

Summary
Entry into host cells and intracellular persistence by invasive bacteria are tightly coupled to the ability of the bacterium to disrupt the eukaryotic cytoskeletal machinery. Herein we review the main strategies used by three intracellular pathogens to harness key modulators of the cytoskeleton. Two of these bacteria, namely Listeria monocytogenes and Salmonella enterica serovar Typhimurium, exhibit quite distinct intracellular lifestyles, and therefore, provide a comprehensive panel for the understanding of the intricate bacteria-cytoskeleton interplay during infections. The emerging intracellular pathogen Vibrio parahaemolyticus is depicted as a developing model for the uncovering of novel mechanisms used to hijack the cytoskeleton.
1. Introduction
The co-evolution between pathogenic bacteria and their hosts has resulted in a number of bacterial strategies used to ensure bacterial persistence during infection. One effective strategy used by some pathogens is to induce their own uptake by non-phagocytic cells. Entry into these cells “hides” the bacteria from the host’s immune system and secures a relatively safe niche for bacterial survival and replication (Cossart & Helenius, 2014).
Following entry into the host cell, bacteria are contained within a membrane-bound compartment (vacuole) that is destined for lysosomal degradation. In order to subvert this bactericidal routing, vacuolar bacteria can “customize” their vacuole to either impede vacuole-lysosome fusion or to convert the fused lysosome-vacuole compartment into a permissive environment for survival. Alternatively, some intracellular bacteria escape from the vacuole into the cytosol (Fredlund & Enninga, 2014). Once in the cytosol, many bacteria recruit actin to move intracellularly and to spread to neighboring cells (Cossart & Helenius, 2014).
Bacterial entry into host cells, vacuolar alterations, and cytosolic motility largely rely on the dynamic rearrangements of the host cytoskeleton. The cytoskeleton of eukaryotes is mainly composed of three types of filaments: actin filaments, microtubules, and intermediate filaments. Actin filaments and microtubules are polymers of actin and tubulin subunits, respectively. The polymerization/depolymerization of these filaments is regulated by proteins such as nucleation-promoting factors, capping and severing proteins (Fletcher & Mullins, 2010). Intracellular bacteria harness many of these regulators and their related factors to disrupt the host cytoskeleton.
Here we review strategies used by intracellular bacteria to disrupt the cytoskeleton. Specifically, we focus on three pathogens: Listeria monocytogenes, Salmonella enterica serovar Typhimurium (S. Typhimurium) and Vibrio parahaemolyticus. While Listeria and Salmonella exhibit very distinct intracellular behaviors and together illustrate a large number of known strategies, Vibrio has recently emerged as an intracellular bacterium and provides a new model for uncovering novel strategies.
Common to all three pathogens is their ability to cause gastroenteritis upon ingestion of contaminated food/water sources. Although the disease seems limited to gastroenteritis in healthy individuals, the infection of immunocompromised patients and extreme-age groups may also lead to bacterial crossing of the intestinal epithelial barrier and consequent bacterial systemic spread (Broberg, et al., 2011, Fabrega & Vila, 2013, Cossart & Lebreton, 2014). Interestingly, it has been shown that the ability of L. monocytogenes and S. Typhimurium to invade non-phagocytic cells is critical for breaching of epithelial barriers (Fabrega & Vila, 2013, Cossart & Lebreton, 2014). Because V. parahaemolyticus has only recently been studied as an intracellular bacterium (Zhang, et al., 2012, de Souza Santos & Orth, 2014), a role for cell invasion during bacterial systemic dissemination remains to be determined.
2. Entry into host cells
2.a. The zipper mechanism
Entry of invasive bacteria into non-phagocytic cells, such as epithelial cells, occurs via two mechanisms known as “zipper” and “trigger”. In the zipper mechanism, ligands on the bacterial surface bind to receptors on the host cell surface, causing the bacterial body to be closely apposed to the host cell plasma membrane. The bacterial activation of host receptors triggers signaling pathways that culminate with moderate rearrangements of the actin cytoskeleton at the site of bacterial entry. Listeria is a canonical model of this strategy. (Lecuit, et al., 1997).
Listeria entry into epithelial cells is mainly regulated by two bacterial surface proteins, namely internalins InlA and InlB (Figure 1). InlA binds E-cadherin, a transmembrane glycoprotein that engages in homophilic interactions with E-cadherin molecules from adjacent cells during formation of adherens junctions (Lecuit, et al., 1997). The initial establishment of cell-cell contact, mediated by E-cadherin, is accompanied by local formation of protrusive forces that allow cells to extend upon one another, bringing them together. The expansion of newly-formed adhesion contact zones is mediated by actin polymerization. Homophilic E-cadherin ligations “mark” sites for actin assembly through a strictly local recruitment of the Arp2/3 complex (Kovacs, et al., 2002). The Arp2/3 complex functions to nucleate new actin filaments from pre-existing ones, creating a branched actin network. However, the Arp2/3 complex exhibits low nucleation activity by itself and requires activation, which in this case is mediated by cortactin (Helwani, et al., 2004). It is proposed that cortactin recruitment is a signaling-regulated process mediated by Rac1 that is locally activated upon E-cadherin ligation (Kovacs, et al., 2002). Rac1 belongs to the family of small Rho-GTPases, molecular switches that cycle between active (GTP-bound), and inactive (GDP-bound) conformations, and are known to regulate many aspects of actin dynamics (Alberts, 2008).
Figure 1.
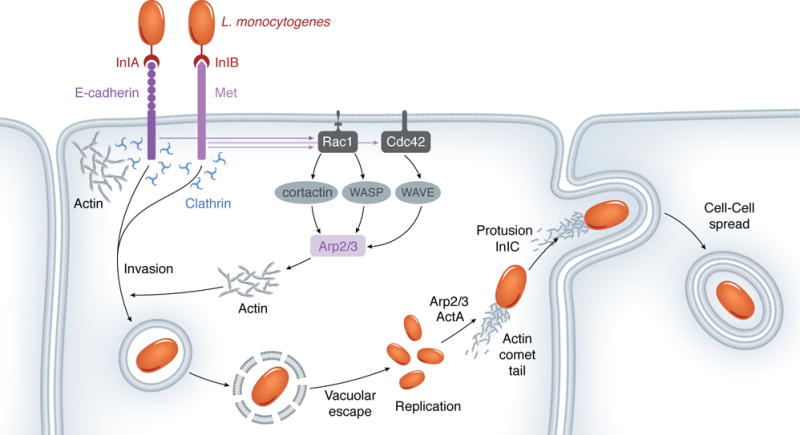
L. monocytogenes induces Arp2/3-dependent actin polymerization in order to enter host cells, move intracellularly, and disseminate.
