

Abstract
Recent research has demonstrated that colon cancer cell proliferation can be suppressed in the cells that overexpress COX-2 via generating 8-hydroxyoctanoic acid (a free radical byproduct) during dihomo-γ-linolenic acid (DGLA, an ω-6 fatty acid) peroxidation from knocking down cellular delta-5-desaturase (D5D, the key enzyme for converting DGLA to the downstream ω-6, arachidonic acid). Here, this novel research finding is extended to pancreatic cancer growth, as COX-2 is also commonly overexpressed in pancreatic cancer. The pancreatic cancer cell line, BxPC-3 (with high COX-2 expression and mutated p53), was used to assess not only the inhibitory effects of the enhanced formation of 8-hydroxyoctanoic acid from cellular COX-2-catalyzed DGLA peroxidation but also its potential synergistic and/or additive effect on current chemotherapy drugs. This work demonstrated that, by inducing DNA damage through inhibition of histone deacetylase, a threshold level of 8-hydroxyoctanoic acid achieved in DGLA-treated and D5D-knockdown BxPC-3 cells subsequently induce cancer cell apoptosis. Furthermore, it was shown that a combination of D5D knockdown along with DGLA treatment could also significantly sensitize BxPC-3 cells to various chemotherapy drugs, likely via a p53-independent pathway through downregulating of anti-apoptotic proteins (e.g., Bcl-2) and activating pro-apoptotic proteins (e.g., caspase 3, −9). This study reinforces the supposition that using commonly overexpressed COX-2 for molecular targeting, a strategy conceptually distinct from the prevailing COX-2 inhibition strategy used in cancer treatment, is an important as well as viable alternative to inhibit cancer cell growth. Based on the COX-2 metabolic cascade, the outcomes presented here could guide the development of a novel ω-6-based dietary care strategy in combination with chemotherapy for pancreatic cancer.
Keywords: Cyclooxygenase-(COX-) catalyzed free radical peroxidation, Delta-5-desaturase mediated ω-6 conversion, Pancreatic cancer cell lines, Targeted and chemotherapeutic drugs, 8-Hydroxyoctanoic acid, a free radical by-product formed from dihomo-γ-linolenic acid metabolism
1. Introduction
Pancreatic cancer is one of the most common causes of cancer death in the United States. A variety of pharmacological and dietary care regimens have been investigated as complementary strategies to improve the efficacy of standard pancreatic cancer chemotherapies. For example, dietary manipulations, such as supplementation with ω-3 fatty acids (ω-3s, normally found in marine products), has been proven to possess anti-cancer effects towards pancreatic cancer cells and tumors [1–8]. Docosahexaenoic acid and eicosapentaenoic acid (two ω-3s) showed some growth inhibitory effects on several pancreatic cancer cell lines, and were also able to sensitize pancreatic cancer cells to chemotherapeutic drugs, such as gemcitabine [9,10]. However, ω-6s, more abundant in our daily diet (e.g., in traditional western diets the ω-6 vs. ω-3 ratio is between ~10:1 and 30:1), have received much less interest from the cancer community, mainly due to the formation of deleterious metabolites (e.g., 2-series prostaglandins) from Cyclooxygenase (COX)-catalyzed arachidonic acid (AA, downstream ω-6 fatty acid) peroxidation [11–17].
COX is a bi-functional membrane bound enzyme that typically catalyzes ω-6s/ω-3s to prostanoids, such as prostaglandins. There are two isoforms of COX, e.g., COX-1, the constitutive form; and COX-2, the inducible form that can be induced by a variety of factors including pathological conditions, stresses and pro-inflammatory signals [18–21]. Overexpressed COX-2 is highly associated with inflammation and many types of cancers, including pancreatic cancer (e.g., ~70% of specimens from pancreatic cancer patients have overexpressed COX-2 [22]). The common elevation of prostaglandin E2 (PGE2) identified in various types of cancers suggests an association between COX-catalyzed AA peroxidation and carcinogenesis [11–17]. While suffering from critical safety issues (e.g., increased risks of cardiovascular disease and gastrointestinal injury [23,24]), many COX-2 inhibitors (e.g., celecoxib and apricoxib) are still tested as a complimentary strategy to enhance the efficacy of chemotherapy against cancers, including pancreatic cancer [25–31]. Alternative strategies for targeting the commonly high COX-2 level in cancer to improve the efficacy of chemotherapy have not yet been considered and developed.
COX-catalyzed lipid peroxidation is a well-known free radical chain reaction [32]. Recently, the Qian lab developed a HPLC/ESR/MS combination approach, along with a spin trapping technique, allowing a thorough characterization of commonly and exclusively generated free radicals from COX-catalyzed AA and dihomo-γ-linolenic acid (DGLA, an intermediate precursor of AA) peroxidation [33,34]. A unique structural moiety in DGLA led to the formation of a novel free radical byproduct, 8-hydroxyoctanoic acid (8-HOA) [35]. More recently, the Qian lab also demonstrated that 8-HOA is the bioactive metabolite responsible for DGLA’s anti-cancer effect [35,36]. Direct treatment with certain level of 8-HOA as well as accumulation of 8-HOA from DGLA-treated cells were also found to be essential to inhibit growth of colon cancer cells (e.g., HCA-7 colony 29, overexpresses COX-2) via arresting the cell cycle in G1 and promoting cell apoptosis in a p53-dependent manner [37]. In the current study, DGLA metabolism in BxPC-3 cells (high COX-2 expression pancreatic cancer cell line) was manipulated via knocking down delta-5-desaturase (D5D) in order to further confirm that 8-HOA accumulated from cellular COX peroxidation can suppress pancreatic cancer growth.
Despite the variety of chemotherapeutics, clinical pancreatic cancer chemotherapy has been hampered by the development of resistance [38–44]. For example, pancreatic cancer cells have shown resistance to gemcitabine, a front-line chemo-drug for pancreatic cancer therapy, due to decreased expression of hENT1 (human equilibrative nucleoside transporter 1, e.g., gemcitabine transporter) [39]. Many complementary strategies have been investigated in order to improve the efficacy and safety of chemotherapy, including COX inhibition [25–31]. In current study, the efficacies of many chemo-drugs for pancreatic cancer treatment can be enhanced by manipulating overexpressed COX-2 and downregulated D5D, along with DGLA treatment. The outcomes of this work could provide a basis for dietary care designs to include utilizing abundant ω-6s to enhance chemotherapeutic efficacy for pancreatic cancer cells overexpressing COX-2.
2. Material and methods
2.1. Cell line and reagent
The human pancreatic cancer cell line BxPC-3 (ATCC, Manassas, VA) was grown in RPMI-1640 medium (Thermo Fisher Scientific, MA, USA) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, MA, USA). Cells were cultured in an incubator containing a 95% humidified atmosphere with 5% CO2 at 37 °C.
DGLA was purchased from Nu-Chek-Prep (MN, USA). CelLytic™ lysis reagent, D5D primary antibody (from rabbit), 8-HOA, and β-actin primary antibody (from mouse) were obtained from Sigma-Aldrich (MO, USA). Erlotinib was acquired from Adooq Biosciences (CA, USA), and gemcitabine and oxaliplatin were purchased from Cayman Chemicals (MI, USA). CellTiter® 96 Aqueous One Solution Reagent was acquired from Promega (Madison, WI, USA). Annexin V Apoptosis Detection Kit I was purchased from BD Pharmingen™ (NJ, USA). Pierce ECL western blot substrates and GlutaMAX™ Opti-MEM reduced serum medium was obtained from Thermo Fisher Scientific (MA, USA). X-ray film was purchased from Phoenix Research Products. (NC, USA). COX-2 primary antibody (from rabbit) was acquired from Abcam (MA, USA). γ-H2AX primary antibody was purchased from Bethyl Laboratories (TX, USA). All other primary antibodies and secondary antibodies were obtained from Cell Signaling (MA, USA).
