

Abstract
Metastatic breast cancer is an incurable disease and identification of novel therapeutic opportunities is vital. Triple negative breast cancer (TNBC) frequently metastasizes and high levels of activated RSK, a downstream MEK-ERK1/2 effector, are found in TNBC. We demonstrate using direct pharmacological and genetic inhibition of RSK1/2 that these kinases contribute to the TNBC metastatic process in vivo. Kinase profiling demonstrated that RSK1 and RSK2 are the predominant kinases targeted by the new inhibitor, which is based on the natural product, SL0101. Further evidence for selectivity was provided by the observations that silencing RSK1 and RSK2 eliminated the ability of the analogue to further inhibit survival or proliferation of a TNBC cell line. In vivo, the new derivative was as effective as the FDA-approved MEK inhibitor, trametinib, in reducing the establishment of metastatic foci. Importantly, inhibition of RSK1/2 did not result in activation of AKT, which is known to limit the efficacy of MEK inhibitors in the clinic. Our results demonstrate that RSK is a major contributor to the TNBC metastatic program and provide preclinical proof-of-concept for the efficacy of the novel SL0101 analogue in vivo.
Keywords: RSK, SL0101, TNBC metastasis
Introduction
Metastatic breast cancer remains incurable, with therapy limited to slowing disease progression (1). In particular, triple negative breast cancer (TNBC) patients have increased probability of death due to metastasis compared with other breast cancer subtypes (2). TNBC is characterized by its lack of currently available targeted markers (3). However, the MEK-ERK1/2 cascade is now considered as a viable drug target for TNBC (4–7). In genetic analysis of basal-like breast cancers, which includes ~ 70% of TNBCs, activated MEK-ERK1/2 signaling is thought to occur in ~ 80% of the tumors (4, 8, 9). Additionally, numerous TNBC cell lines possess an activated RAS-transcriptional program and enhanced sensitivity to MEK inhibition (10, 11). In support of these pre-clinical observations, a complete response was observed in a phase Ib trial using a combination of trametinib, a MEK inhibitor, and gemcitabine, a nucleotide analogue, in a TNBC patient who had failed multiple therapies (5). Based on these data various MEK inhibitors are being tested in clinical trials, which include TNBC patients (12).
However, treating patients with drugs that inhibit “global regulators” such as MEK causes a number of side effects that result in limited efficacy (12). We postulate that inhibiting downstream effectors of MEK like the Ser/Thr protein kinase, p90RSK (RSK), will have fewer side effects because it controls a more limited set of targets. RSK phosphorylates various substrates that control diverse cellular processes, including metastasis (13–18). Approximately, 85% of TNBC patient samples have activated RSK, which is identified by the presence of phosphorylated residues critical for its activity (19). Taken together, these observations suggest that RSK is a viable target for TNBC.
RSK contains two non-identical functional kinase domains referred to as the N-terminal (NTKD) and C-terminal (CTKD) (13). The CTKD functions to regulate RSK activation whereas the NTKD, which belongs to the AGC kinase family, is responsible for substrate phosphorylation (13). In a screen of botanical extracts we identified the first RSK inhibitor, SL0101 (1a), which was isolated from Forsteronia refracta (20). SL0101 is an extremely specific allosteric inhibitor for the NTKD (14, 20–22).
In addition to SL0101, other RSK inhibitors have been described. However, the currently available NTKD inhibitors are not RSK specific (21, 23–26) or demonstrate poor pharmacokinetics (27, 28). Covalent inhibitors of the RSK CTKD (29–31), targeting autoactivation, are also available and have limited off-target effects. However, CTKD inhibitors do not inhibit an activated kinase and the autoactivation mechanism can be bypassed (29), suggesting that the clinical utility of CTKD inhibitors is limited.
Because of the selectivity of SL0101 for RSK we continue to improve its drug-like properties through extensive structure-activity-relationship (SAR) analysis. We have now identified a SL0101 analogue, C6″-n-propyl cyclitol SL0101 (1b), which retains specificity for RSK1/2 and is more potent in in vitro and cell-based assays than the parent compound. This improved analogue inhibits proliferation, survival in a non-adherent environment, and migration of TNBC lines but, unlike MEK inhibitors, does not activate the AKT pathway. Inhibition of RSK1/2 using (1b) or silencing RSK1 or RSK2 inhibited TNBC metastatic colonization in vivo. Moreover, (1b) was as effective as the FDA-approved MEK inhibitor, trametinib. Taken together, these results indicate that RSK1/2 are viable drug targets for TNBC metastasis.
