

Abstract
Keratinocytes are routinely subjected to both internal and external stimulation. This study investigates the effects of interferon gamma, interleukin-4, tumor necrosis factor alpha, and the synthetic immunomodulator muramyl dipeptide on the human keratinocyte cell line, HaCaT. Following HaCaT stimulation with cytokines or muramyl dipeptide for different time periods, changes in the expression of different cell surface receptors, cell proliferation, and cell apoptosis were evaluated by flow cytometry, tritiated thymidine uptake, and annexin-V staining, respectively. A significant decrease in the expression of CD49d was found upon treatment with interleukin-4. Interferon gamma and tumor necrosis factor alpha increased the expression of intercellular adhesion molecule 1 and major histocompatibility complex class I, whereas major histocompatibility complex class II and CD1b were only upregulated by interferon gamma. Interferon gamma and tumor necrosis factor alpha had opposite effects regarding CD119 expression, with the former downregulating, while the latter upregulating its expression. Of the stimuli tested, only interferon gamma and tumor necrosis factor alpha significantly inhibited proliferation of HaCaT cells, yet only interferon gamma played a significant role in inducing HaCaT cell apoptosis. Our data demonstrate differential effects of the three tested cytokines on keratinocytes and reveal that the absence of HaCaT cell responses to muramyl dipeptide is associated with undetectable levels of its cytoplasmic receptor, nucleotide-binding oligomerization domain–containing protein 2.
Keywords: apoptosis, CD molecules, cytokines, keratinocytes, muramyl dipeptide, surface receptors
Introduction
Keratinocytes comprise 95% of the cells within the epidermis. While once believed to function primarily as a passive barrier, it has now been established that keratinocytes play an active and dynamic role in the first line of defense of an organism. Keratinocytes not only facilitate the immune response through the production and secretion of cytokines, chemokines, and host defense peptides but may also be integral in triggering the immune response. Epidermal keratinocytes generally commit to one of two pathways, differentiation or activation. In the normal healthy state, keratinocytes undergo differentiation; it is only when these cells are subjected to physical injury, stimulation, or an internal pathological condition that they embark on the alternative pathway of activation in an attempt to maintain a balanced immune response.1,2 Once activated, keratinocytes undergo a breadth of alterations which encompasses, but is not limited to, changes in the rate of proliferation, the production of cytokines/chemokines, and the expression of cell surface receptors.3 Growth factors and cytokines, produced by keratinocytes themselves and/or the surrounding cells, are able to initiate the activation process.4 The variation of surface receptors inherently expressed on keratinocytes translates to wide-ranging biological effects. In fact, dysregulation and/or abnormal expression of keratinocyte receptors has been correlated with the pathogenesis of chronic inflammatory skin diseases.5–7 The intercellular adhesion molecule 1 (ICAM-1/CD54) and the major histocompatibility complex molecule (MHC II/HLA-DR) have weak or no expression on the surface of resting keratinocytes; however, an elevation in the aforementioned markers is considered a hallmark of activation.8,9
Inflammatory skin diseases such as psoriasis and atopic dermatitis (AD), while highly varied in their respective, all involve the activation of keratinocytes and the infiltration of T lymphocytes through released cytokines that play a pivotal role in guiding the immune response.10,11 The pro-inflammatory cytokine interferon gamma (IFN-γ), a T-helper (Th) 1 cytokine, is considered the most effective activator of keratinocytes since it modulates the expression of a diverse number of membrane receptors as well as the production of cytokines and chemokines. IFN-γ has been linked to a number of skin diseases, namely, Th1 dominant, yet it has also been found to be present in the late stages of chronic AD, a Th2 classified disease.12,13 Interleukin-4 (IL-4) and tumor necrosis factor alpha (TNF-α) are also established activators of keratinocytes, however, to a lesser extent than IFN-γ.14
IL-4 is a pleiotropic Th2-derived cytokine amply expressed in chronic inflammatory skin diseases.14,15 Keratinocytes express functional IL-4 receptor, which shares a common subunit with IL-13.16 IL-4 is known to play a key pathogenic role in Th2-dominant diseases and, similar to IL-13, was found capable of blocking the TNF-α- or IFN-γ-induced chemokine release in keratinocytes.17 In the skin, TNF-α wields numerous biological effects including an upregulation in the expression of adhesion receptors in keratinocytes,3 as well as an increase in their production of pro-inflammatory cytokines such as IL-6, IL-8, and IL-1β.18
In addition to endogenous cytokines, immunomodulators may be of exogenous origin. The peptidoglycan derivative muramyl dipeptide (MDP), a component of the cell wall of many Gram-positive and Gram-negative bacteria, is one such immunomodulator.19 MDP is recognized by the cytosolic pathogen recognition receptor nucleotide-binding oligomerization domain 2 (NOD2/CARD15).20 MDP has been found to elicit adjuvant activity through a variety of mechanisms including increased expression of surface markers essential for adhesion and antigen presentation as well cytokine release.21 Since keratinocytes are routinely exposed to commensal bacteria, it follows that they may be activated by exogenous components like MDP, to aid in recognizing the microorganisms.22
The HaCaT cell line is a well-established immortalized, non-tumorigenic cell line, used extensively in dermatological research as an in vitro skin model owing to its inherent phenotype, which closely resembles that of normal human keratinocytes.23 HaCaT cells, similar to normal keratinocytes, maintain their epidermal differentiation capacity and reform a regularly structured and differentiated epidermis when transplanted onto nude mice.23–25 The goal of our study was to elucidate the influence of three cytokines (IFN-γ, IL-4, and TNF-α) and an exogenous immunomodulator (MDP) on HaCaT cells from two perspectives: the expression of three classes of cell surface receptors and the regulation of cell proliferation/apoptosis.
Materials and methods
Antibodies for flow cytometry
The following mouse monoclonal anti-human antibodies were used for flow cytometry experiments: fluorescein isothiocyanate (FITC)-conjugated CD1b (clone M-T101), CD95 (clone DX2), and HLA-DPDQDR (clone Tu39); PE-conjugated CD119 (clone GIR-208), CD124 (clone hIL4R-M57), and CD132 (clone AG184); APC-conjugated CD1a (clone HI149), CD40 (clone 5C3), CD49d (clone 9F10), and HLA-ABC (clone G46-2.6; BD Biosciences, San Jose, CA, USA); FITC-conjugated CD54 (clone RR1/1); PE-conjugated CD147 (clone 8D12) (eBioscience, San Diego, CA, USA). The following isotype-matched control antibodies were also included in all experiments: FITC-conjugated mouse IgG1 (clone MOPC-21), IgG2a (clone G155-178); PE-conjugated mouse IgG1 (clone MOPC-21); and APC-conjugated mouse IgG1 (clone MOPC-21) (BD Biosciences).
Cell culture
The HaCaT immortalized human keratinocyte cell line (kindly provided by Dr J Usta, Department of Biochemistry and Molecular Genetics, American University of Beirut, Lebanon) was cultured in Dulbecco’s modified Eagle’s medium (DMEM; Lonza, Slough, UK) supplemented with l-glutamine (Sigma-Aldrich, St. Louis, MO, USA), penicillin–streptomycin (Sigma), sodium pyruvate (Sigma), and 10% heat inactivated fetal bovine serum (FBS; Sigma). Cells were maintained in a humidified atmosphere at 37°C and 5% CO2. Cells were passaged regularly with trypsin-EDTA (Sigma) upon reaching 70%–80% confluence and routinely checked for morphology. Cell viability was determined using the standard trypan blue dye exclusion method.