Strikingly, L. monocytogenes hijacks the E-cadherin-mediated actin reorganization to gain entry into host cells. The association of InlA with E-cadherin functions as a “mimic” of homophilic E-cadherin ligations and causes local activation of Rac1. It is proposed that Rac1 regulates the activation of the Arp2/3 complex by mediating cortactin translocation to bacterial entry sites (Sousa, et al., 2007). Activated actin polymerization creates phagocytic cups around the entering bacterium, which are subsequently resolved into vacuoles/phagosomes through the contractile force provided by myosin VII, an actin-binding molecular motor (Sousa, et al., 2004).
While InlA displays selective binding to E-cadherin expressing cells, InlB is sufficient to promote bacterial entry in a variety of cells. This is because the major cell receptor for InlB, Met, is ubiquitously expressed. Met is a receptor tyrosine kinase known to mediate key signaling events during cell proliferation, motility, and morphogenesis (Ferraris, et al., 2010). The endogenous ligand of Met is the hepatocyte growth factor (HGF), and InlB is considered a functional mimic of that ligand (Figure 1) (Seveau, et al., 2007).
InlB association with Met stimulates receptor dimerization and autophosphorylation. Met phosphorylated tyrosine residues act as docking sites for the recruitment of adaptor proteins such as Gab1, which in turn help to recruit the type IA phosphatidylinositol 3-kinase (PI3K). PI3K generates phosphatidylinositol (3,4,5)-triphosphate (PI(3,4,5,)P3), which mediates activation of small Rho GTPases, such as Rac1 (Ireton, et al., 1999, Seveau, et al., 2007).
Rac1 and a second small GTPase, Cdc42, contribute to reshaping the actin cytoskeleton during InlB/Met-dependent entry: while Cdc42 mediates downstream activation of WASP (Wiskott-Aldrich syndrome family protein members), Rac1 activates WAVE (WASP family Verprolin-homologous) proteins. Similar to cortactin, WASP/WAVE proteins activate the Arp2/3 complex to nucleate new actin filaments. Interestingly, the involvement of WASP/WAVE and related proteins during InlB-mediated entry seems to be cell-type specific. For instance, while WAVE2 alone is recruited to phagocytic cups in Vero cells, WAVE1, WAVE2, and N-WASP cooperate to cause Listeria entry into HeLa cells (Bierne, et al., 2005). Cofilin, an actin-depolymerizing factor, is also recruited to bacterial entry sites and is proposed to serve two distinct roles aiding in bacterial entry: its severing activity creates new filament ends for actin polymerization during phagocytic cup formation, followed by depolymerization of filaments to facilitate phagocytic cup closure (Figure 1) (Bierne, et al., 2001).
Interestingly, Listeria also exploits the clathrin-dependent endocytic machinery, normally deployed during recycling of activated receptors, in order to enter the host cell. InlA/InlB association with E-cadherin/Met triggers receptor phosphorylation, which is followed by its ubiquitination via Cbl-like ubiquitin ligases (Veiga & Cossart, 2005, Bonazzi, et al., 2008). Receptor ubiquitination induces the recruitment of clathrin and other components of the endocytic machinery, leading to assembly of clathrin-coated pits, whose reduced size precludes bacterial compartimentalization into those pits. Instead, it is proposed that the clathrin lattices function as platforms for the recruitment of actin, via the actin-binding protein Hip1R, and for the recruitment of myosin VI, which drives bacterial internalization (Bonazzi, et al., 2011).
Together, InlA and InlB exploit E-cadherin- and Met-mediated signaling pathways, as well as their recycling pathways, to cause actin polymerization at bacterial entry sites (Figure 1).
2.b. The trigger mechanism
The trigger mechanism is based on the delivery of bacterial proteins (effectors) across the host plasma membrane through a dedicated Type III Secretion System (T33S) – a needle-like secretion apparatus found in many Gram-negative bacteria (Figure 2) (Cornelis, 2006). Following translocation into the cytosol, many T3SS effectors target components of the actin cytoskeleton machinery to induce formation of extensive actin projections (lamellipodia/ruffles) used to engulf loosely cell-associated bacterium into spacious macropinosomes (Cossart & Sansonetti, 2004). This mode of entry is well characterized in S. Typhimurium.
Figure 2.
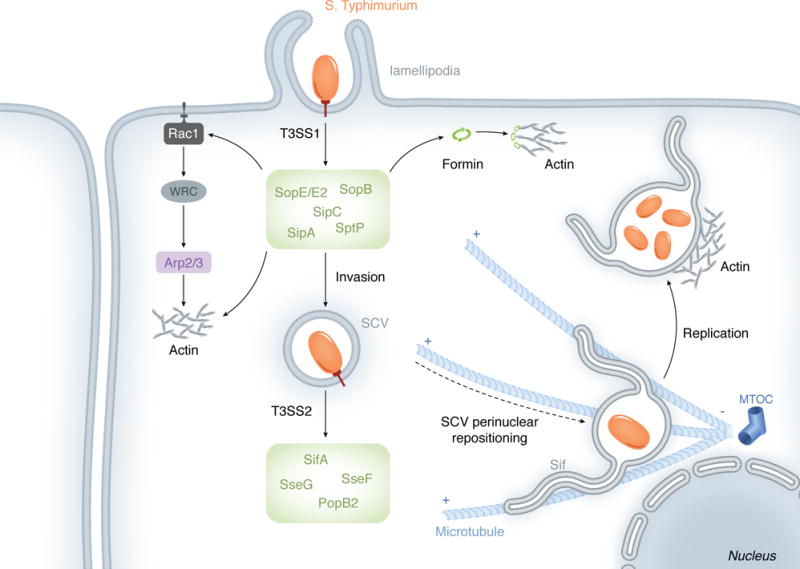
Using a set of T3SS1/T3SS2 effectors, S. Typhimurium hijacks both the actin and the microtubule cytoskeletons in order to invade host cells, translocate the SCV near the nucleus, and develop Sifs.
