

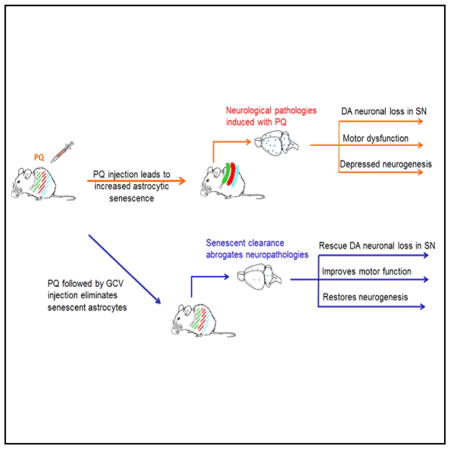
SUMMARY
Exposure to the herbicide paraquat (PQ) is associated with an increased risk of idiopathic Parkinson’s disease (PD). Therapies based on PQ’s presumed mechanisms of action have not, however, yielded effective disease therapies. Cellular senescence is an anticancer mechanism that arrests proliferation of replication-competent cells and results in a pro-inflammatory senescence-associated secretory phenotype (SASP) capable of damaging neighboring tissues. Here, we demonstrate that senescent cell markers are preferentially present within astrocytes in PD brain tissues. Additionally, PQ was found to induce astrocytic senescence and an SASP in vitro and in vivo, and senescent cell depletion in the latter protects against PQ-induced neuropathology. Our data suggest that exposure to certain environmental toxins promotes accumulation of senescent cells in the aging brain, which can contribute to dopaminergic neurodegeneration. Therapies that target senescent cells may constitute a strategy for treatment of sporadic PD, for which environmental exposure is a major risk factor.
In Brief
Chinta et al. argue that eliminating senescent cells could hold therapeutic promise for halting progression of sporadic PD, where environmental exposure is a major risk factor. They find that senescence cell depletion abrogates development of neurodegenerative phenotypes associated with systemic PQ exposure, indicating that senescent cells can contribute to neurodegeneration.
INTRODUCTION
Although genetic mutations that result in the development of familial Parkinson’s disease (PD) have been identified, these account for a minority of disease cases. The majority of cases are sporadic in nature and likely involve the contribution of various environmental exposures over the course of the lifespan (Wirdefeldt et al., 2011). A better understanding of the mechanisms involved in the contribution of these factors to neuropathology is a vital missing piece of the puzzle that will help unravel the underlying causes of idiopathic disease. Exposure to the herbicide paraquat (PQ) is strongly associated with increased lifetime risk for the development of sporadic PD. Its systemic administration in mice results in several hallmark neuropathological features of the disorder. However, therapies based on the agent’s presumed mechanisms of action have yet to yield effective disease therapies (Le et al., 2014; Wirdefeldt et al., 2011). Preventive and therapeutic treatments for idiopathic PD may require uncovering of disease mechanisms and targets distinct from those identified to date.
We report the contribution of a factor thought to contribute to aging in many tissues: cellular senescence (Campisi, 2013; Chinta et al., 2013, 2015; Salminen et al., 2011). Cellular senescence is an anticancer mechanism occurring in cells capable of division, and it results in irreversible arrest of cell proliferation in response to stress that puts cells at risk for malignant transformation (Campisi, 2013; Prieur and Peeper, 2008). Senescent cells express a senescence-associated secretory phenotype (SASP)—the robust secretion of numerous inflammatory cytokines, chemokines, growth factors, and proteases (Coppé et al., 2008). Senescent cells accumulate with age in many human and mouse tissues. There, presumably by virtue of the SASP, they have been shown to contribute to the age-related loss of tissue structure and function (Baker et al., 2016; Campisi, 2013; Dimri et al., 1995; Muñoz-Espín and Serrano, 2014; van Deursen, 2014). Cellular senescence may be an example of evolutionary antagonistic pleiotropy—protecting organisms from cancer early in life but driving aging phenotypes at advanced age.
Although senescent cells have been demonstrated to contribute to age-related pathologies outside the brain, evidence to date for their contribution to age-related neurodegenerative diseases is only correlative (Bhat et al., 2012; Chinta et al., 2015; Mombach et al., 2015; Salminen et al., 2011; Turnquist et al., 2016). Here, we report that postmortem tissue from sporadic PD patients displays increased expression of senescent markers, particularly within astrocytes. PQ is also able to induce senescence in cultured human astrocytes and in vivo. Significantly, inducible depletion of in vivo senescence reduces the development of neurodegenerative phenotypes associated with systemic PQ exposure, demonstrating that senescent cells contribute to neurodegeneration in an in vivo neurodegenerative disease model. Our data suggest that strategies aimed at eliminating senescent cells hold promise as potential therapies to mitigate the development of sporadic PD, for which environmental stress is a major contributing factor.
RESULTS
Senescent Astrocytes and SASP Factors Are Elevated in Parkinsonian Substantia Nigra Pars Compacta
Astrocytes expressing senescence markers increase in human brain tissue during normal aging (Kang et al., 2015) and further in affected brain regions of individuals with Alzheimer’s disease (AD) and amyotrophic lateral sclerosis (ALS) (Bhat et al., 2012; Turnquist et al., 2016), prompting us to ask whether senescent astrocytes are also present in PD (Figures 1A and 1B). Compared to control tissues, Parkinsonian SNpc tissues displayed elevated expression of the senescence marker p16INK4a, and several SASP factors included the protease MMP-3 and the pro-inflammatory cytokines interleukin-6 (IL-6), IL-1α, and IL-8 (Figure 1A). Elevated p16INK4a and MMP-3 expression has also been reported in cortical tissue from AD patients (Bhat et al., 2012).
Figure 1. Senescence Markers in PD SNpc versus Age-Matched Controls.

(A) RNA isolated from autopsied SNpc tissues from PD versus controls (n = 5) were analyzed for p16INK4a and SASP factor (IL-6, IL-1α, IL-8, and MMP-3) mRNA levels by qPCR. Transcripts were normalized to actin and are shown as fold change over control levels; *p < 0.05 and ***p < 0.005.
(B) Comparison of lamin B1 levels in PD versus age-matched control tissues. Representative immunofluorescence images (left side) showing lamin B1 protein levels (green) in GFAP+ (red) astrocytes (red arrows) in the PD SNpc (right panels) compared to age-matched controls (left panels). Neighboring GFAP− cells (yellow arrows) retain lamin B1 expression in PD tissue. Quantification of the image data (right side) using mean pixel values (MPVs) showing that compared to controls (black bar; n = 5 individuals), PD tissues (gray bar; n = 5 individuals) contained significantly less Lamin B1 protein in GFAP+, but not GFAP−, cells; *p < 0.05 (paired t test).
We next determined whether SNpc astrocytes in particular display evidence of cellular senescence in PD tissues. A reduced nuclear level of lamin B1, detectable by immunostaining in intact tissue, is an established senescence-associated marker (Freund et al., 2012). Astrocytes (GFAP+ cells) were found to be lamin B1 deficient in PD SNpc tissues while no significant difference was detected in PD versus aged-matched control tissues in non-astrocytic (GFAP−) neighboring cells (Figures 1B and S1). These findings suggest the elevated presence of senescence in affected PD tissues and that astrocytes may preferentially undergo senescence in diseased brain tissues.
