

Abstract
The disciplines of Borrelia (Borreliella) burgdorferi microbiology and Lyme disease pathogenesis have come to depend on the genetic manipulation of the spirochete. Generating mutants in these recalcitrant bacteria, while not straightforward, is routinely accomplished in numerous laboratories, although there are several crucial caveats to consider. This chapter describes the design of basic molecular genetic experiments as well as the detailed methodologies to prepare and transform competent cells, select for and isolate transformants, and complement or genetically restore mutants.
Keywords: Borrelia burgdorferi, Lyme disease, Spirochete, Molecular genetics, Electroporation, Transformation, Mutagenesis, Complementation, Shuttle vector, Antibiotic resistance
1 Introduction
The first genetic transformation of the spirochete Borrelia (Borreliella) burgdorferi was published in 1994 [1] and a detailed protocol was described in this Methods in Molecular Biology series a year later [2]. We came a long way in the subsequent two decades [3–8]. Landmark advances include the genome sequence [9, 10], improved selectable markers [11–13], shuttle vectors [14–16], transposon mutagenesis [17–19] (see Chapter 16), inducible promoter systems [20–22] (see Chapter 17), gene reporters [14, 20, 23–25] (see Chapter 18), and a counter-selectable marker [26]. However, the genetic manipulation of the serpentine bacterium remains rather labyrinthine due to its extraordinarily complex genome composed of a ~950-kb linear chromosome and about 20–25 linear and circular plasmids encoding several paralogs [9, 10] as well as its fastidiousness and the lack of a defined culture medium [27, 28].
The electrotransformation protocol for B. burgdorferi [1, 2] is not unlike that of other bacteria [29]: A high-voltage electric pulse produces pores in the membrane and DNA enters the cell [30–32]. This chapter presents an updated, detailed protocol for transformation of B. burgdorferi by electroporation in addition to the approach for designing allelic exchange and complementation experiments. The most important parameter for successful electrotransformation is probably the growth phase or culture cell density during competent cell preparation [1, 33]. The other key consideration is maintaining and tracking the profusion of plasmids, many of which can be lost during in vitro passage and genetic manipulation, particularly complementation [34–37]. The methodologies detailed in this chapter have laid a foundation upon which borreliologists have continued to forge a formidable genetic understanding of the microbial physiology of the spirochete and the molecular pathogenesis of Lyme disease.
2 Materials
A few strains of B. burgdorferi are available from ATCC (http://www.atcc.org), including B31 and 297, although they may not be low-passage, infectious isolates (see Note 1). Several low-passage, infectious isolates have been genetically manipulated, including strain B31 [35, 37, 38], strain 297 [14, 20, 39–41], strain N40 [42, 43], and Borrelia afzelii (a closely related genospecies) PKo [44] (see Note 2).
Barbour-Stoenner-Kelly (BSK) II medium (without gelatin): 8% (v/v) 10× CMRL-1066 (without L-glutamine and sodium bicarbonate), 4 g/L Neopeptone, 40 g/L bovine serum albumin (BSA; fraction V), 1.6 g/L Yeastolate (TC), 4.8 g/L N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), 4 g/L glucose, 0.56 g/L sodium citrate, 0.64 g/L sodium pyruvate, 0.32 g/L N-acetyl-D-glucosamine, 1.76 g/L sodium bicarbonate, and 6.6% rabbit serum (non-hemolyzed). Adjust to pH 7.6 with 10 N NaOH, stir slowly for 2–3 h, and sterilize by filtration (0.22-μm filter). Store at −20 °C (see Note 3).
Dulbecco’s phosphate-buffered saline (dPBS): 8 g/L NaCl, 0.2 g/L KCl, 1.15 g/L Na2HPO4, and 0.2 g/L KH2PO4. Sterilize by filtration and store at 4 °C.
Electroporation solution (EPS): 93 g/L sucrose and 15% (v/v) glycerol. Sterilize by filtration and store at 4 °C.
Plating-BSK (P-BSK) medium: Add 83 g BSA, 8.3 g Neopeptone, 10 g HEPES, 1.2 g sodium citrate, 8.3 g glucose, 1.3 g sodium pyruvate, 0.7 g N-acetyl-D-glucosamine, 3.7 g sodium bicarbonate, and 4.2 g Yeastolate to 1 L of water (18 MΩ cm). Adjust to pH 7.5 with 1 N NaOH, stir slowly for 2–3 h, and sterilize by filtration (0.22-μm filter). Store at −20 °C.
1.7% agarose: We use LE (low electro-endosmosis) agarose.
Antibiotic stock solutions (for selection of transformants): 50 mg/mL streptomycin, 40 mg/mL gentamicin, and 50 mg/mL kanamycin (all dissolved in water and filter-sterilized) or 10 mg/mL erythromycin (dissolved in 95% ethanol) (see Notes 4–6).
5% sodium bicarbonate: prepared fresh and filter-sterilized.
3 Methods
3.1 Designing and Generating Transformation Substrates for Gene Disruption by Allelic Exchange (See Note 7)
Screen all relevant DNA sequences for appropriate restriction enzyme sites, including the targeted gene, flanking regions, selectable marker, and cloning vector (see Note 8).
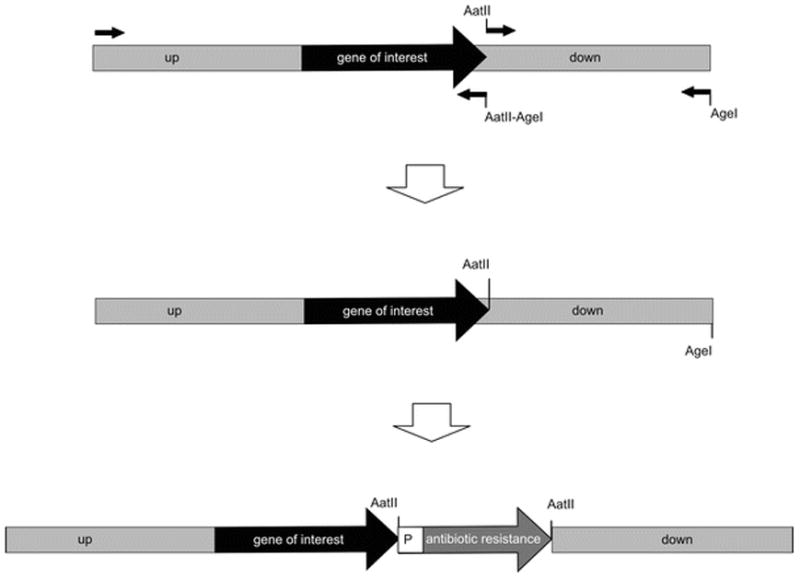
Design four primers to amplify two regions flanking the sequence to mutate with convenient restriction sites on the ends (see Note 8). The 5′ end of the upstream piece will not have any synthetic restriction sites. The 3′ end of the upstream piece will have two adjacent restriction sites (we usually use an AatII site on the inside and an AgeI site on the outside) with a suitable spacer sequence between them (three to five nucleotides), incorporated into the primer. The 5′ end of the downstream piece will have the inside restriction site of the upstream piece (AatII) and the 3′ end will have the outside restriction site of the upstream piece (AgeI), incorporated into the two primers (Fig. 1A).