Immunophenotyping of HaCaT cells
HaCaT cells were seeded 1 day prior to stimulation at a density of 0.5 × 105 viable cells per 25 cm2 flask. The following day, cells were either left unstimulated or stimulated with IFN-γ (50 ng/mL; R&D Systems, Abingdon, UK), IL-4, TNF-α (50 ng/mL; CellGenix, Freiburg, Germany), or MDP (20 µg/mL; kindly provided by ISTAC-SA, Lille, France) for 3, 24, 48, and 72 h at 37°C in 5% CO2 in a humidified incubator, unless otherwise mentioned. All stimulants were resuspended in Dulbecco’s phosphate-buffered saline (DPBS). At the end of each culture period, cells were washed twice with DPBS and then detached with Accutase solution (Gibco, Invitrogen, Karlsruhe, Germany). HaCaT cell suspensions were washed twice with staining buffer consisting of cell wash solution (BD Biosciences) supplemented with 2% FBS. A minimum of 1 × 105 cells/100 µL were incubated with optimized concentrations of fluorochrome-conjugated monoclonal antibodies for 30 min at 4°C in the dark. After washing with 2 mL staining buffer at 300g for 5 min, cells were fixed for 20 min in 4% paraformaldehyde (Sigma). Cells were washed again and resuspended in a final volume of 500 µL staining buffer to be then analyzed on a FACSCalibur flow cytometer (BD Biosciences). Flow cytometry data were analyzed by CellQuest Pro software (BD Biosciences) and for each sample, a minimum of 10,000 events were recorded. The expression of cell surface receptors was measured as total geometric mean fluorescence intensity (MFI) and was presented in histogram plots. Single color stained cells and Calibrite beads (BD Biosciences) were used to adjust fluorescence intensity and color compensation. An isotype control antibody was used for each monoclonal antibody employed.
Proliferation assay
HaCaT cells were seeded, in quadruplets, in 96-well flat bottom plates (Corning, Tewksbury, MA, USA) at a density of 104 cells/well and were either left unstimulated or stimulated with IFN-γ (50 or 100 ng/mL), IL-4 (50 or 100 ng/mL), TNF-α (50 or 100 ng/mL), or MDP (20 µg/mL). Cultures were maintained for 24, 48, and 72 h at 37°C in 5% CO2 in a humidified incubator. The cultures were pulsed with tritiated thymidine (Perkin Elmer, San Jose, CA, USA) at a concentration of 0.5 µCi/well for 18 h prior to cell harvesting. Cells were then transferred onto glass fiber filter disks (Connectorate AG, Dietikon, Switzerland) by means of a cell harvester (Inotech Biotechnologies, Basel, Switzerland) and the amount of incorporated radioactivity was measured in a liquid scintillation counter (Packard). Unstimulated samples served as a control. The radioactivity of incorporated tritiated thymidine was reported as counts per minute (cpm) and results are represented as mean cpm of quadruplicate wells.
Detection of apoptosis
Apoptosis was detected using the annexin V-FITC apoptosis detection kit II (BD Biosciences) following the manufacture’s protocol. Briefly, cells were washed twice with cold DPBS then resuspended in 1× binding buffer at a concentration of 1 × 106 cells/mL. A total of 100 µL of the cell suspension containing 1 × 105 cells was incubated with 5 µL of each annexin V-FITC and propidium iodide (PI) for 15 min at room temperature in the dark. Following incubation, 400 µL of 1× binding buffer was added to each sample and cells were analyzed on a FACSCalibur flow cytometer within 30 min. Unstained cells and single stained cells with either annexin V-FITC or PI were used as controls. Cells incubated at 70°C for 15 min served as a positive control for cell death, while cells treated with betulinic acid (16 µg/mL; Sigma) for 24 h served as a positive control for apoptosis induction.26
Detection of NOD2
The detection of NOD2 protein expression in HaCaT cells was attempted by western blot using either total cell lysate or immunoprecipitated NOD2. For protein extraction, cells were washed twice with PBS, detached with Accutase solution, and counted. The cell pellet was resuspended in lysis buffer at a concentration of 106 cells/50 µL of lysis buffer supplemented with protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN, USA); the tube was left on ice for 20 min with periodic vortexing. Lysates were then passed through a 21-gauge needle up to 10 times, followed by centrifugation at 10,000g for 20 min at 4°C. The supernatant was carefully transferred to a new tube and the concentration was determined by DC Protein Assay Kit (Bio-Rad, Hercules, CA, USA). For western blot detection, a range (5–50 µg) of HaCaT total protein lysate was resolved on 4%–10% sodium dodecyl sulfate (SDS) Tris-glycine gels. Proteins were transferred onto a protran-nitrocellulose membrane (GE Healthcare Life Sciences, Freiburg, Gemany), blocked in tris-buffered saline (TBS) containing 5% bovine serum albumin (BSA; Sigma) and 0.05% Tween 20 (Sigma) for 1.5 h, and incubated overnight in TBS containing 1% BSA, 0.05% Tween 20, and 0.25 µg/mL of anti-NOD2 mouse monoclonal antibody (clone NOD-15) or its matching isotype control (BioLegend, San Diego, CA, USA). The membrane was washed six times in TBS with 0.1% Tween 20 and incubated for 1 h in 1% BSA containing 1:20,000 rabbit anti-mouse IgG horseradish peroxidase conjugate (Santa Cruz Biotechnology, Dallas, TX, USA). The membrane was washed once again and incubated for 5 min with chemiluminescent peroxidase substrate (GE Healthcare Life Sciences) and detected on film (GE Healthcare Life Sciences). For immunoprecipitation, protein extracts from 2 or 3 × 106 HaCaT cells were precipitated with 3-µL NOD2-specific rabbit anti-serum (Cayman Chemical, Ann Arbor, MI, USA) or NOD2-specific mouse monoclonal antibody (BioLegend) using Protein G immunoprecipitation kit (Roche Diagnostics), following the manufacturer’s instructions. Immunoprecipitated proteins were resolved by western blot analysis and probed using 3:10,000 of rabbit NOD2 anti-serum (Cayman Chemical) or 5 µg/mL mouse monoclonal antibody (BioLegend). RAW 264.7 total cell lysate (Santa Cruz Biotechnology) was used as a positive control for NOD2 detection.
Statistical analysis
Data are presented as mean ± standard error of the mean (SEM) from, at least, three independent experiments. Statistical analyses were performed with GraphPad Prism software (version 6) using Student’s t-test for unpaired samples. Differences were considered statistically significant at P values <0.05.
Results
Cell surface receptor expression on HaCaT cells following stimulation with IFN-γ, IL-4, TNF-α, or MDP
The effect of IFN-γ, IL-4, TNF-α, and MDP on the phenotype of HaCaT cells was evaluated through the analysis of the variations in expression of a battery of cell surface receptors after stimulation with the above-mentioned immunomodulators at four time points: 3, 24, 48, and 72 h. The employed concentrations of the immunomodulators are adopted from previous, well-established, and optimized protocols.27–29 The chosen panel of receptors fall under three categories: adhesion, antigen presentation, and cytokine receptors. A representative flow cytometry chart of the MFI for three receptors, before and after stimulation, is shown in Figure 1. In the absence of stimulation, HaCaT cells expressed low to moderate levels of the two adhesion receptors tested, CD49d and CD54 (ICAM-1). Stimulation with IL-4 notably decreased the level of CD49d below baseline expression, while IFN-γ, TNF-α, and MDP did not trigger a significant variation (Figure 2). The level of expression of CD49d began to decrease in response to stimulation with IL-4 at 24 h with a 1.5-fold decrease eventually leading to a complete loss of its expression at 72 h. In contrast, the expression of ICAM-1 on HaCaT cells increased in response to stimulation with IFN-γ or TNF-α at all four time points tested, with IFN-γ being the more potent inducer. A maximum increase in ICAM-1 expression was achieved at 48-h stimulation with IFN-γ or TNF-α (*P < 0.05). However, while IFN-γ led to a staggering 58-fold increase in ICAM-1 level, TNF-α trailed behind with a significantly lower (*P < 0.05) 17-fold increase in expression. In contrast, IL-4 and MDP did not significantly affect the level of ICAM-1 expression. Among the antigen presentation receptors analyzed, only IFN-γ brought forth a significant increase (*P < 0.05) in the expression of CD1b, at 48 and 72 h. The expression levels of MHC I and MHC II were also significantly elevated (*P < 0.05) upon stimulation with IFN-γ starting at 24 h (Figure 2). MHC I was upregulated by TNF-α at 48 and 72 h, but not at 24 h (Figure 2). The effect of TNF-α was significantly lower (*P < 0.05) than that of IFN-γ, reaching a maximum of 1.6-fold increase as opposed to a 2.2-fold increase in expression induced by IFN-γ at 72 h. IL-4, TNF-α, and MDP had no detectable effect on resting levels of CD1b and MHC II. With regard to the cytokine receptors tested, the expression of CD95 (FAS receptor) was elevated by IFN-γ and IL-4 at 24, 48, and 72 h and by TNF-α at 48 and 72 h (Figure 2). At 48 h, a significant difference (*P < 0.05) between the effects of IFN-γ (2.2-fold increase) and TNF-α (1.5-fold increase) was present. CD119 expression on HaCaT cells was significantly reduced by IFN-γ at 24, 48, and 72 h. In contrast, TNF-α significantly increased the level of CD119 at 24, 48, and 72 h, with a maximum of 2.7-fold induction at 72 h (Figure 2). IL-4, TNF-α, and MDP had no detectable effect on resting levels of CD132 on HaCaT cells, while IFN-γ resulted in a significant increase in expression of CD132 at 48 and 72 h. Additional receptors (CD1a, CD40, CD124, and CD147) were also tested under the same conditions, yet all exhibited no significant variation in expression in response to any of the three tested cytokines (Table 1).
