

Summary
PAHSAs are endogenous lipids with anti-diabetic and anti-inflammatory effects. PAHSA levels are reduced in serum and adipose tissue of insulin-resistant people and high-fat-diet (HFD)-fed mice. Here, we investigated whether chronic PAHSA treatment enhances insulin sensitivity and which receptors mediate their effects. Chronic PAHSA administration in chow and HFD-fed-mice raises serum and tissue PAHSA levels ~1.4–3-fold. This improves insulin sensitivity and glucose tolerance without altering body weight. PAHSA administration in chow, but not HFD-fed-mice, augments insulin and glucagon-like peptide (GLP-1) secretion. PAHSAs are selective agonists for GPR40, increasing Ca2+ flux but not intracellular cAMP. Blocking GPR40 reverses improvements in glucose tolerance and insulin sensitivity in PAHSA-treated chow and HFD-fed-mice and directly inhibits PAHSA augmentation of glucose-stimulated insulin secretion in human islets. In contrast, GLP-1 receptor blockade in PAHSA-treated chow-mice reduces PAHSA effects on glucose tolerance but not on insulin sensitivity. Thus, PAHSAs activate GPR40 which is involved in their beneficial metabolic effects.
eTOC blurb
PAHSA levels are reduced in serum and subcutaneous WAT of insulin-resistant people and HFD-fed-mice. Restoring PAHSA levels has high therapeutic potential to treat patients with T2D. Syed, Lee et al. show that chronic PAHSA treatment improves glucose homeostasis in chow and HFD-fed-mice, and identify GPR40 as a novel PAHSA target.
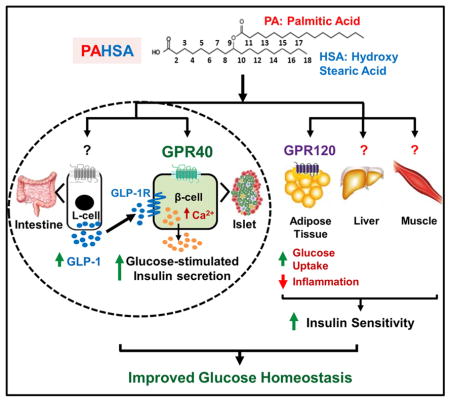
Introduction
Type 2 Diabetes (T2D) is a global epidemic (International Diabetes Federation Atlas 2013) characterized by hyperglycemia due to impaired islet function and insulin resistance in peripheral tissues. Despite advances in understanding the molecular mechanisms contributing to T2D and the development of new treatment modalities, the medical management of T2D remains inadequate (Aroda et al., 2012). At present, many available treatment strategies including analogues of the incretin, GLP-1, and Dipeptidyl peptidase 4 inhibitors work primarily by increasing insulin secretion. Other effective agents promote glucose excretion (Campbell and Drucker 2013; Meier 2012). But there is still a need for effective and safe agents that enhance insulin sensitivity to improve glucose control and prevent diabetic complications.
We discovered a novel class of endogenous lipids, branched fatty acid esters of hydroxy fatty acids (FAHFAs), with beneficial metabolic and anti-inflammatory effects (Yore et al., 2014). More than 16 FAHFA family members have been identified (Yore et al., 2014; Ma et al., 2015). Levels of one of the FAHFA family members, Palmitic Acid Hydroxy Stearic Acid (PAHSAs), are markedly lower in serum and adipose tissue (AT) of insulin-resistant humans. PAHSA levels correlate strongly with insulin sensitivity as measured by euglycemic clamps in humans (Yore et al., 2014). Acute oral treatment with 5- or 9-PAHSA isomer in chow and HFD mice improves glucose tolerance and augments insulin and GLP-1 secretion in vivo. In vitro, PAHSAs directly enhance GLP-1 secretion from enteroendocrine cells and glucose-stimulated insulin secretion (GSIS) from human islets (Yore et al., 2014). Furthermore, PAHSAs have anti-inflammatory effects including decreasing AT inflammation in HFD mice and attenuating LPS-induced dendritic cell activation and cytokine production. Although we reported that a single dose of PAHSAs acutely improves glucose tolerance, whether PAHSAs have effects on insulin sensitivity has not been investigated. Therefore, the first aim of this study was to determine whether PAHSA treatment enhances insulin sensitivity in vivo, and the second aim was to determine whether the beneficial effects of PAHSAs are sustained with chronic treatment.
The receptors responsible for PAHSA effects on insulin secretion and insulin action in vivo have not been identified. The G-protein coupled receptor (GPCR), GPR120, mediates PAHSA effects to enhance insulin-stimulated glucose transport in adipocytes (Yore et al., 2014), but PAHSAs are likely to activate other GPCRs because of their diverse actions in multiple tissues. Major pharmaceutical companies have had high-priority programs to screen for GPR120 and GPR40 activators to treat T2D. Molecules that activate both of these GPCRs with naturally-evolved relative affinities may be more effective for T2D treatment than agonists for single receptors.
Recent research has focused on identifying key agonists and receptors mediating nutrient-induced GLP-1 and insulin secretion. The long-chain fatty acid receptor, GPR40, is the most abundant GPCR expressed in islet β-cells and is also expressed on intestinal L-cells where it contributes to GLP-1 release along with GPR120 (Itoh et al., 2003; Edfalk et al., 2008). GPR40 activation augments GSIS and improves glycemia in rodent T2D models (Itoh et al., 2003; Steneberg et al., 2005), and the GPR40 agonist TAK-875 lowers fasting and postprandial blood glucose and HbA1c levels in humans (Leifke et al., 2012; Burant et al., 2012). Because PAHSAs acutely augment GSIS directly from human islets (Yore et al., 2014), the third aim of this study was to determine if PAHSAs activate GPR40 and whether this contributes to their beneficial effects in vivo. Here we show that PAHSAs directly activate GPR40 which is important for PAHSA effects on glucose homeostasis in both chow and HFD mice.
Since PAHSAs augment GLP-1 secretion in insulin-resistant mice (Yore et al., 2014), we also investigated the role of the GLP-1 Receptor (GLP-1R) in mediating PAHSA effects on glucose metabolism. GLP-1 potentiates insulin secretion and suppresses glucagon release (Holst 2007) but its beneficial actions are not limited to the endocrine pancreas (Ayala et al., 2009; Christensen et al., 2015; Villanueva-Penacarrillo et al., 2011). Here we show that the GLP-1R contributes to the beneficial PAHSA effects on insulin secretion and glucose tolerance but not on insulin sensitivity in chow mice. As well, PAHSA-stimulated augmentation of GSIS in chow mice is directly mediated by GPR40 but their effects on GLP-1 secretion do not involve GPR40. Thus, PAHSAs improve glucose tolerance and insulin sensitivity in chow mice and these effects are sustained for 5 months. In HFD mice, glucose tolerance and insulin sensitivity are also improved by PAHSA treatment but the effects are more modest. Furthermore, this study shows that PAHSAs directly activate GPR40 and this contributes to their beneficial metabolic effects in mice on both chow and HFD.