The two pieces are amplified using a high-fidelity DNA polymerase (we use KOD polymerase from Novagen) and cloned (we TA-clone in pCR2.1-TOPO or, for larger pieces, TOPO-XL from Invitrogen). All cloned PCR products are confirmed by sequencing and comparison to the published genome sequence.
The downstream piece is restriction digested out of the vector with the two restriction sites (AatII and AgeI), gel-purified (see Note 9), and ligated into the vector with the upstream piece, which has also been digested with these two restriction enzymes and gel-purified. This creates a fusion in which the sequence to be mutated has been replaced with a restriction site (AatII) (Fig. 1A).
One (or more) of the selectable markers (see Note 7), which itself is flanked on both sides with this engineered restriction site (AatII), is cloned into this new, unique site. The result is that the region to be mutated has been swapped with the antibiotic resistance cassette (Fig. 1A).
Linearize the DNA by restriction digestion (usually with AhdI, which destroys the ampicillin resistance in pCR2.1 for biosafety reasons) and check by agarose gel electrophoresis to ensure the plasmid is completely linearized (see Note 10).
Purify and concentrate the linearized transformation substrate by ethanol precipitation.
Fig. 1.

Molecular cloning strategies for generating transformation substrates used to disrupt genes by allelic exchange. (a) Mutagenesis by replacing all or most of a gene of interest with an antibiotic resistance cassette transcriptionally driven by a constitutive B. burgdorferi promoter (P). (b) Mutagenesis by replacing all or most of a gene of interest with a promoterless antibiotic resistance gene fused to the promoter for the gene of interest at a synthetic NdeI site. Small arrows represent primers with the relevant restriction enzyme sites (AatII, AgeI, and NdeI); the large black arrow represents the gene of interest to be mutated; the large white arrows represent the genes conferring antibiotic resistance; and the large gray boxes represent the flanking upstream (up) and downstream (down) sequences for recombination.
3.2 Preparation of Competent Cells
Inoculate 500 mL of BSK II medium in a 500-mL screw-top bottle with 5 mL of a late-log-phase culture (see Note 11). Incubate at 32–34 °C (without agitation) until the culture reaches a density of about 5 × 107 cells/mL (see Notes 12 and 13). This requires 36–96 h.
Transfer culture to four sterile 250-mL screw-top centrifuge bottles and cap.
Centrifuge at 4000 × g for 20 min at 4 °C. Decant the supernatant fraction and resuspend each cell pellet in 15 mL of cold, sterile dPBS (see Note 14).
Transfer cells to two sterile 50-mL screw-top centrifuge tubes and cap.
Centrifuge at 3000 × g for 10 min at 4 °C. Decant the supernatant fraction, and resuspend each cell pellet in 30 mL of cold dPBS.
Centrifuge at 3000 × g for 10 min at 4 °C. Decant the supernatant fraction, and resuspend each cell pellet in 10 mL of cold EPS.
Transfer cells to two sterile 14-mL polypropylene tubes and cap.
Centrifuge at 2000 × g for 10 min at 4 °C. Decant the supernatant fraction, and resuspend each cell pellet in 10 mL of cold EPS. Repeat.
Centrifuge at 2000 × g for 10 min at 4 °C. Decant the supernatant fraction, and pool the cell pellets in 0.6 mL of cold EPS (see Note 15).
Distribute 50-μL aliquot fractions of the cell suspension into sterile 1.7-mL tubes pre-chilled on ice (see Notes 16 and 17).
3.3 Electrotransformation
Cool electroporation cuvettes (0.2-cm electrode gap) to 4 °C (see Note 18).
Transfer 1–10 μL of a solution containing ≥5 μg of DNA in water (see Notes 19 and 20) to the cell suspension, mix gently, and incubate on ice for about 5 min.
Transfer the cell/DNA mixture to a chilled electroporation cuvette. Cap the cuvette and gently tap the cell/DNA mixture to the bottom of the cuvette or otherwise ensure that the solution spans the two electrodes, being careful not to introduce bubbles.
Place the cuvette in the pulse generator and deliver a single exponential decay pulse of 2.5 kV, 25 μF, and 200 Ω. This should produce a time constant of 4–5 ms (see Note 20).
Immediately (within 1 min) add 1 mL of BSK II medium (pre-warmed to 34 °C) without antibiotics to gently resuspend the cells.
Transfer the entire mixture to a sterile 14-mL tube that contains an additional 9 mL of BSK II medium (at 34 °C) and incubate (without agitation) at 32–34 °C for 20 h (see Note 21).
3.4 Selection of Transformants by Limiting Dilution (See Note 22)
Recovered transformants are diluted into 90 mL of liquid BSK II medium with antibiotics (see Note 4) and 250 μL of culture is distributed with an 8-channel repeat pipettor into each well of four 96-well plates (see Note 22). Antibiotic final concentrations are 50 μg/mL streptomycin (see Note 5), 40 μg/mL gentamicin (see Note 6), 400 μg/mL kanamycin, or 0.06 μg/mL erythromycin.
The plates are incubated at 32–34 °C in humidified atmosphere containing 1.5% to 5% CO2 and positive wells containing putative mutants are detected by the change in color of the medium from red to yellow (see Note 23).
Isolate clones by transferring the putative mutants in the well to 10 mL of BSK II in the presence of antibiotics. Cultures will reach late-log phase in 3–5 days of growth at ~35 °C.
Screen clones for desired mutation and plasmid profile by PCR (see Note 24).
3.5 Selection of Transformants by Plating (See Note 22)
Mix 240 mL of P-BSK medium, 38 mL of 10× CMRL-1066, and 12 mL of rabbit serum. Equilibrate the mixture at 42 °C in a water bath. Autoclave 200 mL of 1.7% agarose, equilibrate to 42 °C, and combine with the medium mixture. Add 20 mL of fresh 5% sodium bicarbonate with antibiotics (the final volume is 510 mL, which is sufficient to pour 12–14 plates) (see Note 4). Antibiotic final concentrations are 50 μg/mL streptomycin (see Note 5), 40 μg/mL gentamicin (see Note 6), 400 μg/mL kanamycin, or 0.06 μg/mL erythromycin.