D5D siRNA (catalog #4390824), negative control siRNA (NC-si, catalog #4390843), and Lipofectamine™ RNAiMAX transfection reagent were purchased from Thermo Fisher Scientific (MA, USA). DNA oligos encoding D5D-targeted shRNA, TGCTGTAATCATCCAGGCCAA-GTCCAGTTTTGGCCACTGACTGACTGGACTTGCTGGATGATTA (top strand) and CCTGTAATCATCCAGCAAGTCCAGTCAGT-CAGTCAGTGGCCAAAACTGGACTTGGCCTGGATGATTAC (bottom strand), were designed (using BLOCK-IT™ RNAi Designer, www.invitrogen.com/rnai) and obtained from Integrated DNA Technologies (IA, USA). pcDNA™ 6.2-GW/EmGFP-miR vector was purchased from Invitrogen (NY, USA).
2.2. SiRNA transfection
BxPC-3 cells were seeded in 6-well plates for transfection (96-well plate for MTS assay). After overnight incubation, the cell culture medium was removed and cells were washed with phosphate buffered saline (PBS). D5D siRNA or NC-si stock solution (final concentration at 150 μM, in RNase free water) were mixed with transfection reagent that was previously diluted into 250 μl Opti-MEM reduced serum medium. Subsequently, cells were added with the mixture and incubated for 6 h, at which time serum-reduced medium was replaced by culture medium. Cells were further incubated for 48 h for complete siRNA transfection prior to treatments (e.g. ω-6s, chemo-drugs) and assessments (e.g. MTS assay, clonogenic assay, western blot, apoptosis assay and GC/MS analysis). Cells transfected with NC-si were used as a control.
2.3. ShRNA transfection
The DNA oilgos encoding D5D-targeted shRNA were cloned into pcDNA™ 6.2-GW/EmGFP-miR vector and transformed into Escherchia coli. The shRNA expressed vector was transfected into BxPC-3 cells using Lipofectamine™ RNAiMAX transfection reagent. Cells transfected with negative control shRNA (NC-sh) were used as a control.
2.4. MTS assay
CellTiter® 96 Aqueous One Solution Reagent was used to assess cell proliferation of BxPC-3 cells upon different treatments according to manufacturer’s instruction. Briefly, after 48 h treatments of ω-6s and/or chemo-drugs, 20 pl CellTiter® 96 Aqueous One Solution Reagent was added to each well and incubated ~2 h. After incubation, the absorbance at 490 nm, which is proportional to the quantity of formazan product, was recorded with 96-well plate reader (SpectraMax M5; Molecular Devices). Cell viability was calculated as a percentage compared to the control group (treated with vehicle).
2.5. Clonogenic assay
A clonogenic assay was used to assess colony formation of NC-si/NC-sh and delta-5-desaturase knockdown (D5D-KD) BxPC-3 cells after treatments (e.g. 8-HOA, ω-6s, and chemo-drugs). After BxPC-3 cells were transfected with D5D siRNA (shRNA as well) or NC-si (NC-sh as well), transfected cells were harvested and counted, and then plated (2000 cells per well in the 6-well plate). Forty-eight hours after treatment with 8-HOA, ω-6s and/or chemo-drugs, siRNA transfected cells were washed with PBS and then incubated for 10 days with cell culture medium. Cells transfected with shRNA were also used for clonogenic assay incubated with refreshed DGLA-supplemented culture medium every 3 days during 10-day incubation. At day 10, the cells were washed with PBS, fixed with 10% neutral buffered formalin solution for 30 min and stained with 0.05% crystal violet solution for 30 min. Colonies containing more than 30 individual cells were counted. Plate efficiency was calculated as number of colonies counted divided by number of cells plated. Note, 2000 untreated cells were plated in 6-well plates and the average plate efficiencies calculated with range from 0.075 to 0.082. Survival fraction was calculated as the plate efficiency of treatment group vs. the plate efficiency of control groups.
2.6. Cell apoptosis assay (Annexin V-FITC/PI staining)
Annexin V Apoptosis Detection Kit I was used to assess cell apoptosis of BxPC-3 cells upon treatments (e.g. 8-HOA, ω-6s, and chemo-drugs) following the manufacturer’s instruction. Briefly, after different treatments, the cells were trypsinized, washed with cold PBS and re-suspended in 1 × binding buffer at a concentration of 1 × 106 cells/mL. After transferring 100 μl of the sample solution to a new tube, 5.0 μl of FITC Annexin V and 5.0 μl PI solution were added. The cells were gently vortexed and incubated for 15 min at 25 °C in the dark. After adding 400 μl of 1 × binding buffer, 10,000 cells of each sample were injected into an Accuri C6 flow cytometer for analysis. Unstained cells, cells stained with FITC Annexin V only and PI only were used as controls for compensation and quadrants. Data was analyzed by FlowJo (TreeStar, Ashland, OR, USA).
2.7. Western blot
Cells were seeded, incubated overnight and subjected to treatments (e.g. 8-HOA, ω-6s and chemo-drugs). Approximately 48 h after treatments, the protein was extracted using CelLytic™ lysis reagent and quantified via a Bradford protein assay (Bio-Rad, USA). Extracted protein was loaded into each well of 10% or 15% SDS-PAGE gels (based on protein sizes) and then transferred to nitrocellulose membranes. Membranes were blocked with 5% (w/v) non-fat milk in Tris buffered saline with tween 20 (0.5%, TBST) and then incubated with primary antibodies (1:1000 dilution), e.g., D5D, COX-2, Bcl-2, procas-3 and -9, etc, overnight at 4 °C with continuous rocking. Membranes were washed three times with TBST and then incubated with a horseradish peroxidase (HRP)-conjugated secondary antibody (1:2000 dilution). After the blot was washed three times in TBST, membranes were incubated with ECL western blot substrate for one min, and exposed to X-ray film. Luminescent signals were captured using a Mini-Medical Automatic Film Processor (Imageworks).
2.8. GC/MS detection of 8-HOA
GC/MS analysis was used to quantify 8-HOA (in its derivative of pentafluorobenzyl bromide, PFB [45]) generated from D5D-KD and NC-si transfected BxPC-3 cells treated with DGLA as described elsewhere [37]. Briefly, after DGLA treatment (48 h), the cells were scraped into ~1.0 mL medium and added to a methanol containing internal standard (hexanoic acid) and 50 μl of 1.0 N HCl. The mixture was added to 3.0 mL dichloromethane and vortexed. Each sample was subsequently centrifuged to extract 8-HOA, and the dichloromethane layer was collected. The extraction process was repeated again with another 3.0 mL of dichloromethane. The dichloromethane layers were combined and evaporated to dryness by a vacuum evaporator and derivatized using diisopropylethylamine and PFB-bromide. After allowing it to react for 20-min at room temperature, the solvent was removed by vacuum evaporator and reconstituted with dichloromethane and subjected to GC/MS analysis.
GC/MS analysis was carried out by injecting each sample into an Agilent 6890A gas chromatograph. The temperature of the GC oven was programmed to increase from 60 to 300 °C at 25 °C/min. The injector and transfer line were kept at 280 °C. Quantitative analysis was performed by a mass selective detector with a source temperature of 230 °C and nebulizer pressure of 15 psi. The quantification of 8-HOA (in PFB derivative form) was calculated by comparing the base peak of 8-HOA-PFB (m/z 181) with the base peak of the internal standard (hexanoic acid-PFB derivative).
2.9. Statistic analysis
All data was assessed using an unpaired student-test with significance at p < 0.05.
