.Val2424Glu, RNASEL:p.Glu265* was inherited through the paternal side. One of the proband’s unaffected sisters who did not carry ATM:p.Val2424Glu was identified as a carrier of RNASEL:p.Glu265*.
Fig. 2.
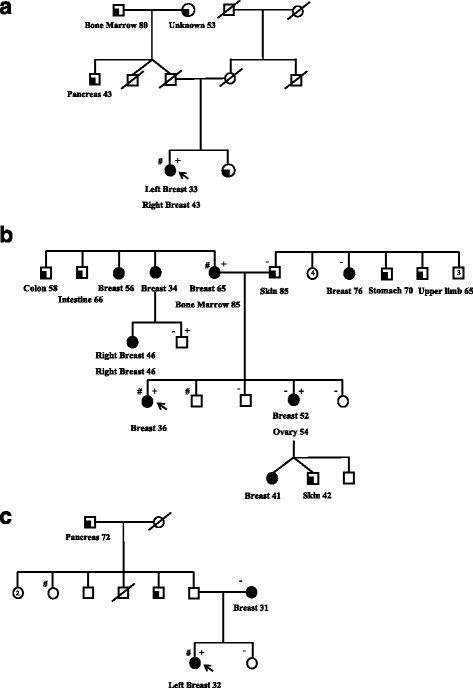
Pedigrees of the families carrying (a) BRCA1:p.Glu1413Aspfs, (b) BRCA1:p.Leu2092Profs and (c) ATM:p.Val2424Glu. +: carriers of the pathogenic mutation; # and -: carriers and non-carriers of RNASEL:p.Glu265*, respectively; arrow: proband. Breast cancer is indicated by black filled symbols, and other cancers are indicated by quarter-filled symbols. Numbers within symbols represent multiple individuals
Pathogenic mutations identified in the carriers and non-carriers of RNASEL:p.Glu265* are reported in Table 2. We identified 17 and 14 carriers of pathogenic mutations in BRCA1 and BRCA2, respectively in affected probands in the ABCFR. One woman carried a mutation in both genes. No other loss-of-function PALB2 mutation was identified in this early-onset breast cancer study.
Table 2.
Pathogenic mutationsa identified by gene-panel testing in probands diagnosed before the age of 40 years in the population-based Australian Breast Cancer Family Registry
| Gene b | HGVS_c c | HGVS_pc | Carriers |
|---|---|---|---|
| BRCA1 | c.5266dupC | p.Gln1756Profs | 4 |
| c.5095C > T | p.Arg1699Trp | 1 | |
| c.4689C > G | p.Tyr1563Ter | 1 | |
| c.4327C > T | p.Arg1443Ter | 1 | |
| c.4239del | p.Glu1413Aspfs | 1 | |
| c.4065_4068delTCAA | p.Asn1355Lysfs | 2 | |
| c.3756_3759delGTCT | p.Ser1253Argfs | 1 | |
| c.3155delA | p.Asn1052Metfs | 1 | |
| c.2681_2682delAA | p.Lys894Thrfs | 2 | |
| c.2475delC | p.Asp825Glufs | 1 | |
| c.1687C > T | p.Gln563Ter | 1 | |
| c.427G > T | p.Glu143Ter | 1 | |
| c.68_69delAG | p.Glu23Valfs | 1e | |
| BRCA2 | c.250C > T | p.Gln84Ter | 1 |
| c.755_758delACAG | p.Asp252Valfs | 2 | |
| c.3847_3848delGT | p.Val1283Lysfs | 1 | |
| c.5576_5579delTTAA | p.Ile1859Lysfs | 1 | |
| c.5946delT | p.Ser1982Argfs | 3 | |
| c.6275_6276delTT | p.Leu2092Profs | 2 | |
| c.8575delC | p.Gln2859Lysfs | 3e | |
| c.8878C > T | p.Gln2960Ter | 1 | |
| c.8904delC | p.Val2969Cysfs | 1 | |
| ATM | c.7271 T > G | p.Val2424Glu | 1 d |
aMutation in BRCA1 and BRCA2 that are classified as pathogenic by the expert panel ENIGMA, PALB2:p.Trp1038* or ATM:p.Val2424Glu
bTranscript sequences are BRCA1: NM_007294.3; BRCA2: NM_00059.3; ATM:NM_000051
cVariant nomenclature according to the Human Genome Variation Society (HGVS), HGVS_c for coding DNA and HGVS_p for protein variants
dData from Chenevix-Trench et al., [10]
eOne woman carried these two mutations
Thus, in probands with early onset disease, the prevalence of RNASEL:p.Glu265* in carriers of a pathogenic mutation in a breast cancer susceptibility gene was 10% (3/30), compared to 0.36% (2/556) in non-carriers (p < 0.002, two-sided Fisher’s Exact test).