Results and Discussion
Chronic PAHSA treatment improves insulin sensitivity and glucose tolerance and reduces adipose tissue inflammation in chow-fed mice without altering food intake or adiposity
5- and 9-PAHSA treatment for up to 18 weeks in chow mice had no effect on body weight, fat mass (Figure 1A), food intake, lean mass, serum triglycerides or free fatty acid (FFA) levels (Supplementary Figure 1A–1B). Serum 5- and 9-PAHSA levels increased 2-fold with 2 months of treatment compared to vehicle mice (Figure 1B). After 5 months of treatment, only 9-PAHSA levels were elevated in serum (Figure 1B). However, 5- and 9-PAHSA levels were increased 1.5- to 3-fold in perigonadal (PG) and subcutaneous (SC) white AT (WAT) and brown adipose tissue. In liver, 9-PAHSA levels were elevated 2.5-fold in PAHSA-treated mice and 5-PAHSA was detected even though this isomer was not found in liver of vehicle-treated mice as expected (Yore et al., 2014) (Figure 1B). 5- and 9-PAHSA levels were not increased in the pancreas or brain with chronic PAHSA treatment (Figure 1B).
Figure 1. Chronic PAHSA treatment improves insulin sensitivity and glucose tolerance, and reduces adipose tissue inflammation without altering food intake or adiposity.

(A) Body weight and fat mass of C57bl6 male chow mice treated with vehicle or 5- and 9- PAHSAs (2mg/kg body weight/day of each) via minipumps. n=16/group. (B) 5- and 9-PAHSA levels in sera at 2 and 5 months, and tissues at 5 months of 5-and 9-PAHSA treatment. n=5– 6/group. (C) Insulin tolerance tests (ITT) and (D) oral glucose tolerance tests (OGTT) in vehicle-and PAHSA-treated mice. n=8–11/group. For A–D, *p<0.05 vs. vehicle. (E) Serum insulin and GLP-1 levels 5 min post-glucose challenge in vehicle- and PAHSA-treated mice. n=14–16/group. *p<0.05 vs. baseline within same treatment, #p<0.05 vs. vehicle at same time point, $p=0.08 vs. vehicle at same time point. (F) ITT, (G) OGTT, and (H–I) serum insulin and GLP-1 levels 5 min post-glucose challenge in vehicle-, palmitate- and PAHSA-treated outbred mice. n=7–9/group. *p<0.05 vs. baseline within same treatment, #p<0.05 vs. vehicle at same time point. (J) Number of AT CD11c+, CD206+, total number of AT macrophages, and macrophages expressing IL-1β and TNFα from PG WAT. n=4–5/group. *p<0.05 vs. vehicle; †p<0.08 vs. vehicle. Statistical significance was evaluated by unpaired two-tailed Student’s t-test or two-way ANOVA with Tukey post-hoc tests. Data are means±SEM.
5- and 9-PAHSA treatment improved insulin sensitivity as early as 13 days of treatment and these effects were sustained for at least 5 months (Figure 1C). Glucose tolerance (Figure 1D) was also improved similar to the effects reported with a single dose (Yore et al. 2014). Loss of first phase insulin response to glucose is one of the major and early impairments of β-cell function in T2D (Luzi and DeFronzo 1989). Restoration of this response is associated with improvements in postprandial glucose excursions (Bruce et al., 1988). Chronic 5- and 9-PAHSA treatment enhances insulin secretion in response to glucose by 40% over vehicle-treated mice (Figure 1E, left panel). In PAHSA-treated mice, GLP-1 levels tended to be reduced at baseline but the stimulation in response to glucose was enhanced 225% over vehicle mice (Figure 1E, right panel). Thus, not only does an acute oral dose of PAHSAs improve first-phase insulin and GLP-1 responses to glucose, but this effect is maintained over 5 months without tachyphylaxis or β-cell exhaustion. To confirm the beneficial effects of PAHSAs on metabolic parameters are distinct from effects of ordinary FFAs, we performed similar studies with palmitate since PAHSAs are made up of palmitate and hydroxystearic acid. Palmitate treatment at the same dose used for PAHSAs did not have favorable effects on glucose tolerance, insulin tolerance, or glucose-stimulated insulin or GLP-1 secretion (Figure 1F–1I). In addition, unlike palmitate which depletes islet insulin content with chronic exposure, PAHSA treatment of human islets for 72 hours results in sustained potentiation of GSIS and no loss of insulin content (data not shown).
In addition, at the same concentration as PAHSAs (20 μM), palmitate did not augment GSIS in pancreatic MIN6 β-cells (Supplementary Figure 1C). This was not because of lower palmitate uptake into cells, since lipid uptake measured as the ratio of labelled PAHSA or 13C16- palmitate within the cell (high density microsomes, low density microsomes, and cytosol fractions) compared to plasma membranes was higher for palmitate than for PAHSAs (data not shown).
We next investigated whether chronic PAHSA treatment reduces AT inflammation. PAHSA treatment reduced the total number of AT CD11c+ (pro-inflammatory) macrophages with no effect on AT CD206+ (anti-inflammatory) macrophages (Figure 1J; Supplementary Figure 1D). The total number of AT macrophages tended to be reduced with PAHSA treatment (Figure 1J) with no change in monocytes or neutrophils (Supplementary Figure 1E–1F). Moreover, PAHSA treatment reduced the number of pro-inflammatory IL-1β and TNF-α AT macrophages (Figure 1J). Together, these data indicate that chronic PAHSA treatment reduces AT Inflammation which could contribute to enhanced glucose homeostasis in chow mice.
PAHSAs activate GPR40 and this plays a role in their effects on insulin secretion
Since FFAs can induce insulin secretion through GPR40, we studied the role of GPR40 in mediating PAHSA effects. 9-PAHSA potentiated GSIS in isolated human islets, and this effect was completely reversed with the GPR40 antagonist, GW1100 (Figure 2A). Similarly, GPR40 knockdown in MIN6 cells completely reversed 9-PAHSA-potentiated GSIS without altering insulin secretion at low glucose (Figure 2B; Supplementary Figure 2A). Thus, both genetic and pharmacologic approaches indicate that PAHSAs augment GSIS by activating GPR40.