PQ Induces Senescence and an SASP in Human iPSC-Derived Astrocytes
We next assessed the ability of human astrocytes derived from induced pluripotent stem cells (iPSCs) to undergo senescence in response to PQ, an environmental insult significantly associated with PD (Pezzoli and Cereda, 2013). In adult mice, chronic, low-dose systemic exposure to PQ recapitulates several important hallmarks of the disease, including selective loss of dopaminergic (DAergic) SNpc neurons and reduced motor function (McCormack and Di Monte, 2003; Pezzoli and Cereda, 2013; Tanner et al., 2011). Cultured human astrocytes exposed to PQ ceased proliferation based on decreased percentage of bromo-deoxyuridine (BrdU)-labeled cells (Figure 2A). They also displayed several additional hallmarks of senescence, including increased senescence-associated β-galactosidase (SA-β-gal) activity (Dimri et al., 1995) (Figure 2B), increased levels of p16INK4a (Beausé jour et al., 2003) mRNA and protein (Figures 2C and 2D), and increased numbers of 53BP1 foci, the latter indicative of DNA-damage signaling, which is a potent stimulator of the SASP (Beausé jour et al., 2003)(Figure 2E). Consistent with development of an SASP, PQ also induced robust secretion of the pro-inflammatory cytokine IL-6 (Figure 2F), a prominent component of the SASP (Coppé et al., 2008).
Figure 2. PQ Induces Senescent Phenotypes in Cultured Human Astrocytes.

Cultured human astrocytes were treated with 100 or 250 μM PQ (or vehicle) for 24 hr, and senescence markers were evaluated 7 days later.
(A) Representative images of BrdU labeling (left), and percentage of BrdU+ cells (right).
(B) Representative images of SA-β-gal activity (left), and percentage of SA-β-gal+ cells (right).
(C) qPCR of p16INK4a mRNA levels.
(D) Representative images of immunostaining for p16INK4a (left), and percentage of p16INK4a-positive cells (right).
(E) Representative images of 53BP1 immunostaining (left), and percentage of cells with >1 53BP1 positive foci (right). Nuclei were counterstained with DAPI (blue) and white arrows denote enlarged cells shown in insets with punctate nuclear foci.
(F) Secreted IL-6 levels measured by ELISA in conditioned media from non-senescent (vehicle) and senescent (PQ) astrocytes.
For (A)–(D), n = 4, where n = experimental replicates; *p < 0.05, **p < 0.01, and ***p < 0.005 (unpaired t test).
Given that genotoxic stress induced by ionizing radiation (IR) initiates senescence with well-documented effects on mitosis and nuclear size (Pospelova et al., 2013), we verified that compared to IR, PQ caused a similar decrease in the percentage of Ki67+ cells (Figure 3A) and an increase in the mean size of astrocyte nuclei (Figure. 3B). Human fibroblasts cultured with PQ for 24 hr also displayed these phenotypes as well as decreased Lamin B1 levels and increased SA-β-gal activity (Figures 3A–3D). These data indicate that PQ is capable of inducing senescence in human astrocytes as well as human fibroblasts (Jung et al., 2009). However, astrocytes were more sensitive than fibroblasts to PQ-induced senescence based on SA-β-gal activity. Interestingly, a concentration of PQ that induced fibroblast senescence (>500 μM PQ) was lethal to astrocytes (Figure 3D).
Figure 3. Paraquat Induces Morphological Features of Senescence and Arrests Proliferation in Both Fibroblasts and Astrocytes.

(A) Human fibroblasts (top panels) and human astrocytes (bottom panels) respond to PQ (1 mM and 300 μM, respectively) by decreasing the percentage of Ki67+ cells (green) compared to vehicle-treated controls. Representative images (left) and quantification (right) are shown. The decreased percentage of Ki67+ cells is similar to the decrease observed when cells are exposed to 10 Gy X-ray (IR, middle panels). For fibroblasts, both IR and 1 mM PQ significantly reduced Ki67 positivity; ***p < 0.00001 (1 mM PQ was not significantly different from IR [p > 0.07]; unpaired t test); n = 4 experiments. For astrocytes, IR and 300 μM PQ significantly reduced Ki67 positivity; **p < 0.01 (300 μM PQ was not significantly different from IR [p > 0.8]; unpaired t test); n = 2 experiments.
(B) Human fibroblasts (top) and human astrocytes (bottom) respond to PQ with a significant increase in nuclear size compared to vehicle-treated controls. Representative images (left) and quantification (right) are shown. Increased nuclear size is similar to the increase obtained with IR exposure. For fibroblasts both conditions (IR and 1mM PQ) are significantly greater than control; ***p < 0.0001 (1 mM PQ is not significantly different from IR [p > 0.30]; unpaired t test); n = 6 experiments. For astrocytes, both conditions (IR; 300 μM PQ) are significantly greater than control; *p < 0.02 (300 μM PQ is not significantly different from IR [p > 0.5]; unpaired t test); n = 5 experiments.
(C) Human fibroblasts respond to PQ (500 μM and 1 mM) with a significant loss of lamin B1 staining in the nuclear lamina relative to vehicle-treated controls. Representative images (left) and quantification (right) are shown. Decreased lamin B1 staining is similar to that obtained with IR. All conditions (IR; 500 μM and 1 mM PQ) are significantly different from control; **p < 0.01 (neither PQ condition differed significantly from IR [p > 0.25]; unpaired t test); n = 2 experiments. (D) Human fibroblasts (1 mM PQ, top), astrocytes derived from iPSCs (40 μM PQ, middle), and murine primary astrocytes from p16-3MR neonates (40 μM PQ, bottom), display obvious increase in SA-β-gal activity, but cell death occurred when human and mouse astrocytes were exposed to higher concentrations of PQ (500 and 250 μM, respectively).
Epidemiological data suggest that, although the risk for developing PD is significant following a short-term (<8 day) environmental exposure to PQ, PD is even more significantly linked with chronic/low-dose PQ exposure (Tanner et al., 2011). We asked whether lower concentrations of PQ for a longer duration could induce senescence in human astrocytes. Cells exposed for 48 hr to 10 μM PQ showed a significant increase in SA-β-gal+ cells, and a 48-hr exposure to 50 μM PQ was nearly as effective as a 24- hr exposure to 100 μM PQ (Figure 4A). All exposures and concentrations reduced cell numbers compared to vehicle-treated cells consistent with proliferative arrest (Figure 4B). Lower more sustained PQ doses increased the percentage of senescent astrocytes to a similar or greater extent than exposure to H202 (150 mM for 2 hr) or the proteasomal inhibitor MG132 (50 nM for 48 hr) (Figure 4A), which are stress conditions reported to induce senescence of cultured human astrocytes (Bitto et al., 2010).
Figure 4. Lower Dosages of PQ Also Induce Cellular Senescence.

(A) Images and quantification percentage of SA-β-gal+ cells of human astrocytes exposed to 10 or 50 μM PQ for 48; also shown are results from exposure to 150 mM H2O2 for 2 hr and 50 nM MG132 for 48 hr; *p < 0.05, **p < 0.01, and ***p < 0.005.