Materials and Methods
Animals
Animal procedures had approval of the Vanderbilt University Institutional Animal Care and Use Committee. For in vivo metastatic models, NOD-SCID-IL2Rγc (NSG) mice (6–8 weeks) (Jackson Laboratory) were injected in the left cardiac ventricle with 1×105 cells/100 μL PBS. Mice injected with MCF-7 cells received a 17β-estradiol pellet (0.36-mg 60-day release; Innovative Research of America). Mice bearing MCF-7 metastasis were injected IP with vehicle (10% (2-hydroxypropyl)-β-cyclodextrin (HPBCD) in 10% DMSO) or (1b) (40 mg/kg) 2h prior to euthanasia (day 50). Mice injected with HDQ-P1-Luc were randomized and at 2 h after injection were treated for 5d with HPBCD, (1b) (40 mg/kg) IP Q12h or trametinib (2 mg/kg, Santa Cruz Biotechnology, Inc) IP Q24 h. For bioluminescence imaging, mice were injected IP with RediJect D-Luciferin (1.5 mg) (PerkinElmer, Inc.) and imaged with a Xenogen IVIS using Living Image acquisition software (Xenogen Corp.). For ex vivo imaging, organs were placed in D-Luciferin (150 mg/mL PBS). After imaging tissue was fixed in 4% buffered formalin and paraffin-embedded.
In vitro cell assays
Cell lines were obtained and cultured as directed by American Type Culture Collection or by German Collection of Microorganisms and Cell Culture. Stocks were prepared within one to two passages after receipt and new stocks thawed frequently and passaged < 6 months. Authentication was based on growth rate, morphology and absence of mycoplasma. Serum-starved cells were pre-treated for 2h with vehicle or inhibitor. MCF-7 cells were treated with phorbol 12-myristate 13-acetate (PMA, Sigma) for 20 min. Cells were lysed as described (32).
For motility assays 2.5×105 cells were plated on fibronectin-coated (5 μg/mL, Corning) 2-chamber Lab Teks (Thermo Fisher Scientific). After 48 h cells were pre-treated with vehicle or inhibitors for 2h and scratched with a P200 pipette tip. After washing, HEPES (50 mM, Thermo Fisher Scientific)-buffered media with vehicle or inhibitor was added and images taken every 20 min using Nikon Eclipse Ti microscope and an Orca R2 digital CCD camera (Hamamatsu). Migration velocity was quantified using Volocity software (PerkinElmer, Inc.). Additional details in Supplementary.
For 2D proliferation assays, 2×105 cells/well in 24-well or 103 cells/well in 96-well were seeded. For 3D proliferation 1.5 × 103 cells/well in 96-well were plated in 2% matrigel (MG; Corning, Inc.) onto 100% MG. Inhibitor or vehicle was added and proliferation was measured at 48–72h using CellTiterGlo reagent (Promega Corp.) with a GloMax Discover luminometer (Promega Corp.).
For survival assays, cells were seeded at 1.5×103 cells/well in 96-well poly-HEMA coated plates (Corning, Inc.) and vehicle or inhibitors added and bioluminescence measured at 48–72h.
The IC50 values for proliferation and survival were determined using non-linear regression analysis (GraphPad Prism version 6.0a).
Immunostaining
Section preparation and antibodies are listed in Supplementary. Fluorescent images were obtained with a laser-scanning microscope (510/Meta/FCS Carl Zeiss, Inc.). Objectives were: mouse tissue 40× Plan-Neofluar oil NA 1.3 (zoom 0.7×); human tissue 20× NA 0.8. Images were acquired using LSM-FCS software (Carl Zeiss, Inc.), quantitated using Openlab 5.5.0 (PerkinElmer, Inc.) and processed in Photoshop version CS6 version 13.0 (Adobe).
In vitro kinase assays
RSK2 kinase assays performed as described (32). The kinase screen was performed using the ZLYTE screen (Thermo Fisher Scientific, Inc.).