S. Typhimurium possesses several gene clusters, referred to as Salmonella pathogenicity islands (SPI), which encode the majority of its virulence factors. It has been generally accepted that the T3SS contained within the SPI-1 (T3SS1) would translocate several effectors required for bacterial invasion, while the T3SS encoded by the SPI-2 (T3SS2) would secrete effectors that regulate the intracellular lifestyle of the pathogen (Figure 2) (Fabrega & Vila, 2013). However, this distinction between the two SPIs has become less clear with the identification of T3SS effectors that not only contribute to bacterial invasion, but also play a role during maturation of the bacterial vacuole (Bakowski, et al., 2010).
Lamellipodia are protrusions composed of Apr2/3-generated actin networks and their formation is regulated by Rac1 (Alberts, 2008). The interconversion between GDP- and GTP-bound states of Rac1 (and other small Rho-GTPases) is controlled by guanine nucleotide exchange factors (GEFs), which catalyze the dissociation of GDP for the binding of GTP, and by GTPase-activating proteins (GAPs), which stimulate GTP hydrolysis (Alberts, 2008). The Salmonella T3SS1 effectors SopE and related SopE2 are considered bona fide GEFs of Rho-GTPases, albeit only SopE facilitates GDP/GTP exchange in Rac1 (Hardt, et al., 1998, Stender, et al., 2000, Friebel, et al., 2001). Activation of Rac1 by SopE determines the enrichment of the WAVE regulatory complex (WRC) around entering bacterium (Humphreys, et al., 2012). The activation of WRC is mediated by Arf (ADP-ribosylation factor) 1, which in turn is activated by ARNO, an Arf GEG (Humphreys, et al., 2012, Humphreys, et al., 2013). Two mechanisms have been proposed for ARNO recruitment to the entry site: via binding of ARNO PH domain to PI(3,4,5)P3 and via Arf6 (Humphreys, et al., 2013). Similarly to WAVE proteins, the WRC activates the Arp2/3 complex to nucleate actin polymerization (Stender, et al., 2000).
Recent studies indicate that while the Arp2/3 complex contributes to bacterial invasion, Arp2/3-independent mechanisms may also be operating during actin assembly at sites of Salmonella invasion (Hanisch, et al., 2010). During Salmonella infections, the activation of Rho kinases induces phosphorylation of FHOD1, a formin family member. SopE/E2 and SopB (discussed below) appear to contribute to Rho kinase activation. In contrast to the Arp2/3-generated branched actin network, formins nucleate linear actin polymerization. It has been suggested that FHOD1 and Arp2/3 regulate distinct, but complementary, aspects of protrusion formation (Truong, et al., 2013).
Another T3SS1 effector, SopB, is also required for Salmonella invasion (Zhou, et al., 2001). SopB contains a phosphatase domain which is necessary for the indirect activation of RhoG (Patel & Galan, 2006). This small Rho-GTPase not only shares identity with Rac1 but has also been suggested to activate Rac1 (Katoh, et al., 2006). The phosphatase activity of SopB is also required for the hydrolysis of phosphatidylinositol 4,5-biphosphate (PI(4,5)P2) located at the base of lamellipodia. It is believed that PI(4,5)P2 depletion at such locations leads to detachment of cortical actin from the plasma membrane, which enhances membrane flexibility and facilitates membrane invaginations and fission during bacterial internalization (Terebiznik, et al., 2002). In addition to a role during ruffling-dependent invasion, SopB participates in a parallel, myosin II-dependent entry pathway. Here, SopB mediates formation of actin stress fibers that colocalize with myosin II. These structures are thought to promote the contractile force that drive membrane invaginations during Salmonella entry (Hanisch, et al., 2011).
Rather than targeting upstream regulators of actin polymerization, the T3SS1 effectors SipC and SipA directly bind to actin. SipC nucleates and bundles actin filaments (Hayward & Koronakis, 1999). SipA binding to actin reduces the critical G-actin (monomeric actin) concentration required for polymerization (Zhou, et al., 1999). SipA also contributes to actin filament stabilization: effector-actin binding precludes actin association with actin depolymerizing factor (ADP) or cofilin. Moreover, SipA inhibits gelsolin-mediated severing of actin filaments and reassembles severed F-actin fragments (McGhie, et al., 2004).
Although several bacterial effectors efficiently manipulate the cytoskeleton to form lamellipodia protrusions, these actin structures eventually subside and the cytoskeleton recovers its regular shape. Lamellipodia disassembly is assisted by the T3SS1 effector SptP, which functions as a GAP protein for both Rac1 and Cdc42 and therefore antagonizes the activity of Salmonella GEFs, such as SopE (Fu & Galan, 1999). Although secreted at amounts equivalent to SptP, SopE is rapidly degraded by a proteasome-dependent pathway (Kubori & Galan, 2003). Consequently, the slower degradation kinetics of SptP allows its GAP activity to prevail over GEFs at later stages of invasion (Kubori & Galan, 2003).
In summary, the T3SS1 effectors from S. Typhimurium appear to work in a concerted and temporal manner to mediate lamellipodia-based bacterial entry (Figure 2). SopE- and SopB-mediated activation of Rac1 results in Arp2/3-generated branched actin networks. These two effectors are also involved in Arp2/3-independent, linear assembly of actin filaments. SipA cooperates with actin assembly by not only facilitating nucleation of actin filaments due to reduction of critical actin concentration, but also by counteracting filament-destabilizing proteins. SopB then uses distinct mechanisms to promote bacterial internalization and SptP reverses lamellipodia formation.
3 L. monocytogenes uses actin to move within and between cells
Shortly after internalization, L. monocytogenes escapes its nascent vacuole to reside in the cytosol, where bacterial replication takes place (Figure 1) (Cossart & Sansonetti, 2004). As with other cytosolic bacteria, such as Shigella flexneri, Listeria stimulates the assembly of short, highly-branched actin microfilaments that protrude from one pole of the bacterium. These structures are named actin comet tails and enable bacterial movement throughout the cell (Lambrechts, et al., 2008). Actin-based motility represents an efficient bacterial strategy for evasion of cytosolic bactericidal factors.