Figure 1.

Representative flow cytometry histogram plots displaying changes in the expression of CD54, CD95, and CD119 on HaCaT cells 48 h post-stimulation, with cytokines or MDP.
Unstim: unstimulated.
Figure 2.

Modulation of surface receptors on HaCaT cells following stimulation with IFN-γ, IL-4, TNF-α, or MDP. Unstimulated cells served as a control. Expression of surface receptors was determined by flow cytometry at 3, 24, 48, and 72 h post-stimulation with 50 ng/mL of each cytokine or 20 µg/mL of MDP. The data shown are representative of three independent experiments (n = 3), values are reported as mean of the total geometric mean fluorescent intensity (MFI) ± SEM. Statistically significant differences were determined by Student’s t-test for unpaired samples (*P < 0.05 vs unstimulated cells).
Table 1.
Surface receptors that were either not induced or did not display a significant variation in expression on HaCaT cells following stimulation with IFN-γ, IL-4, or TNF-α at 3, 24, 48, and 72 h.
| Surface receptor expression on HaCaT Cells (MFI) | ||||
|---|---|---|---|---|
| CD1a | CD40 | CD124 | CD147 | |
| 3 h | ||||
| Unstim | 0 ± 0 | 0.2 ± 0.1 | 0.2 ± 0 | 253 ± 36 |
| IFN-γ | 0 ± 0 | 0.1 ± 0.1 | 0 ± 0 | 250 ± 41 |
| IL-4 | 0.1 ± 0 | 0.1 ± 0.1 | 0 ± 0 | 261 ± 49 |
| TNF-α | 0 ± 0 | 0.1 ± 0.1 | 0 ± 0 | 250 ± 45 |
| 24 h | ||||
| Unstim | 0.1 ± 0.1 | 0.3 ± 0.1 | 0 ± 0 | 282 ± 44 |
| IFN-γ | 0.3 ± 0.1 | 0.6 ± 0.2 | 0 ± 0 | 203 ± 29 |
| IL-4 | 0 ± 0 | 0.1 ± 0.1 | 0 ± 0 | 274 ± 42 |
| TNF-α | 0.2 ± 0 | 0.5 ± 0.1 | 0 ± 0 | 250 ± 28 |
| 48 h | ||||
| Unstim | 0 ± 0 | 0.1 ± 0.1 | 0 ± 0 | 251 ± 15 |
| IFN-γ | 0.3 ± 0.1 | 0.5 ± 0.2 | 0 ± 0 | 197 ± 31 |
| IL-4 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 245 ± 3 |
| TNF-α | 0.1 ± 0 | 0.3 ± 0 | 0 ± 0 | 216 ± 15 |
| 72 h | ||||
| Unstim | 0 ± 0 | 0 ± 0 | 0 ± 0 | 185 ± 9 |
| IFN-γ | 0 ± 0 | 0.1 ± 0.1 | 0 ± 0 | 149 ± 15 |
| IL-4 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 162 ± 5 |
| TNF-α | 0 ± 0 | 0 ± 0 | 0 ± 0 | 167 ± 5 |
IFN-γ: interferon gamma; IL-4: interleukin-4; TNF-α: tumor necrosis factor alpha; SEM: standard error of the mean.
Unstimulated cells (Unstim) served as a control. Data are expressed as mean ± SEM (n = 3) of the total geometric mean fluorescent intensity (MFI) in logU.
IFN-γ and TNF-α inhibit proliferation of HaCaT cells
In parallel to the evaluation of receptor expression, the proliferative response of HaCaT cells to the same set of stimulants was also conducted. Cells were seeded at an appropriate density to maintain an exponential growth state throughout the experiment, and proliferation was monitored after 24, 48, and 72 h post-stimulation with IFN-γ, IL-4, TNF-α, or MDP (Figure 3(a)). Both IFN-γ and TNF-α proved to be substantial inhibitors of HaCaT cell proliferation. The effect of IFN-γ started at 24 h with a significant 36% inhibition (*P < 0.05), remaining stable at 72 h with a 39% inhibition of proliferation (*P < 0.05) (Figure 3). The inhibitory effect of TNF-α on HaCaT proliferation was highest at 24 h, with a 48% inhibition, decreasing to 23% at 48 h and eventually plateauing at 72 h. Stimulation with IL-4 or MDP did not yield a significant influence on the proliferative rate of HaCaT cells. A similar pattern of inhibition was observed when a higher concentration (100 ng/mL) of the cytokines was tested (Figure 3(b)).
Figure 3.
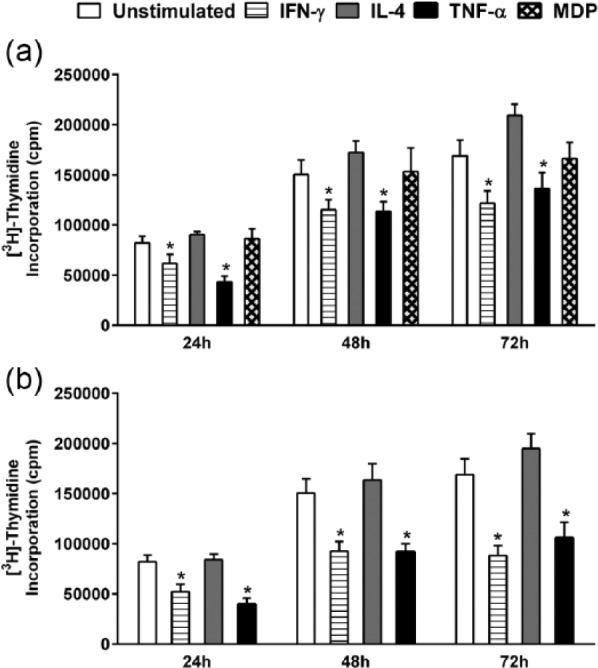
Regulation of [3H] thymidine uptake by HaCaT cells following stimulation with IFN-γ, IL-4, TNF-α, or MDP. HaCaT cells were stimulated with (a) 50 ng/mL of each cytokine and 20 µg/mL MDP or (b) 100 ng/mL of each cytokine for 24, 48, and 72 h. Unstimulated cells served as a control. Data are reported as counts per minute (cpm) of tritiated thymidine uptake. The data shown are from three independent experiments. All values are reported as mean ± SEM. Statistically significant differences were determined by Student’s t-test for unpaired samples (*P < 0.05 vs unstimulated cells).