Transfer 0.1 mL of BSK II medium containing the electroporated cells to a 50-mL tube. Add 35 mL of the molten medium (at 42 °C), and mix by pipetting up and down once. Transfer the mixture to the 100-mm culture dishes and allow to solidify at room temperature.
Centrifuge the remaining 9.9 mL of culture at 8000 × g for 5 min, resuspend in 1 mL of supernatant fraction, and plate as above.
Incubate the plates at 32–34 °C in a humidified 5% CO2 atmosphere. Colonies will appear in about 7–14 days.
Isolate single colonies by picking with a plugged 15-cm sterile Pasteur pipette (with bulb). Transfer to 10 mL of BSK II in the presence of antibiotics. Cultures will reach late-log phase in 6–9 days of growth at ~35 °C.
Screen clones for desired mutation and plasmid profile by PCR (see Note 24).
3.6 Trans-Complementation Using a Shuttle Vector (See Note 25)
Screen all relevant DNA sequences for appropriate restriction enzyme sites, including the gene to be complemented, the relevant flanking regions to be cloned, and the shuttle vector (see Note 8).
Choose a shuttle vector containing a selectable marker different from what was used to generate the mutant. We typically use pKFSS1 [13], which carries spectinomycin/streptomycin resistance, and pBSV2 [16], which carries kanamycin resistance. Other choices include pBSV2G [12], which carries gentamicin resistance, pCE323, which carries kanamycin resistance [45], and pGK12 [15], which confers erythromycin resistance.
Design two primers to amplify the complementing gene and incorporate an appropriate restriction site at both ends of the product (again, we typically use AatII). The amplification product should include the promoter if the gene is a singleton or the first gene in an operon (see Note 26). Fuse a promoter to the ORF if one is not directly upstream (see Note 27).
Clone the gene or gene fusion into the shuttle vector and electrotransform (see Note 19).
3.7 Cis-Complementation by Genetic Reconstitution (See Note 25)
Screen all relevant DNA sequences for appropriate restriction enzyme sites, including the gene to be complemented, the flanking regions, the selectable marker, and the cloning vector (see Note 8).
Design four primers to amplify two regions, one of which will include the gene to be complemented with convenient restriction sites on the ends (we use AatII and AgeI, as described above for gene disruption) (see Notes 8 and 28). The strategy is similar to the mutagenesis described above, except the synthetic restriction site should be located between two genes outside of the operon. The two pieces are amplified and cloned as described above (Fig. 2).
The downstream piece is restriction digested out of the vector with the two restriction sites (AatII and AgeI), gel-purified, and ligated into the vector with the upstream piece, which has also been digested with these two restriction enzymes (AatII and AgeI) and gel-purified. This creates a new restriction site (AatII).
One (or more) of the selectable markers (see Note 7), which itself is flanked on both sides with this new restriction site (AatII), is cloned into this new, unique site. The result is that a marker has been inserted either upstream or downstream from the wild-type sequence.
Linearize the DNA by restriction digestion (usually with AhdI, which destroys the ampicillin resistance in pCR2.1 for biosafety reasons) (see Note 10).
Purify and concentrate the transformation substrate by ethanol precipitation.
Electrotransform into the mutant.
Fig. 2.
Molecular cloning strategy for generating transformation substrates used to cis-complement mutated genes by genetic reconstitution. Small arrows represent primers with the relevant restriction enzyme sites (AatII and AgeI); the large black arrow represents the gene of interest to be complemented; the large dark gray arrow represents the gene conferring antibiotic resistance, which must be different than the antibiotic resistance gene used to mutate the gene of interest, although the promoter (P) driving expression can be the same; and the large light gray boxes represent the flanking upstream (up) and downstream (down) sequences for recombination.
4 Notes
B31 is the type strain [46] and its genome was the first to be completely sequenced [9, 10]; however, genomes of many other B. burgdorferi strains and related genospecies (as well as other Borrelia species) are available [47]. A complete genome, including all plasmids, is useful, particularly for transcriptome studies of mutants (see Chapter 12). Note that there is conserved synteny among chromosomes of different strains, but the plasmids have considerable variability in gene composition between strains [48]. We have used strains B31-A3 [38] and 297 [40], but now rely on strain B31-5A4 [37].
Low-passage, infectious strains are more difficult to transform than high-passage, attenuated strains. The transformation barrier is likely to be restriction-modification systems [34, 36, 49, 50]. Two linear plasmids, lp25 and lp56 (in strain B31), carry genes that are predicted to encode type IV restriction-modification enzymes and are correlated with low transformation efficiency; transformants often lose lp25, which carries bbe02, encoding one of these putative enzymes [36]. Transformation efficiency increases about fortyfold in bbe02 mutants [34], although our laboratory has not observed this level of enhancement [21]. Alas, lp25 also carries the essential gene pncA [51] that is required for infectivity [35, 37]. Transformation efficiency can be increased by either first transforming the DNA into a strain with only one of the putative restriction-modification systems [50, 52] or modifying shuttle vectors in vitro with the methylase M. SssI [49]. However, the principal procedure for overcoming the transformation barrier is the use of large amounts (10 μg or more, up to ~50 μg) of DNA [14, 16, 33, 38, 40].
The quality of BSA varies by source and lot. We have found Gemini Bio-Products to be a reliable source. We reserve 5- or 10-kg lots and test samples for the ability to support growth, transformation, and temperature-induced ospC gene induction. Pretested BSK II medium without gelatin can be purchased from Sigma (BSK-H), but it is expensive (and homemade is always best).
The original antibiotic for selection of resistant mutants was coumermycin A1 [53, 54]; however, the coumermycin A1-resistant gyrB marker has not been routinely used in many years because it requires extensive screening of transformants, due to homologous recombination into the chromosomal gyrB locus [55, 56], and it causes pleiotropic effects, due to anomalous DNA supercoiling [57, 58]. The ermC gene confers erythromycin resistance in B. burgdorferi [15], but it is not widely used. There are both biosafety and physiological limitations on the use of many other antibiotics. Some antibiotic resistance genes do not function in B. burgdorferi: The cat and pac genes do not confer resistance to chloramphenicol and puromycin, respectively [12], probably because of esterases in the medium [59].
When selecting for the aadA gene, which confers resistance to spectinomycin and streptomycin [13], use 50 μg/mL spectinomycin in E. coli (when constructing transformation substrates) and 50 μg/mL streptomycin in B. burgdorferi. Selection with spectinomycin fails in B. burgdorferi because of the high frequency of background-resistant mutants [60] and the vast majority of E. coli laboratory strains are resistant to streptomycin.
The aacC1 gene, which confers resistance to gentamicin [38], does not confer sufficient resistance in E. coli in our hands: we first select E. coli transformants with another antibiotic (typically kanamycin because we usually clone DNA into pCR2.1-TOPO, which carries kanamycin resistance) and then replicaplate to gentamicin.