Figure 2. PAHSAs directly activate GPR40, and GPR40 antagonism reverses PAHSA-mediated improvements in glucose tolerance, insulin sensitivity and insulin secretion.

(A) Human islets treated with DMSO, 9-PAHSA (20μM), GW1100 (10μM), or 9-PAHSA+GW1100 for 45 min during glucose stimulation. 75 islets/condition. *p<0.05 vs. respective 2.5mM glucose, #p<0.05 vs. DMSO 20mM glucose, †p<0.05 vs. 9-PAHSA 20mM glucose. (B) MIN6 cells transfected with scrambled or GPR40 siRNA, treated with DMSO or 9-PAHSA (20μM) for 45 min during glucose stimulation. n=10 wells/condition. *p<0.05 vs. respective 2.5mM glucose, #p<0.05 vs. DMSO 20mM glucose scrambled siRNA, $ p<0.05 vs. 9-PAHSA 20mM glucose scrambled siRNA. (C) Reporter assay in HEK293 cells transfected with mouse GPR40 and SRE-luc treated with 9-PAHSA. n=3 wells/condition. (D) GPR40 reporter assay in HEK293 cells treated with either DMSO or 9-PAHSA with GW1100. n=3/condition.
(E) Calcium flux assay in mGPR40 stably transfected cells treated with DMSO, linoleic acid, 9-PAHSA, or GW1100. n = 3/condition. *p < 0.05 versus buffer and #p < 0.05 versus linoleic acid and 9-PAHSA alone.
(F) cAMP assay in mGPR40 stably transfected cells in presence of DMSO, cAMP agonist control, or 9-PAHSA. n = 3/condition. *p < 0.05 versus all other conditions.
(G and H) Five hours after food removal, PAHSA-treated mice were intraperitoneally injected with DMSO or DC260126 followed by insulin intraperitoneal injection or oral glucose after 30 min for an ITT (G) or OGTT (H). For (G) and (H), *p < 0.05 versus PAHSA+DMSO group.
(I) Glycemia, insulin and GLP-1 levels were measured in PAHSA-treated mice before and 5 min post-glucose gavage. n=8–10/group. *p<0.05 vs. baseline within same treatment, #p<0.05 vs. Vehicle DMSO at same time point. Statistical significance was evaluated by unpaired one-tailed Student’s t-test or two-way ANOVA with Tukey post-hoc tests. Data are means±SEM.
To determine whether PAHSAs directly activate GPR40, we transfected HEK293 cells with mouse GPR40 and SRE-luc. 9-PAHSA dose-dependently activates GPR40 (Figure 2C). 5-PAHSA also activates GPR40 (Supplementary Figure 2B). PAHSAs also directly activate human GPR40 (data not shown). 9-PAHSA-induced GPR40 activation was attenuated by GW1100 in a dose-dependent manner (Figure 2D). We next determined whether PAHSAs are full or selective GPR40 agonists. 9-PAHSA increased Ca2+ flux similar to the positive control Linoleic acid and GW1100 attenuated this effect (Figure 2E). However, 9-PAHSA had no effect on intracellular cAMP levels (Figure 2F). Thus, PAHSAs are selective, not full, agonists for GPR40. We also tested whether GPR40 directly mediates PAHSA effects on GLP-1 secretion in STC-1 enteroendocrine cells. Both 5- and 9-PAHSAs augmented GLP-1 secretion in STC-1 cells by 1.5–2-fold, but GPR40 antagonism did not reduce this effect (Supplementary Figure 2C). This suggests that PAHSA augmentation of GLP-1 secretion in enteroendocrine cells is independent of GPR40.
Inhibition of GPR40 reverses the beneficial effects of 5- and 9-PAHSA on both glucose tolerance and insulin sensitivity
To determine whether PAHSA effects on GSIS are mediated by GPR40 in vivo, vehicle- and 5- and 9-PAHSA-treated chow mice were injected with DC260126, a GPR40 antagonist. DC260126 attenuated the beneficial effects of PAHSAs on insulin sensitivity and glucose tolerance (Figure 2G–2H, compared to Figure 1C–1D), but had no effect in vehicle-treated mice (Supplementary Figure 2D–2F). There was no effect of GPR40 inhibition on glycemia at baseline or 5 min post-glucose gavage (Figure 2I; Supplementary Figure 2G). However, GPR40 inhibition lowered baseline insulinemia (30 min after DC260126) and completely blocked GSIS in PAHSA-treated mice (Figure 2I). In vehicle-treated mice, DC260126 also tended to impair GSIS (Supplementary Figure 2G). The fact that DC260126 blocked the glucose effect on insulin secretion as well as the PAHSA effects may be because DC260126 inhibits an increase in intracellular Ca+2 (Hu et al., 2009), possibly by modifying ligand-independent GPR40 activity (Milligan et al., 2017). In PAHSA- and vehicle-treated mice, GPR40 inhibition did not affect glucose-stimulated GLP-1 secretion (Figure 2I; Supplementary Figure 2G). Thus, PAHSAs stimulate insulin secretion through GPR40 but their effects on GLP-1 secretion do not involve GPR40. The conclusion that DC260126 inhibition of PAHSA-mediated potentiation of insulin secretion in vivo (Figure 2I, middle panel) involves GPR40 is supported by the in vitro data using another GPR40 antagonist, GW1100 (Figure 2D), and genetic knockdown of GPR40 in β-cells (Figure 2B). In terms of the specificity of DC260126, it had no effect on PAHSA activation of another lipid-responsive GPCR, GPR43 (Data not shown). Specificity was also tested by Hu et al., (2009) using a calcium mobilization assay with GPR40 or Melanin Concentrating Hormone Receptor 2 (MCHR2), another GPCR of the same subtype Gαq/11. DC260126 dose-dependently inhibited FFA-induced GPR40 activation, but had no effect on MCHR2 activation by its ligand, melanin concentrating hormone (Hu et al., 2009). Extensive testing for the specificity of DC260126 has not been done but the lack of effect in vehicle-treated mice (Supplemental Figure 2E–2F) reduces the possibility of off-target effects.
We also determined whether the effects of chronic PAHSA treatment in vivo are sustained in islets ex vivo. There was no augmentation of GSIS in islets from PAHSA-treated mice ex vivo compared to islets from vehicle mice (Supplementary Figure 2H). This suggests that the continuous presence of PAHSAs is required to trigger the GPR40 activity.