3. RESULTS
3.1. 8-HOA inhibits cancer cell growth and enhances the cytotoxicity of gemcitabine
BxPC-3 cells were used to test whether direct treatment of 8-HOA (e.g., free radical byproduct formed from COX-2 catalyzed DGLA peroxidation) could inhibit the growth of pancreatic cancer cells overexpressing COX-2. Upon treatment with 8-HOA (1.0 μM), BxPC-3 colony formation was inhibited with the survival fraction ~73.0% (Fig. 1A). Subsequently, 8-HOA was delivered with gemcitabine, a front-line chemo-drug used for pancreatic cancer therapy, and the survival fraction was reduced to ~31.1% compared to cells treated with gemcitabine alone, which has a surviving fraction of ~50.6% (Fig. 1A). Furthermore, FITC-Annexin V and PI staining indicated that direct treatment 8-HOA induced apoptosis, increasing the early apoptotic cell population from ~2.35% (without 8-HOA) to ~6.89% for 8-HOA treatment. Treatment with 8-HOA also promoted gemcitabine-induced cell apoptosis (from 11.8% to 16.1%, Fig. 1B). Expression of acetyl histone H3 and the DNA damage marker γH2AX were both increased in BxPC-3 cells treated by 8-HOA, suggesting that 8-HOA might inhibit histone deacetylase thereby leading to DNA damage [46] (Fig. 1C).
Fig. 1.
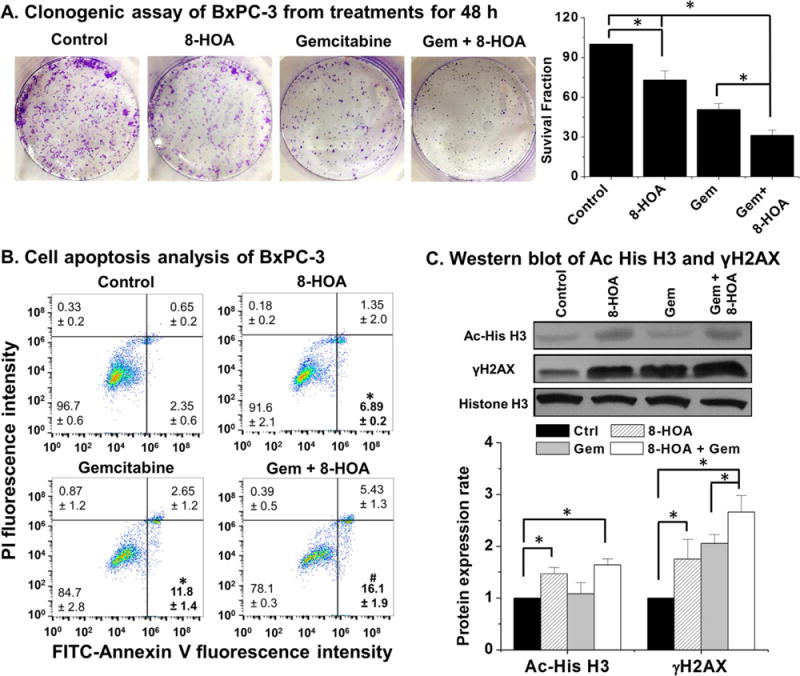
8-HOA’s growth inhibitory effects on BxPC-3 cells. (A) Clonogenic assay of BxPC-3 cells at 10 days after 48 h of 8-HOA treatment (1.0 μM), gemcitabine (0.1 μM), and 8-HOA+gemcitabine. The BxPC-3 cells treated with vehicle only were used as control; (B) Cell apoptosis analysis of BxPC-3 cells. After 48 h treatment of vehicle (control), 8-HOA (1.0 μM), gemcitabine (0.1 μM) and gemcitabine +8-HOA, cells were double stained with FITC-Annexin V/PI and subjected to flow cytometry. (*: significant difference vs. control with p < 0.05; and #: significant difference vs. gemcitabine group with p < 0.05 from more than three experiments); and (C) Western blot and protein expression level of acetyl histone H3 and γH2AX in BxPC-3 cells treated with vehicle, 8-HOA (1.0 μM), gemcitabine (0.1 μM), and 8-HOA plus gemcitabine. Protein expression rate was normalized using histone H3 as loading control (*: significant difference p < 0.05 from more than three experiments).
3.2. Promoted 8-HOA formation from COX-DGLA peroxidation suppresses cancer cell growth
To confirm the promoted formation of 8-HOA from COX-catalyzed DGLA free radical peroxidation could also suppress pancreatic cancer cell growth, BxPC-3 cells were transfected with siRNA (shRNA as well) to knock down D5D and manipulate DGLA metabolism. About 75% D5D expression suppressed in siRNA transfected BxPC-3 cells was observed compared to the cells transfected with the negative control siRNA (NC-si, Fig. 2A, similar inhibition effect also achieved in shRNA, data not shown). Subsequently, DGLA treatment (48 h) significantly inhibited colony formation in D5D knockdown (D5D-KD) BxPC-3 cells (~survival fraction of 70.6%, Fig. 2B), but had no effect on cell growth of the cells transfected with NC-si. Moreover, more cancer cell killing was observed in shRNA transfected BxPC-3 cells when DGLA-supplemented culture medium was used to incubate cells (refreshed 3 times in 10 days, ~ survival fraction of 53.9%, Fig. 2B).
Fig. 2.
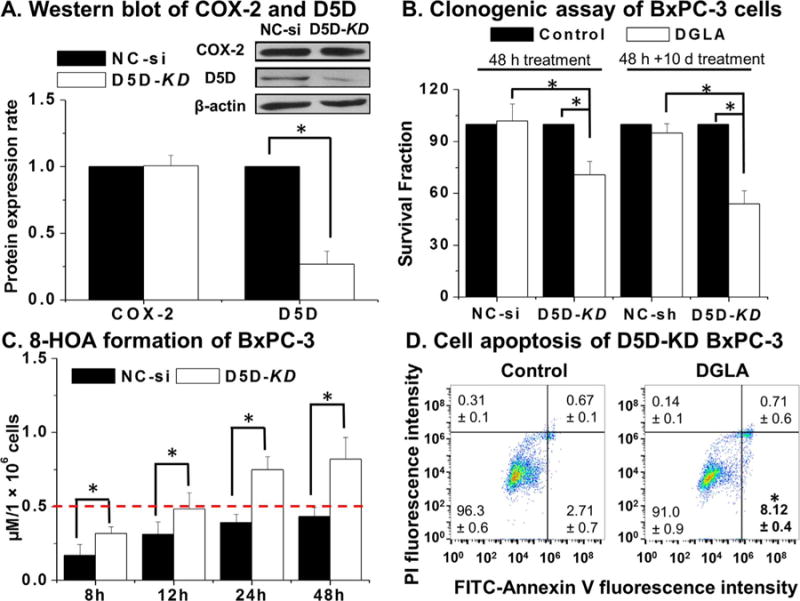
D5D-KD improved DGLA’s growth inhibitory effect in BxPC-3 cells. (A) Western blot and protein expression level of COX-2 and D5D expression in NC-si transfected vs. D5D-KD cells. Protein expression rate was normalized using β-actin as a loading control; (B) Clonogenic assays of NC-si and D5D-KD BxPC-3 cells at day 10 after DGLA (100 μM, 48 h) treatment vs. control (without DGLA, four columns on the left); clonogenic assay of NC-sh and shRNA transfected D5D-KD BxPC-3 cells at day 10 along with culture medium supplemented with/without DGLA (refreshed every three days during 10-day period, four columns on the right); (C) GC/MS quantification of 8-HOA from NC-si transfected or D5D-KD BxPC-3 cells treated with 100 μM DGLA. Data represent as mean ± SD from 3 experiments; (D) Cell apoptosis analysis of D5D-KD cells treated with vehicle (control) or DGLA (100 μM). (*: significant difference with p < 0.05 from more than three experiments).