The Listeria surface protein ActA is necessary and sufficient to mediate actin-based motility (Figure 1) (Kocks, et al., 1992). This protein exhibits a polarized distribution on the bacterial surface and accumulates at sites of comet tail formation. Functionally, ActA mimics eukaryotic nucleation-promoting factors and activates the Arp2/3 complex. Specifically, the N-terminal domain of ActA constitutes the minimal functional region for Ap2/3-binding and activation. This region shares sequence similarity with the VCA (Verprolin-Cofilin-Acidic) domain of WASP and N-WASP (Boujemaa-Paterski, et al., 2001). In vitro reconstitution of the actin-based motility revealed that, in addition to the Ap2/3 complex, propulsion also requires ADF/cofilin and capping proteins. These proteins were inferred as necessary for the maintenance of a steady pool of G-actin required for continuous polymerization (Figure 2) (Loisel, et al., 1999).
The apposition of motile L. monocytogenes to the inner leaflet of the plasma membrane forms finger-like protrusions that extend toward the adjacent host cell. The bacterium localizes to the tip of such protrusions and, therefore, becomes surrounded by two plasma membranes, which resolve into a new double-membrane vacuole within the neighboring cell. This vacuole is ruptured by the bacterium, which has now transferred into the cytoplasm of a new cell, where another infection cycle begins (Figure 2) (Kocks, et al., 1992, Pust, et al., 2005). This process of cell-cell spread represents an efficient dissemination strategy that avoids bacterial exposure to humoral immune factors.
The ERM (ezrin/radixin/moesin) protein family, which localize to the cortical actin, has been suggested to be exploited by the bacterium during protrusion formation as a link between the actin tails and the plasma membrane (Pust, et al., 2005). Efficient cell-cell spread was shown to require the secreted virulence protein internalin C (InlC). InlC binds the sixth SH3 domain of Tuba, an eukaryotic protein that localizes to cell junctions and has been implicated in the maintenance of the cortical actin tension through its binding to N-WASP. It has been proposed that InlC outcompetes N-WASP for Tuba binding. This results in relaxation of the cortical tension and loosening of the junctional membrane, which allow Listeria to form membranse protrusions and spread to the adjacent cell (Rajabian, et al., 2009). Recently, it was demonstrated that recycling of Arp2/3 from the distal to the proximal actin network of bacterial protrusions is necessary for continuous de novo filament assembly that supports resolution of protrusions into vacuoles. This process contributes to efficient bacterial dissemination (Talman, et al., 2014).
4. Manipulation of the microtubule cytoskeleton is essential for the biogenesis of Salmonella-containing vacuoles
Unlike L. monocytogenes, S. Typhimurium has developed mechanisms that allow the bacterium to remain within a vacuole while avoiding intravacuolar killing. Therefore, S. Typhimurium resides and replicates within a modified vacuole referred to as Salmonella-containing vacuole (SCV) (Cossart & Helenius, 2014). It has been reported that a subpopulation of S. Typhimurium can escape the SCV and hyperreplicate in the cytosol (Knodler, et al., 2010). Cytosolic Salmonella is also motile but its motility is mediated by flagellar structures (Knodler, et al., 2010) and therefore will not be reviewed here.
The biogenesis of the SCV involves the acquisition of early endosome markers, such as EEA1 (early endosome antigen 1) and Rab5, which are replaced with late endosomal markers such as Rab7 and Lamp-1 (lysosomal-associated membrane protein 1) during SCV maturation. The maturation of the SCV is accompanied by vacuole translocation to a juxtanuclear position, near the microtubule organizing center (MTOC). At this position, S. Typhimurium initiates intravacuolar replication (Steele-Mortimer, 2008).
Rab7 enrichment on the SCV allows recruitment of its effector RILP (Rab7-interacting lysosomal protein), which associates with the microtubule-based motor dynein to direct SCV cargo transport along cytoskeletal microtubules towards the minus-end of the microtubule, which points to the MTCO (cell center) (Harrison, et al., 2004). At least three T3SS2 effectors contribute to the maintenance of the SCV perinuclear localization: SifA, SseF and SseG. SifA associates with the host protein SKIP (SifA kinesin interacting protein), which in turn down-regulates the recruitment of kinesin to the SCV. Contrary to dynein, kinesin directs transport toward the plus-end of the microtubule (cell periphery). Therefore, SKIP depletion leads to SCV scattering (Boucrot, et al., 2005). SseF and SseG contribute to balance the recruitment of dynein and the SCV and for Golgi-associated subcellular localization of the vacuole (Salcedo & Holden, 2003, Abrahams, et al., 2006).
The later stages of the SCV modification, which overlap with the onset of bacterial replication (4-6h post invasion) include the formation of elongated tubular extensions from the surface of the SCV named Salmonella-induced filaments (Sifs). These structures are thought to be derived from late endocytic compartments that are enriched in Rab7 and Lamp-1 (Brumell, et al., 2002). Sifs are elongated along the microtubule network toward the filament plus-end. SifA is essential for Sif formation; at later SCVs, this effector is predominantly found along Sifs (Brumell, et al., 2002). Although SifA counteracts kinesin recruitment via SKIP, this effector is also suggested to bind Rab7 in Sifs. SifA-Rab7 binding precludes Rab7-association with RILP. As a result, dynein recruitment to the Sifs is down-regulated (Harrison, et al., 2004). At the same time, kinesin recruitment to the Sifs is enhanced by the T3SS2 effector PipB2 (Henry, et al., 2006). The overall kinesin/dynein recruitment balance appears to favor the centrifugal (kinesin-mediated) extension of the filaments. Impairment of Sif formation leads to SCV rupture and defective bacterial replication (Beuzon, et al., 2000).
Several hours following invasion, the Salmonella T3SS2 also organizes the actin cytoskeleton into a meshwork that surrounds the SCV, but the mechanisms used to manipulate actin polymerization and as the specific contributions of T3SS2-effectors to this process remain elusive.
In summary, S. Typhimurium utilizes T3SS2 effectors to usurp functions of both the microtubule and actin cytoskeletons during the later stages of the SCV biogenesis: microtubule-motor proteins mediate SCV translocation near the MTOC, the microtubule filaments provide the framework for Sif elongation, and an actin meshwork wraps around late SCVs (Figure 2). Together, these events enable the establishment of a safe replicative niche for this bacterium.
5. V. parahaemolyticus as an emerging model for the study of pathogen-cytoskeleton interactions
Like S. Typhimurium, V. parahaemolyticus invades the host cell through the trigger mechanism. This bacterium contains two T3SSs (T3SS1 and T3SS2), harbored on different chromosomes. While the T3SS1-encoded effectors conduct a sequence of events to cause host cell toxicity and death in tissue culture cells, the T3SS2 is required for the development of gastroenteritis and cell invasion in mammalian animals and cells, respectively (Ritchie, et al., 2012, Zhang, et al., 2012).