GLP-1R blockade reverses the beneficial effects of chronic PAHSA treatment on glucose tolerance but not insulin sensitivity
To determine whether PAHSA-induced improvements in glucose tolerance and insulin sensitivity are mediated through enhanced GLP-1 secretion, we injected vehicle- and 5- and 9- PAHSA-treated chow mice with the GLP-1R antagonist Exendin(Ex)(9-39) (Figure 3A). Ex(9-39) completely reversed PAHSA-induced improvements in glucose tolerance (Figure 3B; Supplementary Figure 3A) but not insulin sensitivity (Figure 3C). In PAHSA-treated mice, GLP- 1R blockade increased glycemia at baseline (30 minutes after Ex(9-39)) (Figures 3B-3D) and at 5 min post-glucose administration (Figure 3D). In addition, Ex(9-39) treatment attenuated the PAHSA-mediated increase in GSIS. Ex(9-39) increased baseline GLP-1 levels, but attenuated the increase in GLP-1 secretion above baseline in response to glucose (Figure 3D). In vehicle-treated mice, Ex(9-39) had no effect on baseline glucose (30 min after Ex(9-39)) or on glucose tolerance except at 45 min after glucose administration (Supplemental Figure 3B). There was also no effect of Ex(9-39) on insulin sensitivity or on glycemia at baseline or 5 min post-glucose administration (Supplementary Figure 3C–3D). Furthermore, GLP-1R antagonism in vehicle-treated mice increased glucose-stimulated insulin and GLP-1 secretion (Supplementary Figure 3D). This increase in GLP-1 secretion may be a compensatory response to GLP-1R blockade and likely stimulates insulin secretion. As expected, PAHSAs do not directly activate GLP-1R (Supplementary Figure 3E) which is activated by peptides (GLP-1), not lipids. These data indicate that GLP-1 action is necessary for the full effects of PAHSAs on glucose tolerance but not insulin sensitivity, and this may be due to PAHSA-mediated induction of GLP-1 secretion. These observations are consistent with a study in T2D people, in which GLP-1 improves glycemic control by increasing insulin secretion and inhibiting glucagon secretion without improving insulin sensitivity (Vella et al., 2000).
Figure 3. GLP-1R blockade reverses the effect of 5- and 9-PAHSAs on improved glucose tolerance.

(A) 5-hrs after food removal, PAHSA-treated mice were injected with either PBS or 5μg Ex(9-39) i.p. followed 30 min later by OGTT (B) or ITT (C). n=14–16/group. *p<0.05 vs. PAHSA PBS. (D) Glycemia, insulin and GLP-1 levels were measured in PAHSA-treated mice before and 5 min post-glucose gavage. n=8–10/group. Ex indicates Ex(9-39). *p<0.05 vs. baseline within same treatment, #p<0.05 vs. Vehicle PBS at same time point. Statistical significance was evaluated by unpaired two-tailed Student’s t-test or two-way ANOVA with Tukey post-hoc tests. Data are means±SEM.
Chronic PAHSA treatment improves insulin sensitivity and glucose tolerance, and reduces adipose tissue inflammation in HFD mice without altering food intake or adiposity
15–18 weeks of 9-PAHSA treatment via subcutaneous minipumps in HFD mice did not alter body weight, fat mass, lean mass, food intake, serum triglycerides or FFA levels (Figure 4A; Supplementary Figure 4A–4B). 9-PAHSA levels were elevated ~2-fold in serum and liver (Figure 4B) and ~33–42% in PG and SQ WAT compared to vehicle-treated HFD mice. 9-PAHSA levels were restored to chow mouse levels in serum and PG WAT but were not completely restored in SQ WAT (Figure 4B and 1B). Chronic PAHSA treatment in HFD mice improved insulin sensitivity (Figure 4C; Supplementary Figure 4C–4F) and glucose tolerance (Figure 4D; Supplementary Figure 4E) compared to vehicle-treated HFD mice and these effects were sustained for at least 4.5 months. We found that 9-PAHSA treatment (12 mg/kg/day) was more effective than a split dose of 5- and 9-PAHSA (6 mg/kg of each) in HFD mice (data not shown), although the combined treatment consistently lowered glycemia 5 hrs after food removal (Supplementary Figure 4D). Further studies showed 9-PAHSA lowered glycemia at 5 minutes post-glucose in HFD mice compared to vehicle-treated HFD mice with similar insulin levels (Figure 4E), supporting enhanced insulin sensitivity. Also in support of this, chronic 9-PAHSA treatment in HFD mice prevented the increase in islet mass seen in vehicle HFD mice (Figure 4F). This suggests that the same insulin levels are more efficient in lowering glucose in PAHSA HFD mice and expansion of islet mass is not necessary. In contrast to PAHSA-treated chow mice (Figure 1E), PAHSA treatment in HFD mice did not enhance glucose-stimulated insulin or GLP-1 secretion compared to vehicle HFD mice (Figure 4E), but similar insulin levels lowered glucose more in PAHSA HFD mice. Thus, the beneficial PAHSA effects on glucose homeostasis in HFD mice are independent of changes in glucose-stimulated insulin or GLP-1 secretion and appear to involve increased insulin sensitivity. Expression of tnf-α in PG WAT was lower in PAHSA-treated HFD mice compared to vehicle-treated HFD mice (Figure 4G). This indicates that reduced WAT inflammation may contribute to the enhanced glucose homeostasis in PAHSA-treated HFD mice. Overall, the beneficial effects of PAHSAs are more moderate in HFD mice than in chow mice. This may be in part due to lack of full restoration of SQ WAT PAHSA levels.
Figure 4. Chronic PAHSA treatment improves insulin sensitivity and glucose tolerance, and reduces adipose tissue inflammation in HFD mice without altering food intake or adiposity.