IFN-γ, but not TNF-α, induces apoptosis in HaCaT cells
To analyze whether the decrease in the rate proliferation of HaCaT cells upon treatment with IFN-γ or TNF-α could be correlated with an increase in cell death, an apoptosis assay was performed (Figure 4(a) and (b)). Cells that stained PI−Annexin+, PI+Annexin+, and PI+Annexin− were considered as early apoptotic cells, late apoptotic cells, and necrotic cells, respectively. As expected, IL-4 did not induce apoptosis in HaCaT cells. On its own, IFN-γ was shown to induce early as well as late stage apoptosis at 48 and 72 h. A maximum increase in the percentage of late apoptotic cells was reached at 72 h with 43%. At 48 h, TNF-α was shown to induce a modest but significant increase (*P < 0.05) in late stage apoptosis, reaching 15%. This increase, however, was fleeting and did not carry on to 72 h, suggesting other mechanisms at work behind the inhibition of proliferation (Figure 4(a) and (b)).
Figure 4.

Cytokine-induced apoptosis in HaCaT cells. HaCaT cells were either left unstimulated or stimulated with IFN-γ, IL-4, or TNF-α at a concentration of 50 ng/mL for 24, 48, and 72 h and apoptosis was determined by annexin V/propidium iodide (PI) staining following the previously indicated stimulation periods. Betulinic acid (BA) was used as a positive control for apoptosis induction. (a) Representative flow cytometry dot plots demonstrating the change in the rate of apoptosis in response to the above-mentioned stimulants. Numbers within the lower right quadrant indicate the percentage of early apoptotic cells, whereas the numbers in the upper right quadrant refer to late apoptotic cells. (b) Changes in the percentage of early (PI−/Annexin+) and late (PI+/Annexin+) apoptotic cells. Bars indicate the mean of three independent experiments ± SEM. Statistically significant differences were determined by Student’s t-test for unpaired samples (*P < 0.05 vs unstimulated cells).
NOD2 protein is undetectable in HaCaT cells
In an effort to explain the unresponsiveness of HaCaT cells to stimulation with MDP, the expression of NOD2 protein was evaluated. Total HaCaT protein lysate was found to be negative for NOD2 expression at 25 and 50 µg when probed with a mouse monoclonal antibody. To verify whether NOD2 protein expression is in fact absent in HaCaT cells, we moved on to perform immunoprecipitation using a protocol described wherein NOD2 protein expression in primary keratinocytes was detected.30 To begin, 2 and 3 × 106 HaCaT cells were immunoprecipitated with NOD2 rabbit polyclonal antibody in accordance with the previously published data. A single band was observed at 63 kDa, yet no band was detected at the expected 110 kDa (Figure 5(a)). The membrane was stripped and re-probed with the mouse monoclonal antibody, yet no bands were observed (Figure 5(b)). We achieved similar results when using a monoclonal antibody for immunoprecipitation (Figure 5(c)). These observations strongly suggest that HaCaT cells do not express detectable levels of NOD2 protein.
Figure 5.

NOD2 expression in HaCaT cells. (a) Total cell lysates from 2 × 106 (lane 1) and 3 × 106 (lane 2) HaCaT cells were immunoprecipitated with NOD2-specific rabbit anti-serum and detected by western blot using the same rabbit NOD2 anti-serum. (b) Membrane in Figure 2(a) was stripped and re-probed with mouse monoclonal NOD2 antibody or its isotype control antibody (Iso). (c) HaCaT total cell lysates were immunoprecipitated with mouse monoclonal NOD2 antibody or its isotype control antibody and detected with NOD2-specific rabbit anti-serum. (d) NOD2 detection in RAW 264.7 total cell lysate (positive control), using mouse monoclonal NOD2 antibody or its Iso.
Discussion
Serving as an interface between the body and its external surroundings, the skin is constantly subjected to dynamic stimulation. In this article, we chose to expand on previous studies by examining the activation of keratinocytes from two angles: modulation of cell surface receptors and changes in proliferation/apoptosis. To our knowledge, this study is the first to investigate the effects of MDP on HaCaT cells from this perspective. We found that HaCaT keratinocytes did not respond to treatment with MDP which is known to exert its effects by binding to the cytoplasmic receptor NOD2.20 While generally expressed in antigen-presenting cells such as monocytes and macrophages,31 NOD2 expression has also been found in other cells.32 Since the precise role of NOD2 in keratinocytes is still up for debate, we hypothesized that the lack of NOD2 expression in HaCaT cells could be the reason behind our failure to obtain a significant response upon stimulation with MDP. NOD2 transcripts have been detected in HaCaT cells,33 yet to our knowledge no studies have been done at the protein level. Our attempts to detect NOD2 protein expression in HaCaT cells via western blot analysis were not successful. However, a previous study on primary keratinocytes had reported both gene and protein expression of NOD2, with an accompanied response following stimulation with MDP.30 Following the same immunoprecipitation protocol described, and through the use of the same polyclonal anti-serum, we were still unable to detect NOD2 protein expression in HaCaT cell lysate. This leads us to believe that HaCaT cells do not express NOD2 protein or express it at a low level that is undetectable by the aforementioned techniques. While this ambiguity prevails, we cannot rule out the absence of functional NOD2 in HaCaT cells as the reason behind the lack of response to MDP. Alternatively, the culprit may lie downstream.
IFN-γ, IL-4, and TNF-α stimulate resident skin cells to produce chemokines and membrane molecules that play an important role in the inflammatory response, including the retention and activation of T cells. Integrins mediate interaction of cells with the extracellular matrix (ECM) and have also been suggested to mediate interaction among cells themselves. The expression of specific β1 integrins in keratinocytes has been shown to be affected by IFN-γ, TNF-α, and transforming growth factor (TGF)-β.34 To date, no study has evaluated the effect of IFN-γ, IL-4, or TNF-α on the surface expression of CD49d (α4 integrin) in HaCaT cells. CD49d associates with either integrin β1 or β7 chain and is known to play a critical role in lymphocyte development, in providing mechanical support for cell adhesion, in acting as signaling receptor, and in mediating leukocyte trafficking and activation.35 Despite extensive studies describing the role of other α integrins in regulating the migration, proliferation, and differentiation of epidermal cells,36 very little is known about the level and relevance of CD49d expression in keratinocytes.29 Increased expression of multiple α integrins, including CD49d, had been reported following treatment of primary keratinocytes with a mixture of sugar and PI, known to accelerate the healing of cutaneous wounds.37 On the contrary, treatment of HaCaT keratinocytes with non-thermal atmospheric-pressure plasma was found to upregulate the surface expression of several α-integrins but not that of α4.38 Our findings on the absence of effect on CD49d expression in HaCaT cells by IFN-γ or TNF-α, and the dramatic decrease or even the absence of constitutive expression induced by IL-4 stimulation, may represent an additional marker of the differences in keratinocyte responses to Th1 and Th2 cytokines.39 Alternatively, the IL-4-mediated inhibition of CD49d expression in HaCaT keratinocytes could be a mechanism to downregulate leukocyte adhesion to keratinocytes to control the inflammatory process, since CD49d is known to play a critical role in leukocyte adhesion to other cell types.40 This phenomenon may also reflect a mechanism by which the keratinocyte response to locally released IL-4 attempts to control excessive inflammatory processes such as AD.