(B) Images and quantification of cell density based on DAPI staining after exposure to 10 or 50 μM PQ for 48 hr; *p < 0.05 and **p < 0.01.
For (A) and (B), n = 4, where n = experimental replicates (unpaired t test).
Soluble Factors Secreted by Senescent Human Astrocytes Compromise the Function of Neurons and Neural Progenitor Cells
Senescent cells can exert powerful paracrine effects on tissue microenvironments (Coppé et al., 2010); SASP factors have been shown to disrupt tissue structure and function (Brkic et al., 2015). We asked whether PQ-induced senescent astrocytes could drive PD-related neuropathologies associated with the neurotoxin, including DAergic neuronal cell death (Blesa and Przedborski, 2014) and reduced neural progenitor cell (NPC) proliferation and migration (Marxreiter et al., 2013). We cultured human DAergic neurons and NPCs in the presence of conditioned media (CM) from human astrocytes. CM from PQ-induced senescent astrocytes, but not non-senescent (vehicle-treated) astrocytes, significantly reduced the viability of DAergic neurons (Figure 5A). Similarly, CM from senescent astrocytes suppressed both NPC proliferation (Figure 5B) and migration (Figure 5C).
Figure 5. Factors Secreted by Senescent Human Astrocytes Have Detrimental Effects.

(A) Conditioned media (CM) from PQ-treated human astrocytes was added to DAergic neurons, and cell viability was assessed via an MTT assay; n = 3 experiments.
(B) CM from PQ-treated human astrocytes was added to neural progenitor cells (NPCs), and proliferation of NPCs was assessed by BrdU positivity. Representative images (left) and quantification (right) are shown; n = 3 experiments.
(C) NPCs migration in the presence of CM from vehicle-treated (black bars) versus PQ-treated (250 μM) (gray bars) astrocytes assayed using the scratch test; images show “cleared areas” of NPCs at 0 hr (far left panels), 24 hr (middle panels), and 48 hr (right panels) post-scratch. Black lines (perpendicular to scratches) indicate the width of the uninvaded area used for the quantification (right side). The percent scratch width >24 and 48 hr was calculated; n = 5 scratches. The experiment was run twice with 2 different sets of CM (2 scratch assays). *p < 0.05 (unpaired t test).
IL-6 is a prominent SASP factor, and elevated serum levels are associated with increased PD risk (Scalzo et al., 2010). Treating cultured DAergic neurons with recombinant IL-6 at the concentration found in the CM of PQ-treated astrocytes (1.5 μM) showed a trend toward reducing DAergic cell viability, but the effect was not statistically significant (Figure S2). This finding suggests that although IL-6 may contribute to neurotoxic SASP effects, additional factors are likely needed to mediate the detrimental impact on DAergic viability.
Depleting Senescent Cells Abrogates PQ-Induced Neuropathology
To test whether PQ-induced senescence contributes to neuropathologies in vivo, we used a mouse model that allows selective depletion of senescent cells (Demaria et al., 2014). These mice (p16-3MR) use the promoter for the senescence marker p16INK4a to drive expression of a modified herpes simplex virus thymidine kinase (HSV-TK), which converts the pro-drug ganci-clovir (GCV) into a DNA chain terminator, causing cells to apoptose (Laberge et al., 2013). GCV eliminates 70%–80% of senescent cells in p16-3MR mice without discernible off-target or bystander effects (Demaria et al., 2014).
We subjected p16-3MR mice to a systemic PQ regimen shown to result in several neuropathological features of PD (McCormack and Di Monte, 2003; Shimizu et al., 2001). PQ increased nigrostriatal expression of p16INK4a and IL-6 (Figure 6A). As was the case for human astrocytes, PQ-treated p16-3MR astrocytes display increased SA-β-gal activity and were more sensitive to PQ-induced toxicity. Similar to astrocytes in human PD tissues, astrocytes from PQ-treated mice also displayed significant loss of nuclear lamin B1 and HMGB1 (high mobility group box 1), the latter necessary for SASP initiation (Figures 6B and 6C) (Davalos et al., 2013). Similar to our findings in the human PD SNpc, PQ did not significantly reduce nuclear lamin B1 in GFAP− cells (Figure 6D). Significantly, all senescent phenotypes in the SNpc were abrogated in mice in which senescent cells were eliminated by systemic GCV administration (Figures 6A–6D). We failed to detect a decline in the proliferation marker Ki67 in astrocytes in PQ-treated mice (Figure S3). However, Ki67 staining in astroglia in saline-treated animals was within the range of post-mitotic neurons and less than half the levels found in Ki67+ cells within the inner granule cell layer of the dentate gyrus, which are presumed to be actively dividing granule cell progenitors (Nicola et al., 2015).
Figure 6. PQ-Induced Senescence Is Prevented by Senescent Cell Ablation.

p16-3MR mice were treated with (1) saline, (2) PQ, (3) GCV alone, or (4) PQ plus GCV and assessed for senescence-associated markers.
(A) Striatal RNA analyzed by qPCR for p16INK4a and IL-6 mRNA levels.
(B) SNpc immunofluorescence of Lamin B1 (green) of DAPI-stained nuclei (blue) within a field of TH+ (magenta)-stained neurites. GFAP+ (red) astrocytes are indicated by yellow arrows. A representative image from each of the 4 conditions (left) and corresponding quantification of nuclear Lamin B1 fluorescence (right). Top left inlays show lamin B1 staining within astrocytes.
(C) HMGB1 immunofluorescence (green) counterstained as in (B); shown are representative images (left) and quantification of nuclear HMGB1 fluorescence (right).
(D) Representative images (left) of lamin B1 immunofluorescence (green) in astrocytes versus non-astrocytic neighbors and quantification (right). Neighboring GFAP− cells marked with yellow arrows.
In all experiments, n = 4 mice for saline, PQ plus GCV, and GCV; and n = 3 mice for PQ. *p < 0.05 (unpaired t test).
Human PD neuropathology is characterized by the preferential loss of DAergic neurons in the SNpc, resulting in impairment of locomotor function (Jankovic, 2008). These phenotypes are recapitulated in the systemic PQ murine model. In the mouse SNpc, PQ caused a significant decline in a marker of DAergic neurons, tyrosine hydroxylase (TH); co-administration of GCV with PQ prevented this decline (Figure 7A). Systemic PQ administration is known to repress adult neurogenesis, but co-GCV administration abrogated this effect (Figure 7B) (Marxreiter et al., 2013). Finally, rearing behavior, a measure of motor neuron function more sensitive to DAergic cell loss than other motor tests (Willard et al., 2015), was significantly curbed in mice treated with PQ alone but normal in mice co-treated with GCV (Figure 7C). Taken together, these results suggest that senescent cell depletion abrogates PQ-mediated neuropathology and motor deficits.
Figure 7. Depletion of Senescent Cells Abrogates PQ-Mediated Neuropathological Phenotypes.

(A) Stereological SN TH+ cell counts were performed on p16-3MR mice treated with (1) saline, (2) PQ, (3) GCV alone, or (4) PQ plus GCV. Representative images (left) and quantification (right) are shown. Data are expressed as total SN TH+ cell numbers per animal; cell counts were verified by Nissl staining (not shown).