Genotyping of RNASEL:P.Glu265*
Taqman-based genotyping identified 9/1445 (0.62%) breast cancer affected women and 6/817 (0.74%) unaffected controls who carry RNASEL:p.Glu265* indicating that the carrier frequency of RNASEL:p.Glu265* in Australian women was similar in affected probands and unaffected controls.
Discussion
Whole-exome sequencing (WES) was performed on four women from Family A. On the basis of these results, we sought to investigate whether RNASEL:p.E265* could be a modifier of breast cancer susceptibility in high-risk mutation carriers.
RNASEL encodes the 2′,5′-oligoisoadenylate synthetase (2-5A)- dependent ribonuclease L (RNase L), an enzyme which has an antiviral role and may regulate the half-life of many mRNAs. The interferon viral response stimulates synthesis of 2-5A, which in turn stimulates activity of RNase L. The ribonuclease activity of RNase L inhibits proliferation of a variety of viruses. Additionally, continued activation of RNase L leads to degradation of 28S and 18S rRNA, which in turn activates a Jun-kinase-dependent apoptosis pathway [12–14]. An animal model of RNase L function showed that mice devoid of RNase L have defects in both interferon-induced apoptosis and antiviral response [12].
Carpten et al. identified RNASEL as a candidate prostate cancer susceptibility gene located within the Hereditary Prostate Cancer 1 (HPC1) linkage peak on chromosome 1q on the basis of evidence that two inactivating mutations in the gene, RNASEL:p.Met1Ile and RNASEL:p.Glu265*, segregated with prostate cancer in chromosome 1q–linked pedigrees [15]. In that study, the reported median age at prostate cancer onset was 11 years less in carriers of RNASEL:p.Glu265*. This variant is classified as pathogenic for prostate cancer susceptibility in ClinVar [16].
In a study of sporadic and familial pancreatic cancer, Bartsch et al. observed RNASEL:p.Glu265* in 1/36 (2.8%) pancreatic cancer cases with a family history of the disease, 1/75 (1.3%) pancreatic cancer cases without family history and none in 108 unaffected controls suggesting a possible association with pancreatic cancer susceptibility [17].
Some missense substitutions in RNASEL have been reported to interact with other genetic and environmental factors to increase early-onset risk of disease, e.g. RNASEL:c.1385G > A (p.Arg462Gln) and early-onset hereditary non-polyposis colorectal cancer in MSH2 or MLH1 pathogenic mutation carriers [18, 19]. Our study did not provide any evidence for a modifying role for RNASEL:p.Arg462Gln in breast cancer predisposition.
Genotyping of RNASEL:p.Glu265* in our case-control breast cancer study did not show an increased carrier frequency in breast cancer cases versus controls. However, our data indicate that in women with early-onset disease (diagnosed under 40 years old) the frequency of carriers of pathogenic mutations in a breast cancer susceptibility gene was significantly higher in RNASEL:p.Glu265* carriers than in RNASEL:p.Glu265* non-carriers.
The pathogenic mutations considered in this report are any BRCA1 or BRCA2 classified as pathogenic by ENIGMA, PALB2:p.Trp1038* and ATM:p.Val2424Glu. In this regard, it is notable that the only confirmed modifier of breast cancer risk, RAD51:c.135G > C, modifies risk only in BRCA2 pathogenic mutation carriers [20]. Our findings suggest that RNASEL:p.Glu265* could be a genetic modifier of cancer predisposition for carriers of high-risk mutations in different breast cancer susceptibility genes.