(A) Body weight and fat mass in chow-fed and HFD mice treated with 9-PAHSA via minipumps. n = 16/group. (B) 9-PAHSA levels in sera and tissues after 4.5 months of 9-PAHSA treatment. n = 5–6/group. (C and D) ITT (98 days of treatment; C) and OGTT (57 days of treatment; D) in vehicle and PAHSA HFD mice. Area under the curve for OGTT. n = 8–14/group. For (B),*p < 0.05 versus vehicle (VEH). For (C), *p < 0.05 versus HFD vehicle and chow, #p < 0.05 versus HFD vehicle and HFD PAHSA. For (D), *p < 0.05 HFD PAHSA versus HFD vehicle, #p < 0.05 chow versus HFD vehicle, $p < 0.05 versus chow and HFD PAHSA. (E) Serum glucose, insulin, and GLP-1 levels 5 min post-glucose challenge in vehicle and PAHSA HFD mice after 70 days of treatment. n = 7–8/group. ∗p < 0.05 versus vehicle at same time point. (F) β cell mass quantification (n = 2–3 sections/mouse, 4–6 mice/group). ∗p < 0.05 versus HFD vehicle; #p < 0.05 versus chow. (G–I) PG WAT tnf-α expression (G) in vehicle and PAHSA HFD mice. n = 8–9/group. ∗p < 0.05 versus HFD vehicle. Five hours after food removal, PAHSA-treated mice were injected intraperitoneally with DMSO or DC260126 followed 30 min later by ITT (H) or OGTT (I). n = 5–6/group. For (H) and (I),*p < 0.05 versus HFD vehicle DMSO and HFD PAHSA DC260126. Statistical significance was evaluated by unpaired one-tailed Student’s t test or two-way ANOVA with Tukey post hoc tests. Data are means ± SEM.
Since PAHSA beneficial effects on glucose homeostasis in chow mice are in part mediated by GPR40 (Figure 2G–2H), we studied the role of GPR40 in mediating PAHSA effects in HFD mice. DC260126, a GPR40 antagonist, reversed PAHSA improvements on insulin and glucose tolerance tests in HFD mice (Figure 4H–4I), but had no effect in vehicle HFD mice (Supplementary Figure 4E–4F). This effect of GPR40 inhibition in PAHSA HFD mice is mainly due to reversing PAHSA effects on lowering glycemia at time “0” min of the OGTT and ITT (Figure 4H–4I; Supplementary Figure 4G–4H). These data suggest that GPR40 mediates at least some of the beneficial effects of PAHSAs in HFD mice similar to the effects in chow mice.
Prior to this study, we had investigated only acute effects of PAHSAs on glucose homeostasis, primarily with a single dose (Yore et al., 2014). Furthermore, we did not report data on insulin sensitivity. A major advance of this study is demonstrating that PAHSAs enhance insulin sensitivity. This is important because there is a major unmet medical need for safe insulin-sensitizing agents. Currently, there are no anti-diabetic drugs that are primarily insulin-sensitizing except thiazolidinediones, which have adverse effects. Safe insulin-sensitizing agents could be used to prevent and treat insulin resistance, T2D, and cardiovascular complications.
Since serum PAHSA levels correlate highly with insulin sensitivity in humans, and PAHSA levels are reduced in serum and SQ WAT of insulin-resistant people and HFD mice, we designed the current study to restore circulating and tissue PAHSA levels within the physiologic range (Yore et al., 2014). Although we achieved this in chow mice and in serum and PG WAT of HFD mice, we did not fully restore levels in SQ WAT of HFD mice. This could contribute to the more modest effects on insulin sensitivity and glucose tolerance in HFD mice. The effects in chow mice are also greater with a mixed genetic background (Fig 1F–I), which is more relevant to human disease than inbred C57bl6 mice. Future studies will investigate the effects of PAHSA treatment in HFD mice on a mixed genetic background.
Another major advance is that we identified a new receptor that is directly activated by PAHSAs, GPR40. GPR40 blockade in chow mice attenuates PAHSA-mediated improvements in glucose tolerance, insulin sensitivity, and GSIS but not glucose-stimulated GLP-1 secretion. The fact that PAHSAs have direct effects on insulin secretion independent of GLP-1 as well as GLP-1- dependent effects, is supported by our data showing they stimulate GSIS in isolated human islets (Figure 2A). GLP-1R plays a role in some but not all PAHSA effects in chow mice since GLP- 1R blockade attenuates PAHSA-mediated improvements in glucose tolerance but not insulin sensitivity. In HFD mice, GPR40 blockade attenuates PAHSA-mediated beneficial effects on glucose tolerance and systemic insulin sensitivity. These observations do not eliminate a role for GPR120 which could mediate the PAHSA effects on GLP-1 secretion, glucose uptake and some anti-inflammatory effects (Yore et al 2014; Lee et al 2016).
In sum, this study expands our understanding of PAHSA biology by demonstrating sustained beneficial effects of chronic treatment without tachyphylaxis or islet exhaustion. In fact, PAHSAs appear to enhance islet function in HFD mice since expansion of islet mass which we observe in vehicle-treated HFD mice, is prevented with PAHSA treatment. Yet the islets produce the same amount of insulin and lower glycemia more in PAHSA-treated HFD mice than vehicle-treated HFD mice. PAHSA-mediated beneficial effects are achieved with subcutaneous delivery via minipumps, indicating that the oral route is not necessary for PAHSA effects on glucose homeostasis. These sustained effects of PAHSAs are in part mediated by GPR40. Because PAHSAs can activate multiple GPCRs which are known to mediate beneficial metabolic effects, and PAHSA levels are reduced in insulin-resistant people, restoring PAHSA levels has high therapeutic potential to prevent and/or treat T2D.
Study Limitations
These studies clearly demonstrate that chronic treatment of chow and HFD-fed mice with PAHSAs enhances insulin sensitivity and glucose tolerance and reduces adipose inflammation. Furthermore, PAHSAs activate GPR40 which mediates some of their beneficial effects. Nevertheless, there are some limitations of these studies. Although we and others (Hu et al., 2009) tested the specificity of the GPR40 inhibitor, DC260126, in assays with several other GPCRs including another receptor of the same subtype, Gαq/11, it is still possible that there could be off target effects of this inhibitor. The fact that DC260126 does not have effects on GTT or ITT in vehicle-treated mice makes that less of a concern. The observation that DC260126 lowers baseline insulinemia in PAHSA-treated mice and impairs glucose-stimulated insulin secretion (as well as the augmentation of insulin secretion by PAHSA treatment) may be unexpected since GPR40 mediates fatty acid-activated insulin secretion but generally not glucose-activated insulin secretion. But DC260126 inhibits the rise in intracellular Ca+2 levels in response to fatty acids (Hu et al., 2009). Since PAHSAs are delivered constantly by minipump, DC260126 may be reducing intracellular Ca+2 even in the basal state (ie before glucose administration) thereby lowering baseline insulinemia. In addition, DC260126 may inhibit an increase in intracellular Ca+2 elicited by glucose or other insulin secretagogues in islet β cells. Another mechanism for the effects of DC260126 on glucose-stimulated insulin secretion is that small molecule inhibitors can bind to GPCRs like GPR40 in a fashion that modulates their baseline activity (Milligan et al., 2017). Future experiments in which we can conditionally knockdown GPR40 in adult mice in a tissue-specific manner will be useful to investigate these mechanisms. In spite of these caveats, the fact that genetic knockdown of GPR40 in a β cell line and that a different GPR40 inhibitor directly blocks PAHSA potentiation of insulin secretion in human islets supports our conclusion that PAHSA effects on insulin secretion are mediated by GPR40.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and request for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Barbara B. Kahn (bkahn@bidmc.harvard.edu).