The infiltration of lymphocytes into the points of inflammation is partly mediated by CD54 (ICAM-1), one of the ligands for lymphocyte function–associated antigen 1 (LFA-1).41 Both primary cultured keratinocytes and HaCaT cells have shown weak to no constitutive expression of ICAM-1, which is in agreement with our results.4,42,43 Nevertheless, an upregulation in the expression of ICAM-1 is observed in skin keratinocytes and endothelial cells during inflammation, ultimately elevating epidermal trafficking of T lymphocytes and extending the inflammatory response.8,44 Upon stimulation with IFN-γ, we detected a significant upregulation of ICAM-1 expression at 24 h that persisted till 72 h. Our results are validated by numerous studies conducted on both primary keratinocytes and HaCaT cells, demonstrating that IFN-γ on its own is able to induce high ICAM-1 surface expression.4,42,43,45,46 Furthermore, we also detected a significant increase in ICAM-1expression starting 24 h after treatment with TNF-α. This particular effect contradicts previous studies done on cultured keratinocytes which reported that, on its own, TNF-α evokes either no change47 or a weak increase in ICAM-1 expression.8,14,42,48 This discrepancy might be explained by an indirect effect involving TNF-α induction of IL-33 release by keratinocytes49,50 and subsequent upregulation of ICAM-1 expression by IL-33.51,52 However, IL-4 did not significantly affect the expression of ICAM-1 in our study, which is in line with studies conducted on primary keratinocytes.16,53 A single study; however, did report that IL-4 downregulates ICAM-1 expression at 48 h in HaCaT cells,43 contrary to all other reported findings and to our results.
In humans, a debate exists as to whether activated keratinocytes can effectively process and present antigens. The inherent expression of MHC I and the induced expression of MHC II upon activation with IFN-γ lends support in favor of this theory, yet the low expression of the co-stimulatory receptors CD80/86 casts doubt.54 In our study, the level of expression of MHC I was upregulated from its basal state upon stimulation with either IFN-γ or TNF-α, but not with IL-4. A previously conducted report on primary keratinocytes demonstrated an increase in MHC I expression upon stimulation with IFN-γ, yet in contrast to what we have obtained, TNF-α on its own was not able to elicit a significant response.14 Upon stimulation with IFN-γ, we observed an induction of MHC II expression. Normal human keratinocytes generally do not express MHC II receptor, yet expression could be induced, a phenomenon known to arise in numerous skin disorders.42 Induction of MHC II has been accredited to IFN-γ released by infiltrating T cells.55 Our results relating to INF-γ are in accordance with numerous studies conducted on cultured keratinocytes as well as HaCaT cells.54–56 Similar to previous studies done on primary keratinocytes, we did not observe a significant effect on the expression of MHC II when HaCaT cells were treated with either TNF-α or IL-4.8,16,42,53 The functional significance of induced MHC II receptor on keratinocytes following activation by IFN-γ is still unclear. Nevertheless, the possibility of peptide presentation by MHC II–positive keratinocytes to epitope-specific CD4+ T cells may occur in a comparable fashion to the reported MHC I–mediated keratinocyte presentation of peptides to CD8+ T cells.45
The expression of CD1b on HaCaT cells was low and increased significantly at 48 and 72 h following stimulation with IFN-γ. CD1b is a member of the CD1 family of transmembrane glycoproteins, which are remotely related to the MHC receptors and are involved in presenting lipids and glycolipids.57 Very few studies have been conducted on the expression of CD1b in keratinocytes. Primary keratinocytes have been shown to lack CD1a expression,58 similar to what we observed in HaCaT cells. In oral epithelium, no CD1b+ cells were found, while expression of CD1a+ keratinocytes was only present in sites of inflammation.59 Our detected increase in CD1b expression, following stimulation with IFN-γ, raises the possibility that presentation of lipids and glycolipids by keratinocytes may take place during states of inflammation.
The IFN-γR/CD119 is abundantly found on both epidermal keratinocytes and on HaCaT cells,43,45,60 and it is necessary for IFN-γ to exert its biological effects. Skin biopsies from patients with psoriasis maintained positive staining for the IFN-γ receptors, yet with altered localization.60 In our study, CD119 was significantly downregulated by IFN-γ at 24, 48, and 72 h, whereas it was upregulated by TNF-α. The observed decrease in expression of CD119 following stimulation with its corresponding cytokine may result from endocytosis of the complex or by shedding of the receptor. Future studies would be required to validate this hypothesis. No studies have so far been conducted on the effect of TNF-α on CD119. The accompanied upregulation we observed could explain the synergistic effect TNF-α has been shown to have on IFN-γ activity. TNF-α possibly enhances the effect of IFN-γ to further activate keratinocytes through the expression of certain receptors, ultimately leading to a positive feedback of the inflammatory response. Indeed, previous reports have indicated that TNF-α synergies with IFN-γ to enhance the expression of ICAM-1 and MHC I in cultured keratinocytes.8,42,48 IL-4 had no effect on CD119 receptor expression, which is consistent with previous findings.14
IL-4 receptors fall under two types composed of either IL-4Rα (CD124) associated with the common gamma chain (γc), CD132, which binds to IL-4, or CD124 associated with IL-13Rα1 chain which binds to both IL-4 and IL-13.61 We found that the expression of CD124 and CD132 on HaCaT cells to be constitutively absent or low. Conflicting data exists regarding the expression of CD124 in keratinocytes. In a study on HaCaT cells, both immunoblot and polymerase chain reaction (PCR) analysis revealed no expression of CD124.62 On the contrary, a study on freshly isolated and cultured keratinocytes demonstrated the expression of CD124 at the messenger RNA (mRNA) as well as the protein level.16,43,63 The low basal expression of CD132 on HaCaT cells was increased significantly after 48 h of stimulation with IFN-γ. While typically expressed on lymphocytes, keratinocytes have occasionally been found to express CD132,64 yet this expression is quite inconsistent.65,66 A study carried out on HaCaT cells has demonstrated their lack of CD132 expression. Moreover, HaCat cell stimulation with IL-4 did not affect their level of CD124 expression.63 The HaCaT response we have seen with IL-4 regarding CD49d and CD95 expression may therefore be unrelated to CD124 and CD132, since both receptors were neither expressed nor regulated by IL-4. On the contrary, the significant increase in CD132 expression may constitute a basis for the previously reported synergy between IL-4 and IFN-γ. Combined stimulation with IL-4 and IFN-γ enhanced the expression of ICAM-1 and MHC II in cultured keratinocytes and HaCaT cells.14,53 This synergy appears to be independent of CD124, since it has been previously reported that IFN-γ selectively reduces IL-4R expression in cultured keratinocytes, a mechanism by which IFN-γ may limit the effects of IL-4.14
It has been proposed that IFN-γ sensitizes keratinocytes for CD95 (Fas)-induced apoptosis by upregulating the Fas receptor, thus allowing Fas to transduce an apoptotic signal.67 Others have suggested that while Fas ligand (FasL) has a synergistic effect on IFN-γ-induced apoptosis, the observed apoptosis is not a secondary occurrence due to Fas/FasL interaction.68 The constitutive expression of Fas which we observed on HaCaT cells agrees with previous reports.67 Interestingly, stimulation with IFN-γ, TNF-α, or IL-4 all significantly increased Fas expression. Yet, only IFN-γ (and TNF-α, to a certain extent) induced apoptosis of HaCaT cells, suggesting that factors other than Fas-mediated apoptosis may be implicated in our detected increase in apoptosis. In keeping with our findings, Fas mRNA, but not FasL, was reported to be expressed in primary keratinocytes and upregulated in the presence of IFN-γ. When treated with IFN-γ, but not with TNF-α or IL-4, keratinocytes were efficiently killed.69 In HaCaT-T cell 48-h co-cultures, surface Fas levels increased on HaCaT cells. This effect was attributed to elevated levels of IFN-γ in the co-cultures. Moreover, enhanced Fas expression on HaCaT cells was associated with induction of apoptosis.70
We investigated the effect of IFN-γ, IL-4, and TNF-α on HaCaT cell proliferation and apoptosis. Although the anti-proliferative effect of IFN-γ was also accompanied by an induction of apoptosis, the same was not observed with TNF-α. A significant inhibition of proliferation was observed as early as 24 h post treatment with IFN-γ, yet apoptosis only became evident after 48 h of stimulation. Therefore, the inhibition of proliferation by IFN-γ could be only partly explained by an accompanied induction of apoptosis. The anti-proliferative and growth arrest effects of IFN-γ have been previously studied on cultured keratinocytes71,72 and on HaCaT cells.73,74 In fact, in HaCaT cells, IFN-γ was shown to be the main mediator of apoptosis; with apoptosis initiated 48-h post-treatment, which is in agreement with our results.68 It should be noted, however, that HaCaT cells possess a mutation in both p53 alleles, which has been linked to a heightened apoptotic response to IFN-γ.72
With respect to TNF-α, we could not find a direct correlation between the observed inhibition of proliferation and apoptosis. At 48 h, TNF-α stimulation of HaCaT cells significantly induced apoptosis, yet this effect was not preserved at 72 h. Conflicting data exists regarding the ability of TNF-α to induce apoptosis. In one study, TNF-α on its own was not sufficient in inducing apoptosis in HaCaT cells, but when combined with IFN-γ, it enhanced the effect of the latter and induced autocrine TNF-α production.68 However, in another study, TNF-α was found to induce apoptosis in HaCaT cells.75 Still, other studies have shown that TNF-α induces apoptosis in HaCaT cells only when previously sensitized with cycloheximide76 or when combined with TNF-related apoptosis-inducing ligand (TRAIL).77 TNF-α was also reported to induce cytotoxic effects on cultured keratinocytes in a time- and concentration-dependent fashion, with the effects being observed only at periods longer than 96 h.78 This might explain why we did not observe a measurable effect of TNF-α on apoptosis induction at the 24 and 72 h, with only a small non-sustained effect at the 48-h period.
