

ABSTRACT
This was a randomized, placebo-controlled, Phase I/II study conducted in a Japanese cohort to assess the safety and immunogenicity of Clostridium difficile vaccine (the same formulation as that used in the ongoing global Phase III study). Healthy Japanese adults aged 40–75 years were randomized to receive either C. difficile vaccine (N = 67) or placebo (N = 34) by intramuscular injection on Days 0, 7, and 30.
Serum IgG specific for toxins A and B was measured by enzyme-linked immunosorbent assay (ELISA) and in vitro functional activity by toxin neutralizing assay (TNA). The seroconversion rate (percentage of participants with a ≥4-fold rise in antibody levels from baseline) was high for both toxin A (ELISA and TNA) and toxin B (ELISA), approaching 100% for each by Day 60. For toxin B assessed by TNA, however, the response was lower, with the seroconversion rate not rising significantly beyond the value of 42.9% seen on Day 14 (44.4% at Day 60). Although the response in the participants who were seronegative at baseline was slower than that in those who were seropositive, seroconversion was seen in nearly all (100%) subjects by Day 60, with the exception of the response to toxin B evaluated using TNA (16–18% on Days 14–60).
The proportion of participants with solicited local reactions, solicited systemic reactions, and vaccine-related unsolicited reactions were 67.6%, 19.1%, and 20.6%, respectively. Most of the adverse reactions were mild to moderate in intensity, occurring within 3 days post-vaccination, and resolving by 3–6 days post-vaccination. There were no withdrawals due to adverse events and no serious adverse events.
These data confirm the safety and immunogenicity of C. difficile vaccine in Japanese adults.
KEYWORDS: Clostridium difficile, immunogenicity, safety, vaccine
Introduction
Clostridium difficile is a Gram-positive, anaerobic, spore-forming bacterium and a leading cause of antibiotic-associated diarrhea and colitis.1 C. difficile infection (CDI) is caused by toxin A and toxin B. CDI includes mild to severe diarrhea that can progress to pseudomembranous colitis and even death.2,3
The incidence of CDI varies from nation to nation, and EU data demonstrate 4.1 cases/10,000 patient days.4 In Japan, CDI awareness is low, and CDI may be incorrectly diagnosed.5 Some data derived from single center show 0.8-6.8 cases/10,000 patient days.6–8
The risk factors for CDI include antibiotics use, aging, and underlying diseases. Additionally, frequent use of antibiotics in Japan put the elderly at risk of CDI. As CDI imposes considerable financial burden9,10), vaccine can prevent the elderly from CDI.
C. difficile vaccine is a novel toxoid vaccine developed by Sanofi Pasteur and currently under evaluation in global Phase III clinical trial. The C. difficile vaccine yielded good safety and immunogenicity in Phase I and Phase II trials in the USA.11–13 This study was Phase I/II trial to assess immunogenicity and safety of C. difficile vaccine in Japanese adults prior to Japan's entry to ongoing global Phase III clinical trial.
This study was conducted according to Good Clinical Practice (GCP), International Conference on Harmonization (ICH) guidelines, the principles of the Declaration of Helsinki, and applicable rules.
Results
Participants
A total of 102 participants were enrolled into the study and randomized in a 2:1 ratio (68 receiving C. difficile vaccine and 34 receiving placebo). Only one participant failed to complete the study, due to voluntary withdrawal prior to the third vaccination in the C. difficile vaccine group. Thus, 102 and 101 participants were included in the safety and immunogenicity analyses, respectively (Fig. 1).
Figure 1.
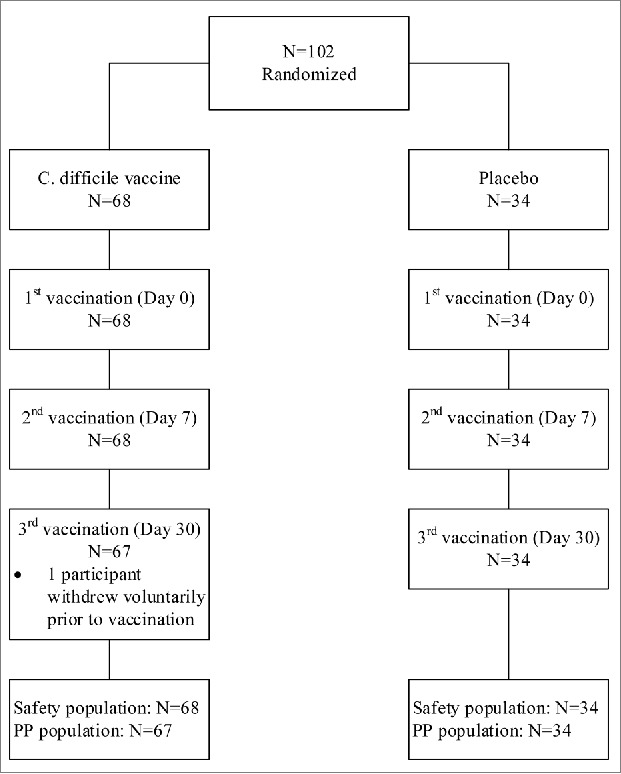
Participant flow chart.