(B) Neurogenesis assessed by quantitation of BrdU+ cells in the subventricular zone (SVZ) (Peng et al., 2008). Representative sections showing BrdU+ cells in the SVZ (left); quantitation of BrdU+ cells in the SVZ (right). Values are reported as BrdU+ cells per field averaged from 5 fields.
(C) Prior to sacrifice, locomotor activity was monitored using the cylinder test as a measurement of spontaneous rearing rate. Still images demonstrating the behavioral phenotype (left) and quantification (right) are shown.
For all experiments, n = 6 per condition; *p < 0.05 (unpaired t test).
DISCUSSION
Late-onset idiopathic PD is thought to result from the combined effects of genetic risk factors, aging, and environmental exposure. Data from several laboratories suggest that humans and animals exposed to certain chemicals can develop neuropathological phenotypes associated with PD (Chinta et al., 2008; McCormack et al., 2005; Peng et al., 2010). One of the best studied of these is PQ. A meta-analysis of >100 epidemiological studies concluded that of several specific pesticides and chemical classes evaluated, only PQ was significantly associated with PD. Moreover, as this study points out, the epidemiologic data are backed by numerous animal studies demonstrating that PQ selectively depletes SNpc DAergic neurons and reduces motor function in adult mice, which are important hallmarks of human PD (Pezzoli and Cereda, 2013).
PQ is a free radical generator that causes oxidative stress (Bus and Gibson, 1984), which has been demonstrated to induce cellular senescence in replication-competent cell types (Campisi, 2013). PQ also results in compromised mitochondrial function, which has recently been tied to senescence induction in human fibroblasts (Wiley et al., 2016). One mechanism by which PQ may contribute to PD is by inducing cellular senescence within the brain (Chinta et al., 2013). Indeed, PQ induced senescence more readily in cultured human and murine astrocytes than in human fibroblasts, and their sensitivity was found to track with baseline SA-β-gal activity. Our studies suggest that PQ, like other inducers of senescence (Park et al., 2013), has a bandwidth of concentrations that induce senescence; above an upper limit, cells die. This finding raises the possibility that astrocytes are particularly prone to undergo senescence in response to chronic low-dose exposure to PQ that is associated with late-onset idiopathic PD. Indeed, administration of PQ to mice appears to preferentially increase senescent markers in astrocytes. Importantly, senescence markers were also elevated in astrocytes in SNpc tissues from autopsied PD brain samples, suggesting that the proclivity for astrocytes to undergo senescence may have physiological disease relevance. To our knowledge, this is the first report of elevated senescent markers in the PD brain.
While our data suggest that senescence occurs preferentially within astrocytes, this does not rule out senescence induction of other brain cell types. While neurons are post-mitotic and therefore unlikely to undergo traditional senescence induction, senescence has reported in other non-astrocytic cell types. For example, in response to repeated lipopolysaccharide administration, microglial BV2 cells display growth arrest, enhanced SA-β-gal activity, and heterochromatic foci (Yum et al., 2012). Based on our own findings that PQ can mediate senescence in disparate cell types (astrocytes and fibroblasts), we anticipate that at some concentrations PQ may induce senescence of other replicative brain cell types.
It is theoretically possible that senescence induction in peripheral tissues could contribute to PQ-mediated brain pathologies, but this seems unlikely, as the chronic, low-dose PQ regimen utilized in our studies is known to result in receptor-mediated uptake and immune response in the brain (McCormack and Di Monte, 2003) without activating peripheral immune cells or disrupting blood-brain barrier integrity (Vilas-Boas et al., 2014; Watson et al., 2013).
How might senescent astrocytes contribute to PD neuropathology? Mature astrocytes rarely divide in the adult brain (Ge et al., 2012; Sohn et al., 2015), consistent with our inability to detect Ki67+ astrocytes in the adult mouse brain; however, astrocytes do divide in response to certain injury (Barreto et al., 2011) and disease states (Boekhoorn et al., 2006; Bondolfi et al., 2002). The inability of senescent astrocytes to proliferate may exacerbate neuropathology when proliferation is an appropriate response to injury or disease. Astrocytes provide structural, metabolic, and trophic support to neurons (Allaman et al., 2011). When astrocytes adopt pro-inflammatory phenotypes, they can compromise neuronal function and contribute to age-related neurodegeneration and decrements in brain function in vivo (Rossi, 2015; Salminen et al., 2011). Factors secreted by senescent astrocytes had detrimental effects on both cultured human DAergic neurons and NPCs. CM from senescent astrocytes was supplemented with growth factors; therefore, trophic factor depletion seems an unlikely explanation. Secretion of IL-6 by cultured senescent human astrocytes was recently reported to decrease the viability of co-cultured motor neurons (Turnquist et al., 2016). We observed a similar increase in IL-6 secretion following PQ treatment in culture or in vivo, but IL-6 alone was not sufficient to induce significant degeneration of cultured DAergic neurons, suggesting that additional SASP factors may be required.
Why do DAergic SNpc neurons selectively degenerate in response to PQ and in PD? This is a pressing question in the field that our study does not explicitly address. However, we hypothesize that DAergic SNpc neurons may be particularly sensitive to the effects of oxidative stress on neighboring astrocytes, including senescence induction. In agreement with this idea, using an inducible astrocyte-specific oxidative stress mouse model, we previously demonstrated selective degeneration of DAergic SNpc neurons, despite astrocytes in other brain regions experiencing similar levels of oxidative stress (Mallajosyula et al., 2008).
In conclusion, our data show that astrocytes undergo cellular senescence in response to a well-established environmental stressor linked to PD and, significantly, that PQ-induced senescence contributes to PD-related neuropathology. This finding provide novel links among environmental exposure, cellular senescence, and PD. Further studies are warranted to identify other environmental and endogenous stressors capable of senescence induction in the brain. Our results suggest that senescent cells might be a novel therapeutic target for sporadic PD and other chronic neurodegenerative diseases in which environmental factors are implicated.
EXPERIMENTAL PROCEDURES
PD Tissues and Ethics Statement
Formalin-fixed paraffin-embedded SNpc sections were obtained from the Neurodegenerative Disease Research Brain Bank, University of Pennsylvania (U Penn) Center for Neurodegenerative Disease Research. Samples were from subjects aged 50–92 years at autopsy with previously diagnosed PD. Control samples were from age- and sex-matched subjects without PD. All samples were de-identified and obtained after approval by the U Penn institutional review board (protocol number 180600). A broker de-identified each specimen, no protected health information was made available to the researcher, and specimens were assigned numbers that could not be traced to the donors. There is no link between donor identifiers and the data; therefore, consent for this protocol was waived.
qPCR of Human Tissues and Cells
Total RNA was prepared from human SNpc or human-iPSC-derived cells using TRIzol (Invitrogen) and reverse transcribed using an Applied Biosystems kit (Carlsbad, CA). qRT-PCR was performed using the Universal Probe Library system (Roche, South San Francisco, CA) with the following primers and probes:
p16INK4a: forward (FW) 5′-gagcagcatggagccttc-3′; RV 5′-cgtaactattcggtg cgttg-3′
IL-6: FW 5′-gcccagctatgaactccttct-3′; RV 5′-gaaggcagcaggcaacac-3′
IL-1α: FW 5′-ggttgagtttaagccaatcca-3′; RV 5′-tgctgacctaggcttgatga-3′
IL-8: FW 5′-agacagcagagcacacaagc-3′; RV 5′-atggttccttccggtggt-3′
MMP-3: FW 5′-caaaacatatttctttgtagaggacaa-3′;RV 5′-ttcagctatttgcttgg gaaa-3′.