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Mice and Husbandry Conditions
Chow-fed (Purina Lab diet, 5008) male C57Bl/6J wild-type mice (Jackson Laboratories) and in-house bred mixed background mice (C57Bl/6J and FVB) were randomized to treatment group based on body weight at 8 wks of age and implanted with subcutaneous minipumps (Alzet, Model2006). Similarly, randomized male C57Bl/6J wild-type mice were placed on HFD (Research Diets, Td93075) at 6 wks of age. After 8 weeks on HFD, insulin resistance was confirmed by an insulin tolerance test (ITT). Minipumps were implanted after 9 weeks on HFD. All mice were singly housed in ventilated cages with ad libitum access to food and water. Mice were kept on a 14 hr light, 10 hr dark schedule at 22–23°C. All animal care and use procedures were in accordance with and approved by the Institutional Animal Care and Use Committee at Beth Israel Deaconess Medical Center, Boston, MA.
Cell Culture
MIN6 cells obtained from ATCC (animals of unknown gender) were maintained at 37°C and cultured in RPMI 1640 medium containing 10 % heat-inactivated fetal bovine serum supplemented with 100 IU/ml penicillin and 100 IU/ml streptomycin, 1 mM sodium pyruvate, 50 μM 2-mercaptoethanol, and 10 mM HEPES [pH 7.4]. The medium was changed twice and cells were subcloned weekly. Endogenous GPR40 expression was depleted by transfecting MIN6 cells using a pool of GPR40 siRNA at a final concentration of 100 nmol/l using Lipofectamine transfection reagent. To assess the specificity of RNA interference method, cells were transfected with non-targeting RNA i.e., scrambled RNA. Efficiency of GPR40 knockdown was determined by qPCR.
STC-1 and HEK293T cells obtained from ATCC were maintained in DMEM supplemented with 10% FCS, 100 IU/ml penicillin and 100 IU/ml streptomycin, and maintained at 37°C and 5% CO2.
Human islets from normal donors obtained from BADERC Islet Core Facility, Massachusetts General Hospital, were cultured in human islet media with 100 IU/ml penicillin and 100 IU/ml streptomycin, and maintained at 37°C and 5% CO2.
METHOD DETAILS
Measurement of metabolic parameters
Minipumps filled with either vehicle (50% PEG-400, 0.5% Tween-80, 49.5% distilled water) or 5- and 9-PAHSAs (Chow: 2mg/kg of each; HFD: 6mg/kg of each or 12mg/kg of 9-PAHSA) or 4mg/kg of palmitate were inserted subcutaneously in 8-wk-old chow and 15-wk-old HFD mice. For in vivo studies, the GLP-1R antagonist, Ex(9-39) (5μg/mouse) and the GPR40 antagonist, DC260126 (5mg/kg; Tocris Bioscience) were administered intraperitoneally. Body weight and food intake were measured weekly and body composition measured by MRI. Insulin tolerance test (ITT) and oral glucose tolerance test (OGTT) were performed as described (Yore et al. 2014) following 5 hr. food removal. 4.5 hr. after food removal (0.5 hr. before initiation of the OGTT or ITT) mice were intraperitoneally injected with either GLP-1 receptor antagonist, Exendin (9-39) or GPR40 antagonist, DC260126. Mice received 1 g/Kg glucose by gavage or 0.5–0.7 IU of insulin intraperitoneally 30 min after antagonist administration and glycemia was monitored over a 2 hr. period. For glucose-stimulated insulin and GLP-1 secretion studies, mice were bled at t=0 (baseline) and 5 min post glucose gavage from the tail vein and blood was transferred into tubes and serum insulin and total GLP-1 levels were measured by ELISA. At the end of the study, mice were sacrificed by decapitation for serum collection and tissues were harvested, snap frozen in liquid nitrogen and stored at −80°C until further processed.
PAHSA extraction and measurements
Lipid extraction was performed as described (Yore et al., 2014). Tissues (60–100mg) were Dounce homogenized on ice in a mixture of 1.5mL MeOH, 1.5mL chloroform and 3mL citric acid buffer. PAHSA standards were added to chloroform prior to extraction. The final mixture was centrifuged and the organic phase containing was dried under N2 and stored at −80°C prior to solid phase extraction (Yore et al., 2014; Zhang et al., 2016).
Anti-inflammatory effects of PAHSAs
PG WAT stromal vascular fraction cells were harvested and surface markers were stained with monoclonal antibodies (Biolegend, E-Bioscience, BD bioscience) for multicolor flow cytometry. Intracellular cytokines were measured as previously described (Moraes-Vieira et al., 2014). In brief, SVFs were stimulated in vitro for 5 h at 37° C in 5% CO2 with phorbol-12-myristate-13- acetate (PMA; 100 ng/mL), ionomycin (1 μg/mL) and brefeldin A (1 μg/mL) (Sigma-Aldrich, St. Louis, MO, USA). Cells were washed and stained with specific antibodies for surface molecules (CD11b, CD45, CD11c, CD206, F4/80) (Biolegend) and permeabilized using the BD Cytofix/Cytoperm Fixation/Permeabilization solution kit (BD Biosciences, San Jose, CA, USA). Intracellular staining was performed with PE-Cy7-conjugated anti-TNF (Biolegend) and PE-conjugated anti-IL-1beta (eBioscience) antibodies. Cells were acquired with an LSR II flow cytometer (BD) and analyzed by FlowJo software.
GPR40 receptor reporter assay
HEK293T cells were transfected with GPR40 and SRE-luc 44-235 and treated with DMSO control, 5-PAHSA (100μM), or 9-PAHSA (0–100μM) with or without GW1100 (0–10μM). At the end of the treatment luciferase activity was measured and relative luciferase units were calculated.
GLP-1 receptor reporter assay
HEK293T cells were transfected with GLP-1 and CRE-luc and treated with DMSO control, 5- PAHSA (20 and 100μM), or 9-PAHSA (20 and 100μM), hydroxystearic acid (100μM), sodium palmitate (100μM) and GLP-1 (10pM). At the end of the treatment luciferase activity was measured and relative luciferase units were calculated.