Since cells from heterozygous carriers of RNASEL:p.Glu265* were shown to contain half the amount of RNase L [15], it is possible that this variant could induce a decreased apoptotic response. However, the mechanisms by which RNASEL could influence the risk of breast cancer are still unknown and should be further investigated.
Further work is required to test the hypothesis raised in this report. Studies of genetic modifiers utilising very large sample sizes can be achieved through the Consortium of Investigators of Modifiers of BRCA1 and BRCA2 (CIMBA) [3] who have collected DNA and epidemiological and clinical data for over 15,000 BRCA1 carriers and 8,000 BRCA2 carriers. Similar future studies related to PALB2 mutation carriers could possibly be achieve within the PALB2 Interest Group www.palb2.org.
Conclusion
Here, we present new data that raises the possibility that RNASEL:p.Glu265* acts as a modifier of risk for carriers of rare high-risk genetic mutations. This case-only study report supports an interesting hypothesis that requires further testing in large case only and case-control studies.
Modifier genes/variants could partly explain inter-individual variation in risk between pathogenic mutation carriers. The identification of modifiers of breast cancer risk will help to refine individual risk estimates and optimise risk management.
Acknowledgements
Not applicable
Funding
The ABCFR is the Australian site of the Breast Cancer Family Registry, and this work was supported by grant UM1 CA164920 from the USA National Cancer Institute. The content of the manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centres in the Breast Cancer Family Registry (BCFR), nor does mention of trade names, commercial products or organisations imply endorsement by the USA Government or the BCFR. The Australian Breast Cancer Family Registry was also supported by the Australian National Health and Medical Research Council (NHMRC), the New South Wales Cancer Council, the Victorian Health Promotion Foundation (Australia) and the Victorian Breast Cancer Research Consortium (VBCRC).
This work was supported by the NHMRC (APP1025145), the USA National Institute of Health (RO1CA155767), the VBCRC and by a Victorian Life Sciences Computation Initiative grant (number VR0182) on its Peak Computing Facility, an initiative of the Victorian Government. TN-D is a National Breast Cancer Foundation (Australia) Career Development Fellow. ZLT was supported by Postgraduate Scholarships provided by the Faculty of Medicine, Dentistry and Health Sciences, The University of Melbourne and the NHMRC (Dora Lush Postgraduate Fellowship). AR was supported by a Bourse de Mobilité from Région Rhône-Alpes, France. JLH is a NHMRC Senior Principal Research Fellow and a VBCRC Group Leader. MCS is an NHMRC Senior Research Fellow and a VBCRC Group Leader. The funding bodies had no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ABCFR
Australian Breast Cancer Family Registry
- ATM
ATM serine/threonine kinas
- BRCA1
BRCA1, DNA repair associated
- BRCA2
BRCA2, DNA repair associated
- ENIGMA
Evidence-based Network for the Interpretation of Germline Mutant Alleles
- HGVS
Human Genome Variation Society
- PALB2
Partner and Localiser of BRCA2
- RNASEL
Ribonuclease L
Authors’ contributions
TN-D contributed to study design, performed the data analysis, contributed to statistical analyses and drafted the manuscript. ZLT performed the Taqman genotyping. FH, AR, MM performed gene-panel testing. HT managed the related bioresources and prepared the DNA samples. DJP, BJP and AL designed the technology for panel testing. MKK, DEG and KM contributed to statistical analyses. IW provided clinical perspective and contributed to the drafting of the manuscript. JLH and GGG contributed to study design and were responsible for subjects ascertained through the ABCFR. MCS was responsible for overall study design and contributed substantially to data analysis and drafting of the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
All participants provided written informed consent for participation in this research program, which was approved by the ABCFR and the University of Melbourne Human Research Ethics Committee, Melbourne, VIC, Australia (Ethics Application #1441420).
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Tú Nguyen-Dumont, Email: tun@unimelb.edu.au.
Zhi L. Teo, Email: Joyce.Teo@petermac.org
Fleur Hammet, Email: fhammet@unimelb.edu.au.
Alexis Roberge, Email: alex.rob@hotmail.fr.