Our approach to generating mutants is to replace the entire open reading frame (ORF) with an antibiotic resistance gene or cassette [61–64]; we believe this is cleaner and more rigorous than an insertional mutation as no remnant of the gene product can be produced. A salient caveat to heed is polar effects on adjacent genes. Ensure that there are no overlapping genes: these could be other ORFs or RNA-encoding genes. We recommend using a promoterless antibiotic resistance gene to disrupt genes in operons [65] (Fig. 1B) or a transcriptional terminator fused to antibiotic resistance cassettes transcribed by strong promoters for singleton genes and terminal genes in operons [61]. For the latter, we prefer antibiotic resistance cassettes transcribed from the B. burgdorferi flgB promoter [11, 66] with the Bacillus subtilis trpL terminator [67]: flgBp-aadA-trpLt conferring streptomycin resistance, flgBp-aacC1-trpLt conferring gentamicin resistance, and flgBp-aphI-trpLt conferring kanamycin resistance. We typically design two different constructs with two different antibiotic resistance markers for two reasons: there is a backup if one marker does not work and there are two independent mutants if both markers work. For site-directed mutations of the genome, the antibiotic resistance cassette is inserted adjacent to the gene targeted for mutation and outside of the operon, preferably between two divergently or two convergently transcribed genes [55, 68–70]; this will require screening for the mutation(s) following selection and confirmation by DNA sequencing. We usually use overlap extension PCR to generate mutated transformation substrates [1, 21, 55, 68, 69], but QuikChange is also effective [51, 64, 70, 71].
We use MacVector software (although many other applications are suitable) to analyze DNA sequences, including finding suitable restriction enzymes; the “List non-cutters” option is particularly useful. The restriction enzyme sites that we have typically used are AatII and AgeI; we use NdeI (CATATG) for fusions of an ORF to a promoter. Again, ensure that these restriction sites are not present elsewhere in the flanking sequences to be amplified, the cloning vector, or the selectable marker.
Large fragments of DNA (>1.5 kb) are more easily cloned using crystal violet to stain the DNA in the gel instead of ethidium bromide.
Following restriction enzyme digestion of the transformation substrate for allelic exchange, analyze a small amount next to the uncut vector by agarose gel electrophoresis to confirm that the DNA is completely linearized. A circular transformation substrate can recombine into the genome and result in a merodiploid rather than a gene disruption.
B. burgdorferi is a class 2 human pathogen and therefore should be handled at BSL-2 (biosafety level 2) in a class II BSC (biological safety cabinet). In addition, BSK II is a rich medium and all procedures should be performed aseptically. Introduction of recombinant DNA into a class 2 pathogen requires approval from the IBC (Institutional Biosafety Committee) before initiation of the experiments according to Section III-D of the NIH Guidelines (https://osp.od.nih.gov/biotechnology/biosafety-and-recombinant-dna-activities/).
The cell density (or growth phase) is a significant factor for successful electrotransformation [1, 33]. The cells will not transform efficiently if the cell density is too high (when the color of the medium is yellow). We have had success in electrotransforming cultures harvested at 1–7 × 107 cells/mL, although a low cell density (1–2 × 107 cells/mL) requires pelleting the cells at a higher g force (up to 5000 × g) and adjusting the final volume of the cell suspension (see Note 15). Cell density should be determined using a Petroff-Hausser Counting Chamber (Hausser Scientific Partnership). Dilute 0.1 mL of the culture with 0.9 mL of cold dPBS and place in the counting chamber. Count cells over all 25 groups of 16 small squares in all planes using a dark-field microscope. Multiply the number of cells counted by 5 × 105 to calculate cells/mL. Alternatively, cell density can be determined by spectrophotometry [53]. Centrifuge 10 mL of the culture at 5000 × g for 10 min. Decant the supernatant fraction, and resuspend the cell pellet in 1 mL of dPBS. Centrifuge at 8000 × g for 5 min. Decant the supernatant fraction, resuspend the cell pellet in 1 mL of dPBS, and measure the OD at 600 nm. Multiply the OD by 1.4 × 108 to calculate cells/mL in the culture.
Cells can be cultured at 23 °C (or ambient temperature) to slow growth until the appropriate density is reached at a convenient time.
Thorough washing is important to remove components of the medium (see Note 20). Cell pellets are resuspended in both dPBS and EPS by pipetting up and down followed by vortex mixing. The two PBS washes can be eliminated [33, 38], although we find that decreases transformation efficiency (unpublished data).
The final cell concentration should be 1–5 × 1010 cells/mL (with a final volume of about 0.9 mL). The volume of EPS used to resuspend the final cell pellet may have to be adjusted to account for initial cell number and efficiency of decanting.
We use presterilized aerosol-resistant pipette tips (with aerosol barriers) to help maintain sterility when handling small volumes of liquid.
Competent cells can be stored on ice for a few hours or at −80 °C for a long time. Transformation efficiency peaks (up to threefold) between 2 and 6 h of incubation on ice before transformation (unpublished data). Freezing cells only slightly decreases transformation efficiently (~50% of peak), but is better than leaving the cells on ice overnight (unpublished data).
We suggest using Electroporation Cuvettes Plus, Model 620, from BTX (part number 45-0125), which includes a disposable sterile pipette that allows for easier transfer of the fragile electroporated cells from the cuvette to the culture tube.
We routinely obtain about five transformants/μg of DNA in allelic exchange experiments, but this depends on the strain and the structure of the electrotransformation substrate. The transformation efficiency of shuttle vectors is higher than allelic exchange with linear DNA [12] (unpublished data). We often use less DNA (1–2 μg) when transforming shuttle vectors than when transforming linear allelic exchange substrates (otherwise, there are too many colonies or positive wells). DNA ranging in size from 27-mer oligonucleotides [30] to 28-kb linear plasmids [72] has been used to transform B. burgdorferi.
Electroporation in the presence of high-ionic-strength solutions causes arcing (and a lower time constant). Two arcs will kill all of the B. burgdorferi cells. We use the Wizard DNA purification system (Promega), ethanol-precipitate eluted DNA, wash with 70% ethanol, and resuspend in a small amount of water: ~10 μg DNA in 10 μL water and electroporate 9 μL for allelic exchange experiments. The ethanol precipitation is important to remove traces of salt from the column and we use no more than 10 μL DNA solution in water. Again, transformation of low-passage, infectious isolates requires large amounts of DNA [14, 16, 33, 38, 40].
Do not vortex or even gently mix the electroporated cells after they have been transferred to the culture medium; vortexing will kill the electroporated cells.