VopC is the only Vibrio T3SS2 effector known to mediate bacterial entry into non-phagocytic cells to date. This effector appears to have hijacked the catalytic domain from cytotoxic necrotizing factor (CNF) toxins by linking it to a T3SS ecretion motif (Zhang, et al., 2012). CNFs are found in Yersinia ssp., Bordetella spp. and Escherichia coli, and induce actin cytoskeleton reorganization to enable bacterial invasion (Aktories & Barbieri, 2005). Like CNFs, VopC deamidates Rac1 and Cdc42, which are rendered constitutively active (Zhang, et al., 2012). Consistent with Rac1 activation, the Arp2/3 complex is also necessary for invasion (Okada, et al., 2014). As a result of stimulated actin polymerization, an entering bacterium becomes surrounded by lamellipodia protrusions (de Souza Santos & Orth, 2014). Following entry, V. parahaemolyticus escapes its vacuole to reside in the cytosol where it replicates prolifically (de Souza Santos & Orth, 2014). The mechanisms governing this intracellular lifestyle are poorly understood.
At least two other Vibrio T3SS2 effectors, VopL and VopV, alter actin assembly but their contributions to bacterial invasion and/or intracellular survival have not been investigated. VopL encodes three tandem WH2 (Wiskott-Aldrich homology 2) domains, which are known to bind actin monomers and to nucleate filament assembly. The tandem WH2 array of VopL is a weak actin-nucleator on its own; nucleation activity is improved through VopL dimerization, which is mediated by the effector’s C-terminal domain (Yu, et al., 2011). Cell expression of VopL causes a dramatic actin phenotype characterized by formation of stress fibers that span the whole cell body (Liverman, et al., 2007). Stress fibers exert tension that allows cell reshaping; this may prove beneficial for bacterial entry or for maintenance of cell structure during bacterial replication.
VopV does not possess any motifs or sequence homology to previously described proteins. Instead, this effector contains a N-terminal, a long repeat (LR), and a C-terminal domain, the last two suggested to directly bind to F-actin. The LR is composed of three types of repeats: the rep1 was shown to be required for actin-binding activity and multiple rep1 sequences enable the LR domain to also bundle actin filaments (Hiyoshi, et al., 2011). During infection, VopV mediates accumulation of actin filaments underneath microcolonies attached to the cell surface. Interestingly, loss of actin accumulation does not preclude microcolony-formation on the infected cell, suggesting that, rather than a role in bacterial-cell adhesion, VopV-mediated actin accumulation may contribute to cell invasion (Hiyoshi, et al., 2011). A subsequent study revealed that the C-terminal domain of VopV contains a filamin-binding region. Filamin is an actin-binding protein responsible for cross-linking of actin filaments into an orthogonal network (Zhou, et al., 2014). This study also proposes that VopV mediates reorganization of the brush border of epithelial cells as a mechanism to enhance bacterial attachment to the host cell (Zhou, et al., 2014). Currently it is not clear what role VopV and actin bundling play in invasion.
6. Conclusions
In this review, we described the main strategies used by the well-established intracellular pathogens L. monocytogenes and S. Typhimurium, as well as the emerging intracellular pathogen V. parahaemolyticus, to disrupt the cytoskeletal machinery and achieve host cell entry, intracellular survival, and dissemination. Listeria and Salmonella are regarded as paradigms of how the co-evolution between the pathogen and its host has allowed the development of distinct approaches for subversion of cytoskeletal signaling pathways. Unravelling the intracellular lifestyle of these bacteria has provided invaluable contributions to the identification and characterization of bacterial factors necessary for control of actin/microtubule assembly in eukaryotic cells. At the same time, the elucidation of bacterial mechanisms deployed during manipulation of the cytoskeleton has been instrumental for the discovery and functional definition of key modulators of the intricate regulation of the cytoskeletal pathway. For instance, the study of L. monocytogenes actin-based motility led to the characterization of the Arp2/3 complex and its contribution during filament nucleation. As demonstrated by the emerging studies with V. parahaemolyticus, this fascinating two-sided field of research that certainly holds many new key discoveries.
Figure 3.
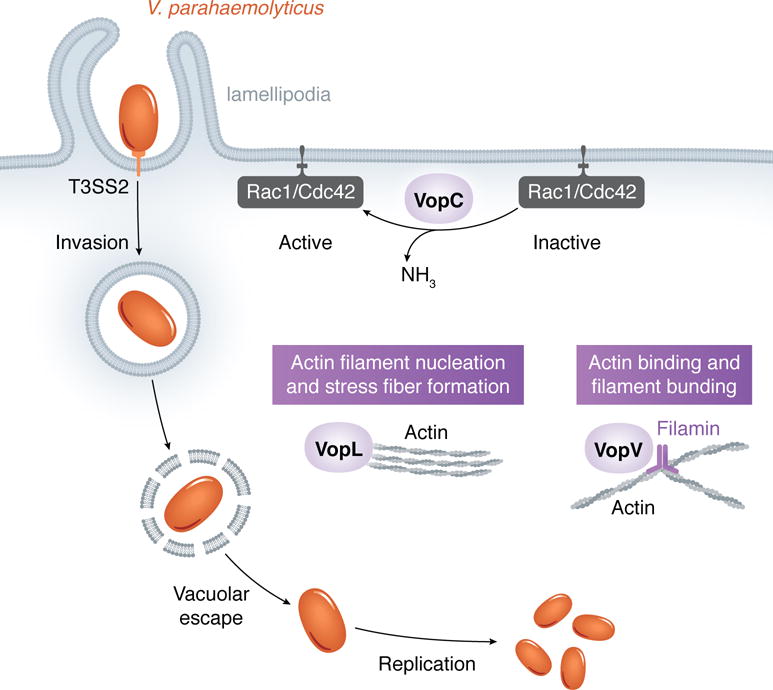
V. parahaemolyticus uses its T3SS2 effector VopC to activate Rac1 and Cdc42, which induce actin polymerization to cause bacterium entry into the host cell. V. parahaemolyticus then escapes its vacuole and replicates in the host cytosol. While VopL nucleates new actin filaments, VopV binds to and bundles actin filaments, their role in bacterial invasion and/or intracellular survival remains to be understood.