Demographic characteristics
There were slightly more males than females (55.9% vs 44.1%), and most participants were aged 40–64 years (77.9% and 67.6% in the C. difficile vaccine and placebo groups, respectively). The median age was 50.2 and 57.8 years in the C. difficile vaccine and placebo groups, respectively (Table 1).
Table 1.
Demographic characteristics at baseline.
| C. difficile toxoid vaccine (N = 68) | Placebo (N = 34) | All (N = 102) | |
|---|---|---|---|
| Sexa | |||
| Male | 38 (55.9%) | 19 (55.9%) | 57 (55.9%) |
| Female | 30 (44.1%) | 15 (44.1%) | 45 (44.1%) |
| Age (years) | |||
| Mean (SD) | 53.8 (9.5) | 57.3 (9.5) | 54.9 (9.6) |
| Median | 50.2 | 57.8 | 51.5 |
| Min-Max | 41.4–73.8 | 42.6–73.2 | 41.4–73.8 |
| Age group (years)a | |||
| ≥40–64 | 53 (77.9%) | 23 (67.6%) | 76 (74.5%) |
| ≥65–75 | 15 (22.1%) | 11 (32.4%) | 26 (25.5%) |
Values represent the number (percentage) of participants
Safety
The proportion of participants with any solicited injection site AEs was 67.6% for the C. difficile vaccine and 14.7% for placebo (Table 2). For systemic AEs, the proportion was 19.1% for the C. difficile vaccine and 11.8% for placebo. No immediate AEs occurred in the 30 minutes after any vaccination. No AE led to discontinuation from the study, and there were no SAEs or deaths in either group.
Table 2.
Solicited injection site and systemic adverse reactions occurring within 7 days and unsolicited adverse events (by system organ class) occurring within 30 days after any dose of vaccine or placebo (safety analysis set).
| C. difficile toxoid vaccine | Placebo | |||
|---|---|---|---|---|
| (N = 68) |
(N = 34) |
|||
| Adverse event | n (%) | 95% CI | n (%) | 95% CI |
| Solicited adverse reactions | ||||
| Any solicited reaction | 46 (67.6%) | 55.2;78.5 | 7 (20.6%) | 8.7;37.9 |
| Injection site reactions | 46 (67.6%) | 55.2;78.5 | 5 (14.7%) | 5.0;31.1 |
| Pain | 46 (67.6%) | 55.2;78.5 | 5 (14.7%) | 5.0;31.1 |
| Erythema | 11 (16.2%) | 8.4;27.1 | 0 | 0.0;10.3 |
| Swelling | 8 (11.8%) | 5.2;21.9 | 0 | 0.0;10.3 |
| Systemic reactions | 13 (19.1%) | 10.6;30.5 | 4 (11.8%) | 3.3;27.5 |
| Fever | 2 (2.9%) | 0.4;10.2 | 0 | 0.0;10.3 |
| Headache | 6 (8.8%) | 3.3;18.2 | 1 (2.9%) | 0.1;15.3 |
| Malaise | 8 (11.8%) | 5.2;21.9 | 2 (5.9%) | 0.7;19.7 |
| Myalgia | 7 (10.3%) | 4.2;20.1 | 3 (8.8%) | 1.9;23.7 |
| Arthralgia | 2 (2.9%) | 0.4;10.2 | 0 | 0.0;10.3 |
| Unsolicited adverse events* | ||||
| Any unsolicited adverse event | 17 (25.0%) | 15.3;37.0 | 2 (5.9%) | 0.7;19.7 |
| Infections and infestations | 1 (1.5%) | 0.0;7.9 | 1 (2.9%) | 0.1;15.3 |
| Musculoskeletal and connective tissue disorders | 2 (2.9%) | 0.4;10.2 | 0 | — |
| General disorders and administration site conditions | 14 (20.6%) | 11.7;32.1 | 1 (2.9%) | 0.1;15.3 |
| Injury, poisoning, and procedural complications | 1 (1.5%) | 0;7.9 | 0 | — |
N = number of participants in safety analysis set.
n = number of participants with available data for the event.
95% CI = 95% confidence interval.
Adverse events are presented by System Organ Class (SOC). SOCs follow the structural hierarchy of the MedDRA (Medical Dictionary for Regulatory Activities) terminology, developed by the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). In this study, the coding of adverse events and reactions was in accordance with MedDRA version 16.0.