Isolating B. burgdorferi mutants by selection as single colonies in a semi-solid medium [73–75] was an experimental breakthrough. However, we have almost exclusively switched to cloning by limiting dilution [41] primarily because of the convenience as the protocol requires considerably less time and effort. In addition, at least one mutant was not able to be isolated in semi-solid medium despite exhaustive efforts [41].
The color change in the medium is due to the production of lactic acid by the bacteria during growth as detected by phenol red. Incubating the 96-well plates at ambient atmosphere for 1–4 h increases the color difference by shifting the bicarbonate-CO2 equilibrium, which results in more basic (pinker) wells where there is no growth. Transformants are isolated from 96-well plates that have fewer than ten positive wells because the probability that a well is inoculated with a single cell is greater than 0.94 [26].
Plasmid content is determined by PCR screening [14, 35, 51]; there is a multiplex primer set [76] that some laboratories find convenient (see Note 2).
Complementation in trans from a shuttle vector is the most convenient approach; however, complementation in cis via either genetic reconstitution or insertion of the wild-type gene into a second genomic site is preferred for three reasons: (1) shuttle vectors can be lost in vivo in the absence of selection [77]; (2) the expression of genes in trans from shuttle vectors can be different than in cis [68]; and (3) the copy number of shuttle vectors is higher than the other genomic elements [77].
Promoters should be empirically mapped by 5′-RACE [64] or primer extension [78]; they are hard to discern by manually examining the sequence due to the low GC content and they can be far upstream from the ORF.
To fuse a promoter to the ORF, amplify a promoter (either the native promoter for the operon or a heterologous promoter, such as the strong constitutive flgB promoter or the inducible flac promoter) with an NdeI site on the 3′ end and amplify the ORF with an NdeI site (CATATG) on the 5′ end, overlapping the start codon. The PCR products are cloned into pCR2.1-TOPO. After the cloned DNA is confirmed to be the correct sequence, the clones are restriction digested with NdeI and a restriction enzyme that only cuts at the multiple cloning site of the vector on the 3′ end of the ORF. The promoter needs to be in the same orientation of the ORF in pCR2.1-TOPO so NdeI and the other restriction enzyme cut adjacent to each other. Thus, the promoter remains with pCR2.1-TOPO and the ORF is excised. The ORF is then ligated to the promoter in pCR2.1-TOPO.
Instead of cis-complementation by genetic reconstitution, the complementing gene can be inserted into the genome at another location. This strategy would entail amplifying the region surrounding the site of insertion and inserting the complementing gene.
Acknowledgments
We thank Phil Stewart for thoughtful review of the manuscript, Rich Marconi, Patti Rosa, Tom Schwan, and Kit Tilly for advice during development of the original protocol in the early 1990s, Frank Yang for suggesting the cloning-by-limiting-dilution protocol, and Christian Eggers, Mike Gilbert, Meghan Lybecker and the other members, past and present, of our laboratory for useful discussions. Genetic transformation and complementation experiments in our laboratory are supported by National Institutes of Health grant AI051486 (to D.S.S.).
References
- 1.Samuels DS, Mach KE, Garon CF. Genetic transformation of the Lyme disease agent Borrelia burgdorferi with coumarin-resistant gyrB. J Bacteriol. 1994;176:6045–6049. doi: 10.1128/jb.176.19.6045-6049.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Samuels DS. Electrotransformation of the spirochete Borrelia burgdorferi. In: Nickoloff JA, editor. Electroporation protocols for microorganisms, Methods in molecular biology. Vol. 47. Humana Press; Totowa, NJ: 1995. pp. 253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brisson D, Drecktrah D, Eggers CH, Samuels DS. Genetics of Borrelia burgdorferi. Annu Rev Genet. 2012;46:515–536. doi: 10.1146/annurev-genet-011112-112140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Groshong AM, Blevins JS. Insights into the biology of Borrelia burgdorferi gained through the application of molecular genetics. Adv Appl Microbiol. 2014;86:41–143. doi: 10.1016/B978-0-12-800262-9.00002-0. [DOI] [PubMed] [Google Scholar]
- 5.Lin T, Troy EB, Hu LT, Gao L, Norris SJ. Transposon mutagenesis as an approach to improved understanding of Borrelia pathogenesis and biology. Front Cell Infect Microbiol. 2014;4:63. doi: 10.3389/fcimb.2014.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosa PA, Cabello F, Samuels DS. Genetic manipulation of Borrelia burgdorferi. In: Samuels DS, Radolf JD, editors. Borrelia: molecular biology, host interaction and pathogenesis. Caister Academic Press; Norfolk, UK: 2010. pp. 189–219. [Google Scholar]
- 7.Rosa PA, Tilly K, Stewart PE. The burgeoning molecular genetics of the Lyme disease spirochaete. Nat Rev Microbiol. 2005;3:129–143. doi: 10.1038/nrmicro1086. [DOI] [PubMed] [Google Scholar]
- 8.Samuels DS. Antibiotic resistance in Borrelia burgdorferi: applications for genetic manipulation and implications for evolution. In: Cabello FC, Hulinska D, Godfrey HP, editors. Molecular biology of spirochetes, NATO science series: life and behavioural sciences. Vol. 373. IOS Press; Amsterdam, Netherlands: 2006. pp. 56–70. [Google Scholar]
- 9.Casjens S, Palmer N, van Vugt R, Huang WM, Stevenson B, Rosa P, Lathigra R, Sutton G, Peterson J, Dodson RJ, Haft D, Hickey E, Gwinn M, White O, Fraser CM. A bacterial genome in flux: the twelve linear and nine circular extrachromosomal DNAs in an infectious isolate of the Lyme disease spirochete Borrelia burgdorferi. Mol Microbiol. 2000;35:490–516. doi: 10.1046/j.1365-2958.2000.01698.x. [DOI] [PubMed] [Google Scholar]
- 10.Fraser CM, Casjens S, Huang WM, Sutton GG, Clayton R, Lathigra R, White O, Ketchum KA, Dodson R, Hickey EK, Gwinn M, Dougherty B, Tomb J-F, Fleischmann RD, Richardson D, Peterson J, Kerlavage AR, Quakenbush J, Salzberg S, Hanson M, van Vugt R, Palmer N, Adams MK, Gocayne J, Weidman J, Utter-back T, Watthey L, McDonald L, Artiach P, Bowman C, Garland S, Fujii C, Cotton MD, Horst K, Roberts K, Hatch B, Smith HO, Venter JC. Genomic sequence of a Lyme disease spirochete, Borrelia burgdorferi. Nature. 1997;390:580–586. doi: 10.1038/37551. [DOI] [PubMed] [Google Scholar]