Ca2+ measurements – Multispan Inc
Multispan’s mouse GPR40 stable cell line, HEK293T cells were grown in DMEM, 10% FBS, and 1 μg/mL puromycin. On the day of assay, cells were lifted with non-enzymatic cell stripper and seeded in 384-well poly-d-lysine coated plate in assay buffer (HBSS + 20mM HEPES) at an appropriate density. The calcium dye loading buffer (Multispan Inc., Cat# MSCA01-1) was added to cells and incubated for 30 minutes at 37°C followed by 30 minutes at room temperature. In antagonist mode, cells were pre-incubated with GW1100 for 15 minutes at room temperature prior to the addition of 9-PAHSA at 100mM concentration. Calcium flux was monitored for 180 sec with 9-PAHSA injected into the well at the 19th second read by FLIPR 384 (Molecular Devices).
Cyclic AMP Assay – Multispan Inc
Cells were lifted with non-enzymatic cell stripper and resuspended in assay buffer at desired concentrations. cAMP assays were performed according to the manufacturer’s protocol using CisBio’s HTRF cAMP HiRange Kit. Cells were incubated with compounds for 30 minutes at 37°C. The reaction was terminated by sequentially adding D2-labeled cAMP and cryptate-labeled anti-cAMP antibody in lysis buffer. The plate was incubated at room temperature for 60 minutes before reading fluorescent emissions at 620 nm and 668 nm with excitation at 314 nm on FlexStation III (Molecular Devices). Cyclic AMP assay results are shown as “Ratio 668/620 × 10,000” (ratio of fluorescence at 668 nm and 620 nm x 10,000). Data in graphs are represented in Mean ± SD. Dose-dependent responses were fitted with sigmoidal dose-response curves allowing variable slopes using GraphPad Prism version 6. The graphs display dose-dependent stimulation of intracellular cAMP level upon treatment with control ligand or compounds.
Glucose-stimulated insulin secretion in human islets
Human islets from normal donor were obtained from BADERC Core facility, MGH, Bosotn. GSIS was assessed as previously outlined (Kowluru et al., 2010). Briefly, human islets were cultured overnight in human islet media. Following a 2 hr incubation in Krebs-Ringer Bicarbonate buffer (KRB) with 0.5% BSA (pH 7.4), islets were stimulated with either 2.5 mM or 20 mM glucose in presence or absence of DMSO, 9-PAHSA (20μM), and GW1100 (10μM) for 45 mins at 37°C. At the end, media was collected and insulin was quantitated by ELISA (Alpco Diagnostics).
GLP-1 secretion from enteroendocrine STC-1 cells
GLP-1 secretion studies were performed as previously described (Yore et al., 2014). In brief, STC-1 cells were plated in 24-well plates 72 hrs before assay initiation. Cells were washed with incubation buffer B (Hanks’ balanced salt solution supplemented with 20mM HEPES) and incubated with 5- or 9-PAHSA or DMSO control at 37°C for 1 hr. Total GLP-1 measurements were performed using supernatant by ELISA (Millipore, Billerica, MA).
Determination of β-cell mass
Pancreata from mice treated chronically with vehicle or 9-PAHSA were fixed in 10% formalin and processed for hematoxylin and eosin staining. 2–3 full pancreas sections with 19–40 islets per section were imaged in bright field to determine β-cell mass as described (Iglesias et. al., 2012). β-cell mass was calculated using the following equation: β-cell mass = pancreas weight (mg) x relative insulin surface (total islet area μm2/total pancreas area μm2). Relative islet surface areas were analyzed using Fiji software.
Quantitative PCR
RNA was extracted using Tri-Reagent (MRC) and cDNA was generated with random hexamers (Clontech). Quantitative real-time PCR was performed with the ABI Prism sequence detection system. Each sample was run in duplicate, and the quantity of GPR40 mRNA in each sample was normalized to mouse TATA-box binding protein (mTBP) mRNA levels. All primers were obtained from IDT DNA.
Analytical procedures
Serum triglycerides were measured by colorimetric enzyme assay, and serum free fatty acids were measured using the NEFA-C kit (Wako, Richmond, Virginia).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as means±SEM. Significance was assessed by Student’s t-tests and/or ANOVA with Tukey post-test for multiple comparisons where appropriate. No data were excluded from the analyses. Statistical analysis were performed with GraphPad Prism and differences were considered significant when p<0.05. Statistical parameters can be found in the figure legends.
Supplementary Material
Highlights.
Chronic PAHSA treatment improves insulin sensitivity in chow- and HFD-fed mice
PAHSAs activate GPR40 which mediates their augmentation of insulin secretion
GPR40 plays a role in the beneficial effects of PAHSAs on glucose homeostasis
GLP-1 receptor indirectly mediates PAHSA effects on glucose tolerance
Acknowledgments
We thank Dr. Ji Lei, the director of BADERC Pancreatic Islet Core for providing human islets; and Dr. Douglas Hanahan for the STC-1 cells. We thank Dr. Susan Bonner-Weir from the Joslin Diabetes Center for her expertise and conversations related to islet biology analyses. The Harvard Digestive Disease Center Core B (NIH Grant # P30DK034854) processed and immunostained samples. Imaging was performed at the Neuro Imaging Facility (NINDS P30 Core Center Grant #NS072030) for islet mass quantification. Supported by grants from the NIH R01 DK43051 and P30 DK57521 (B.B.K.); R01 DK106210 (B.B.K. and A.S.); a grant from the JPB foundation (B.B.K.). T32DK07516 (B.B.K. and J.L.).
B.B.K., A.S., I.S., J.L., P.M.M.-V. and M.M.Y. are inventors on patents related to the fatty acid hydroxy-fatty acids.
Footnotes
Author Contributions
I.S. and J.L. conceived of, designed, performed, and interpreted experiments and made figures. P.M.M.-V. designed, performed and interpreted the immunology experiments and made the figures. C.J.D., A.S. and B.B.K. designed and performed the receptor activation experiments. M.J.K. performed the uptake studies. A.T.N., D.S. and A.S. designed and performed chemical synthesis of the lipids. A. Sontheimer, P.A., O.D.P., K.W., and J.J.Z., assisted with animal studies. J.M. and S.F. participated in interpreting results of GPR40 assays. M.M.Y designed and performed GLP-1 assay in vitro. B.B.K. and A.S. conceived of, designed and supervised the experimental plan and interpreted experiments. B.B.K., I.S., and J.L. wrote the manuscript. A.S. and P.M.M.V. edited the manuscript.
Declaration
“The authors declare no competing interests.”