The most common solicited injection site AE and systemic AE in the C. difficile vaccine group was pain (67.6%) and malaise (11.8%), respectively (Table 2). Most solicited AEs were Grade 1 or 2 in intensity; Grade 3 injection site AEs occurred in 8.8% of the C. difficile vaccine group (there were none in the placebo group) and there were no Grade 3 systemic AEs in either group. Most solicited AEs occurred within 3 days post-vaccination and resolved within 3 days (injection site AEs) and 6 days (systemic AEs) of vaccination.
The proportion of participants with any unsolicited AE in the 30 days after any vaccination was higher for the C. difficile vaccine group (25.0%) than for placebo (5.9%) (Table 2). Of these unsolicited AEs, 20.6% (C. difficile vaccine) and 2.9% (placebo) were considered to be related to the investigational medicinal product.
Immunogenicity
Geometric mean concentration (GMC)
In the C. difficile vaccine group, GMCs at Day 0 and Day 60 were 0.91 EU/mL and 96.06 EU/mL for toxin A, and 1.36 EU/mL and 94.04 EU/mL for toxin B (Table 3). For both toxins A and B, the overall GMCs increased throughout the study, being highest at Day 60. The percentage of participants seropositive at baseline for toxin A (≥1.5 EU/mL) and toxin B (≥0.8 EU/mL) was, respectively, 16.4% and 52.2% in the C. difficile vaccine group. Of these, peak GMCs were observed at Day 14 (600.22 EU/mL) for toxin A and at Day 60 (219.12 EU/mL) for toxin B (Table 3). On the other hand, in the seronegative group, peak GMCs were observed at Day 60 (75.00 EU/mL for toxin A, 37.28 EU/mL for toxin B), being lower than those of seropositive group. We observed no meaningful differences between age or sex sub-groups.
Table 3.
Summary of ELISA GMCs for toxin A and toxin B on Days 0, 14, 30, and 60 (PP set).
|
C. difficile toxoid vaccine (N = 67) |
||||||||
|---|---|---|---|---|---|---|---|---|
| Parameter | Overall |
Seropositivea |
Seronegativea |
Placebo (N = 34) |
||||
| Visit | GMC | 95% CI | GMC | 95% CI | GMC | 95% CI | GMC | 95% CI |
| Toxin A IgG (EU/mL) | ||||||||
| Day 0 | 0.91 | 0.81;1.01 | 2.37 | 1.88;3.00 | 0.75 | NC | 0.90 | 0.76;1.06 |
| Day 14 | 15.53 | 8.73;27.63 | 600.22 | 226.41;1591.19 | 7.58 | 4.73;12.15 | 0.91 | 0.76;1.08 |
| Day 30 | 48.10 | 33.05;70.00 | 472.18 | 238.53;934.70 | 30.71 | 22.36;42.17 | 0.88 | 0.76;1.02 |
| Day 60 | 96.06 | 78.79;117.11 | 338.66 | 226.79;505.72 | 75.00 | 64.04;87.82 | 0.87 | 0.75;1.02 |
| Toxin B IgG (EU/mL) | ||||||||
| Day 0 | 1.36 | 0.97;1.93 | 4.19 | 2.90;6.07 | 0.40 | NC | 1.06 | 0.72;1.55 |
| Day 14 | 11.70 | 5.28;25.92 | 144.90 | 66.20;317.16 | 0.75 | 0.45;1.24 | 1.03 | 0.71;1.49 |
| Day 30 | 38.64 | 21.88;68.25 | 194.12 | 110.80;340.09 | 6.61 | 3.74;11.69 | 1.05 | 0.72;1.53 |
| Day 60 | 94.04 | 66.81;132.38 | 219.12 | 150.27;319.52 | 37.28 | 25.24;55.06 | 1.10 | 0.75;1.61 |
Participants who were seropositive by ELISA (N = 11 and 35 for toxins A and B, respectively) or seronegative by ELISA (N = 56 and 32 for toxins A and B, respectively) at baseline.
NC = not calculated.
Geometric mean titer (GMT)
For toxin A, the overall GMT response profile was similar to the GMC response, peaking at Day 60 (834.77 [1/dil]) (Table 4). However, for toxin B, the overall GMT response peaked at Day 14 (139.59 [1/dil]) with no further change to Day 60 (126.58 [1/dil]). The percentage of participants seropositive at baseline for toxin A (≥16 [1/dil]) and toxin B (≥16 [1/dil]) was, respectively, 26.9% and 31.7% in the C. difficile vaccine group. The GMT response profile was similar to that for GMCs for toxin A (i.e. peak response was earlier [Day 14] and higher for participants who were seropositive at baseline) (Table 4). In the seronegative group, peak GMTs were observed at Day 60 for toxins A and B. There were no significant differences between age or sex sub-groups.
Table 4.