Acknowledgments
We apologize to those whose work could not be cited owing to space limitations. We thank the Andrew Woolery. Dor Salomon and other members of the Orth lab for valuable edits and suggestions. K.O. and M.S.S. are supported by NIH grant R01-AI056404 and Grant I-1561 from the Welch Research Foundation. M.S.S. is supported by NIH grant 5T32DK007745-17. K.O. is a Burroughs Welcome Investigator in Pathogenesis of Infectious Disease, Beckman Young Investigator, a W.W. Caruth, Jr. Biomedical Scholar and has an Earl A. Forsythe Chair in Biomedical Science.
References
- Abrahams GL, Muller P, Hensel M. Functional dissection of SseF, a type III effector protein involved in positioning the salmonella-containing vacuole. Traffic. 2006;7:950–965. doi: 10.1111/j.1600-0854.2006.00454.x. [DOI] [PubMed] [Google Scholar]
- Aktories K, Barbieri JT. Bacterial cytotoxins: targeting eukaryotic switches. Nat Rev Microbiol. 2005;3:397–410. doi: 10.1038/nrmicro1150. [DOI] [PubMed] [Google Scholar]
- Alberts A, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular Biology of the Cell. Garland Science; New York: 2008. [Google Scholar]
- Bakowski MA, Braun V, Lam GY, et al. The phosphoinositide phosphatase SopB manipulates membrane surface charge and trafficking of the Salmonella-containing vacuole. Cell Host Microbe. 2010;7:453–462. doi: 10.1016/j.chom.2010.05.011. [DOI] [PubMed] [Google Scholar]
- Beuzon CR, Meresse S, Unsworth KE, et al. Salmonella maintains the integrity of its intracellular vacuole through the action of SifA. EMBO J. 2000;19:3235–3249. doi: 10.1093/emboj/19.13.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierne H, Gouin E, Roux P, Caroni P, Yin HL, Cossart P. A role for cofilin and LIM kinase in Listeria-induced phagocytosis. J Cell Biol. 2001;155:101–112. doi: 10.1083/jcb.200104037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierne H, Miki H, Innocenti M, Scita G, Gertler FB, Takenawa T, Cossart P. WASP-related proteins, Abi1 and Ena/VASP are required for Listeria invasion induced by the Met receptor. J Cell Sci. 2005;118:1537–1547. doi: 10.1242/jcs.02285. [DOI] [PubMed] [Google Scholar]
- Bonazzi M, Veiga E, Pizarro-Cerda J, Cossart P. Successive post-translational modifications of E-cadherin are required for InlA-mediated internalization of Listeria monocytogenes. Cell Microbiol. 2008;10:2208–2222. doi: 10.1111/j.1462-5822.2008.01200.x. [DOI] [PubMed] [Google Scholar]
- Bonazzi M, Vasudevan L, Mallet A, et al. Clathrin phosphorylation is required for actin recruitment at sites of bacterial adhesion and internalization. J Cell Biol. 2011;195:525–536. doi: 10.1083/jcb.201105152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucrot E, Henry T, Borg JP, Gorvel JP, Meresse S. The intracellular fate of Salmonella depends on the recruitment of kinesin. Science. 2005;308:1174–1178. doi: 10.1126/science.1110225. [DOI] [PubMed] [Google Scholar]
- Boujemaa-Paterski R, Gouin E, Hansen G, et al. Listeria protein ActA mimics WASp family proteins: it activates filament barbed end branching by Arp2/3 complex. Biochemistry. 2001;40:11390–11404. doi: 10.1021/bi010486b. [DOI] [PubMed] [Google Scholar]
- Broberg CA, Calder TJ, Orth K. Vibrio parahaemolyticus cell biology and pathogenicity determinants. Microbes Infect. 2011;13:992–1001. doi: 10.1016/j.micinf.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumell JH, Goosney DL, Finlay BB. SifA, a type III secreted effector of Salmonella typhimurium, directs Salmonella-induced filament (Sif) formation along microtubules. Traffic. 2002;3:407–415. doi: 10.1034/j.1600-0854.2002.30604.x. [DOI] [PubMed] [Google Scholar]
- Cornelis GR. The type III secretion injectisome. Nat Rev Microbiol. 2006;4:811–825. doi: 10.1038/nrmicro1526. [DOI] [PubMed] [Google Scholar]
- Cossart P, Sansonetti PJ. Bacterial invasion: the paradigms of enteroinvasive pathogens. Science. 2004;304:242–248. doi: 10.1126/science.1090124. [DOI] [PubMed] [Google Scholar]
- Cossart P, Helenius A. Endocytosis of Viruses and Bacteria. Cold Spring Harb Perspect Biol. 2014;6 doi: 10.1101/cshperspect.a016972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart P, Lebreton A. A trip in the “New Microbiology” with the bacterial pathogen Listeria monocytogenes. FEBS Lett. 2014;588:2437–2445. doi: 10.1016/j.febslet.2014.05.051. [DOI] [PubMed] [Google Scholar]
- de Souza Santos M, Orth K. Intracellular Vibrio parahaemolyticus Escapes the Vacuole and Establishes a Replicative Niche in the Cytosol of Epithelial Cells. MBio. 2014;5 doi: 10.1128/mBio.01506-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrega A, Vila J. Salmonella enterica serovar Typhimurium skills to succeed in the host: virulence and regulation. Clin Microbiol Rev. 2013;26:308–341. doi: 10.1128/CMR.00066-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraris DM, Gherardi E, Di Y, Heinz DW, Niemann HH. Ligand-mediated dimerization of the Met receptor tyrosine kinase by the bacterial invasion protein InlB. J Mol Biol. 2010;395:522–532. doi: 10.1016/j.jmb.2009.10.074. [DOI] [PubMed] [Google Scholar]
- Fletcher DA, Mullins RD. Cell mechanics and the cytoskeleton. Nature. 2010;463:485–492. doi: 10.1038/nature08908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredlund J, Enninga J. Cytoplasmic access by intracellular bacterial pathogens. Trends Microbiol. 2014;22:128–137. doi: 10.1016/j.tim.2014.01.003. [DOI] [PubMed] [Google Scholar]