In conclusion, our data on the differences in the effects of IFN-γ, IL-4, and TNF-α on keratinocyte receptor expression, proliferation, and apoptosis induction support and extend previous reports and could pave the way for selected cytokine therapy in different skin pathologies. Moreover, the lack of responses of HaCaT keratinocytes to the synthetic immunomodulator MDP was found to be associated with undetectable levels of NOD2 protein in HaCaT cells. To overcome the limitations of our study, future studies are planned to evaluate and compare the effects of the three tested cytokines and MDP on primary human keratinocytes and on additional human keratinocyte cell lines, an approach which could reveal differences in cell responsiveness to stimulation with IFN-γ, IL-4, TNF-α, or MDP.
Footnotes
Author contribution: G.M.B conceived and designed the experiments. E.E. and S.B. performed the major part of the experimental work and S.D. a minor part. E.E., S.B., K.E.S, and G.M.B. analyzed the data. K.E.S. contributed reagents. E.E., S.B., and G.M.B. wrote the paper.
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: This work was supported by an internal research grant from the University of Balamand to G.M.B.
References
- 1. Kupper TS, Fuhlbrigge RC. (2004) Immune surveillance in the skin: Mechanisms and clinical consequences. Nature Reviews Immunology 4: 211–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Freedberg IM, Tomic-Canic M, Komine M, et al. (2001) Keratins and the keratinocyte activation cycle. Journal of Investigative Dermatology 116: 633–640. [DOI] [PubMed] [Google Scholar]
- 3. Uchi H, Terao H, Koga T, et al. (2000) Cytokines and chemokines in the epidermis. Journal of Dermatological Science 24(Suppl. 1): S29–S38. [DOI] [PubMed] [Google Scholar]
- 4. Caughman SW, Li LJ, Degitz K. (1990) Characterization and functional analysis of interferon-gamma-induced intercellular adhesion molecule-1 expression in human keratinocytes and A-431 cells. Journal of Investigative Dermatology 94: 22S–26S. [DOI] [PubMed] [Google Scholar]
- 5. Valins W, Amini S, Berman B. (2010) The expression of toll-like receptors in dermatological diseases and the therapeutic effect of current and newer topical toll-like receptor modulators. The Journal of Clinical and Aesthetic Dermatology 3: 20–29. [PMC free article] [PubMed] [Google Scholar]
- 6. Wei-yuan M, Wen-ting L, Chen Z, et al. (2011) Significance of DC-LAMP and DC-SIGN expression in psoriasis vulgaris lesions. Experimental and Molecular Pathology 91: 461–465. [DOI] [PubMed] [Google Scholar]
- 7. Gaspar K, Kukova G, Bunemann E, et al. (2013) The chemokine receptor CCR3 participates in tissue remodeling during atopic skin inflammation. Journal of Dermatological Science 71: 12–21. [DOI] [PubMed] [Google Scholar]
- 8. Griffiths CE, Voorhees JJ, Nickoloff BJ. (1989) Characterization of intercellular adhesion molecule-1 and HLA-DR expression in normal and inflamed skin: Modulation by recombinant gamma interferon and tumor necrosis factor. Journal of the American Academy of Dermatology 20: 617–629. [DOI] [PubMed] [Google Scholar]
- 9. Strange P, Skov L, Baadsgaard O. (1994) Interferon gamma-treated keratinocytes activate T cells in the presence of superantigens: Involvement of major histocompatibility complex class II molecules. Journal of Investigative Dermatology 102: 150–154. [DOI] [PubMed] [Google Scholar]
- 10. Bos JD, de Rie MA, Teunissen MB, et al. (2005) Psoriasis: Dysregulation of innate immunity. British Journal of Dermatology 152: 1098–1107. [DOI] [PubMed] [Google Scholar]
- 11. Grewe M, Bruijnzeel-Koomen CA, Schopf E, et al. (1998) A role for Th1 and Th2 cells in the immunopathogenesis of atopic dermatitis. Immunology Today 19: 359–361. [DOI] [PubMed] [Google Scholar]
- 12. Werfel T, Morita A, Grewe M, et al. (1996) Allergen specificity of skin-infiltrating T cells is not restricted to a type-2 cytokine pattern in chronic skin lesions of atopic dermatitis. Journal of Investigative Dermatology 107: 871–876. [DOI] [PubMed] [Google Scholar]
- 13. Rebane A, Zimmermann M, Aab A, et al. (2012) Mechanisms of IFN-gamma-induced apoptosis of human skin keratinocytes in patients with atopic dermatitis. Journal of Allergy and Clinical Immunology 129: 1297–1306. [DOI] [PubMed] [Google Scholar]
- 14. Albanesi C, Cavani A, Girolomoni G. (1999) IL-17 is produced by nickel-specific T lymphocytes and regulates ICAM-1 expression and chemokine production in human keratinocytes: Synergistic or antagonist effects with IFN-gamma and TNF-alpha. Journal of Immunology 162: 494–502. [PubMed] [Google Scholar]
- 15. Szepietowski JC, McKenzie RC, Keohane SG, et al. (1997) Atopic and non-atopic individuals react to nickel challenge in a similar way. A study of the cytokine profile in nickel-induced contact dermatitis. British Journal of Dermatology 137: 195–200. [DOI] [PubMed] [Google Scholar]
- 16. Junghans V, Jung T, Neumann C. (1996) Human keratinocytes constitutively express IL-4 receptor molecules and respond to IL-4 with an increase in B7/BB1 expression. Experimental Dermatology 5: 316–324. [DOI] [PubMed] [Google Scholar]
- 17. Xiao T, Kagami S, Saeki H, et al. (2003) Both IL-4 and IL-13 inhibit the TNF-alpha and IFN-gamma enhanced MDC production in a human keratinocyte cell line, HaCaT cells. Journal of Dermatological Science 31: 111–117. [DOI] [PubMed] [Google Scholar]
- 18. Park K, Lee JH, Cho HC, et al. (2010) Down-regulation of IL-6, IL-8, TNF-alpha and IL-1beta by glucosamine in HaCaT cells, but not in the presence of TNF-alpha. Oncology Letters 1: 289–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bahr GM, Darcissac E, Bevec D, et al. (1995) Immunopharmacological activities and clinical development of muramyl peptides with particular emphasis on murabutide. International Journal of Immunopharmacology 17: 117–131. [DOI] [PubMed] [Google Scholar]
- 20. Girardin SE, Boneca IG, Viala J, et al. (2003) Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. Journal of Biological Chemistry 278: 8869–8872. [DOI] [PubMed] [Google Scholar]
- 21. Darcissac EC, Bahr GM, Pouillart PR, et al. (1996) Selective potentiation of cytokine expression in human whole blood by murabutide, a muramyl dipeptide analogue. Cytokine 8: 658–666. [DOI] [PubMed] [Google Scholar]
- 22. Corridoni D, Arseneau KO, Cifone MG, et al. (2014) The dual role of nod-like receptors in mucosal innate immunity and chronic intestinal inflammation. Frontiers in Immunology 5: 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Boukamp P, Petrussevska RT, Breitkreutz D, et al. (1988) Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. Journal of Cell Biology 106: 761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Breitkreutz D, Schoop VM, Mirancea N, et al. (1998) Epidermal differentiation and basement membrane formation by HaCaT cells in surface transplants. European Journal of Cell Biology 75: 273–286. [DOI] [PubMed] [Google Scholar]