- 11.Bono JL, Elias AF, Kupko JJ, III, Stevenson B, Tilly K, Rosa P. Efficient targeted mutagenesis in Borrelia burgdorferi. J Bacteriol. 2000;182:2445–2452. doi: 10.1128/jb.182.9.2445-2452.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elias AF, Bono JL, Kupko JJ, 3rd, Stewart PE, Krum JG, Rosa PA. New antibiotic resistance cassettes suitable for genetic studies in Borrelia burgdorferi. J Mol Microbiol Biotechnol. 2003;6:29–40. doi: 10.1159/000073406. [DOI] [PubMed] [Google Scholar]
- 13.Frank KL, Bundle SF, Kresge ME, Eggers CH, Samuels DS. aadA confers streptomycin-resistance in Borrelia burgdorferi. J Bacteriol. 2003;185:6723–6727. doi: 10.1128/JB.185.22.6723-6727.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eggers CH, Caimano MJ, Clawson ML, Miller WG, Samuels DS, Radolf JD. Identification of loci critical for replication and compatibility of a Borrelia burgdorferi cp32 plasmid and use of a cp32-based shuttle vector for expression of fluorescent reporters in the Lyme disease spirochaete. Mol Microbiol. 2002;43:281–296. doi: 10.1046/j.1365-2958.2002.02758.x. [DOI] [PubMed] [Google Scholar]
- 15.Sartakova M, Dobrikova E, Cabello FC. Development of an extrachromosomal cloning vector system for use in Borrelia burgdorferi. Proc Natl Acad Sci U S A. 2000;97:4850–4855. doi: 10.1073/pnas.080068797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stewart P, Thalken R, Bono J, Rosa P. Isolation of a circular plasmid region sufficient for autonomous replication and transformation of infectious Borrelia burgdorferi. Mol Microbiol. 2001;39:714–721. doi: 10.1046/j.1365-2958.2001.02256.x. [DOI] [PubMed] [Google Scholar]
- 17.Lin T, Gao L, Zhang C, Odeh E, Jacobs MB, Coutte L, Chaconas G, Philipp MT, Norris SJ. Analysis of an ordered, comprehensive STM mutant library in infectious Borrelia burgdorferi: insights into the genes required for mouse infectivity. PLoS One. 2012;7:e47532. doi: 10.1371/journal.pone.0047532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morozova OV, Dubytska LP, Ivanova LB, Moreno CX, Bryksin AV, Sartakova ML, Dobrikova EY, Godfrey HP, Cabello FC. Genetic and physiological characterization of 23S rRNA and ftsJ mutants of Borrelia burgdorferi isolated by mariner transposition. Gene. 2005;357:63–72. doi: 10.1016/j.gene.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 19.Stewart PE, Hoff J, Fischer E, Krum JG, Rosa PA. Genome-wide transposon mutagenesis of Borrelia burgdorferi for identification of phenotypic mutants. Appl Environ Microbiol. 2004;70:5973–5979. doi: 10.1128/AEM.70.10.5973-5979.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blevins JS, Revel AT, Smith AH, Bachlani GN, Norgard MV. Adaptation of a luciferase gene reporter and lac expression system to Borrelia burgdorferi. Appl Environ Microbiol. 2007;73:1501–1513. doi: 10.1128/AEM.02454-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gilbert MA, Morton EA, Bundle SF, Samuels DS. Artificial regulation of ospC expression in Borrelia burgdorferi. Mol Microbiol. 2007;63:1259–1273. doi: 10.1111/j.1365-2958.2007.05593.x. [DOI] [PubMed] [Google Scholar]
- 22.Whetstine CR, Slusser JG, Zückert WR. Development of a single-plasmid-based regulatable gene expression system for Borrelia burgdorferi. Appl Environ Microbiol. 2009;75:6553–6558. doi: 10.1128/AEM.02825-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carroll JA, Stewart PE, Rosa P, Elias AF, Garon CF. An enhanced GFP reporter system to monitor gene expression in Borrelia burgdorferi. Microbiology. 2003;149:1819–1828. doi: 10.1099/mic.0.26165-0. [DOI] [PubMed] [Google Scholar]
- 24.Hayes BM, Jewett MW, Rosa PA. lacZ reporter system for use in Borrelia burgdorferi. Appl Environ Microbiol. 2010;76:7407–7412. doi: 10.1128/AEM.01389-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hyde JA, Weening EH, Chang M, Trzeciakowski JP, Höök M, Cirillo JD, Skare JT. Bioluminescent imaging of Borrelia burgdorferi in vivo demonstrates that thefibronectin-binding-protein BBK32 is required for optimal infectivity. Mol Microbiol. 2011;82:99–113. doi: 10.1111/j.1365-2958.2011.07801.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Drecktrah D, Douglas JM, Samuels DS. Use of rpsL as a counterselectable marker in Borrelia burgdorferi. Appl Environ Microbiol. 2010;76:985–987. doi: 10.1128/AEM.02172-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barbour AG. Isolation and cultivation of Lyme disease spirochetes. Yale J Biol Med. 1984;57:521–525. [PMC free article] [PubMed] [Google Scholar]
- 28.Burgdorfer W, Barbour AG, Hayes SF, Benach JL, Grunwaldt E, Davis JP. Lyme disease—a tick-borne spirochetosis? Science. 1982;216:1317–1319. doi: 10.1126/science.7043737. [DOI] [PubMed] [Google Scholar]
- 29.Nickoloff JA, editor. Methods in molecular biology. Vol. 47. Humana Press; Totowa, NJ: 1995. Electroporation protocols for microorganisms. [Google Scholar]
- 30.Samuels DS, Garon CF. Oligonucleotide-mediated genetic transformation of Borrelia burgdorferi. Microbiology. 1997;143:519–522. doi: 10.1099/00221287-143-2-519. [DOI] [PubMed] [Google Scholar]
- 31.Shigekawa K, Dower WJ. Electroporation of eukaryotes and prokaryotes: a general approach to the introduction of macromolecules into cells. Biotechniques. 1988;6:742–751. [PubMed] [Google Scholar]
- 32.Trevors JT, Chassy BM, Dower WJ, Blaschek HP. Electrotransformation of bacteria by plasmid DNA. In: Chang DC, Chassy BM, Saunders JA, Sowers AE, editors. Guide to electroporation and electrofusion. Academic Press; San Diego: 1992. pp. 265–290. [Google Scholar]
- 33.Tilly K, Elias AF, Bono JL, Stewart P, Rosa P. DNA exchange and insertional inactivation in spirochetes. J Mol Microbiol Biotechnol. 2000;2:433–442. [PubMed] [Google Scholar]