Statistical Analysis
Statistical analyses (GraphPad Prism 6.0a) using the Mann-Whitney test (two-sided) unless indicated. *p<0.05 was statistically significant.
Results
SL0101 analogue with improved in vitro and cell-based efficacy
In prior SAR studies of the flavonoid glycoside, SL0101 (1a), we determined that replacement of the C5-methyl group on the pyran with an n-propyl moiety (1c), improved the IC50 by > twenty-five -fold but that the compound had limited aqueous solubility (32). Additionally, we determined that exchanging the rhamnose with a cyclitol (1d), improved the cell-based efficacy for inhibition of proliferation but this compound was not RSK specific (33). We hypothesized that combining the modifications would improve the potency for RSK inhibition while maintaining specificity for RSK. Consistent with our hypothesis C6″-n-propyl cyclitol SL0101 (1b) has a six-fold improved IC50 in an in vitro kinase assay for RSK inhibition compared to SL0101 (1a) (Fig. 1A). Furthermore, (1b) inhibited the proliferation in 2D culture of the ER+ breast cancer line, MCF-7, with an IC50 of ~ 8 μM versus ~ 50 μM for SL0101 (Fig. 1B) (20). Previously, we found that the proliferation of the immortalized but untransformed breast line, MCF-10A, is less dependent on RSK for proliferation than the MCF-7 line (33). Consistent with these observations, only a slight decrease in MCF-10A proliferation occurred at the highest concentrations of (1b) (Fig. 1B). The efficacy of SL0101 diminishes after > 48h in in vitro culture (20). One advantage in replacing the rhamnose with a cyclitol moiety is that the cyclitol should be resistant to acid catalyzed anomeric bond hydrolysis, which should increase stability. To test this possibility we incubated MCF-7 cells with (1b) (25 μM) for varying lengths of time. In agreement with our rationale, only a minor increase in proliferation over a 96h time course was observed when MCF-7 cells were incubated with (1b) (Fig. 1C). In contrast, there was a 100% increase in proliferation from 48–96h in the presence of SL0101 (100 μM). These data indicate that the modifications to generate the SL0101 analogue (1b) resulted in a more potent RSK inhibitor than the parent compound.
Figure 1. C6″-n-propyl cyclitol SL0101 (1b) shows improved potency compared to the parent compound.

(A) Structure and IC50 for selected SL0101 analogues. (B) Efficacy of (1a) and (1b) in inhibiting proliferation of MCF-7 and MCF-10A cells. Symbol, mean ± S.D. (n ≥ 2, triplicate; *p < 0.01 compared to vehicle). (C) The in vitro stability of (1b) (25 μM) is increased in comparison to (1a) (100 μM). Bar, mean (n=2, quadruplicate; *p < 0.0001). (D) Analysis of lysates from MCF-7 cells pre-treated with (1a), (1b) or DMSO for 2h and treated with or without 500 nM PMA (20 min). (E) Representative images of MCF-7 cells treated as in D. Scale bar = 10 μm. Bar graph showing the decrease in pS6. (n ≥ 30 cells). (F) Representation of (1b) specificity in a kinase screen indicating % inhibition at 10 μM compared to RSK2. (G) Bioluminescence images of NSG mice at day 50 after IC injection with MCF-7-Luc cells. (H) Representative paraffin-embedded tibia sections from mice in (G) treated with (40 mg/kg) or vehicle 2h prior to euthanasia. Scale bar = 40 μm. Bar graph showing the decrease in pS6. (n= 6 sections/mouse).