- 25. Ryle CM, Breitkreutz D, Stark HJ, et al. (1989) Density-dependent modulation of synthesis of keratins 1 and 10 in the human keratinocyte line HACAT and in ras-transfected tumorigenic clones. Differentiation 40: 42–54. [DOI] [PubMed] [Google Scholar]
- 26. Galgon T, Wohlrab W, Drager B. (2005) Betulinic acid induces apoptosis in skin cancer cells and differentiation in normal human keratinocytes. Experimental Dermatology 14: 736–743. [DOI] [PubMed] [Google Scholar]
- 27. Bahr GM, Modabber FZ, Morin A, et al. (1984) Regulation by muramyl dipeptide (MDP) of the lymphoproliferative responses and polyclonal activation of human peripheral blood mononuclear cells. Clinical & Experimental Immunology 57: 178–186. [PMC free article] [PubMed] [Google Scholar]
- 28. Schreck R, Bevec D, Dukor P, et al. (1992) Selection of a muramyl peptide based on its lack of activation of nuclear factor-kappa B as a potential adjuvant for AIDS vaccines. Clinical & Experimental Immunology 90: 188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Danussi C, Petrucco A, Wassermann B, et al. (2011) EMILIN1-alpha4/alpha9 integrin interaction inhibits dermal fibroblast and keratinocyte proliferation. Journal of Cell Biology 195: 131–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Voss E, Wehkamp J, Wehkamp K, et al. (2006) NOD2/CARD15 mediates induction of the antimicrobial peptide human beta-defensin-2. Journal of Biological Chemistry 281: 2005–2011. [DOI] [PubMed] [Google Scholar]
- 31. Pauleau AL, Murray PJ. (2003) Role of nod2 in the response of macrophages to toll-like receptor agonists. Molecular and Cellular Biology 23: 7531–7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lala S, Ogura Y, Osborne C, et al. (2003) Crohn’s disease and the NOD2 gene: A role for paneth cells. Gastroenterology 125: 47–57. [DOI] [PubMed] [Google Scholar]
- 33. Koller B, Muller-Wiefel AS, Rupec R, et al. (2011) Chitin modulates innate immune responses of keratinocytes. PLoS ONE 6: e16594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hakkinen L, Westermarck J, Johansson N, et al. (1998) Suprabasal expression of epidermal alpha 2 beta 1 and alpha 3 beta 1 integrins in skin treated with topical retinoic acid. British Journal of Dermatology 138: 29–36. [DOI] [PubMed] [Google Scholar]
- 35. Rose DM, Han J, Ginsberg MH. (2002) Alpha4 integrins and the immune response. Immunological Reviews 186: 118–124. [DOI] [PubMed] [Google Scholar]
- 36. Rippa AL, Vorotelyak EA, Vasiliev AV, et al. (2013) The role of integrins in the development and homeostasis of the epidermis and skin appendages. Acta Naturae 5: 22–33. [PMC free article] [PubMed] [Google Scholar]
- 37. Nakao H, Yamazaki M, Tsuboi R, et al. (2006) Mixture of sugar and povidone–iodine stimulates wound healing by activating keratinocytes and fibroblast functions. Archives of Dermatological Research 298: 175–182. [DOI] [PubMed] [Google Scholar]
- 38. Haertel B, Strassenburg S, Oehmigen K, et al. (2013) Differential influence of components resulting from atmospheric-pressure plasma on integrin expression of human HaCaT keratinocytes. BioMed Research International 2013: 761451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Di ZH, Ma L, Qi RQ, et al. (2016) T helper 1 and T helper 2 cytokines differentially modulate expression of filaggrin and its processing proteases in human keratinocytes. Chinese Medical Journal 129: 295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Eppihimer MJ, Sushkova N, Lavigne MC. (2004) Relative contributions of alpha4 and alphaL integrins to IL-4-induced leukocyte rolling and adhesion. Microcirculation 11: 655–668. [DOI] [PubMed] [Google Scholar]
- 41. Roebuck KA, Finnegan A. (1999) Regulation of intercellular adhesion molecule-1 (CD54) gene expression. Journal of Leukocyte Biology 66: 876–888. [DOI] [PubMed] [Google Scholar]
- 42. Dustin ML, Singer KH, Tuck DT, et al. (1988) Adhesion of T lymphoblasts to epidermal keratinocytes is regulated by interferon gamma and is mediated by intercellular adhesion molecule 1 (ICAM-1). Journal of Experimental Medicine 167: 1323–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fujii-Maeda S, Kajiwara K, Ikizawa K, et al. (2004) Reciprocal regulation of thymus and activation-regulated chemokine/macrophage-derived chemokine production by interleukin (IL)-4/IL-13 and interferon-gamma in HaCaT keratinocytes is mediated by alternations in E-cadherin distribution. Journal of Investigative Dermatology 122: 20–28. [DOI] [PubMed] [Google Scholar]
- 44. De Vries IJ, Langeveld-Wildschut EG, van Reijsen FC, et al. (1998) Adhesion molecule expression on skin endothelia in atopic dermatitis: Effects of TNF-alpha and IL-4. Journal of Allergy and Clinical Immunology 102: 461–468. [DOI] [PubMed] [Google Scholar]
- 45. Black AP, Ardern-Jones MR, Kasprowicz V, et al. (2007) Human keratinocyte induction of rapid effector function in antigen-specific memory CD4+ and CD8+ T cells. European Journal of Immunology 37: 1485–1493. [DOI] [PubMed] [Google Scholar]
- 46. Wu C, Feng D, Ma H, et al. (2009) Effect of Pinus massoniana bark extract on IFN-gamma-induced ICAM-1 expression in HaCaT human keratinocytes. Journal of Ethnopharmacology 122: 48–53. [DOI] [PubMed] [Google Scholar]
- 47. Bito T, Roy S, Sen CK, et al. (2002) Flavonoids differentially regulate IFN gamma-induced ICAM-1 expression in human keratinocytes: Molecular mechanisms of action. FEBS Letters 520: 145–152. [DOI] [PubMed] [Google Scholar]
- 48. Barker JN, Sarma V, Mitra RS, et al. (1990) Marked synergism between tumor necrosis factor-alpha and interferon-gamma in regulation of keratinocyte-derived adhesion molecules and chemotactic factors. The Journal of Clinical Investigation 85: 605–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Balato A, Di Caprio R, Canta L, et al. (2014) IL-33 is regulated by TNF-alpha in normal and psoriatic skin. Archives of Dermatological Research 306: 299–304. [DOI] [PubMed] [Google Scholar]
- 50. Taniguchi K, Yamamoto S, Hitomi E, et al. (2013) Interleukin 33 is induced by tumor necrosis factor alpha and interferon gamma in keratinocytes and contributes to allergic contact dermatitis. Journal of Investigational Allergology & Clinical Immunology 23: 428–434. [PubMed] [Google Scholar]
- 51. Demyanets S, Konya V, Kastl SP, et al. (2011) Interleukin-33 induces expression of adhesion molecules and inflammatory activation in human endothelial cells and in human atherosclerotic plaques. Arteriosclerosis, Thrombosis, and Vascular Biology 31: 2080–2089. [DOI] [PubMed] [Google Scholar]