Immunofluorescence of Human SNpc Tissue
SNpc sections (6 μM) were rehydrated as follows: 2 × 7 min, xylene; 4 min each; 100%, 95%, 80%, and 70% ETOH; rinsed in dH20; and washed in Tris-buffered saline (TBS), 10 min. Slides were then microwaved in 10 mM citrate buffer (pH 6.0) for 5 min at 40% power, cooled for 20 min, washed 10 min in TBS, and then blocked with 10% normal goat serum (NGS) in TBS + 0.05% Triton X-100 for 1 hr at room temperature. Sections were incubated at 4°C overnight with primary antibodies in blocking buffer (chicken TH: Novus, NB100-1613, 1:200 dilution; lamin B1: Abcam, ab16048, 1:200 dilution). Slides were washed three times in TBS followed by TBS, 0.025% Tween-20, 5% normal goat serum, and fluorescent conjugated antibodies (goat anti-rabbit-488: Life Technologies, A31566, 1:500 dilution; goat-anti chicken 633: Life Technologies, A21103, 1:500 dilution; and GFAP-cy3: Sigma, C9205, 1:2,000 dilution). Slides were washed three times in TBS and mounted with ProLong Gold plus DAPI (Life Technologies, P36934).
Lamin B1 Quantitation in Human SNpc Astrocytes
Immunostained sections were imaged with a Zeiss LSM 780 using an EC Plan-Neofluar 40×/1.30 oil Ph3 objective. 10× bright-field microscopy was used to locate GFAP+ cells in the vicinity of clustered pigmented neurons. A z stack spanning the cell body was acquired at 1,024 × 1,024 −12 bit at 3× zoom using 1-μm steps. Fluorescence intensity was assessed using ImageJ. The optical section with the greatest green intensity (lamin B1) was identified. In the DAPI channel, the edge of the DAPI signal was outlined (defining the nuclear lamina), and the mean pixel value (MPV) on the green channel (lamin B1) was calculated; this signal was divided by a signal outside all nuclei in order to normalize for background staining and normalized fluorescence intensity averaged across 5 GFAP+ cells within a single section. Neighboring GFAP− cells were chosen based on being closest and in the same Z section as the analyzed astrocyte. Significance was calculated using a paired t test pairing age- and sex-matched individuals from PD and control groups (n = 5 PD samples, 5 controls).
Treatment of Human Cultures
Human astrocytes derived from iPSCs purchased from XCell Science (Novato CA) grown in astrocyte media were cultured at 37°C, 3% O2, and 5% CO2 in DMEM supplemented with 10% horse serum, 10% fetal bovine serum (FBS), and penicillin/streptomycin. Cells were seeded at 1 × 104 cells/cm2 and cultured to 70%–80% confluence. WI-38 primary human fibroblasts were obtained from the Coriell Cell Repository. Cell culture and irradiation (IR, 10 Gy X-rays) were carried out as described previously (Freund et al., 2012). Cells were treated with indicated PQ concentrations for 24 or 48 hr and washed twice with culture media; analyses were performed 7–8 days later.
BrdU Incorporation
Cell proliferation was determined by measuring DNA synthesis using a modified BrdU labeling kit (Roche). Cells were seeded in 4-well chamber slides and incubated with BrdU for 1 hr before washing and fixing with 4% paraformaldehyde. Fixed cells were washed with PBS and BrdU immunofluorescence quantified.
SA-β-gal Assay
SA-β-gal staining was performed as described previously (Dimri et al., 1995). Cells were plated at 1 × 104 cells/cm2 and assessed for SA-β-gal activity 24–48 hr after seeding in a minimum of 400 cells. Positive (blue) cells are expressed as a percentage of total cell number.
Immunofluorescence of Cultured Cells
Cells were fixed with 4% formaldehyde, permeabilized with 0.5% Triton, and blocked with 10% goat or donkey serum before incubating overnight with primary antibody at 4°C. After washing in PBS, cells were incubated with Alexa-conjugated secondary antibodies for 30–45 min at room temperature. Nuclear DNA was stained with DAPI in mounting media (Vectashield). CellProfiler image analysis software was used to score cells. Primary antibodies were anti-p16 (clone JC8: Santa Cruz Biotechnology, sc-56330, 1:200 dilution), anti-53BP1 (clone BP13: EMD Millipore, MAB3802, 1:200 dilution), anti-lamin B1 (Abcam, ab16048, 1:200 dilution), and anti-Ki67 (Abcam, ab15580; 1:200 dilution). For the latter, image data were analyzed using CellProfiler to quantify the number of Ki67+ (% Ki67+) DAPI-stained nuclei. The mean pixel area of DAPI-stained nuclei was obtained for each condition (IR and PQ) and normalized to the mean pixel area of nuclei under control conditions (for relative nuclear size).
IL-6 ELISA
CM was prepared by washing cells three times in PBS followed by incubation in serum-free DMEM for 24 hr. CM was filtered and stored at −80°C. Cell numbers were determined by counting. ELISAs were performed using the alphaLISA IL-6 Immunoassay Research kit (Perkin Elmer). Data were normalized to cell number and expressed as picograms per 1,000 cells.
Preparation of Primary 3MR Astrocytes
Primary cortical astrocytes were prepared as described previously (Schildge et al., 2013). Cortices were dissected from post-natal day 2 mice and dissociated, plated, and cultured for 2 weeks with removal of microglia by orbital shaking prior to conducting experiments. Cells were treated with PQ as indicated and after 24 hr washed twice with culture media. SA-β-gal activity was assessed 8 days after PQ treatment as described above.
Viability of Human DAergic Neurons
Primary human DAergic neurons were obtained from Celprogen (San Pedro, CA) and cultured as instructed by the manufacturer. Cells were maintained in complete medium and subcultured every 24–48 hr on human dopamine neuronal primary cell culture extracellular matrix. Briefly, 1 × 104 cells were plated onto flat-bottomed 96-well plates. CM from astrocytes was concentrated using spin columns with a 10-kDa cutoff and reconstituted in DA-complete media. After 24 hr, cells were exposed to CM astrocytic CM for 72 hr. Cells were treated with 50 μL MTT (3-[4, 5-dimethylthiazol-2-yl]-2,5-diphenyl-tertra-zolium bromide) solution (5 mg/mL) for 3 hr at 37°C. 200 μL dimethyl sulfoxide (Sigma, USA) was added to each well and incubated for 30 min at 37°C. Color intensity was quantified at 570 nm. The experiment was run three times in 3 different sets of CM, and each condition was run in quadruplicate.