Summary of TNA GMTs for toxin A and toxin B on Days 0, 14, 30, and 60 (PP set).
|
C. difficile toxoid vaccine (N = 67a) |
||||||||
|---|---|---|---|---|---|---|---|---|
| Parameter | Overall |
Seropositiveb |
Seronegativeb |
Placebo (N = 34c) |
||||
| Visit | GMT | 95% CI | GMT | 95% CI | GMT | 95% CI | GMT | 95% CI |
| Toxin A | ||||||||
| Day 0 | 14.86 | 11.31;19.53 | 80.29 | 52.26;123.34 | 8.00 | NC | 12.83 | 8.99;13.83 |
| Day 14 | 251.19 | 113.33;556.73 | 12932.65 | 3362.61;49739.12 | 59.05 | 33.07;105.45 | 13.07 | 9.19;18.59 |
| Day 30 | 361.67 | 185.97;703.33 | 9068.94 | 2888.30;28475.47 | 110.74 | 67.19;182.51 | 13.25 | 9.24;19.01 |
| Day 60 | 834.77 | 536.46;1298.94 | 7839.07 | 3375.77;18203.53 | 366.65 | 277.96;483.62 | 13.89 | 9.78;19.73 |
| Toxin B | ||||||||
| Day 0 | 17.12 | 12.57;23.33 | 87.92 | 57.98;133.34 | 8.00 | NC | 15.91 | 10.94;23.13 |
| Day 14 | 139.59 | 59.87;325.46 | 11104.54 | 6315.81;19524.13 | 18.47 | 10.50;32.46 | 15.00 | 10.42;21.58 |
| Day 30 | 135.84 | 59.26;311.38 | 9817.25 | 5878.55;16394.93 | 18.88 | 10.69;33.32 | 15.50 | 10.61;22.64 |
| Day 60 | 126.58 | 58.15;275.51 | 7619.48 | 4583.79;12665.60 | 22.82 | 12.92;40.29 | 16.24 | 11.24;23.46 |
N = 63 for Toxin B (Day 0) and N = 66 for Toxin B (Days 14, 30, 60).
Participants who were seropositive by TNA (N = 18 and 20 for toxins A and B, respectively) or seronegative by TNA (N = 49 and 43 for toxins A and B, respectively) at baseline.
N = 33 for Toxin B (Days 0 and 30).
NC = not calculated.
Seroconversion
The seroconversion rate (percentage of participants with a ≥4-fold increase from Day 0) for each toxin measured by ELISA showed the same trends as the GMC data, with the highest percentage of seroconverted participants being at Day 60 (Table 5). The seroconversion rate for both toxins (composite response) was highest at Day 60. While relatively higher seroconversion rates for both toxins A and B were observed at Days 14 and 30 for participants who were seropositive at baseline (i.e. the seroconversion occurred earlier if seropositive) compared to those who were seronegative at baseline, at Day 60 there was no difference in seroconversion rate for either toxin based on baseline seropositivity (approximately 100% for both toxins in each sub-group) (Table 5).
Table 5.
Summary of ELISA seroconversion rates for toxin A, toxin B, and composite response on Days 14, 30 and 60 (PP set).
|
C. difficile toxoid vaccine (N = 67) |
||||||||
|---|---|---|---|---|---|---|---|---|
| Parameter | Overall |
Seropositivea |
Seronegativea |
Placebo (N = 34) |
||||
| Visit | % | 95% CI | % | 95% CI | % | 95% CI | % | 95% CI |
| Toxin A IgG | ||||||||
| Day 14 | 49.3 | 36.8;61.8 | 90.9 | 58.7;99.8 | 41.1 | 28.1;55.0 | 0 | 0.0;10.3 |
| Day 30 | 94.0 | 85.4;98.3 | 100 | 71.5;100.0 | 92.9 | 82.7;98.0 | 0 | 0.0;10.3 |
| Day 60 | 100 | 94.6;100.0 | 100 | 71.5;100.0 | 100 | 93.6;100.0 | 0 | 0.0;10.3 |
| Toxin B IgG | ||||||||
| Day 14 | 46.3 | 34.0;58.9 | 82.9 | 66.4;93.4 | 6.3 | 0.8;20.8 | 0 | 0.0;10.3 |
| Day 30 | 83.6 | 72.5;91.5 | 97.1 | 85.1;99.9 | 68.8 | 50.0;83.9 | 0 | 0.0;10.3 |
| Day 60 | 97.0 | 89.6;99.6 | 97.1 | 85.1;99.9 | 96.9 | 83.8;99.9 | 0 | 0.0;10.3 |
| Composite responseb | ||||||||
| Day 14 | 37.3 | 25.8;50.0 | 90.9 | 58.7;99.8 | 3.1 | 0.1;16.2 | 0 | 0.0;10.3 |
| Day 30 | 80.6 | 69.1;89.2 | 100 | 71.5;100.0 | 65.6 | 46.8;81.4 | 0 | 0.0;10.3 |
| Day 60 | 97.0 | 89.6;99.6 | 100 | 71.5;100.0 | 96.9 | 83.8;99.9 | 0 | 0.0;10.3 |
Participants who were seropositive by ELISA (N = 11 and 35 for toxins A and B, respectively) or seronegative by ELISA (N = 56 and 32 for toxins A and B, respectively) at baseline.