- 52. Numata T, Ito T, Maeda T, et al. (2016) IL-33 promotes ICAM-1 expression via NF-kB in murine mast cells. Allergology International 65: 158–165. [DOI] [PubMed] [Google Scholar]
- 53. Blume C, Foerster S, Gilles S, et al. (2009) Human epithelial cells of the respiratory tract and the skin differentially internalize grass pollen allergens. Journal of Investigative Dermatology 129: 1935–1944. [DOI] [PubMed] [Google Scholar]
- 54. Bal V, McIndoe A, Denton G, et al. (1990) Antigen presentation by keratinocytes induces tolerance in human T cells. European Journal of Immunology 20: 1893–1897. [DOI] [PubMed] [Google Scholar]
- 55. Czernielewski JM, Bagot M. (1986) Class II MHC antigen expression by human keratinocytes results from lympho-epidermal interactions and gamma-interferon production. Clinical & Experimental Immunology 66: 295–302. [PMC free article] [PubMed] [Google Scholar]
- 56. Van der Fits L, van der Wel LI, Laman JD, et al. (2004) In psoriasis lesional skin the type I interferon signaling pathway is activated, whereas interferon-alpha sensitivity is unaltered. Journal of Investigative Dermatology 122: 51–60. [DOI] [PubMed] [Google Scholar]
- 57. Huttinger R, Staffler G, Majdic O, et al. (1999) Analysis of the early biogenesis of CD1b: Involvement of the chaperones calnexin and calreticulin, the proteasome and beta(2)-microglobulin. International Immunology 11: 1615–1623. [DOI] [PubMed] [Google Scholar]
- 58. Meunier L, Vian L, Lagoueyte C, et al. (1996) Quantification of CD1a, HLA-DR, and HLA class I expression on viable human Langerhans cells and keratinocytes. Cytometry 26: 260–264. [DOI] [PubMed] [Google Scholar]
- 59. Lundqvist C, Hammarstrom ML. (1993) T-cell receptor gamma delta-expressing intraepithelial lymphocytes are present in normal and chronically inflamed human gingiva. Immunology 79: 38–45. [PMC free article] [PubMed] [Google Scholar]
- 60. Scheynius A, Fransson J, Johansson C, et al. (1992) Expression of interferon-gamma receptors in normal and psoriatic skin. Journal of Investigative Dermatology 98: 255–258. [DOI] [PubMed] [Google Scholar]
- 61. Ratthe C, Ennaciri J, Garces Goncalves DM, et al. (2009) Interleukin (IL)-4 induces leukocyte infiltration in vivo by an indirect mechanism. Mediators of Inflammation 2009: 193970 ( 10 pp.). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Seto K, Shoda J, Horibe T, et al. (2013) Interleukin-4 receptor alpha-based hybrid peptide effectively induces antitumor activity in head and neck squamous cell carcinoma. Oncology Reports 29: 2147–2153. [DOI] [PubMed] [Google Scholar]
- 63. David M, Ford D, Bertoglio J, et al. (2001) Induction of the IL-13 receptor alpha2-chain by IL-4 and IL-13 in human keratinocytes: Involvement of STAT6, ERK and p38 MAPK pathways. Oncogene 20: 6660–6668. [DOI] [PubMed] [Google Scholar]
- 64. Wang S, Teng Q, Jia L, et al. (2014) Infectious bursal disease virus influences the transcription of chicken gammac and gammac family cytokines during infection. PLoS ONE 9: e84503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yasumura S, Lin WC, Weidmann E, et al. (1994) Expression of interleukin 2 receptors on human carcinoma cell lines and tumor growth inhibition by interleukin 2. International Journal of Cancer 59: 225–234. [DOI] [PubMed] [Google Scholar]
- 66. Wery-Zennaro S, Letourneur M, David M, et al. (1999) Binding of IL-4 to the IL-13Ralpha(1)/IL-4Ralpha receptor complex leads to STAT3 phosphorylation but not to its nuclear translocation. FEBS Letters 464: 91–96. [DOI] [PubMed] [Google Scholar]
- 67. Aragane Y, Kulms D, Metze D, et al. (1998) Ultraviolet light induces apoptosis via direct activation of CD95 (Fas/APO-1) independently of its ligand CD95L. Journal of Cell Biology 140: 171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Konur A, Schulz U, Eissner G, et al. (2005) Interferon (IFN)-gamma is a main mediator of keratinocyte (HaCaT) apoptosis and contributes to autocrine IFN-gamma and tumour necrosis factor-alpha production. British Journal of Dermatology 152: 1134–1142. [DOI] [PubMed] [Google Scholar]
- 69. Trautmann A, Akdis M, Kleemann D, et al. (2000) T cell-mediated Fas-induced keratinocyte apoptosis plays a key pathogenetic role in eczematous dermatitis. The Journal of Clinical Investigation 106: 25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Daehn IS, Varelias A, Rayner TE. (2010) T-lymphocyte-induced, Fas-mediated apoptosis is associated with early keratinocyte differentiation. Experimental Dermatology 19: 372–380. [DOI] [PubMed] [Google Scholar]
- 71. Federici M, Giustizieri ML, Scarponi C, et al. (2002) Impaired IFN-gamma-dependent inflammatory responses in human keratinocytes overexpressing the suppressor of cytokine signaling 1. Journal of Immunology 169: 434–442. [DOI] [PubMed] [Google Scholar]
- 72. Chaturvedi V, Bodner B, Qin JZ, et al. (2006) Knock down of p53 levels in human keratinocytes increases susceptibility to type I and type II interferon-induced apoptosis mediated by a TRAIL dependent pathway. Journal of Dermatological Science 41: 31–41. [DOI] [PubMed] [Google Scholar]
- 73. Hattori N, Komine M, Yano S, et al. (2002) Interferon-gamma, a strong suppressor of cell proliferation, induces upregulation of keratin K6, one of the inflammatory- and proliferation-associated keratins. Journal of Investigative Dermatology 119: 403–410. [DOI] [PubMed] [Google Scholar]
- 74. Chaturvedi V, Qin JZ, Denning MF, et al. (1999) Apoptosis in proliferating, senescent, and immortalized keratinocytes. Journal of Biological Chemistry 274: 23358–23367. [DOI] [PubMed] [Google Scholar]
- 75. Liang PF, Huang XY, Chen BF, et al. (2007) Inhibitory effect of epidermal growth factor on apoptosis in HaCaT keratinocytes induced by TNF-alpha. Zhonghua Shao Shang Za Zhi 23: 284–287. [PubMed] [Google Scholar]
- 76. Reinartz J, Bechtel MJ, Kramer MD. (1996) Tumor necrosis factor-alpha-induced apoptosis in a human keratinocyte cell line (HaCaT) is counteracted by transforming growth factor-alpha. Experimental Cell Research 228: 334–340. [DOI] [PubMed] [Google Scholar]
- 77. Sun J, Han J, Zhao Y, et al. (2012) Curcumin induces apoptosis in tumor necrosis factor-alpha-treated HaCaT cells. International Immunopharmacology 13: 170–174. [DOI] [PubMed] [Google Scholar]
- 78. Faurschou A, Gniadecki R, Calay D, et al. (2008) TNF-alpha impairs the S-G2/M cell cycle checkpoint and cyclobutane pyrimidine dimer repair in premalignant skin cells: Role of the PI3K-Akt pathway. Journal of Investigative Dermatology 128: 2069–2077. [DOI] [PubMed] [Google Scholar]