In order to confirm the similar threshold level of endogenous 8-HOA proposed for colon cancer cells would be also essential for inhibiting pancreatic cancer cell growth, GC/MS was performed to measure the PFB-derivative form generated from both D5D-KD and NC-si BxPC-3 cells treated with DGLA for 48 h [37]. Endogenous 8-HOA continually accumulated in D5D-KD BxPC-3 cells and maintained the threshold level 0.5 μM most of time during 48 h (Fig. 2C). However, endogenous 8-HOA in NC-si transfected cells never reached 0.5 μM during the 48 h treatment with DGLA, revealing that concentrations less than 0.5 μM were insufficient to exert an effect on cancer cell growth. With FITC-Annexin V-PI double staining study, we observed that DGLA treatment significantly increased the early apoptotic cell population in D5D-KD BxPC-3 cells (8.12%, Fig. 2D) vs. the control (e.g., D5D-KD cells without DGLA treatment, ~2.71%).
3.3. D5D-KD and DGLA treatment enhances the efficacy of gemcitabine
Gemcitabine, a nucleoside analog, has been used as a front-line chemo-drug for pancreatic cancer treatment. However, a variety of pancreatic cancer cell lines (including BxPC-3) have been reported resistant to gemcitabine [39–44]. When cells were co-treated with DGLA and gemcitabine, DGLA significantly enhanced the efficacy of gemcitabine on D5D-KD BxPC-3 cells as indicated by the MTS assay for cell proliferation (41.3% cell viability for co-treatment vs. 54.6% for gemcitabine only, Fig. 3A). Colony formation was also significantly inhibited in D5D-KD BxPC-3 cells upon co-treatment with gemcitabine and DGLA (survival fraction ~30.8% vs. 51.3% for gemcitabine only, Fig. 3B). For NC-si BxPC-3 cells, DGLA treatment has no effect on drug efficacy as shown in cell proliferation and colony formation assays (Fig. 3A–B). Gemcitabine-induced cell apoptosis was further promoted by DGLA treatment on D5D-KD BxPC-3 cells (population of early apoptotic cells, ~18.0% vs. gemcitabine treatment alone ~12.6%, Fig. 3C).
Fig. 3.
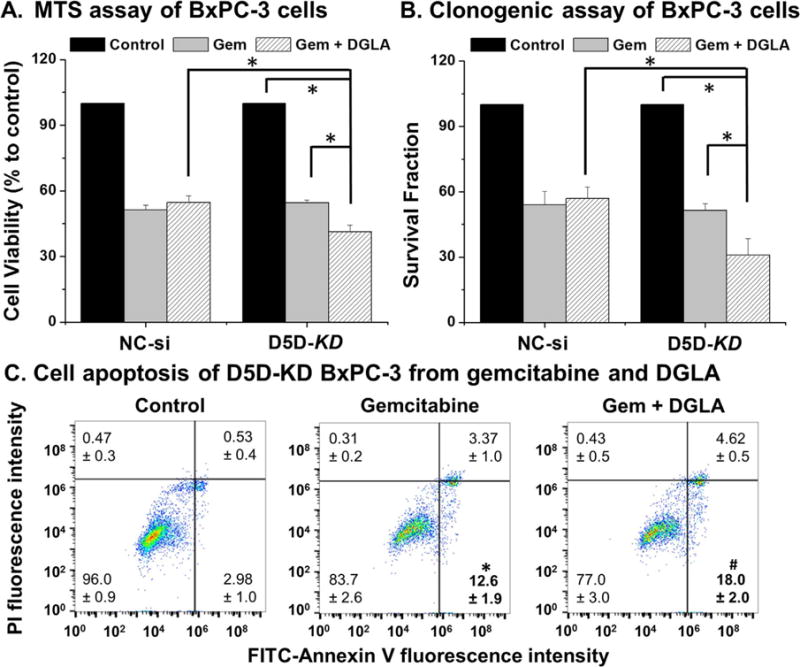
D5D-KD and DGLA treatment enhanced efficacy of gemcitabine to kill BxPC-3 cells. (A) MTS assay of NC-si transfected and D5D-KD BxPC-3 cells upon treatment with gemcitabine (1.0 μM) alone or gemcitabine (1.0 μM)+DGLA (100 μM). The NC-si transfected and D5D-KD cells without fatty acid and drug treatment were used as controls; (B) Clonogenic assay of NC-si and D5D-KD BxPC-3 cells at 10 days with treatment of DGLA (100 μM), gemcitabine (0.1 μM) or gemcitabine (0.1 μM)+DGLA (100 μM) for 48 h. The NC-si transfected and D5D-KD cells without fatty acid and drug treatment were used as controls; (C) Cell apoptosis of D5D-KD BxPC-3 cells treated with vehicle (control), gemcitabine (0.1 μM) alone or gemcitabine (0.1 μM)+DGLA (100 μM). (*: significant difference vs. control with p < 0.05; and #: significant difference vs. gemcitabine group with p < 0.05 from more than three experiments).
Increased expression of acetyl histone H3 and γH2AX was observed in D5D-KD BxPC-3 cells treated with DGLA, consistent with Fig. 1C and confirmed again that 8-HOA can act as a DNA damaging agent. Gemcitabine is known to suppress cancer cell growth by inducing p53-dependent as well as p53-independent cell apoptosis [47,48]. In the current study, gemcitabine down-regulated Bcl-2 and Bcl-XL (anti-apoptotic proteins) as well as activated procaspase 9 and procaspase 3 (pro-apoptotic proteins, Fig. 4). Considering the mutated p53 background in D5D-KD BxPC-3 cells, apoptosis seems proceed according to a p53-independent pathway. Further increased expression of γH2AX and down-regulated expressions of Bcl-2, procaspase 9 and procaspase 3 (Fig. 4) were observed when D5D-KD BxPC-3 cells were co-treated with DGLA and gemcitabine. In addition, we would like to point it out that there is no statistically different effect between DGLA+/− gem on Ac-his-H3 as gemcitabine itself did not inhibit histone deacetylase.
Fig. 4.
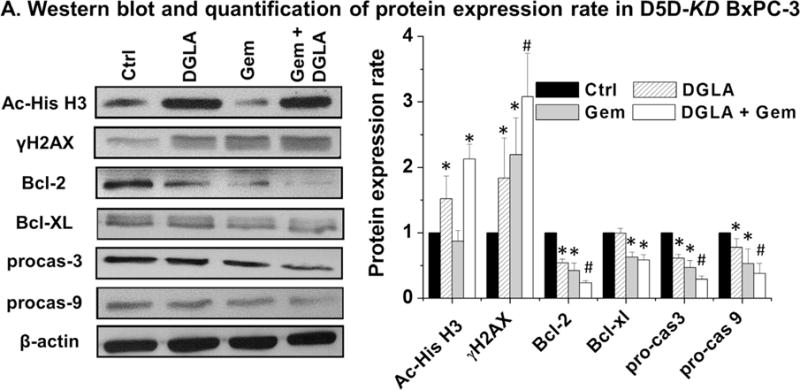
D5D-KD and DGLA treatment promoted gemcitabine-induced apoptosis via p53 independent pathway. (A) Western blot and protein expression level of acetyl histone H3, γH2AX, Bcl-2, Bcl-XL, procaspase-3 and procaspase-9 from D5D-KD BxPC-3 cells treated with vehicle (control), DGLA (100 μM), gemcitabine (0.1 μM) or gemcitabine (0.1 μM)+DGLA (100 μM) for 48h. Protein expression rate was normalized using β-actin as loading control. (*: significant difference vs. control with p <0.05; and #: significant difference vs. gemcitabine group with p < 0.05 from more than three experiments).