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aroda VR, Henry R, Han J, Huang W, DeYoung MB, Darsow T, Hoogwerf BJ. Efficacy of GLP-1R agonists and DPP-4 inhibitors: meta-analysis and systematic review. Clin Ther. 2012;34:1247– 1258. doi: 10.1016/j.clinthera.2012.04.013. [DOI] [PubMed] [Google Scholar]
- Ayala JE, Bracy DP, James FD, Julien BM, Wasserman DH, Drucker DJ. The glucagon-like peptide-1 receptor regulates endogenous glucose production and muscle glucose uptake independent of its incretin action. Endocrinology. 2009;150(3):1155–1164. doi: 10.1210/en.2008-0945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burant CF, Viswanathan P, Marcinak J, Cao C, Vakilynejad M, Xie B, Leifke E. TAK-875 versus placebo or glimepiride in T2D mellitus: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet. 2012;379:1403–1411. doi: 10.1016/S0140-6736(11)61879-5. [DOI] [PubMed] [Google Scholar]
- Campbell JE, Drucker DJ. Pharmacology physiology and mechanisms of incretin hormone action. Cell Metab. 2013;17:819–837. doi: 10.1016/j.cmet.2013.04.008. [DOI] [PubMed] [Google Scholar]
- Christensen LW, Kuhre RE, Janus C, Svendsen B, Holst JJ. Vascular, but not luminal, activation of FFAR1 (GPR40) stimulates GLP-1 secretion from isolated perfused rat small intestine. Physiol Rep. 2015;3(9):e12551. doi: 10.14814/phy2.12551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edfalk S, Steneberg P, Edlund H. Gpr40 is expressed in enteroendocrine cells and mediates free fatty acid stimulation of incretin secretion. Diabetes. 2008;57(9):2280–22877. doi: 10.2337/db08-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev. 2007;87:1409–1439. doi: 10.1152/physrev.00034.2006. [DOI] [PubMed] [Google Scholar]
- Hu H, He LY, Gong Z, Li N, Lu YN, Liu H, Jiang HL, Zhu WL, Wang HY. A novel class of antagonists for the FFAs receptor GPR40. Biochem Biophys Res Commun. 2009;390(3):557–563. doi: 10.1016/j.bbrc.2009.10.004. [DOI] [PubMed] [Google Scholar]
- International Diabetes Federation. IDF Diabetes Atlas. 6. International Diabetes Federation; Brussels, Belgium: 2013. [Google Scholar]
- Iglesias J, Barg S, Vallois D, Lahiri S, Roger C, Yessoufou A, Pradevand S, McDonald A, Bonal C, Reimann F, et al. PPARβ/δ affects pancreatic β-cell mass and insulin secretion in mice. J Clin Invest. 2012;122(11):4105–4117. doi: 10.1172/JCI42127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, Ogi K, Hosoya M, Tanaka Y, Uejima H, et al. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature. 2003;422(6928):173–176. doi: 10.1038/nature01478. [DOI] [PubMed] [Google Scholar]
- Kowluru A, Veluthakal R, Rhodes CJ, Kamath V, Syed I, Koch BJ. Protein farnesylation-dependent Raf/extracellular signal-related kinase signaling links to cytoskeletal remodeling to facilitate glucose-induced insulin secretion in pancreatic β-cells. Diabetes. 2010;59(4):967–977. doi: 10.2337/db09-1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leifke E, Naik H, Wu J, Viswanathan P, Demanno D, Kipnes M, Vakilynejad M. A multiple ascending-dose study to evaluate safety, pharmacokinetics, and pharmacodynamics of a novel GPR40 agonist, TAK-875, in subjects with type 2 diabetes. Clin Pharmacol Ther. 2012;92:29–39. doi: 10.1038/clpt.2012.43. [DOI] [PubMed] [Google Scholar]
- Ma Y, Kind T, Vaniya A, Gennity I, Fahrmann JF, Fiehn O. An in silico MS/MS library for automatic annotation of novel FAHFA lipids. J Cheminform. 2015;7:53. doi: 10.1186/s13321-015-0104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier JJ. GLP-1 receptor agonists for individualized treatment of type 2 diabetes mellitus. Nat Rev Endocrinol. 2012;8:728–742. doi: 10.1038/nrendo.2012.140. [DOI] [PubMed] [Google Scholar]
- Milligan G, Shimpukade B, Ulven T, Hudson BD. Complex Pharmacology of Free Fatty Acid Receptors. Chem Rev. 2017;117(1):67–110. doi: 10.1021/acs.chemrev.6b00056. [DOI] [PubMed] [Google Scholar]
- Moraes-Vieira PM, Yore MM, Dwyer PM, Syed I, Aryal P, Kahn BB. RBP4 activates antigen-presenting cells, leading to adipose tissue inflammation and systemic insulin resistance. Cell Metab. 2014;19(3):512–526. doi: 10.1016/j.cmet.2014.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steneberg P, Rubins N, Bartoov-Shifman R, Walker MD, Edlund H. The FFA receptor GPR40 links hyperinsulinemia, hepatic steatosis, and impaired glucose homeostasis in mouse. Cell Metab. 2005;1:245–258. doi: 10.1016/j.cmet.2005.03.007. [DOI] [PubMed] [Google Scholar]
- Vella A, Shah P, Basu R, Basu A, Holst JJ, Rizza RA. Effect of glucagon-like peptide 1(7–36) amide on glucose effectiveness and insulin action in people with type 2 diabetes. Diabetes. 2000;49(4):611–617. doi: 10.2337/diabetes.49.4.611. [DOI] [PubMed] [Google Scholar]
- Villanueva-Peñacarrillo ML, Martin-Duce A, Ramos-Alvarez I, Gutierrez-Rojas I, Moreno P, Nuche-Berenguer B, Acitores A, Sancho V, Valverde I, Gonzalez N. Characteristic of GLP-1 effects on glucose metabolism in human skeletal muscle from obese patients. Regul Pept. 2011;168(1–3):39–44. doi: 10.1016/j.regpep.2011.03.002. [DOI] [PubMed] [Google Scholar]
- Yore MM*, Syed I*, Moraes-Vieira PM, Zhang T, Herman MA, Homan EA, Patel RT, Lee J, Chen S, Peroni OD, et al. Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell. 2014;159(2):318–332. doi: 10.1016/j.cell.2014.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Chen S, Syed I, Stahlman M, Kolar MJ, Homan EA, Chu Q, Smith U, Boren J, Kahn BB, et al. A LC-MS–based workflow for measurement of branched fatty acid esters of hydroxy fatty acids. Nature Protocols. 2016;11(4):747–763. doi: 10.1038/nprot.2016.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.