Composite response indicates a ≥4-fold increase for both toxins.
The seroconversion rate measured by TNA showed the highest percentage of seroconverted participants being at Day 60 for toxin A (94.0%) and the response increasing for toxin B to Day 60 (44.4%) with little further change from Day 14 (42.9%) (Table 6). The composite response increased from 33.3% at Days 14 and 30 to 42.9% at Day 60. The seroconversion response for toxin A was earlier for participants who were seropositive at baseline but by Day 60 approximately 100% of participants seroconverted in each group (Table 6). For toxin B, however, whereas 100% of seropositive participants seroconverted by Day 14, only 18.6% of seronegative participants seroconverted by Day 60. No important differences were found between age or sex sub-groups.
Table 6.
Summary of TNA seroconversion rates for toxin A, toxin B, and composite response on Days 14, 30 and 60 (PP set).
|
C. difficile toxoid vaccine (N = 67a) |
||||||||
|---|---|---|---|---|---|---|---|---|
| Parameter | Overall |
Seropositiveb |
Seronegativeb |
Placebo (N = 34c) |
||||
| Visit | % | 95% CI | % | 95% CI | % | 95% CI | % | 95% CI |
| Toxin A | ||||||||
| Day 14 | 49.3 | 36.8;61.8 | 83.3 | 58.6;96.4 | 36.7 | 23.4;51.7 | 0 | 0.0;10.3 |
| Day 30 | 62.7 | 50.0;74.2 | 88.9 | 65.3;98.6 | 53.1 | 38.3;67.5 | 0 | 0.0;10.3 |
| Day 60 | 94.0 | 85.4;98.3 | 94.4 | 72.7;99.9 | 93.9 | 83.1;98.7 | 0 | 0.0;10.3 |
| Toxin B | ||||||||
| Day 14 | 42.9 | 30.5;56.0 | 100 | 83.2;100.0 | 16.3 | 6.8;30.7 | 0 | 0.0;10.6 |
| Day 30 | 42.9 | 30.5;56.0 | 100 | 83.2;100.0 | 16.3 | 6.8;30.7 | 0 | 0.0;10.6 |
| Day 60 | 44.4 | 31.9;57.5 | 100 | 83.2;100.0 | 18.6 | 8.4;33.4 | 0 | 0.0;10.6 |
| Composite responsed | ||||||||
| Day 14 | 33.3 | 22.0;46.3 | 91.7 | 61.5;99.8 | 13.2 | 4.4;28.1 | 0 | 0.0;10.6 |
| Day 30 | 33.3 | 22.0;46.3 | 100 | 73.5;100.0 | 10.5 | 2.9;24.8 | 0 | 0.0;10.6 |
| Day 60 | 42.9 | 30.5;56.0 | 100 | 73.5;100.0 | 15.8 | 6.0;31.3 | 0 | 0.0;10.6 |
N = 63 for Toxin B and composite response.
Participants who were seropositive by TNA (N = 18 and 20 for toxins A and B, respectively) or seronegative by TNA (N = 49 and 43 for toxins A and B, respectively) at baseline.
N = 33 for Toxin B and composite response.
Composite response indicates a ≥4-fold increase for both toxins.
Discussion
There are some vaccines in clinical development for CDI prevention.15,16 The C. difficile vaccine used in the present study is a formaldehyde-inactivated vaccine that contains toxoids A and B purified from anaerobic cultures of C. difficile ATCC 43255 strain.
The early clinical development studies i.e. Phase I & II for C. difficile vaccine by Sanofi Pasteur were conducted in the USA to define the optimal formulation and schedule in adults.11–13 These trials provided good safety and immunogenicity, leading to bringing the C. difficile vaccine into Japan.
This was the first clinical study assessing the C. difficile vaccine in Japan to assess safety and immunogenicity in adults. This study followed the dose (0.5 mL), vaccination route (intramuscularly), and schedule (Day 0, 7, 30) confirmed in the USA trials.
As to immunogenicity, peak GMC was observed at Day 60 in the C. difficile vaccine group. The immune response was stronger in participants who were seropositive at baseline than in those seronegative at baseline, with the highest GMCs at Day 60 in both sub-groups.
The peak GMTs were observed at Day 60 for toxin A and at Day 14 for toxin B in C. difficile vaccine group. The immune response was highest at Day 14 in participants seropositive at baseline and at Day 60 in those seronegative at baseline.