3.4. D5D-KD and DGLA treatment improves the cytotoxicities of other chemotherapy drugs
It was apparent that D5D-KD and DGLA treatment could sensitize BxPC-3 cells to gemcitabine, we also assessed the potential synergistic and/or additive effect on other chemotherapeutic drugs, e.g., oxaliplatin (classic chemo-drug used for pancreatic cancer therapy [49]) and erlotinib (targeted therapy drug used for pancreatic cancer [50–53]). DGLA treatment improved the cytotoxicity of erlotinib and oxaliplatin on D5D-KD BxPC-3 cells as shown via MTS assays (~39.6% and 34.3% cell viability for treatment DGLA plus chemo-drugs vs. 52.1% and 46.9% for only erlotinib and oxaliplatin respectively Fig. 5A). Clonogenic assays also demonstrated that co-treatment of DGLA with erlotinib and oxaliplatin (using relative lower drug concentration compared to MTS assays) also inhibited colony formation (~34.5% and 36.7% surviving fraction for treatment DGLA plus chemo-drugs vs. ~52.2% and 62.9% for erlotinib and oxaliplatin alone, respectively Fig. 5B). In NC-si transfected BxPC-3 cells, treatment of DGLA has no influence on the efficacy of either erlotinib or oxaliplatin.
Fig. 5.
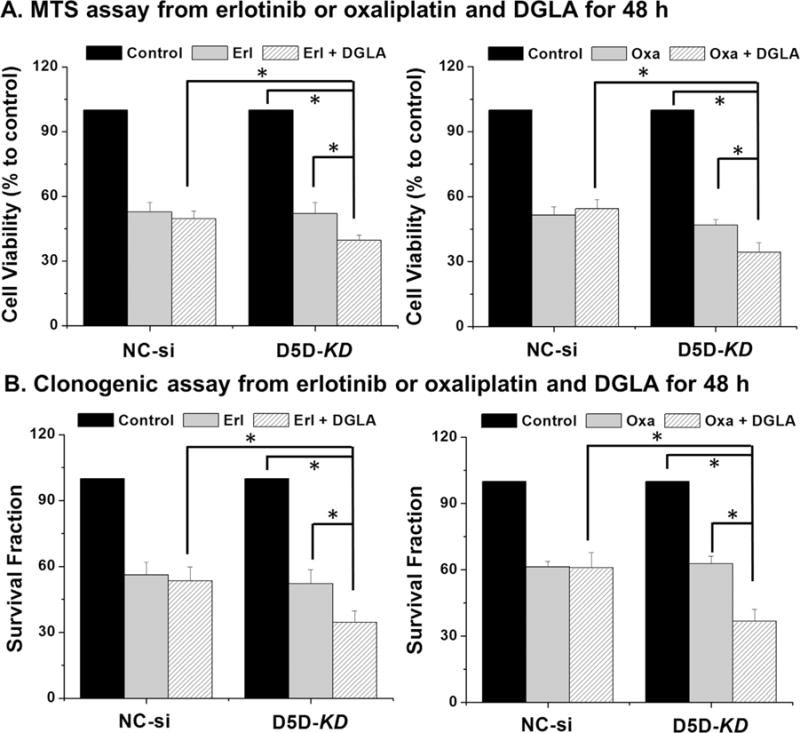
Improved cytotoxicity of erlotinib and oxaliplatin by DGLA in D5D-KD BxPC-3 cells. (A) MTS assay of NC-si transfected and D5D-KD BxPC-3 cells upon treatment of erlotinib (10 μM) alone or erlotinib (10 μM)+DGLA (100 μM) as well as oxaliplatin (20 μM) alone or oxaliplatin (20 μM)+DGLA (100 μM) for 48 h. The NC-si transfected and D5D-KD cells without fatty acid and drug treatment were used as controls; (B) Clonogenic assay of NC-si transfected and D5D-KD BxPC-3 cells at 10 days with treatment of erlotinib (2.0 μM) or erlotinib (2.0 μM)+DGLA (100 μM) as well as oxaliplatin (2.0 μM) or oxaliplatin (2.0 μM)+DGLA (100 μM) for 48 h. The NC-si transfected and D5D-KD cells without fatty acid and drug treatment were used as controls. Note, relative lower amount of chemo-drugs (vs. MTS assay) were used for clonogenic assay in order to allow enough colony to be formed. (*: significant difference with p < 0.05 from more than three experiments).
4. Discussion
Previous studies in the colon cancer cell line HCA-7 showed that direct treatment of 8-HOA as well as accumulated endogenous formation of 8-HOA in D5D knockdown and COX-catalyzed DGLA peroxidation can not only inhibit cell growth, but also enhanced the cytotoxicity of the chemotherapy drug 5-FU, likely via a p53-dependent pathway [36,37]. The current study aimed to extend the supposition that D5D knockdown in conjunction with DGLA treatment can also be used to inhibit growth of pancreatic cancer cells via p53 independent pathway, using pancreatic cancer cell line BxPC-3 which has both high COX-2 expression and mutated p53.
Considering that a variety of aliphatic acid compounds have been reported to inhibit histone deacetylase [54,55], we found that 8-HOA could also act as a histone deacetylase inhibitor (Fig. 1C). Increased expression of acetyl histone H3 and γH2AX were also observed when D5D-KD BxPC-3 cells were treated with DGLA (Fig. 4), analogous to promoting the endogenous formation and accumulation of 8-HOA (Fig. 2C), suggesting that the observed growth inhibitory effect of DGLA is derived from generation of certain amount of 8-HOA and 8-HOA-induced DNA damage.
A threshold level of endogenous 8-HOA (> 0.5 μM) was determined essential to inhibit the growth of pancreatic cancer as previously reported for colon cancer [37]. In addition to measuring levels of 8-HOA (via GC/MS), LC/MS was used to detect levels of DGLA, AA and their metabolites PGE1 and PGE2 in both D5D-KD and NC-si BxPC-3 cells treated with DGLA for 48 h. Higher levels of free DGLA and formation of PGE1, and decreased level of free AA and formation of PGE2, were observed in D5D-KD BxPC-3 cells when compared to NC-si transfected cells (Supplement Table 1) as the conversion from DGLA to AA was suppressed in D5D-KD cells. Note, the basal levels of undetected 8-HOA and negligible DGLA in D5D-KD and NC-si BxPC-3 cells (e.g., without DGLA treatment) were also observed as we previously noted for colon cancer cells. Unlike single dose DGLA treatment, however, the continued supplement of DGLA (aiming to keep a steady state concentration of 8-HOA) to mimic availability in dietary care could further inhibit colony formation of D5D-KD BxPC-3 cells (Fig. 2B).
Gemcitabine, acting as a nucleoside analog to interfere DNA replication [44], has been used as front line chemo-drug to treat patients with advanced pancreatic cancer. However, pancreatic cancer cells can develop resistance to gemcitabine for a variety of factors, such as a decrease of hENT1 expression [39]. Various regimens such as COX-2 inhibitors (e.g., celecoxib) as well as ω-3 fatty acids have been used to improve the efficacy of gemcitabine on pancreatic cancer cells [10,29]. The current study has demonstrated that DGLA treatment along with D5D-KD could also enhance the efficacy of gemcitabine to control pancreatic cancer cell growth by further promoting gemcitabine-induced cell apoptosis (Fig. 3). Additionally, this study also indicates that DGLA treatment in the presence of D5D-KD can improve the cytotoxicity of oxaliplatin (classic chemotherapy drug [49]) or erlotinib (FDA-approved targeted therapeutic drug [50–53], Fig. 5). Although only additive effects of DGLA (Fig. 5) as well as 8-HOA (Fig. 1) on chemo-drug efficacy was observed, we would like to point out that the synergic effect could occur on the situation of a diet (supplemented with DGLA) continually fed to the animals, consistent with our unpublished in vivo study.