- 34.Kawabata H, Norris SJ, Watanabe H. BBE02 disruption mutants of Borrelia burgdorferi B31 have a highly transformable, infectious phenotype. Infect Immun. 2004;72:7147–7754. doi: 10.1128/IAI.72.12.7147-7154.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Labandeira-Rey M, Skare JT. Decreased infectivity in Borrelia burgdorferi strain B31 is associated with loss of linear plasmid 25 or 28-1. Infect Immun. 2001;69:446–455. doi: 10.1128/IAI.69.1.446-455.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lawrenz MB, Kawabata H, Purser JE, Norris SJ. Decreased electroporation efficiency in Borrelia burgdorferi containing linear plasmids lp25 and lp56: impact on transformation of infectious B. burgdorferi. Infect Immun. 2002;70:4798–4804. doi: 10.1128/IAI.70.9.4798-4804.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Purser JE, Norris SJ. Correlation between plasmid content and infectivity in Borrelia burgdorferi. Proc Natl Acad Sci U S A. 2000;97:13865–13870. doi: 10.1073/pnas.97.25.13865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elias AF, Stewart PE, Grimm D, Caimano MJ, Eggers CH, Tilly K, Bono JL, Akins DR, Radolf JD, Schwan TG, Rosa P. Clonal polymorphism of Borrelia burgdorferi strain B31 MI: implications for mutagenesis in an infectious strain background. Infect Immun. 2002;70:2139–2150. doi: 10.1128/IAI.70.4.2139-2150.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Caimano MJ, Eggers CH, Hazlett KRO, Radolf JD. RpoS is not central to the general stress response in Borrelia burgdorferi but does control expression of one or more essential virulence determinants. Infect Immun. 2004;72:6433–6445. doi: 10.1128/IAI.72.11.6433-6445.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hübner A, Yang X, Nolen DM, Popova TG, Cabello FC, Norgard MV. Expression of Borrelia burgdorferi OspC and DbpA is controlled by a RpoN-RpoS regulatory pathway. Proc Natl Acad Sci U S A. 2001;98:12724–12729. doi: 10.1073/pnas.231442498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang XF, Pal U, Alani SM, Fikrig E, Norgard MV. Essential role for OspA/B in the life cycle of the Lyme disease spirochete. J Exp Med. 2004;199:641–648. doi: 10.1084/jem.20031960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chan K, Alter L, Barthold SW, Parveen N. Disruption of bbe02 by insertion of a luciferase gene increases transformation efficiency of Borrelia burgdorferi and allows live imaging in Lyme disease susceptible C3H mice. PLoS One. 2015;10:e0129532. doi: 10.1371/journal.pone.0129532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parveen N, Cornell KA, Bono JL, Chamberland C, Rosa P, Leong JM. Bgp, a secreted glycosaminoglycan-binding protein of Borrelia burgdorferi strain N40, displays nucleosidase activity and is not essential for infection of immunodeficient mice. Infect Immun. 2006;74:3016–3020. doi: 10.1128/IAI.74.5.3016-3020.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fingerle V, Goettner G, Gern L, Wilske B, Schulte-Spechtel U. Complementation of a Borrelia afzelii OspC mutant highlights the crucial role of OspC for dissemination of Borrelia afzelii in Ixodes ricinus. Int J Med Microbiol. 2007;297:97–107. doi: 10.1016/j.ijmm.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 45.Eggers CH, Caimano MJ, Radolf JD. Sigma factor selectivity in Borrelia burgdorferi: RpoS recognition of the ospE/ospF/elp promoters is dependent on the sequence of the −10 region. Mol Microbiol. 2006;59:1859–1875. doi: 10.1111/j.1365-2958.2006.05066.x. [DOI] [PubMed] [Google Scholar]
- 46.Johnson RC, Schmid GP, Hyde FW, Steigerwalt AG, Brenner DJ. Borrelia burgdorferi sp. nov.: etiologic agent of Lyme disease. Int J Syst Bacteriol. 1984;34:496–497. [Google Scholar]
- 47.Di L, Pagan PE, Packer D, Martin CL, Akther S, Ramrattan G, Mongodin EF, Fraser CM, Schutzer SE, Luft BJ, Casjens SR, Qiu W-G. BorreliaBase: a phylogeny-centered browser of Borrelia genomes. BMC Bioinformatics. 2014;15:233. doi: 10.1186/1471-2105-15-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Casjens SR, Mongodin EF, Qiu W-G, Luft BJ, Schutzer SE, Gilcrease EB, Huang WM, Vujadinovic M, Aron JK, Vargas LC, Freeman S, Radune D, Weidman JF, Dimitrov GI, Khouri HM, Sosa JE, Halpin RA, Dunn JJ, Fraser CM. Genome stability of Lyme disease spirochetes: comparative genomics of Borrelia burgdorferi plasmids. PLoS One. 2012;7:e33280. doi: 10.1371/journal.pone.0033280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen Q, Fischer JR, Benoit VM, Dufour NP, Youderian P, Leong JM. In vitro CpG methylation increases the transformation efficiency of Borrelia burgdorferi strains harboring the endogenous linear plasmid lp56. J Bacteriol. 2008;190:7885–7891. doi: 10.1128/JB.00324-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rego ROM, Bestor A, Rosa PA. Defining the plasmid-borne restriction-modification systems of the Lyme disease spirochete Borrelia burgdorferi. J Bacteriol. 2011;193:1161–1171. doi: 10.1128/JB.01176-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Purser JE, Lawrenz MB, Caimano MJ, Howell JK, Radolf JD, Norris SJ. A plasmid-encoded nicotinamidase (PncA) is essential for infectivity of Borrelia burgdorferi in a mammalian host. Mol Microbiol. 2003;48:753–764. doi: 10.1046/j.1365-2958.2003.03452.x. [DOI] [PubMed] [Google Scholar]
- 52.Jewett MW, Lawrence K, Bestor AC, Tilly K, Grimm D, Shaw P, VanRaden M, Gherardini F, Rosa PA. The critical role of the linear plasmid lp36 in the infectious cycle of Borrelia burgdorferi. Mol Microbiol. 2007;64:1358–1374. doi: 10.1111/j.1365-2958.2007.05746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Samuels DS, Garon CF. Coumermycin A1 inhibits growth and induces relaxation of supercoiled plasmids in Borrelia burgdorferi, the Lyme disease agent. Antimicrob Agents Chemother. 1993;37:46–50. doi: 10.1128/aac.37.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Samuels DS, Marconi RT, Huang WM, Garon CF. gyrB mutations in coumermycin A1-resistant Borrelia burgdorferi. J Bacteriol. 1994;176:3072–3075. doi: 10.1128/jb.176.10.3072-3075.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Knight SW, Kimmel BJ, Eggers CH, Samuels DS. Disruption of the Borrelia burgdorferi gac gene, encoding the naturally synthesized GyrA C-terminaldomain. J Bacteriol. 2000;182:2048–2051. doi: 10.1128/jb.182.7.2048-2051.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rosa P, Samuels DS, Hogan D, Stevenson B, Casjens S, Tilly K. Directed insertion of a selectable marker into a circular plasmid of Borrelia burgdorferi. J Bacteriol. 1996;178:5946–5953. doi: 10.1128/jb.178.20.5946-5953.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alverson J, Samuels DS. groEL expression in gyrB mutants of Borrelia burgdorferi. J Bacteriol. 2002;184:6069–6072. doi: 10.1128/JB.183.21.6069-6072.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alverson J, Bundle SF, Sohaskey CD, Lybecker MC, Samuels DS. Transcriptional regulation of the ospAB and ospC promoters from Borrelia burgdorferi. Mol Microbiol. 2003;48:1665–1677. doi: 10.1046/j.1365-2958.2003.03537.x. [DOI] [PubMed] [Google Scholar]