Maryam Mahmoodi, Email: maryamm@unimelb.edu.au.
Helen Tsimiklis, Email: htsi@unimelb.edu.au.
Daniel J. Park, Email: djp@unimelb.edu.au
Bernard J. Pope, Email: bjpope@unimelb.edu.au
Andrew Lonie, Email: alonie@unimelb.edu.au.
Miroslav K. Kapuscinski, Email: kmk@unimelb.edu.au
Khalid Mahmood, Email: khalid.mahmood@unimelb.edu.au.
David E. Goldgar, Email: david.goldgar@hsc.utah.edu
Graham G. Giles, Email: graham.giles@cancervic.org.au
Ingrid Winship, Email: Ingrid.Winship@mh.org.au.
John L. Hopper, Email: j.hopper@unimelb.edu.au
Melissa C. Southey, Phone: +61 3 8559 7003, Email: msouthey@unimelb.edu.au
References
- 1.Easton DF, Ford D, Bishop DT. Breast and ovarian cancer incidence in BRCA1-mutation carriers. Breast cancer linkage consortium. Am J Hum Genet. 1995;56(1):265–271. [PMC free article] [PubMed] [Google Scholar]
- 2.Ford D, Easton DF, Stratton M, Narod S, Goldgar D, Devilee P, Bishop DT, Weber B, Lenoir G, Chang-Claude J, et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium Am J Hum Genet. 1998;62(3):676–689. doi: 10.1086/301749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chenevix-Trench G, Milne RL, Antoniou AC, Couch FJ, Easton DF, Goldgar DE, CIMBA An international initiative to identify genetic modifiers of cancer risk in BRCA1 and BRCA2 mutation carriers: the consortium of investigators of modifiers of BRCA1 and BRCA2 (CIMBA) Breast Cancer Res. 2007;9(2):104. doi: 10.1186/bcr1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.John EM, Hopper JL, Beck JC, Knight JA, Neuhausen SL, Senie RT, Ziogas A, Andrulis IL, Anton-Culver H, Boyd N, et al. The breast cancer family registry: an infrastructure for cooperative multinational, interdisciplinary and translational studies of the genetic epidemiology of breast cancer. Breast Cancer Res. 2004;6(4):R375–R389. doi: 10.1186/bcr801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Southey MC, Teo ZL, Dowty JG, Odefrey FA, Park DJ, Tischkowitz M, Sabbaghian N, Apicella C, Byrnes GB, Winship I, et al. A PALB2 mutation associated with high risk of breast cancer. Breast Cancer Res. 2010;12(6):R109. doi: 10.1186/bcr2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park DJ, Lesueur F, Nguyen-Dumont T, Pertesi M, Odefrey F, Hammet F, Neuhausen SL, John EM, Andrulis IL, Terry MB, et al. Rare mutations in XRCC2 increase the risk of breast cancer. Am J Hum Genet. 2012;90(4):734–739. doi: 10.1016/j.ajhg.2012.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nguyen-Dumont T, Hammet F, Mahmoodi M, Pope B, Giles G, Hopper G, Southey M, Park D. Abridged adapter primers increase the target scope of hi-Plex. BioTechniques. 2015;58(1):33–36. doi: 10.2144/000114247. [DOI] [PubMed] [Google Scholar]
- 8.Pope BJ, Nguyen-Dumont T, Hammet F, Park DJ. ROVER variant caller: read-pair overlap considerate variant-calling software applied to PCR-based massively parallel sequencing datasets. Source Code Biol Med. 2014;9(1):3. doi: 10.1186/1751-0473-9-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spurdle AB, Healey S, Devereau A, Hogervorst FB, Monteiro AN, Nathanson KL, Radice P, Stoppa-Lyonnet D, Tavtigian S, Wappenschmidt B, et al. ENIGMA--evidence-based network for the interpretation of germline mutant alleles: an international initiative to evaluate risk and clinical significance associated with sequence variation in BRCA1 and BRCA2 genes. Human Mutat. 2012;33(1):2–7. doi: 10.1002/humu.21628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chenevix-Trench G, Spurdle AB, Gatei M, Kelly H, Marsh A, Chen X, Donn K, Cummings M, Nyholt D, Jenkins MA, et al. Dominant negative ATM mutations in breast cancer families. Journal of the Natl Cancer Inst. 2002;94(3):205–215. doi: 10.1093/jnci/94.3.205. [DOI] [PubMed] [Google Scholar]