Previously, D5D knockdown and DGLA treatment were reported to be able to inhibit growth of colon cancer cell line HT-29 (p53 mutated), likely through a p53-independent pathway [37]. Here, DGLA treatment could further promote gemcitabine-induced cell apoptosis in D5D-KD BxPC-3 (mutated p53). This effect is hypothesized to be mediated by the formation and accumulation of DGLA’s free radical byproduct (e.g., 8-HOA) that could (1) damage DNA, (2) decrease expression of anti-apoptotic proteins (e.g., Bcl-2), and (3) activate pro-apoptotic proteins (e.g., caspase-9 and caspase-3), via a p53-independent mechanism (Fig. 4). Given that the p53 mutation can be found in all types of cancers [56], this strategy might be useful for improving the efficacy of chemotherapy in pancreatic cancer commonly carrying p53 mutation.
To determine whether the level of COX-2 expression could affect the growth inhibition of cancer cells, we also knocked down D5D via siRNA transfection in PANC-1 (another pancreatic cancer cell line with deficient COX-2 expression), as well as doubly knocked down D5D and COX-2 in BxPC-3 cells. The data showed that DGLA has no inhibitory effects on D5D-KD PANC-1 or COX/D5D double KD BxPC-3 cells (Supplement Fig. 1), indicating that overexpressed COX-2 can be exploited to increase formation of 8-HOA from DGLA peroxidation to control pancreatic cancer cell growth. Note, when D5D-KD PANC-1 and COX-2/D5D double KD BxPC-3 were treated with 8-HOA, the growth of both cell lines were also inhibited (data not shown), demonstrating that 8-HOA could suppress growth of pancreatic cancer cells irrespective of their COX-2 and D5D expression level. These results suggested that in a tumor environment, the growth of cancer cells which have low or deficient COX-2 level (e.g., not preferred killing by our strategy) might also be inhibited by the 8-HOA that is continually generated from nearby cancer cells (~70%, overexpressing COX-2) in a paracrine manner. Our strategy should preferentially inhibit the growth of pancreatic cancer cells and tumors, while having less influence on normal cells due to their lower intake of fatty acids as well as much lower COX expression.
In the present study, high COX-2 expression could be used to promote formation of 8-HOA from DGLA peroxidation to inhibit growth of pancreatic cancer cell lines as well as improve the efficacy of multiple chemo-drugs. By targeting high COX-2 expression in conjunction with the development of an ω-6-based diet (more pervasive than ω-3 in daily diet), the proliferation of pancreatic cancer cells could be inhibited. Since no effective and selective D5D inhibitors have so far been used for cancer treatments, as well as D5D knockdown may have limited clinical value, we are now working on two workable approaches of D5D down-regulation: (1) to screen and select small compounds or molecules used as effective D5D inhibitor(s) for drug development; and (2) to apply RNA nanotechnology for cancer therapy, e.g, to construct a thermodynamically and chemically stable RNA nanoparticles harboring D5D siRNA and allow specific delivery of D5D siRNA to cancer cells/tumors [57]. The outcomes of this work will support our translational efforts to modify ω-6 dietary supplements and COX-2-mediated free radical peroxidation, and therefore enhancing the efficacy of chemotherapy in pancreatic cancer treatment.
Supplementary Material
Acknowledgments
This work was supported by NIH Grant 1R15CA140833 (S Qian) and R01CA186100 (B Guo).
Abbreviations
- AA
arachidonic acid
- COX
Cyclooxygenase
- DGLA
dihomo-γ-linolenic acid
- D5D
delta-5-desaturase
- D5D-KD
delta-5-desaturase knockdown
- GC
gas chromatography
- NC-si
negative control siRNA transfected
- NC-sh
negative control shRNA transfected
- MS
mass spectrometry
- PBS
phosphate buffered saline
- PFB
pentafluorobenzyl
- PGE1
2, prostaglandin E1, E2
- PUFAs
polyunsaturated fatty acids
- 8-HOA
8-hydroxyoctanoic acid
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.freeradbiomed.2016.06.028.
Footnotes
Conflict of interest
The authors claim no conflict of interest.
References
- 1.Mohammed A, Janakiram NB, Brewer M, Duff A, Lightfoot S, Brush RS, Anderson RE, Rao CV. Endogenous n-3 polyunsaturated fatty acids delay progression of pancreatic ductal adenocarcinoma in Fat-1-p48 (Cre/+)-LSL-Kras (G12D/+) mice. Neoplasia. 2012;14:1249–1259. doi: 10.1593/neo.121508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fukui M, Kang KS, Okada K, Zhu BT. EPA, an omega-3 fatty acid, induces apoptosis in human pancreatic cancer cells: role of ROS accumulation, caspase-8 activation, and autophagy induction. J Cell Biochem. 2013;114:192–203. doi: 10.1002/jcb.24354. [DOI] [PubMed] [Google Scholar]
- 3.Song KS, Jing K, Kim JS, Yun EJ, Shin S, Seo KS, Park JH, Heo JY, Kang JX, Suh KS, Wu T, Park JI, Kweon GR, Yoon WH, Hwang BD, Lim K. Omega-3-polyunsaturated fatty acids suppress pancreatic cancer cell growth in vitro and in vivo via downregulation of Wnt/Beta-catenin signaling. Pancreatology. 2011;11:574–584. doi: 10.1159/000334468. [DOI] [PubMed] [Google Scholar]
- 4.Shirota T, Haji S, Yamasaki M, Iwasaki T, Hidaka T, Takeyama Y, Shiozaki H, Ohyanagi H. Apoptosis in human pancreatic cancer cells induced by eicosapentaenoic acid. Nutrition. 2005;21:1010–1017. doi: 10.1016/j.nut.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 5.Merendino N, Loppi B, D’Aquino M, Molinari R, Pessina G, Romano C, Velotti F. Docosahexaenoic acid induces apoptosis in the human PaCa-44 pancreatic cancer cell line by active reduced glutathione extrusion and lipid peroxidation. Nutr Cancer. 2005;52:225–233. doi: 10.1207/s15327914nc5202_12. [DOI] [PubMed] [Google Scholar]
- 6.D’Eliseo D, Manzi L, Merendino N, Velotti F. Docosahexaenoic acid inhibits invasion of human RT112 urinary bladder and PT45 pancreatic carcinoma cells via down-modulation of granzyme B expression. J Nutr Biochem. 2012;23:452–457. doi: 10.1016/j.jnutbio.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 7.Merendino N, Molinari R, Loppi B, Pessina G, D’Aquino M, Tomassi G, Velotti F. Induction of apoptosis in human pancreatic cancer cells by docosahexaenoic acid. Ann N Y Acad Sci. 2003;1010:361–364. doi: 10.1196/annals.1299.143. [DOI] [PubMed] [Google Scholar]
- 8.Park KS, Lim JW, Kim H. Inhibitory mechanism of omega-3 fatty acids in pancreatic inflammation and apoptosis. Ann N Y Acad Sci. 2009;1171:421–427. doi: 10.1111/j.1749-6632.2009.04887.x. [DOI] [PubMed] [Google Scholar]
- 9.Haqq J, Howells LM, Garcea G, Dennison AR. Targeting pancreatic cancer using a combination of gemcitabine with the omega-3 polyunsaturated fatty acid emulsion, Lipidem™. Mol Nutr Food Res. 2015 doi: 10.1002/mnfr.201500755. http://dx.doi.org/10.1002/mnfr.201500755. [DOI] [PubMed]