The seroconversion rate measured by ELISA was almost 100% for each toxin and both toxins (composite response) at Day 60 in the C. difficile vaccine group. It is also noteworthy that although the response was slower in participants seronegative at baseline, the percentage that seroconverted by Day 60 was similar (nearly 100%) to those seropositive at baseline.
On the other hand, the seroconversion rate measured by TNA was nearly 100% for each toxin at Day 60 in participants seropositive at baseline. However, in those seronegative at baseline, the seroconversion rate was 90% for toxin A at Day 60, while the highest seroconversion rates for toxin B was less than 20%. As a consequence, seroconversion rates for toxin B and both toxins (composite response) were less than 50%.
The immunogenic response was slower and lower in participants seronegative at baseline than in those seropositive at baseline. While one of the C. difficile strains circulating in Japan is ATCC 43598 (tcdA-tcdB+; Toxinotype VIII; Ribotype 017)17), the vaccine strain, ATCC 43255 (tcdA+tcdB+; Toxinotype 0; Ribotype 087), has been shown to cross-protect ATCC 43598 strain in both in vitro studies and in vivo hamster challenge studies (manuscript in preparation). As the high seroconversion rates even for toxin A were observed in baseline seronegatives, the difference between the vaccine and circulating strains may not be the best explanation. One possible explanation is that higher immunogenicity of toxin A vs toxin B is attributed to innate response to toxin A. The Phase II trial conducted in the USA resulted in the same trend as the present study with low immunogenicity for toxin B.13 There is no published literature linking specific levels of vaccine-induced antibody in either of the two assays to clinical outcomes. In the absence of efficacy trial, there is no correlate of protection. However, data indicate that essentially all participants are able to mount an antibody response against both Toxin A and Toxin B. As this Phase I/II study recruited relatively small number of subjects, the immunogenicity for toxin B may be validated in a large-scale Phase III clinical trial.
In terms of safety, major (≥10% of participants) solicited injection site AEs and solicited systemic AEs included pain, erythema, swelling, malaise, and myalgia with Grade 1–2 in intensity. Most solicited injection site AEs and solicited systemic AEs occurred within 3 days post-vaccination, lasted 1–5 days, and resolved within 6 days post-vaccination. There were no differences based on participant age or sex. Overall, the safety profile was similar to that of Phase II data.13
These data demonstrate the safety and immunogenicity of the C. difficile vaccine, given intramuscularly to healthy Japanese adults aged 40–75 years in a 0, 7, 30 day schedule.
Materials and methods
This study took place at a single center between July to October 2013, sponsored by Sanofi K.K. (Tokyo, Japan). The protocol was agreed with Pharmaceuticals and Medical Devices Agency (PMDA) and approved by IRB prior to study start. The trial was registered on ClinicalTrials.gov (NCT01896830).
Participants
Participants aged 40 to 75 years were eligible. The main exclusion criteria were pregnancy or lack of effective contraception for women of child-bearing age; participation in a clinical study within 4 weeks prior to the study or planned participation in another clinical study during the duration of this study; receipt of any vaccination (except influenza and pneumococcal vaccines) in the 4 weeks prior to the study; previous vaccination against C. difficile; current or prior CDI episode; receipt of blood products in the previous 3 months; congenital or acquired immunodeficiency or receipt of immunosuppressive therapy in the previous 6 months, or corticosteroid therapy for >2 consecutive weeks in the previous 3 months; hepatitis B, hepatitis C, or human immunodeficiency virus seropositivity; hypersensitivity to a vaccine component; convulsions, thrombocytopenia, intestinal bleeding, surgery for gastrointestinal malignancy in the previous 3 months, or any chronic/acute condition or addiction that could interfere with compliance with study procedures.
Informed consent
Written informed consent was obtained from each participant prior to enrolment into the study.
Vaccines
The C. difficile vaccine (batch number UD16108) was in a lyophilized preparation that was reconstituted prior to intramuscular injection. Each dose of reconstituted vaccine (0.5 mL/dose) contained 100 µg toxoids A and B, and aluminum adjuvant. The C. difficile Toxoid Vaccine consists of formaldehyde-inactivated toxin A and toxin B isolated from anaerobic cultures of C. difficile by diafiltration and chromatographic purification. The complete C. difficile Toxoid Vaccine manufacturing process of the Drug Substance and Drug Product is performed at Sanofi Pasteur, Inc., Swiftwater, PA, USA. The manufacturing facilities and processes are designed in compliance with current Good Manufacturing Practices (GMP) and the European Union Clinical Trial Directive. The strain of C. difficile vaccine contains American Type Culture Collection (ATCC) 43255.
The placebo (0.5 mL/dose) was 0.9% normal saline (batch number UD16109), which has been licensed as part of a yellow fever vaccine (Stamaril®, multi-dose vial presentation) in France for over 30 years.
Study design
Following registration, each participant was randomized to receive either C. difficile vaccine or placebo in a 2:1 ratio on Days 0, 7 and 30 intramuscularly into the upper arm.