Derivation of Human NPCs
Human embryonic stem cell colonies were harvested after collagenase treatment, cultured as embryoid bodies for 8 days in DMEM with 20% FBS, and cultured in neural induction medium (DMEM/F12) N2 supplement (Invitrogen, 25 ng/mL basic fibroblast growth factor [bFGF]) for 7 days on plates coated with Geltrex. Rosettes were manually isolated, dissociated into single cells and expanded in neural expansion medium. NPCs were then incubated for 48 hr in astrocytic CM. Cell proliferation was determined a using a modified BrdU labeling kit (Roche) as above.
Scratch Migration Assay
As described elsewhere (Liang et al., 2007), NPCs were cultured to confluence in 60-mm dishes. The monolayer was scraped with a p200 pipet tip to create a “scratch.” Debris was removed and astrocytic CM was added for 48 hr. Using phase contrast microscopy, images were acquired at 0, 24, and 48 hr after the scratch. The mean width of each scratch was calculated by averaging 6 width measurements taken at even intervals along the scratch length. The relative rate of migration was determined by calculating the percentage of scratch width invaded = 100% − (100% (mean width at tx − mean width at t0). Quantification was carried out using ImageJ.
PQ Administration
p16-3MR mice were generated in the C57Bl6/J mouse strain as described previously (Demaria et al., 2014). Animals were housed according to standard animal care protocols on a 12-hr light/dark cycle in a pathogen-free environment in the vivarium at the Buck Institute. 8-month-old male and female mice were injected intraperitoneally (i.p.) with saline or 7 mg/kg PQ (in saline) at 2-day intervals for a total of 6 doses. GCV (25 mg/kg body weight) was injected i.p. after the second PQ injection and every other day for a total of 5 doses. Animals were sacrificed 7 d after the last PQ administration as described previously (Peng et al., 2010). Animal care and use were in accordance with the National Institutes of Health guidelines for use of live animals and approved by the Animal Care and Use Committee at the Buck Institute.
qPCR of Mouse Striatal Tissues
RNA from PQ- and saline-treated mice was reverse transcribed using an Applied Biosystems kit (Carlsbad CA). qRT-PCR reactions were performed using the following primers and probes:
p16INK4a: FW, 5′-aatctccgcgaggaaagc-3′, RV, 5′-gtctgcagcggactccat-3′
mRFP: FW, 5′–gacctcggcgtcgtagtg-3′, RV 5′–aagggcgagatcaagatgag-3′
IL-6: FW, 5′-gctaccaaactggatataatcagga-3′, RV, 5′-ccaggtagctatggtac tccagaa-3′
tubulin: FW, 5′-ctggaacccacggtcatc-3′ and RV, 5′-gtggccacgagcatagtt att-3′.
Immunofluorescence of Mouse Brain Tissue
Whole brains were dissected and placed in cold 4% paraformaldehyde in PBS for 12 hr, washed in cold TBS, cut using a Leica VT 1000S Vibratome (collecting 50-μm horizontal sections in cryogenic buffer [0.1 M potassium acetate, 40% ethylene glycol, and 1% polyvinyl pyrrolidone]), and stored at −20°C. Free-floating sections containing the SN were transferred to 1 mL unmasking buffer (10 mM Tris and 1 mM EDTA [pH 9]) in 24-well dishes and microwaved 3 × 5 s at 100% power. Sections were washed in TBS, permeabilized in TBS plus 0.2% Triton X-100 for 1 hr, blocked in 5% normal goat serum in permeabilization buffer for 1 hr, and then bathed in blocking buffer at 4°C overnight with primary antibodies (chicken anti-TH with either rabbit anti-lamin B1 or rabbit anti-HMGB1, Abcam, ab18256, 1:200). Section were washed three times with TBS plus 0.1% Tween-20 for 1 hr and bathed in TBS, 0.1% Tween-20, 5% NGS, and fluorescent-conjugated antibodies (goat anti-rabbit immunoglobulin G [IgG]-Alexa 488 at 1:500; goat anti-chicken IgG/Alexa 633 at 1:500, and GFAP-cy3 at 1:2,000). Primary and secondary antibodies are the same as those listed for human IF. Sections were washed three times with TBS plus 0.1% Tween-20 for 2 hr before mounting on glass slides with ProLong Gold plus DAPI.
Analysis of Lamin B1 and HMGB1 in Mouse SNpc Astrocytes
Image acquisition and analysis of Lamin B1 and HMGB1 immunostaining in mouse astrocytes was conducted similar to that described for human brain tissues, except GFAP+ cells were chosen in fields containing TH staining. For HMGB1 analysis, the region of interest was drawn in the center of DAPI signal (in the nucleoplasm). Signals were calculated by averaging fluorescence from 5 astrocytes per section and subsequently averaging 3 sections per animal. Significance was calculated using non-paired t test with n = 4 animals in each condition.
Stereological SN TH+ Counts
Mouse tissues were fixed by perfusion as described and cryostat-cut sections (40 μm) were taken through the entire midbrain (Mallajosyula et al., 2008). TH+ neurons were immune-labeled by incubating the sections successively with a rabbit polyclonal anti-TH antibody (1:200) and biotinylated horse anti-rabbit IgG (1:200, Vector Laboratories) in combination with 3,3′-diaminobenzidine (DAB) reagents. Total # TH+ SNpc neurons were counted from 4 to 5 litter-mates per group using the optical fractionator method (Peng et al., 2010).
Cylinder Test of Motor Behavior
Mice were placed individually in a cylinder to determine the number of rearing events over 1 min (Willard et al., 2015). Each rearing session was video recorded. The average number of rears over two trials was taken.
In Vivo BrdU Labeling and Quantitation
BrdU (50 mg/kg; Sigma) in saline was given i.p. two times daily at 8-hr intervals on consecutive days (days 1–3 after PQ or saline administration). Mice were sacrificed on day 14 after the last injection. Brains were removed following 4% paraformaldehyde in PBS. Adjacent 50-μm sections were cut and stored at −80°C. BrdU detection was performed as described previously (Peng et al., 2008). Briefly, sections were incubated with 2 μg/mL mouse monoclonal anti-BrdU antibody (1:200, Roche) at 4°C overnight, washed with PBS, incubated with Alexa 488 anti-mouse secondary antibody (1:250) in blocking solution (Life Technologies) for 1 hr, and then washed and mounted on gelatin-coated slides with Fluoromount. SVZ BrdU+ cells were counted blinded in five 50-μm coronal sections per mouse spaced 200 μm apart under high power (200×) using a Nikon E300 microscope with a Magnifire digital camera. Results are expressed as the average number of BrdU+ cells per section.
Statistical Methods
For experiments using post-mortem human tissues (Figure 1), n represents individuals; for all experiments using cultured cells (Figures 2, 3, 4, and 5), n represents experimental replicates conducted on separate days; and for all mouse studies (Figures 6 and 7), n represents individual mice. See figure legends for exact numbers for each individual experiment. All error bars in figures display SEM. Results were reported as significant when p values were <0.05, running an unpaired Student’s t test, unless noted otherwise in figure legends.
Supplementary Material
Highlights.