- 10.Hering J, Garrean S, Dekoj TR, Razzak A, Saied A, Trevino J, Babcock TA, Espat NJ. Inhibition of proliferation by omega-3 fatty acids in chemoresistant pancreatic cancer cells. Ann Surg Oncol. 2007;14:3620–3628. doi: 10.1245/s10434-007-9556-8. [DOI] [PubMed] [Google Scholar]
- 11.Funahashi H, Satake M, Hasan S, Sawai H, Newman RA, Reber HA, Hines OJ, Eibl G. Opposing effects of n-6 and n-3 polyunsaturated fatty acids on pancreatic cancer growth. Pancreas. 2008;36:353–362. doi: 10.1097/MPA.0b013e31815ccc44. [DOI] [PubMed] [Google Scholar]
- 12.Ito H, Duxbury M, Benoit E, Clancy TE, Zinner MJ, Ashley SW, Whang EE. Prostaglandin E2 enhances pancreatic cancer invasiveness through an Ets-1-dependent induction of matrix metalloproteinase-2. Cancer Res. 2004;64:7439–7446. doi: 10.1158/0008-5472.CAN-04-1177. [DOI] [PubMed] [Google Scholar]
- 13.Dufour M, Faes S, Dormond-Meuwly A, Demartines N, Dormond O. PGE2-induced colon cancer growth is mediated by mTORC1. Biochem Biophys Res Commun. 2014;451:587–591. doi: 10.1016/j.bbrc.2014.08.032. [DOI] [PubMed] [Google Scholar]
- 14.Reader J, Holt D, Fulton A. Prostaglandin E2 EP receptors as therapeutic targets in breast cancer. Cancer Metastas Rev. 2011;30:449–463. doi: 10.1007/s10555-011-9303-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eibl G, Bruemmer D, Okada Y, Duffy JP, Law RE, Reber HA, Hines OJ. PGE (2) is generated by specific COX-2 activity and increases VEGF production in COX-2-expressing human pancreatic cancer cells. Biochem Biophys Res Commun. 2003;306:887–897. doi: 10.1016/s0006-291x(03)01079-9. [DOI] [PubMed] [Google Scholar]
- 16.Bu X, Zhao C, Dai X. Involvement of COX-2/PGE (2) Pathway in the Upregulation of MMP-9 Expression in Pancreatic Cancer. Gastroenterol Res Pract. 2011;2011:214269. doi: 10.1155/2011/214269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takahashi H, Li A, Dawson DW, Hines OJ, Reber HA, Eibl G. Cyclooxygenase-2 confers growth advantage to syngeneic pancreatic cancer cells. Pancreas. 2011;40:453–459. doi: 10.1097/MPA.0b013e31820b9733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim JB, Han AR, Park EY, Kim JY, Cho W, Lee J, Seo EK, Lee KT. Inhibition of LPS-induced iNOS, COX-2 and cytokines expression by poncirin through the NF-kappaB inactivation in RAW 264.7 macrophage cells. Biol Pharm Bull. 2007;30:2345–2351. doi: 10.1248/bpb.30.2345. [DOI] [PubMed] [Google Scholar]
- 19.Eliopoulos AG, Dumitru CD, Wang CC, Cho J, Tsichlis PN. Induction of COX-2 by LPS in macrophages is regulated by Tpl2-dependent CREB activation signals. EMBO J. 2002;21:4831–4840. doi: 10.1093/emboj/cdf478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitchell JA, Belvisi MG, Akarasereenont P, Robbins RA, Kwon OJ, Croxtall J, Barnes PJ, Vane JR. Induction of cyclooxygenase-2 by cytokines in human pulmonary epithelial cells: regulation by dexamethasone. Br J Pharmacol. 1994;113:1008–1014. doi: 10.1111/j.1476-5381.1994.tb17093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akarasereenont P, Bakhle YS, Thiemermann C, Vane JR. Cytokine-mediated induction of cyclooxygenase-2 by activation of tyrosine kinase in bovine endothelial cells stimulated by bacterial lipopolysaccharide. Br J Pharmacol. 1995;115:401–408. doi: 10.1111/j.1476-5381.1995.tb16347.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yip-Schneider MT, Barnard DS, Billings SD, Cheng L, Heilman DK, Lin A, Marshall SJ, Crowell PL, Marshall MS, Sweeney CJ. Cyclooxygenase-2 expression in human pancreatic adenocarcinomas. Carcinogenesis. 2000;21:139–146. doi: 10.1093/carcin/21.2.139. [DOI] [PubMed] [Google Scholar]
- 23.Hawkey CJ, Langman MJS. Non-steroidal anti-inflammatory drugs: overall risks and management. Complementary roles for COX-2 inhibitors and proton pump inhibitors. Gut. 2003;52:600–608. doi: 10.1136/gut.52.4.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greenberg JD, Fisher MC, Kremer J, Chang H, Rosenstein ED, Kishimoto M, Lee S, Yazici Y, Kavanaugh A, Abramson SB. CORRONA Investigators. The COX-2 inhibitor market withdrawals and prescribing patterns by rheumatologists in patients with gastrointestinal and cardiovascular risk. Clin Exp Rheumatol. 2009;27:395–401. [PubMed] [Google Scholar]
- 25.Kirane A, Toombs JE, Ostapoff K, Carbon JG, Zaknoen S, Braunfeld J, Schwarz RE, Burrows FJ, Brekken RA. Apricoxib, a novel inhibitor of COX-2, markedly improves standard therapy response in molecularly defined models of pancreatic cancer. Clin Cancer Res. 2012;18:5031–5042. doi: 10.1158/1078-0432.CCR-12-0453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yip-Schneider MT, Wu H, Hruban RH, Lowy AM, Crooks PA, Schmidt CM. Efficacy of dimethylaminoparthenolide and sulindac in combination with gemcitabine in a genetically engineered mouse model of pancreatic cancer. Pancreas. 2013;42:160–167. doi: 10.1097/MPA.0b013e318254f455. [DOI] [PubMed] [Google Scholar]
- 27.Rosendahl AH, Gundewar C, Said K, Karnevi E, Andersson R. Celecoxib synergizes human pancreatic ductal adenocarcinoma cells to sorafenib-induced growth inhibition. Pancreatology. 2012;12:219–226. doi: 10.1016/j.pan.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 28.Al-Wadei HA, Al-Wadei MH, Ullah MF, Schuller HM. Celecoxib and GABA cooperatively prevent the progression of pancreatic cancer in vitro and in xenograft models of stress-free and stress-exposed mice. PLoS One. 2012;7:e43376. doi: 10.1371/journal.pone.0043376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lipton A, Campbell-Baird C, Witters L, Harvey H, Ali S. Phase II trial of gemcitabine, irinotecan, and celecoxib in patients with advanced pancreatic cancer. J Clin Gastroenterol. 2010;44:286–288. doi: 10.1097/MCG.0b013e3181cda097. [DOI] [PubMed] [Google Scholar]
- 30.Ding N, Cui XX, Gao Z, Huang H, Wei X, Du Z, Lin Y, Shih WJ, Rabson AB, Conney AH, Hu C, Zheng X. A triple combination of atorvastatin, celecoxib and tipifarnib strongly inhibits pancreatic cancer cells and xenograft pancreatic tumors. Int J Oncol. 2014;44:2139–2145. doi: 10.3892/ijo.2014.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Molina MA, Sitja-Arnau M, Lemoine MG, Frazier ML, Sinicrope FA. Increased cyclooxygenase-2 expression in human pancreatic carcinomas and cell lines: growth inhibition by nonsteroidal anti-inflammatory drugs. Cancer Res. 1999;59:4356–4362. [PubMed] [Google Scholar]
- 32.Gardner HW. Oxygen radical chemistry of polyunsaturated fatty acids. Free Radic Biol Med. 1989;7:65–86. doi: 10.1016/0891-5849(89)90102-0. [DOI] [PubMed] [Google Scholar]