- 59.Sohaskey CD, Barbour AG. Esterases in serum-containing growth media counteract chloramphenicol acetyltransferase activity in vitro. Antimicrob Agents Chemother. 1999;43:655–660. doi: 10.1128/aac.43.3.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Criswell D, Tobiason VL, Lodmell JS, Samuels DS. Mutations conferring aminoglycoside and spectinomycin resistance in Borrelia burgdorferi. Antimicrob Agents Chemother. 2006;50:445–452. doi: 10.1128/AAC.50.2.445-452.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Drecktrah D, Lybecker M, Popitsch N, Rescheneder P, Hall LS, Samuels DS. The Borrelia burgdorferi RelA/SpoT homolog and stringent response regulate survival in the tick vector and global gene expression during starvation. PLoS Pathog. 2015;11:e1005160. doi: 10.1371/journal.ppat.1005160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hoon-Hanks LL, Morton EA, Lybecker MC, Battisti JM, Samuels DS, Drecktrah D. Borrelia burgdorferi malQ mutants utilize disaccharides and traverse the enzootic cycle. FEMS Immunol Med Microbiol. 2012;66:157–165. doi: 10.1111/j.1574-695X.2012.00996.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lybecker MC, Abel CA, Feig AL, Samuels DS. Identification and function of the RNA chaperone Hfq in the Lyme disease spirochete Borrelia burgdorferi. Mol Microbiol. 2010;78:622–635. doi: 10.1111/j.1365-2958.2010.07374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lybecker MC, Samuels DS. Temperature-induced regulation of RpoS by a small RNA in Borrelia burgdorferi. Mol Microbiol. 2007;64:1075–1089. doi: 10.1111/j.1365-2958.2007.05716.x. [DOI] [PubMed] [Google Scholar]
- 65.Sultan SZ, Pitzer JE, Miller MR, Motaleb MA. Analysis of a Borrelia burgdorferi phosphodiesterase demonstrates a role for cyclic-diguanosine monophosphate in motility and virulence. Mol Microbiol. 2010;77:128–142. doi: 10.1111/j.1365-2958.2010.07191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ge Y, Old IG, Saint Girons I, Charon NW. Molecular characterization of a large Borrelia burgdorferi motility operon which is initiated by a consensus σ70 promoter. J Bacteriol. 1997;179:2289–2299. doi: 10.1128/jb.179.7.2289-2299.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Babitzke P, Yealy J, Campanelli D. Interaction of the trp RNA-binding attenuation protein (TRAP) of Bacillus subtilis with RNA: effects of the number of GAG repeats, the nucleotides separating adjacent repeats, and RNA secondary structure. J Bacteriol. 1996;178:5159–5163. doi: 10.1128/jb.178.17.5159-5163.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Drecktrah D, Hall LS, Hoon-Hanks LL, Samuels DS. An inverted repeat in the ospC operator is required for induction in Borrelia burgdorferi. PLoS One. 2013;8:e68799. doi: 10.1371/journal.pone.0068799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Earnhart CG, LeBlanc DV, Alix KE, Desrosiers DC, Radolf JD, Marconi RT. Identification of residues within ligand-binding domain 1 (LBD1) of the Borrelia burgdorferi OspC protein required for function in the mammalian environment. Mol Microbiol. 2010;76:393–408. doi: 10.1111/j.1365-2958.2010.07103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang XF, Alani SM, Norgard MV. The response regulator Rrp2 is essential for the expression of major membrane lipoproteins in Borrelia burgdorferi. Proc Natl Acad Sci U S A. 2003;100:11001–11006. doi: 10.1073/pnas.1834315100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Beaurepaire C, Chaconas G. Mapping of essential replication functions of the linear plasmid lp17 of B. burgdorferi by targeted deletion walking. Mol Microbiol. 2005;57:132–142. doi: 10.1111/j.1365-2958.2005.04688.x. [DOI] [PubMed] [Google Scholar]
- 72.Grimm D, Eggers CH, Caimano MJ, Tilly K, Stewart PE, Elias AF, Radolf JD, Rosa PA. Experimental assessment of the roles of linear plasmids lp25 and lp28-1 of Borrelia burgdorferi throughout the infectious cycle. Infect Immun. 2004;72:5938–5946. doi: 10.1128/IAI.72.10.5938-5946.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bundoc VG, Barbour AG. Clonal polymorphisms of outer membrane protein OspB of Borrelia burgdorferi. Infect Immun. 1989;57:2733–2741. doi: 10.1128/iai.57.9.2733-2741.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kurtti TJ, Munderloh UG, Johnson RC, Ahlstrand GG. Colony formation and morphology in Borrelia burgdorferi. J Clin Microbiol. 1987;25:2054–2058. doi: 10.1128/jcm.25.11.2054-2058.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rosa PA, Hogan DM. Colony formation by Borrelia burgdorferi in solid medium: clonal analysis of osp locus variants. In: Munderloh UG, Kurtti TJ, editors. First international conference on tick-borne pathogens at the host-vector Interface: an agenda for research. University of Minnesota; St. Paul: 1992. pp. 95–103. [Google Scholar]
- 76.Bunikis I, Kutschan-Bunikis S, Bonde M, Bergström S. Multiplex PCR as a tool for validating plasmid content of Borrelia burgdorferi. J Microbiol Methods. 2011;86:243–247. doi: 10.1016/j.mimet.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 77.Tilly K, Krum JG, Bestor A, Jewett MW, Grimm D, Bueschel D, Byram R, Dorward D, Vanraden MJ, Stewart P, Rosa P. Borrelia burgdorferi OspC protein required exclusively in a crucial early stage of mammalian infection. Infect Immun. 2006;74:3554–3564. doi: 10.1128/IAI.01950-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Marconi RT, Samuels DS, Garon CF. Transcriptional analyses and mapping of the ospC gene in Lyme disease spirochetes. J Bacteriol. 1993;175:926–932. doi: 10.1128/jb.175.4.926-932.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]