- 11.Southey MC, Goldgar DE, Winqvist R, Pylkas K, Couch F, Tischkowitz M, Foulkes WD, Dennis J, Michailidou K, van Rensburg EJ, et al. PALB2, CHEK2 and ATM rare variants and cancer risk: data from COGS. J Med Genet. 2016; [DOI] [PMC free article] [PubMed]
- 12.Zhou A, Paranjape J, Brown TL, Nie H, Naik S, Dong B, Chang A, Trapp B, Fairchild R, Colmenares C, et al. Interferon action and apoptosis are defective in mice devoid of 2′,5′-oligoadenylate-dependent RNase L. EMBO J. 1997;16(21):6355–6363. doi: 10.1093/emboj/16.21.6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li G, Xiang Y, Sabapathy K, Silverman RH. An apoptotic signaling pathway in the interferon antiviral response mediated by RNase L and c-Jun NH2-terminal kinase. J Biol Chem. 2004;279(2):1123–1131. doi: 10.1074/jbc.M305893200. [DOI] [PubMed] [Google Scholar]
- 14.Malathi K, Paranjape JM, Ganapathi R, Silverman RH. HPC1/RNASEL mediates apoptosis of prostate cancer cells treated with 2′,5′-oligoadenylates, topoisomerase I inhibitors, and tumor necrosis factor-related apoptosis-inducing ligand. Cancer Res. 2004;64(24):9144–9151. doi: 10.1158/0008-5472.CAN-04-2226. [DOI] [PubMed] [Google Scholar]
- 15.Carpten J, Nupponen N, Isaacs S, Sood R, Robbins C, Xu J, Faruque M, Moses T, Ewing C, Gillanders E, et al. Germline mutations in the ribonuclease L gene in families showing linkage with HPC1. Nat Genet. 2002;30(2):181–184. doi: 10.1038/ng823. [DOI] [PubMed] [Google Scholar]
- 16.Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, Maglott DR. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42(Database issue):D980–D985. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bartsch DK, Fendrich V, Slater EP, Sina-Frey M, Rieder H, Greenhalf W, Chaloupka B, Hahn SA, Neoptolemos JP, Kress R. RNASEL germline variants are associated with pancreatic cancer. Int J Cancer. 2005;117(5):718–722. doi: 10.1002/ijc.21254. [DOI] [PubMed] [Google Scholar]
- 18.Kruger S, Silber AS, Engel C, Gorgens H, Mangold E, Pagenstecher C, Holinski-Feder E, von Knebel Doeberitz M, Moeslein G, Dietmaier W, et al. Arg462Gln sequence variation in the prostate-cancer-susceptibility gene RNASEL and age of onset of hereditary non-polyposis colorectal cancer: a case-control study. Lancet Oncol. 2005;6(8):566–572. doi: 10.1016/S1470-2045(05)70253-9. [DOI] [PubMed] [Google Scholar]
- 19.Kruger S, Engel C, Bier A, Silber AS, Gorgens H, Mangold E, Pagenstecher C, Holinski-Feder E, von Knebel Doeberitz M, Royer-Pokora B, et al. The additive effect of p53 Arg72Pro and RNASEL Arg462Gln genotypes on age of disease onset in lynch syndrome patients with pathogenic germline mutations in MSH2 or MLH1. Cancer Lett. 2007;252(1):55–64. doi: 10.1016/j.canlet.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 20.Antoniou AC, Sinilnikova OM, Simard J, Leone M, Dumont M, Neuhausen SL, Struewing JP, Stoppa-Lyonnet D, Barjhoux L, Hughes DJ, et al. RAD51 135G-->C modifies breast cancer risk among BRCA2 mutation carriers: results from a combined analysis of 19 studies. Am J Hum Genet. 2007;81(6):1186–1200. doi: 10.1086/522611. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.