- Friebel A, Ilchmann H, Aepfelbacher M, Ehrbar K, Machleidt W, Hardt WD. SopE and SopE2 from Salmonella typhimurium activate different sets of RhoGTPases of the host cell. J Biol Chem. 2001;276:34035–34040. doi: 10.1074/jbc.M100609200. [DOI] [PubMed] [Google Scholar]
- Fu Y, Galan JE. A salmonella protein antagonizes Rac-1 and Cdc42 to mediate host-cell recovery after bacterial invasion. Nature. 1999;401:293–297. doi: 10.1038/45829. [DOI] [PubMed] [Google Scholar]
- Hanisch J, Kolm R, Wozniczka M, Bumann D, Rottner K, Stradal TE. Activation of a RhoA/myosin II-dependent but Arp2/3 complex-independent pathway facilitates Salmonella invasion. Cell Host Microbe. 2011;9:273–285. doi: 10.1016/j.chom.2011.03.009. [DOI] [PubMed] [Google Scholar]
- Hanisch J, Ehinger J, Ladwein M, et al. Molecular dissection of Salmonella-induced membrane ruffling versus invasion. Cell Microbiol. 2010;12:84–98. doi: 10.1111/j.1462-5822.2009.01380.x. [DOI] [PubMed] [Google Scholar]
- Hardt WD, Chen LM, Schuebel KE, Bustelo XR, Galan JE. S. typhimurium encodes an activator of Rho GTPases that induces membrane ruffling and nuclear responses in host cells. Cell. 1998;93:815–826. doi: 10.1016/s0092-8674(00)81442-7. [DOI] [PubMed] [Google Scholar]
- Harrison RE, Brumell JH, Khandani A, et al. Salmonella impairs RILP recruitment to Rab7 during maturation of invasion vacuoles. Mol Biol Cell. 2004;15:3146–3154. doi: 10.1091/mbc.E04-02-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward RD, Koronakis V. Direct nucleation and bundling of actin by the SipC protein of invasive Salmonella. EMBO J. 1999;18:4926–4934. doi: 10.1093/emboj/18.18.4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helwani FM, Kovacs EM, Paterson AD, et al. Cortactin is necessary for E-cadherin-mediated contact formation and actin reorganization. J Cell Biol. 2004;164:899–910. doi: 10.1083/jcb.200309034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry T, Couillault C, Rockenfeller P, et al. The Salmonella effector protein PipB2 is a linker for kinesin-1. Proc Natl Acad Sci U S A. 2006;103:13497–13502. doi: 10.1073/pnas.0605443103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiyoshi H, Kodama T, Saito K, et al. VopV, an F-actin-binding type III secretion effector, is required for Vibrio parahaemolyticus-induced enterotoxicity. Cell Host Microbe. 2011;10:401–409. doi: 10.1016/j.chom.2011.08.014. [DOI] [PubMed] [Google Scholar]
- Humphreys D, Davidson A, Hume PJ, Koronakis V. Salmonella virulence effector SopE and Host GEF ARNO cooperate to recruit and activate WAVE to trigger bacterial invasion. Cell Host Microbe. 2012;11:129–139. doi: 10.1016/j.chom.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys D, Davidson AC, Hume PJ, Makin LE, Koronakis V. Arf6 coordinates actin assembly through the WAVE complex, a mechanism usurped by Salmonella to invade host cells. Proc Natl Acad Sci U S A. 2013;110:16880–16885. doi: 10.1073/pnas.1311680110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireton K, Payrastre B, Cossart P. The Listeria monocytogenes protein InlB is an agonist of mammalian phosphoinositide 3-kinase. J Biol Chem. 1999;274:17025–17032. doi: 10.1074/jbc.274.24.17025. [DOI] [PubMed] [Google Scholar]
- Katoh H, Hiramoto K, Negishi M. Activation of Rac1 by RhoG regulates cell migration. J Cell Sci. 2006;119:56–65. doi: 10.1242/jcs.02720. [DOI] [PubMed] [Google Scholar]
- Knodler LA, Vallance BA, Celli J, Winfree S, Hansen B, Montero M, Steele-Mortimer O. Dissemination of invasive Salmonella via bacterial-induced extrusion of mucosal epithelia. Proc Natl Acad Sci U S A. 2010;107:17733–17738. doi: 10.1073/pnas.1006098107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocks C, Gouin E, Tabouret M, Berche P, Ohayon H, Cossart P. L. monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell. 1992;68:521–531. doi: 10.1016/0092-8674(92)90188-i. [DOI] [PubMed] [Google Scholar]
- Kovacs EM, Ali RG, McCormack AJ, Yap AS. E-cadherin homophilic ligation directly signals through Rac and phosphatidylinositol 3-kinase to regulate adhesive contacts. J Biol Chem. 2002;277:6708–6718. doi: 10.1074/jbc.M109640200. [DOI] [PubMed] [Google Scholar]
- Kovacs EM, Goodwin M, Ali RG, Paterson AD, Yap AS. Cadherin-directed actin assembly: E-cadherin physically associates with the Arp2/3 complex to direct actin assembly in nascent adhesive contacts. Curr Biol. 2002;12:379–382. doi: 10.1016/s0960-9822(02)00661-9. [DOI] [PubMed] [Google Scholar]
- Kubori T, Galan JE. Temporal regulation of salmonella virulence effector function by proteasome-dependent protein degradation. Cell. 2003;115:333–342. doi: 10.1016/s0092-8674(03)00849-3. [DOI] [PubMed] [Google Scholar]
- Lambrechts A, Gevaert K, Cossart P, Vandekerckhove J, Van Troys M. Listeria comet tails: the actin-based motility machinery at work. Trends Cell Biol. 2008;18:220–227. doi: 10.1016/j.tcb.2008.03.001. [DOI] [PubMed] [Google Scholar]
- Lecuit M, Ohayon H, Braun L, Mengaud J, Cossart P. Internalin of Listeria monocytogenes with an intact leucine-rich repeat region is sufficient to promote internalization. Infect Immun. 1997;65:5309–5319. doi: 10.1128/iai.65.12.5309-5319.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liverman AD, Cheng HC, Trosky JE, et al. Arp2/3-independent assembly of actin by Vibrio type III effector VopL. Proc Natl Acad Sci U S A. 2007;104:17117–17122. doi: 10.1073/pnas.0703196104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loisel TP, Boujemaa R, Pantaloni D, Carlier MF. Reconstitution of actin-based motility of Listeria and Shigella using pure proteins. Nature. 1999;401:613–616. doi: 10.1038/44183. [DOI] [PubMed] [Google Scholar]