Specificity of C6″-n-propyl cyclitol SL0101 (1b) for RSK1/2
SL0101 (1a) is highly selective for RSK (14, 21), which is most likely due to the fact that SL0101 inhibits RSK by an allosteric mechanism (22). Therefore, to evaluate the specificity of (1b) we compared its ability to inhibit RSK substrates in comparison to SL0101 (1a). In agreement with previous results, SL0101 induces an increase in the phosphorylation of eukaryotic elongation factor 2 (p-eEF2) in MCF-7 cells, which also occurred with (1b) (Fig. 1D). This increase is due to the activation of eEF2 kinase, which is inhibited by RSK (34). Furthermore, both RSK inhibitors decreased the phosphorylation of Ser167-ERα, an important marker for anti-estrogen responsiveness (35). SL0101 and (1b) also decreased the phosphorylation of the ribosomal protein, S6 (pS6), a known RSK downstream effector (Fig. 1E) (36). Previously, we identified that silencing RSK2 reduced cyclin D1 levels (37), and consistent with these results RSK inhibition decreased cyclin D1 levels. In a more global analysis, in vitro kinase assays were performed against a panel of 247 purified kinases, which contained representatives from all kinase families (Supplementary Fig. S1). At 10 μM of (1b), RSK1 and RSK2 were the top hits, with colony stimulating factor 1 receptor (CSF1R) and mitogen-activated protein kinase kinase kinase kinase (MAP4K4) being inhibited by ~ 37 % compared to RSK2 (Fig. 1F). CSF1R regulates macrophage function, and inhibitors are currently in development as cancer therapies (38). MAP4K4 is an endothelial protein kinase, and inhibitors are being developed as anti-diabetic drugs (39). Thus, the off target effects of (1b) are very limited. Neither of these off-target effects is viewed as problematic for further drug development. Taken together, these data demonstrate that (1b) is very specific for RSK1/2.
RSK Inhibition in vivo
The overall goal of our studies is to develop a RSK inhibitor for in vivo use. To evaluate the ability of (1b) to inhibit RSK1/2 in vivo we used an MCF-7 metastatic model because most of our prior characterization of SL0101 was performed using this line. MCF-7 cells that stably express luciferase (MCF7-Luc) were introduced by intracardiac (IC) injection into NSG mice, and metastasis were established for ~ 50 days. Before treatment we determined that the tumor burden between animals was equivalent (Fig. 1G). Two hours after treatment with (1b) or vehicle the animals were euthanized and the tibia isolated, as ER+ tumors frequently metastasize to the bone. Moreover, MCF-7 cells within the tibia were easily identified by their positive staining with cytokeratin 8 (K8) (Fig. 1H). The levels of the RSK target, pS6, were decreased by > 2.5-fold with (1b). These results demonstrate that (1b) is able to attain a sufficient concentration to induce pharmacodynamic changes in vivo.
RSK as a drug target for TNBC
RSK has been proposed as a drug target for TNBC based on observations that ~ 85% of TNBC tumors have activated RSK (19). In agreement with these observations we found that the levels of activated RSK (pRSK) were higher in TNBC tumors than normal tissue (Fig. 2A, 2B and Supplementary Table S1). The levels of activated RSK varied considerably within and between tumors. Moreover, in TNBC tumor tissue activated RSK could be present in the nucleus, cytoplasm or both whereas it was mainly cytoplasmic in normal breast cells. The differences in subcellular localization suggest that the substrates regulated by RSK differ between normal and TNBC tissue. Taken together, these results are consistent with RSK as a viable drug target for TNBC.
Figure 2. Active RSK in TNBC.

(A) Activated RSK levels are increased in TNBC. Bar, median ± quartile (n ≥ 5 field/tissue sample). (B) Representative paraffin-embedded sections of normal breast and TNBC tissue stained for the cytokeratins 8 (K8), 14 (K14) and phospho-Thr359/phospho-Ser363 RSK (pRSK). Scale bar = 20 μm. (C) Analysis of TNBC cell lysates normalized using the housekeeping protein, RAN. (D) Quantitation of the levels of pRSK normalized to RAN. (n=3) (E) Comparison of pRSK relative to total RSK1 and RSK2 for various TNBC lines. To control for antibody sensitivity the levels of RSK1 and RSK2 were determined using purified, recombinant protein.
To investigate whether activated RSK was functionally important in TNBC we chose a panel of 8 cell lines representing 5 different TNBC subtypes (40). We observed that activated RSK was present at different levels in these lines (Fig. 2C and 2D). In 2D culture the proliferation of all the TNBC lines was inhibited at a lower concentration of (1b) than SL0101 (1a) (Supplementary Fig. S2A). The lines from the mesenchymal subtype, CAL-120 and MDA-MB-231, were relatively resistant whereas the basal-like 2 (BL2), HDQ-P1 and HCC70 were among the most sensitive (Fig. 3A). The BL2 lines are of interest clinically because this subtype is correlated with the poorest response to neoadjuvant chemotherapy (41). To better understand these observations we compared the levels of activated RSK normalized to total RSK1 and RSK2, which should reflect RSK1/2 specific activity (Fig. 2E). The anti-RSK2 antibody is less sensitive than the anti-RSK1 antibody and this difference was accounted for by normalizing to recombinant proteins. We observed an inverse relationship between the IC50 for (1b) and the specific activity of the combined isoforms (Supplementary Fig. S2B), consistent with the hypothesis that higher RSK specific activity increases sensitivity to the inhibitor. In a separate analysis active RSK was normalized to RSK2 or RSK1 separately and a statistically significant inverse correlation was observed for RSK2 (Fig. 3B).