Endpoints
Immunogenicity
Blood samples were collected on Days 0, 14, 30, and 60, and analyzed for (i) serum antibody concentrations against C. difficile toxins A and B (measurement of immunoglobulin G [IgG] using enzyme-linked immunosorbent assay [ELISA]), and (ii) serum antibody titers against toxins A and B (measurement of neutralizing capacity by toxin neutralizing assay [TNA]). All analyses were conducted centrally at the Sponsor's Global Clinical Immunology (PA, USA) as described previously.13 Briefly, for ELISA, ELISA plates were coated with C. difficile toxin A or toxin B, and then incubated at 37°C after addition of controls, reference, and samples, followed by incubation with goat anti-human IgG conjugated to horseradish peroxidase. After exposure to peroxidase enzyme substrate, the plates were read using SoftMax Pro software.
For TNA, 2-fold serial dilutions of sera were mixed with C. difficile challenge toxin for 60–75 min at 37°C. Toxin-sensitive Vero cells were added to toxin-serum mixtures and grown for 6 days. The optical density (OD) was determined at 562 nm. The neutralizing antibody titers were interpolated using SoftMax Pro software as the reciprocal dilution corresponding to the 50% specific signal, i.e., the OD value of the cell control plus one half the difference between the OD values of toxin and cell controls.
Immunogenicity assessment was based on seroconversion rate and geometric mean concentration (GMC) as measured by ELISA or geometric mean titer (GMT) as measured by TNA. Seroconversion rate is defined as percentage of participants with a ≥4-fold increase from Day 0.
Safety
All participants were monitored for any immediate adverse events (AEs) that occurred in the 30 minutes after each vaccination. AEs were defined any untoward medical occurrence following immunization irrespective of causal relationship with vaccination. Solicited injection site AEs (pain, erythema, and swelling) and systemic AEs (fever, headache, malaise, myalgia, and arthralgia) were collected on the day of vaccination and for 6 days after each vaccination. The solicited injection site AEs and systemic AEs were collected through diary and assessed by the investigator.
Assessment of the intensity of solicited injection site reactions was graded as follows: pain: grade 1 (no interference with activity); grade 2 (some interference with activity); grade 3 (significant and prevents daily activity), erythema and swelling: grade 1 (≥25 to ≤50 mm); grade 2 (≥51 to ≤100 mm); grade 3 (>100 mm). Solicited systemic reactions were graded as follows: fever: grade 1 (≥38.0°C to ≤38.4°C); grade 2 (≥38.5°C to ≤38.9°C); grade 3 (≥39.0°C), headache, malaise, and myalgia: grade 1 (no interference with activity); grade 2 (some interference with activity); grade 3 (significant and prevents daily activity), arthralgia: grade 1 (free range of motion but complains of pain or discomfort); grade 2 (decreased range of motion due to pain or discomfort); grade 3 (unwilling to move due to pain).
Statistical analysis
The safety population (all participants who received at least one dose of vaccine) was used for the safety analyses, and the per protocol (PP) population (participants without a pre-defined relevant protocol deviation) was used for the immunogenicity analyses. For the main parameters, 95% confidence intervals (CIs) of point estimates were calculated using the normal approximation for quantitative data and the exact binomial distribution for proportions.14
Sub-group descriptive analyses of immunogenicity were also performed to examine the effect of age (40 to 64 years and 65 to 75 years), sex, and baseline seropositivity (defined for ELISA as ≥1.5 EU/mL and ≥0.8 EU/mL for toxins A and B, respectively, and for TNA as ≥16 [1/dil] for toxins A and B).
Disclosure of potential conflicts of interest
OM is the study investigator. DMP, SS, HO, TS, PJP, TL, AB, JM and GdeB are employees of Sanofi.
Acknowledgments
The authors would like to acknowledge all study participants and all study staff for their contributions to the study. In particular, the authors thank Kumiko Kato, Tazuko Ishikawa, Toshihiro Emori, Yusuke Hatanaka, and Ginamarie Foglia. The authors also acknowledge Global Clinical Immunology (Sanofi Pasteur) for serological testing, and Dr Andrew Lane (Lane Medical Writing, France) for providing medical writing assistance for the preparation and editing of the manuscript in accordance with the European Medical Writers Association guidelines and Good Publication Practice.
Funding
This work was supported by Sanofi Pasteur (PA, USA).