Postmortem PD brain samples display increased astrocytic senescence
Cultured human astrocytes exposed to paraquat (PQ) become senescent
Clearance of senescent cells mitigates neurodegeneration in a mouse model of PD
Senescent Rastrocytes may contribute to the development of sporadic PD
Acknowledgments
We thank Dr. John Trojanowski for providing materials from the University of Pennsylvania Brain Bank and Chris Lieu for assistance with behavioral studies. This work was funded by an Ellison Senior Scholar in Aging Award AG-SS-2620-11 (J.K.A.), Buck Impact circle (J.K.A and J.C), Michael J Fox Foundation, Target validation grant MJFF-12113 (J.K.A), NIH grant AG009909 (J.C.), an NIH Institutional Postdoctoral Training Grant T32-AG000266 (G.W.), a CIRM training grant TG2-01155 (S.J.C.), and the American-Italian Cancer Foundation (M.D.).
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and three figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2017.12.092.
AUTHOR CONTRIBUTIONS
S.J.C., G.W., J.K.A., and J.C. conceived the experiments and wrote the paper. S.J.C. and G.W., with the help of M.D., A.R., Y.Z., A.M., S.R., C.L., and D.T.M., conducted experiments and gathered data. S.J.C. and G.W. analyzed the data and prepared the figures.
DECLARATION OF INTERESTS
J.C. a scientific founder of Unity Biotechnology, which is developing “senolytics” therapies for age-related pathologies. She own shares in Unity but does not receive consulting fees. Unity supports basic research in her laboratory, but not the research described in this publication. The remaining authors declare no competing interests.
References
- Allaman I, Bélanger M, Magistretti PJ. Astrocyte-neuron metabolic relationships: for better and for worse. Trends Neurosci. 2011;34:76–87. doi: 10.1016/j.tins.2010.12.001. [DOI] [PubMed] [Google Scholar]
- Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016;530:184–189. doi: 10.1038/nature16932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreto GE, Sun X, Xu L, Giffard RG. Astrocyte proliferation following stroke in the mouse depends on distance from the infarct. PLoS ONE. 2011;6:e27881. doi: 10.1371/journal.pone.0027881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauséjour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 2003;22:4212–4222. doi: 10.1093/emboj/cdg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat R, Crowe EP, Bitto A, Moh M, Katsetos CD, Garcia FU, Johnson FB, Trojanowski JQ, Sell C, Torres C. Astrocyte senescence as a component of Alzheimer’s disease. PLoS ONE. 2012;7:e45069. doi: 10.1371/journal.pone.0045069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitto A, Sell C, Crowe E, Lorenzini A, Malaguti M, Hrelia S, Torres C. Stress-induced senescence in human and rodent astrocytes. Exp Cell Res. 2010;316:2961–2968. doi: 10.1016/j.yexcr.2010.06.021. [DOI] [PubMed] [Google Scholar]
- Blesa J, Przedborski S. Parkinson’s disease: animal models and dopaminergic cell vulnerability. Front Neuroanat. 2014;8:155. doi: 10.3389/fnana.2014.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boekhoorn K, Joels M, Lucassen PJ. Increased proliferation reflects glial and vascular-associated changes, but not neurogenesis in the presenile Alzheimer hippocampus. Neurobiol Dis. 2006;24:1–14. doi: 10.1016/j.nbd.2006.04.017. [DOI] [PubMed] [Google Scholar]
- Bondolfi L, Calhoun M, Ermini F, Kuhn HG, Wiederhold KH, Walker L, Staufenbiel M, Jucker M. Amyloid-associated neuron loss and gliogenesis in the neocortex of amyloid precursor protein transgenic mice. J Neurosci. 2002;22:515–522. doi: 10.1523/JNEUROSCI.22-02-00515.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brkic M, Balusu S, Libert C, Vandenbroucke RE. Friends or foes: matrix metalloproteinases and their multifaceted roles in neurodegenerative diseases. Mediators Inflamm. 2015;2015:620581. doi: 10.1155/2015/620581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bus JS, Gibson JE. Paraquat: model for oxidant-initiated toxicity. Environ Health Perspect. 1984;55:37–46. doi: 10.1289/ehp.845537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685–705. doi: 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinta SJ, Rane A, Poksay KS, Bredesen DE, Andersen JK, Rao RV. Coupling endoplasmic reticulum stress to the cell death program in dopaminergic cells: effect of paraquat. Neuromolecular Med. 2008;10:333–342. doi: 10.1007/s12017-008-8047-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinta SJ, Lieu CA, Demaria M, Laberge RM, Campisi J, Andersen JK. Environmental stress, ageing and glial cell senescence: a novel mechanistic link to Parkinson’s disease? J Intern Med. 2013;273:429–436. doi: 10.1111/joim.12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinta SJ, Woods G, Rane A, Demaria M, Campisi J, Andersen JK. Cellular senescence and the aging brain. Exp Gerontol. 2015;68:3–7. doi: 10.1016/j.exger.2014.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppé JP, Patil CK, Rodier F, Krtolica A, Beauséjour CM, Parrinello S, Hodgson JG, Chin K, Desprez PY, Campisi J. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS ONE. 2010;5:e9188. doi: 10.1371/journal.pone.0009188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos AR, Kawahara M, Malhotra GK, Schaum N, Huang J, Ved U, Beausejour CM, Coppe JP, Rodier F, Campisi J. p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J Cell Biol. 2013;201:613–629. doi: 10.1083/jcb.201206006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, Laberge RM, Vijg J, Van Steeg H, Dollé ME, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell. 2014;31:722–733. doi: 10.1016/j.devcel.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund A, Laberge RM, Demaria M, Campisi J. Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell. 2012;23:2066–2075. doi: 10.1091/mbc.E11-10-0884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge WP, Miyawaki A, Gage FH, Jan YN, Jan LY. Local generation of glia is a major astrocyte source in postnatal cortex. Nature. 2012;484:376–380. doi: 10.1038/nature10959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankovic J. Parkinson’s disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry. 2008;79:368–376. doi: 10.1136/jnnp.2007.131045. [DOI] [PubMed] [Google Scholar]
- Jung T, Höhn A, Catalgol B, Grune T. Age-related differences in oxidative protein-damage in young and senescent fibroblasts. Arch Biochem Biophys. 2009;483:127–135. doi: 10.1016/j.abb.2008.12.007. [DOI] [PubMed] [Google Scholar]
- Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, Lu T, Yankner BA, Campisi J, Elledge SJ. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. 2015;349:aaa5612. doi: 10.1126/science.aaa5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laberge RM, Adler D, DeMaria M, Mechtouf N, Teachenor R, Cardin GB, Desprez PY, Campisi J, Rodier F. Mitochondrial DNA damage induces apoptosis in senescent cells. Cell Death Dis. 2013;4:e727. doi: 10.1038/cddis.2013.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le W, Sayana P, Jankovic J. Animal models of Parkinson’s disease: a gateway to therapeutics? Neurotherapeutics. 2014;11:92–110. doi: 10.1007/s13311-013-0234-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2:329–333. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- Mallajosyula JK, Kaur D, Chinta SJ, Rajagopalan S, Rane A, Nicholls DG, Di Monte DA, Macarthur H, Andersen JK. MAO-B elevation in mouse brain astrocytes results in Parkinson’s pathology. PLoS ONE. 2008;3:e1616. doi: 10.1371/journal.pone.0001616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marxreiter F, Regensburger M, Winkler J. Adult neurogenesis in Parkinson’s disease. Cell Mol Life Sci. 2013;70:459–473. doi: 10.1007/s00018-012-1062-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack AL, Di Monte DA. Effects of L-dopa and other amino acids against paraquat-induced nigrostriatal degeneration. J Neurochem. 2003;85:82–86. doi: 10.1046/j.1471-4159.2003.01621.x. [DOI] [PubMed] [Google Scholar]
- McCormack AL, Atienza JG, Johnston LC, Andersen JK, Vu S, Di Monte DA. Role of oxidative stress in paraquat-induced dopaminergic cell degeneration. J Neurochem. 2005;93:1030–1037. doi: 10.1111/j.1471-4159.2005.03088.x. [DOI] [PubMed] [Google Scholar]
- Mombach JC, Vendrusculo B, Bugs CA. A Model for p38MAPK-Induced Astrocyte Senescence. PLoS ONE. 2015;10:e0125217. doi: 10.1371/journal.pone.0125217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15:482–496. doi: 10.1038/nrm3823. [DOI] [PubMed] [Google Scholar]
- Nicola Z, Fabel K, Kempermann G. Development of the adult neurogenic niche in the hippocampus of mice. Front Neuroanat. 2015;9:53. doi: 10.3389/fnana.2015.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Jo YH, Cho CH, Choe W, Kang I, Baik HH, Yoon KS. ATM-deficient human fibroblast cells are resistant to low levels of DNA double-strand break induced apoptosis and subsequently undergo drug-induced premature senescence. Biochem Biophys Res Commun. 2013;430:429–435. doi: 10.1016/j.bbrc.2012.11.040. [DOI] [PubMed] [Google Scholar]
- Peng J, Xie L, Jin K, Greenberg DA, Andersen JK. Fibroblast growth factor 2 enhances striatal and nigral neurogenesis in the acute 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Neuroscience. 2008;153:664–670. doi: 10.1016/j.neuroscience.2008.02.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Oo ML, Andersen JK. Synergistic effects of environmental risk factors and gene mutations in Parkinson’s disease accelerate age-related neurodegeneration. J Neurochem. 2010;115:1363–1373. doi: 10.1111/j.1471-4159.2010.07036.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezzoli G, Cereda E. Exposure to pesticides or solvents and risk of Parkinson disease. Neurology. 2013;80:2035–2041. doi: 10.1212/WNL.0b013e318294b3c8. [DOI] [PubMed] [Google Scholar]
- Pospelova TV, Chitikova ZV, Pospelov VA. An integrated approach for monitoring cell senescence. Methods Mol Biol. 2013;965:383–408. doi: 10.1007/978-1-62703-239-1_26. [DOI] [PubMed] [Google Scholar]
- Prieur A, Peeper DS. Cellular senescence in vivo: a barrier to tumorigenesis. Curr Opin Cell Biol. 2008;20:150–155. doi: 10.1016/j.ceb.2008.01.007. [DOI] [PubMed] [Google Scholar]
- Rossi D. Astrocyte physiopathology: At the crossroads of intercellular networking, inflammation and cell death. Prog Neurobiol. 2015;130:86–120. doi: 10.1016/j.pneurobio.2015.04.003. [DOI] [PubMed] [Google Scholar]
- Salminen A, Ojala J, Kaarniranta K, Haapasalo A, Hiltunen M, Soininen H. Astrocytes in the aging brain express characteristics of senescence-associated secretory phenotype. Eur J Neurosci. 2011;34:3–11. doi: 10.1111/j.1460-9568.2011.07738.x. [DOI] [PubMed] [Google Scholar]
- Scalzo P, Kümmer A, Cardoso F, Teixeira AL. Serum levels of interleukin-6 are elevated in patients with Parkinson’s disease and correlate with physical performance. Neurosci Lett. 2010;468:56–58. doi: 10.1016/j.neulet.2009.10.062. [DOI] [PubMed] [Google Scholar]
- Schildge S, Bohrer C, Beck K, Schachtrup C. Isolation and culture of mouse cortical astrocytes. J Vis Exp. 2013;71:50079. doi: 10.3791/50079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu K, Ohtaki K, Matsubara K, Aoyama K, Uezono T, Saito O, Suno M, Ogawa K, Hayase N, Kimura K, Shiono H. Carrier-mediated processes in blood–brain barrier penetration and neural uptake of paraquat. Brain Res. 2001;906:135–142. doi: 10.1016/s0006-8993(01)02577-x. [DOI] [PubMed] [Google Scholar]
- Sohn J, Orosco L, Guo F, Chung SH, Bannerman P, Mills Ko E, Zarbalis K, Deng W, Pleasure D. The subventricular zone continues to generate corpus callosum and rostral migratory stream astroglia in normal adult mice. J Neurosci. 2015;35:3756–3763. doi: 10.1523/JNEUROSCI.3454-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner CM, Kamel F, Ross GW, Hoppin JA, Goldman SM, Korell M, Marras C, Bhudhikanok GS, Kasten M, Chade AR, et al. Rote-none, paraquat, and Parkinson’s disease. Environ Health Perspect. 2011;119:866–872. doi: 10.1289/ehp.1002839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnquist C, Horikawa I, Foran E, Major EO, Vojtesek B, Lane DP, Lu X, Harris BT, Harris CC. p53 isoforms regulate astrocyte-mediated neuroprotection and neurodegeneration. Cell Death Differ. 2016;23:1515–1528. doi: 10.1038/cdd.2016.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Deursen JM. The role of senescent cells in ageing. Nature. 2014;509:439–446. doi: 10.1038/nature13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilas-Boas V, Silva R, Guedes-de-Pinho P, Carvalho F, Bastos ML, Remião F. RBE4 cells are highly resistant to paraquat-induced cyto-toxicity: studies on uptake and efflux mechanisms. J Appl Toxicol. 2014;34:1023–1030. doi: 10.1002/jat.2926. [DOI] [PubMed] [Google Scholar]
- Watson MB, Nobuta H, Abad C, Lee SK, Bala N, Zhu C, Richter F, Chesselet MF, Waschek JA. PACAP deficiency sensitizes nigrostriatal dopaminergic neurons to paraquat-induced damage and modulates central and peripheral inflammatory activation in mice. Neuroscience. 2013;240:277–286. doi: 10.1016/j.neuroscience.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, Shirakawa K, Lim HW, Davis SS, Ramanathan A, et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016;23:303–314. doi: 10.1016/j.cmet.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willard AM, Bouchard RS, Gittis AH. Differential degradation of motor deficits during gradual dopamine depletion with 6-hydroxydopamine in mice. Neuroscience. 2015;301:254–267. doi: 10.1016/j.neuroscience.2015.05.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirdefeldt K, Adami HO, Cole P, Trichopoulos D, Mandel J. Epidemiology and etiology of Parkinson’s disease: a review of the evidence. Eur J Epidemiol. 2011;26(Suppl 1):S1–S58. doi: 10.1007/s10654-011-9581-6. [DOI] [PubMed] [Google Scholar]
- Yum HM, Zhao YM, Luo XG, Feng Y, Ren Y, Shang H, He ZY, Luo XM, Chen SD, Wang XY. Repeated LPS stimulation induces cellular senescence in BV2 cells. Neuroimmunomod. 2012;19:131–136. doi: 10.1159/000330254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.