Figure 3. RSK is required for TNBC proliferation, survival and motility.

(A) IC50s for (1b) in MCF-7 and TNBC lines. Bar, median ± range (n ≥ 2, ≥ quadruplicate). (B) Correlation of IC50 for inhibition of proliferation by (1b) of TNBC lines versus activated RSK normalized to total RSK2 levels. (C) Analysis of lysates from MDA-MB-231 cells transduced with scramble (scrbl) or double transduced with RSK1/2 targeting shRNAs. Bar: non-relevant lanes removed. ns:nonspecific (D) Efficacy of (1b) in inhibiting proliferation of MDA-MB-231 cells transduced as in (C). Symbol, mean ± S.D. (n ≥ 2, triplicate; *p < 0.03 compared to vehicle). (E) Bar graph showing (1b) IC50 for MDA-MB-231 proliferation in 2D and 3D. Bar, median ± range (n ≥ 2, ≥ quadruplicate). (F) IC50s for (1b) for survival of TNBC lines. Bar, median ± range (n ≥ 2, triplicate). (G) Efficacy of (1b) in inhibiting survival of MDA-MB-231 cells transduced as in (B). Symbol, mean ± S.D (n ≥ 2, triplicate; *p < 0.01 compared to vehicle). Scatter plots showing efficacy of (1a) and (1b) in inhibiting motility of (H) MDA-MB-231, (I) HCC70 and (J) HDQ-P1. Each circle represents a cell trace. Bar, median (n ≥ 2, 30 cells/treatment).
To evaluate specificity we investigated the efficacy of (1b) in the context of RSK1/2 silencing. As expected, loss of RSK1/2 decreased 2D proliferation by ~ 60% in MDA-MB-231 cells (Figs. 3C, D). Importantly, silencing RSK1/2 resulted in loss of sensitivity to (1b) (Fig. 3D). These results support the conclusion that (1b) is specific for RSK1/2 and also demonstrate that RSK1/2 are primarily responsible for regulating the proliferation of MDA-MB-231 cells.
Proliferation of the MDA-MB-231 line is reported to be more sensitive to RSK inhibition in 3D versus 2D (27). In agreement we observed that the IC50 for (1b) is ~ 8 μM in 3D and ~ 50 μM in 2D (Fig. 3E). Surprisingly, HDQ-P1 and HCC70 were unable to proliferate in 3D, suggesting that these lines have more stringent requirements for proliferation than MDA-MB-231.
The ability of cancer cells to survive in circulation is an important step in metastasis (42) and therefore, we analyzed survival in ultra low adhesion plates. Survival of HDQ-P1, HCC70, and MDA-MB-231 was dependent on RSK and the IC50 for inhibition of survival by (1b) was ~ 30, 15 and 3 μM, respectively (Fig. 3F). Silencing RSK1/2 in MDA-MB-231 cells decreased survival by ~ 75% and was not further inhibited by (1b) (Fig. 3G). These results demonstrate that the survival of some TNBC lines depends on RSK and confirm that (1b) is a very specific RSK inhibitor.
RSK has been implicated in regulating motility (14) and we investigated this possibility using the scratch assay. In all lines tested, (1b) reduced cell velocity to the same extent as SL0101 but at lower concentrations (Fig. 3H–J and Supplementary Fig. S3A–C and Supplementary Movies S1–3). The motility of HCC70 was reduced by ~ 50%, and in MDA-MB-231 and HDQ-P1 cells motility was decreased by at least 75%. Apoptosis was not detected with the doses and time course used in the scratch assay (Supplementary Fig. S3D). Taken together, our results demonstrate that inhibition of RSK by (1b) reduces proliferation, survival in a non-adherent environment and motility, which are essential components of the metastatic process.