References
- 1.Lessa FC, Mu Y, Bamberg WM, Beldavs ZG, Dumyati GK, Dunn JR, Farley MM, Holzbauer SM, Meek JI, Phipps EC, et al.. Burden of Clostridium difficile infectin in the United States. N Engl J Med. 2015;372(9):825–34. doi: 10.1056/NEJMoa1408913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kyne L, Farrell FJ, Kelly CP. Clostridium difficile. Gastroenterol Clin North Am. 2001;30(3):753–77, ix-x. doi: 10.1016/S0889-8553(05)70209-0. [DOI] [PubMed] [Google Scholar]
- 3.Vaishnavi C. Clinical spectrum & pathogenesis of Clostridium difficile associated diseases. Indian J Med Res. 2010;131:487–99. [PubMed] [Google Scholar]
- 4.Bauer MP, Notermans DW, van Benthem BH, Brazier JS, Wilcox MH, Rupnik M, Monnet DL, van Dissel JT, Kuijper EJ; ECDIS Study Group . Clostridium difficile infection in Europe: a hospital-based survey. Lancet 2011;377(9759):63–73. doi: 10.1016/S0140-6736(10)61266-4. [DOI] [PubMed] [Google Scholar]
- 5.Mori N, Yoshizawa S, Saga T, Ishii Y, Murakami H, Iwata M, Collins DA, Riley TV, Tateda K. Incorrect diagnosis of Clostridium difficile infection in a university hospital in Japan. J Infect Chemother. 2015;21(10):718–22. doi: 10.1016/j.jiac.2015.06.009. [DOI] [PubMed] [Google Scholar]
- 6.Hikone M, Ainoda Y, Tago S, Fujita T, Hirai Y, Takeuchi K, Totsaku K. Risk factors for recurrent hospital-acquired Clostridium difficile infection in a Japanese university hospital. Clin Exp Gastroenterol. 2015;8:191–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Honda H, Yamazaki A, Sato Y, Erik Dubberke. Incidence and mortality associated with Clostridium difficile infection at a Japanese tertiary care center. Anaerobe. 2014;25:5–10. doi: 10.1016/j.anaerobe.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 8.Yoshida J, Kikuchi T, Asano I, Ueno T. Clostridium difficile infection relationship to ward-monthly antimicrobial use density and days of treatment: a three-year study. Jpn J Infect Prev Control. 2016;31(2):92–9. doi: 10.4058/jsei.31.92. [DOI] [Google Scholar]
- 9.Dubberke ER, Olsen MA. Burden of Clostridium difficile on the healthcare system. Clin Infect Dis. 2012;55(Suppl 2):S88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yasunaga H, Horiguchi H, Hashimoto H, Matsuda S, Fushimi K. The burden of Clostridium difficile-associated disease following digestive tract surery in Japan. J Hosp Infect. 2012;82(3):175–80. doi: 10.1016/j.jhin.2012.07.023. [DOI] [PubMed] [Google Scholar]
- 11.Greenberg RN, Marbury TC, Foglia G, Warny M. Phase I dose finding studies of an adjuvanted Clostridium diffcile toxoid vaccine. Vaccine. 2012;30(13):2245–9. doi: 10.1016/j.vaccine.2012.01.065. [DOI] [PubMed] [Google Scholar]
- 12.Foglia G, Shah S, Luxemburger C, Pietrobon PJ. Clostridium difficile: development of a novel candidate vaccine. Vaccine. 2012;30(29):4307–9. doi: 10.1016/j.vaccine.2012.01.056. [DOI] [PubMed] [Google Scholar]
- 13.de Bruyn G, Saleh J, Workman D, Pollak R, Elinoff V, Fraser NJ, Lefebvre G, Martens M, Mills RE, Nathan R, et al.. Defining the optimal formulation and schedule of a candidate toxoid vaccine agrainst Clostridium difficile infection: A randomized Phase 2 clinical trial. Vaccine. 2016;34(19):2170–8. doi: 10.1016/j.vaccine.2016.03.028. [DOI] [PubMed] [Google Scholar]
- 14.Newcombe RG. Two-sided confidence intervals for the single proportion: comparison of seven methods. Statistics in Medicine. 1998;17(8):857–72. doi: 10.1002/(SICI)1097-0258(19980430)17:8%3c857::AID-SIM777%3e3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 15.Sheldon E, Kitchin N, Peng Y, Eiden J, Gruber W, Johnson E, Jansen KU, Pride MW, Pedneault L. A phase 1, placebo-controlled, randomized study of the safety, tolerability, and immunogenicity of a Clostridium difficile vaccine administered with or without aluminum hydroxide in healthy adults. Vaccine. 2016;34(18):2082–91. doi: 10.1016/j.vaccine.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 16.Bezay N, Ayad A, Dubischar K, Firbas C, Hochreiter R, Kiermayr S, Kiss I, Pinl F, Jilma B, Westritschnig K. Safety, immunogenicity and dose response of VLA84, a new vaccine candidate against Clostridium difficile, in healthy volunteers. Vaccine. 2016;34(23):2585–92. doi: 10.1016/j.vaccine.2016.03.098. [DOI] [PubMed] [Google Scholar]
- 17.Burke KE, Lamont JT. Clostridium difficile infection: a worldwide disease. Gut Liver. 2014;8(1):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]