- McGhie EJ, Hayward RD, Koronakis V. Control of actin turnover by a salmonella invasion protein. Mol Cell. 2004;13:497–510. doi: 10.1016/s1097-2765(04)00053-x. [DOI] [PubMed] [Google Scholar]
- Okada R, Zhou X, Hiyoshi H, et al. The Vibrio parahaemolyticus effector VopC mediates Cdc42-dependent invasion of cultured cells but is not required for pathogenicity in an animal model of infection. Cell Microbiol. 2014;16:938–947. doi: 10.1111/cmi.12252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JC, Galan JE. Differential activation and function of Rho GTPases during Salmonella-host cell interactions. J Cell Biol. 2006;175:453–463. doi: 10.1083/jcb.200605144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pust S, Morrison H, Wehland J, Sechi AS, Herrlich P. Listeria monocytogenes exploits ERM protein functions to efficiently spread from cell to cell. EMBO J. 2005;24:1287–1300. doi: 10.1038/sj.emboj.7600595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajabian T, Gavicherla B, Heisig M, Muller-Altrock S, Goebel W, Gray-Owen SD, Ireton K. The bacterial virulence factor InlC perturbs apical cell junctions and promotes cell-to-cell spread of Listeria. Nat Cell Biol. 2009;11:1212–1218. doi: 10.1038/ncb1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie JM, Rui H, Zhou X, et al. Inflammation and disintegration of intestinal villi in an experimental model for Vibrio parahaemolyticus-induced diarrhea. PLoS Pathog. 2012;8:e1002593. doi: 10.1371/journal.ppat.1002593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salcedo SP, Holden DW. SseG, a virulence protein that targets Salmonella to the Golgi network. EMBO J. 2003;22:5003–5014. doi: 10.1093/emboj/cdg517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seveau S, Pizarro-Cerda J, Cossart P. Molecular mechanisms exploited by Listeria monocytogenes during host cell invasion. Microbes Infect. 2007;9:1167–1175. doi: 10.1016/j.micinf.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Seveau S, Tham TN, Payrastre B, Hoppe AD, Swanson JA, Cossart P. A FRET analysis to unravel the role of cholesterol in Rac1 and PI 3-kinase activation in the InlB/Met signalling pathway. Cell Microbiol. 2007;9:790–803. doi: 10.1111/j.1462-5822.2006.00832.x. [DOI] [PubMed] [Google Scholar]
- Sousa S, Cabanes D, El-Amraoui A, Petit C, Lecuit M, Cossart P. Unconventional myosin VIIa and vezatin, two proteins crucial for Listeria entry into epithelial cells. J Cell Sci. 2004;117:2121–2130. doi: 10.1242/jcs.01066. [DOI] [PubMed] [Google Scholar]
- Sousa S, Cabanes D, Bougneres L, Lecuit M, Sansonetti P, Tran-Van-Nhieu G, Cossart P. Src, cortactin and Arp2/3 complex are required for E-cadherin-mediated internalization of Listeria into cells. Cell Microbiol. 2007;9:2629–2643. doi: 10.1111/j.1462-5822.2007.00984.x. [DOI] [PubMed] [Google Scholar]
- Steele-Mortimer O. The Salmonella-containing vacuole: moving with the times. Curr Opin Microbiol. 2008;11:38–45. doi: 10.1016/j.mib.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stender S, Friebel A, Linder S, Rohde M, Mirold S, Hardt WD. Identification of SopE2 from Salmonella typhimurium, a conserved guanine nucleotide exchange factor for Cdc42 of the host cell. Mol Microbiol. 2000;36:1206–1221. doi: 10.1046/j.1365-2958.2000.01933.x. [DOI] [PubMed] [Google Scholar]
- Talman AM, Chong R, Chia J, Svitkina T, Agaisse H. Actin network disassembly powers dissemination of Listeria monocytogenes. J Cell Sci. 2014;127:240–249. doi: 10.1242/jcs.140038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terebiznik MR, Vieira OV, Marcus SL, et al. Elimination of host cell PtdIns(4,5)P(2) by bacterial SigD promotes membrane fission during invasion by Salmonella. Nat Cell Biol. 2002;4:766–773. doi: 10.1038/ncb854. [DOI] [PubMed] [Google Scholar]
- Truong D, Brabant D, Bashkurov M, et al. Formin-mediated actin polymerization promotes Salmonella invasion. Cell Microbiol. 2013;15:2051–2063. doi: 10.1111/cmi.12173. [DOI] [PubMed] [Google Scholar]
- Veiga E, Cossart P. Listeria hijacks the clathrin-dependent endocytic machinery to invade mammalian cells. Nat Cell Biol. 2005;7:894–900. doi: 10.1038/ncb1292. [DOI] [PubMed] [Google Scholar]
- Yu B, Cheng HC, Brautigam CA, Tomchick DR, Rosen MK. Mechanism of actin filament nucleation by the bacterial effector VopL. Nat Struct Mol Biol. 2011;18:1068–1074. doi: 10.1038/nsmb.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Krachler AM, Broberg CA, Li Y, Mirzaei H, Gilpin CJ, Orth K. Type III effector VopC mediates invasion for Vibrio species. Cell Rep. 2012;1:453–460. doi: 10.1016/j.celrep.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Mooseker MS, Galan JE. Role of the S. typhimurium actin-binding protein SipA in bacterial internalization. Science. 1999;283:2092–2095. doi: 10.1126/science.283.5410.2092. [DOI] [PubMed] [Google Scholar]
- Zhou D, Chen LM, Hernandez L, Shears SB, Galan JE. A Salmonella inositol polyphosphatase acts in conjunction with other bacterial effectors to promote host cell actin cytoskeleton rearrangements and bacterial internalization. Mol Microbiol. 2001;39:248–259. doi: 10.1046/j.1365-2958.2001.02230.x. [DOI] [PubMed] [Google Scholar]
- Zhou X, Massol RH, Nakamura F, et al. Remodeling of the intestinal brush border underlies adhesion and virulence of an enteric pathogen. MBio. 2014;5 doi: 10.1128/mBio.01639-14. [DOI] [PMC free article] [PubMed] [Google Scholar]