- 33.Yu Q, Purwaha P, Ni K, Sun C, Mallik S, Qian SY. Characterization of novel radicals from COX-catalyzed arachidonic acid peroxidation. Free Radic Biol Med. 2009;47:568–576. doi: 10.1016/j.freeradbiomed.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiao Y, Gu Y, Purwaha P, Ni K, Law B, Mallik S, Qian SY. Characterization of free radicals formed from COX-catalyzed DGLA peroxidation. Free Radic Biol Med. 2011;50:1163–1170. doi: 10.1016/j.freeradbiomed.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gu Y, Xu Y, Law B, Qian SY. The first characterization of free radicals formed from cellular COX-catalyzed peroxidation. Free Radic Biol Med. 2013;57:49–60. doi: 10.1016/j.freeradbiomed.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu Y, Qi J, Yang X, Wu E, Qian SY. Free radical derivatives formed from cyclooxygenase-catalyzed dihomo-γ-linolenic acid peroxidation can attenuate colon cancer cell growth and enhance 5-fluorouracil’s cytotoxicity. Redox Biol. 2014;2:610–618. doi: 10.1016/j.redox.2014.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu Y, Yang X, Zhao P, Yang Z, Yan C, Guo B, Qian SY. Knockdown of delta-5-desaturase promotes the anti-cancer activity of dihomo-γ-linolenic acid and enhances the efficacy of chemotherapy in colon cancer cells expressing COX-2. Free Radic Biol Med. 2016;96:67–77. doi: 10.1016/j.freeradbiomed.2016.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gresham GK, Wells GA, Gill S, Cameron C, Jonker DJ. Chemotherapy regimens for advanced pancreatic cancer: a systematic review and network meta-analysis. BMC Cancer. 2014;14:471. doi: 10.1186/1471-2407-14-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andersson R, Aho U, Nilsson BI, Peters GJ, Pastor-Anglada M, Rasch W, Sandvold ML. Gemcitabine chemoresistance in pancreatic cancer: molecular mechanisms and potential solutions. Scand J Gastroenterol. 2009;44:782–786. doi: 10.1080/00365520902745039. [DOI] [PubMed] [Google Scholar]
- 40.Shi X, Liu S, Kleeff J, Friess H, Büchler MW. Acquired resistance of pancreatic cancer cells towards 5-Fluorouracil and gemcitabine is associated with altered expression of apoptosis regulating genes. Oncology. 2002;62:354–362. doi: 10.1159/000065068. [DOI] [PubMed] [Google Scholar]
- 41.Zheng C, Jiao X, Jiang Y, Sun S. ERK1/2 activity contributes to gemcitabine resistance in pancreatic cancer cells. J Int Med Res. 2013;41:300–306. doi: 10.1177/0300060512474128. [DOI] [PubMed] [Google Scholar]
- 42.Horiguchi S, Shiraha H, Nagahara T, Kataoka J, Iwamuro M, Matsubara M, Nishina S, Kato H, Takaki A, Nouso K, Tanaka T, Ichimura K, Yagi T, Yamamoto K. Loss of runt-related transcription factor 3 induces gemcitabine resistance in pancreatic cancer. Mol Oncol. 2013;7:840–849. doi: 10.1016/j.molonc.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen Q, Wang Z, Zhang K, Liu X, Cao W, Zhang L, Zhang S, Yan B, Wang Y, Xia C. Clusterin confers gemcitabine resistance in pancreatic cancer. World J Surg Oncol. 2011;9:59. doi: 10.1186/1477-7819-9-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Sousa Cavalcante L, Monteiro G. Gemcitabine: metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. Eur J Pharmacol. 2014;741:8–16. doi: 10.1016/j.ejphar.2014.07.041. [DOI] [PubMed] [Google Scholar]
- 45.Quehenberger O, Armando A, Dumlao D, Stephens DL, Dennis EA. Lipidomics analysis of essential fatty acids in macrophages. Prostaglandins Leukot Essent Fat Acids. 2008;79:123–129. doi: 10.1016/j.plefa.2008.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li Y, Li X, Guo B. Chemopreventive agent 3,3′-diindolylmethane selectively induces proteasomal degradation of class I histone deacetylases. Cancer Res. 2010;70:646–654. doi: 10.1158/0008-5472.CAN-09-1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chang GC, Hsu SL, Tsai JR, Wu WJ, Chen CY, Sheu GT. Extracellular signalregulated kinase activation and Bcl-2 downregulation mediate apoptosis after gemcitabine treatment partly via a p53-independent pathway. Eur J Pharmacol. 2004;502:169–183. doi: 10.1016/j.ejphar.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 48.Elnaggar M, Giovannetti E, Peters GJ. Molecular targets of gemcitabine action: rationale for development of novel drugs and drug combinations. Curr Pharm Des. 2012;18:2811–2829. doi: 10.2174/138161212800626175. [DOI] [PubMed] [Google Scholar]
- 49.Xiong HQ, Varadhachary GR, Blais JC, Hess KR, Abbruzzese JL, Wolff RA. Phase 2 trial of oxaliplatin plus capecitabine (XELOX) as second-line therapy for patients with advanced pancreatic cancer. Cancer. 2008;113:2046–2052. doi: 10.1002/cncr.23810. [DOI] [PubMed] [Google Scholar]
- 50.Lu YY, Jing DD, Xu M, Wu K, Wang XP. Anti-tumor activity of erlotinib in the BxPC-3 pancreatic cancer cell line. World J Gastroenterol. 2008;14:5403–5411. doi: 10.3748/wjg.14.5403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Diep CH, Munoz RM, Choudhary A, Von Hoff DD, Han H. Synergistic effect between erlotinib and MEK inhibitors in KRAS wild-type human pancreatic cancer cells. Clin Cancer Res. 2011;17:2744–2756. doi: 10.1158/1078-0432.CCR-10-2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Iwai T, Moriya Y, Shirane M, Fujimoto-Ouchi K, Mori K. Continuous inhibition of epidermal growth factor receptor phosphorylation by erlotinib enhances antitumor activity of chemotherapy in erlotinib-resistant tumor xenografts. Oncol Rep. 2012;27:923–928. doi: 10.3892/or.2011.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ali S, Banerjee S, Ahmad A, El-Rayes BF, Philip PA, Sarkar FH. Apoptosis-inducing effect of erlotinib is potentiated by 3, 3′-diindolylmethane in vitro and in vivo using an orthotopic model of pancreatic cancer. Mol Cancer Ther. 2008;7:1708–1719. doi: 10.1158/1535-7163.MCT-08-0354. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 54.Chen JS, Faller DV, Spanjaard RA. Short-chain fatty acid inhibitors of histone deacetylases: promising anticancer therapeutics? Curr Cancer Drug Targets. 2003;3:219–236. doi: 10.2174/1568009033481994. [DOI] [PubMed] [Google Scholar]
- 55.Shi P, Yin T, Zhou F, Cui P, Gou S, Wang C. Valproic acid sensitizes pancreatic cancer cells to natural killer cell-mediated lysis by upregulating MICA and MICB via the PI3K/Akt signaling pathway. BMC Cancer. 2014;14:370. doi: 10.1186/1471-2407-14-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deer EL, González-Hernández J, Coursen JD, Shea JE, Ngatia J, Scaife CL, Firpo MA, Mulvihill SJ. Phenotype and genotype of pancreatic cancer cell lines. Pancreas. 2010;39:425–435. doi: 10.1097/MPA.0b013e3181c15963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cui D, Zhang C, Liu B, Shu Y, Du T, Shu D, Wang K, Dai F, Liu Y, Li C, Pan F, Yang Y, Ni J, Li H, Brand-Saberi B, Guo P. Regression of gastric cancer by systemic injection of RNA nanoparticles carrying both ligand and siRNA. Sci Rep. 2015;5:10726. doi: 10.1038/srep10726. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.