Silencing RSK decreases TNBC metastasis in vivo
To identify the contributions of RSK1 and RSK2 to metastasis we used an in vivo metastatic MDA-MB-231 model in which luciferase was stably expressed (MDA-MB-231-Luc). MDA-MB-231-Luc cells were transduced with control, RSK1 or RSK2 specific shRNAs (Supplementary Fig. S4A). The cells were quality controlled for their luciferase signal, and equal numbers of cells were introduced by IC injection into female NSG mice (Supplementary Fig. S4B). This model will identify whether RSK1 or RSK2 contribute to the metastatic processes that includes metastatic colonization and proliferation at the metastatic site. At day 19 silencing RSK1 or RSK2 decreased the total metastatic burden, as determined by bioluminescence, by > three-fold (Fig. 4A, B and Supplementary Fig. S4C). This decrease in metastatic burden is further supported by the observations that silencing RSK1 or RSK2 increased survival by ~ 40–60% (Fig. 4C). Silencing RSK1 or RSK2 reduced the number of metastatic foci by ~ half (Fig. 4D), and remained constant over the duration of the experiment. The number of bioluminescent foci was linearly correlated with the number of metastatic foci as determined by histology (Supplementary Fig. S4D). Therefore, we conclude that the increased whole animal bioluminescence from day 5 onwards reflects proliferation at the metastatic sites (Fig. 4E). Thus, silencing RSK1 or RSK2 decreased proliferation from day twelve to day nineteen > three-fold compared to the control. We also conclude that the decrease in metastatic foci reflects that RSK1 or RSK2 is necessary for metastatic colonization. This decrease in metastatic foci was not organ dependent (Fig. 4F). Ex vivo analysis of bioluminescence was also performed as it improved the resolution for determining individual metastatic foci. These results of the ex vivo bioluminescence (Fig. 4G, H) and the histological analysis (Supplementary Fig. S4E, F) were consistent. We conclude that RSK1/2 regulate numerous steps that comprise the metastatic process, which results in improved survival.
Figure 4. RSK1 and RSK2 contribute to the metastatic phenotype.

(A) Representative bioluminescence images of NSG mice injected IC with MDA-MB-231-Luc cells transduced with scramble (scrbl), RSK1- or RSK2-targeting shRNAs (t=19 d). (B) RSK1 and RSK2 decreased the metastatic burden in mice from (A) (t=19 d). (n = 8 mice/group). (C) Kaplan-Meier curves from (A). (n= 8 mice/group; test = log-rank). (D) The number of metastatic foci is constant in mice from (A). Symbol, mean. (E) Total bioluminescence in mice from (A) is decreased with RSK1 or RSK2 silencing. Each line represents a mouse; the data are fold change over day 6. (F) Loss of RSK1 or RSK2 decreased the number of metastatic foci in numerous organs in mice from (A) (t=19 d). Bar, mean ± SD (n = 8 mice/group). (G) Representative ex vivo bioluminescence images of livers of mice from (A). (H) Ex vivo analysis confirms that silencing RSK1 or RSK2 decreased the number of metastatic foci in the livers from (A). Bar, median ± quartile (n=4 mice/group).
Inhibition of RSK decreases metastatic colonization
We investigated whether (1b) would be sufficiently efficacious to decrease metastatic colonization in vivo. For these experiments we used the HDQ-P1 model because of the clinical importance of the BL2 subtype. This model has not previously been used as an in vivo metastatic model. To validate the model, HDQ-P1 cells were transduced with luciferase (HDQ-P1-Luc) and introduced by IC injection into male NSG mice. At 24 h after injection, the cells were widespread through the animal, but by day five the cells were primarily localized to the liver, adrenal glands and testes (Supplementary Fig. S5). This model is in contrast to the widely used MDA-MB-231 IC metastatic model, in which the cells primarily metastasize to the bone. TNBC primarily metastasizes to lymph nodes and viscera more often than to the bone, and the HDQ-P1 model better recapitulates these clinical observations.
To evaluate the efficacy of (1b), we compared it to a drug that is in the same class as (1b) and therefore, would be expected to generate a similar phenotype. The MEK inhibitor, trametinib, is approved for melanoma and is currently in multiple clinical trials including those for breast cancer (43). MEK inhibition will decrease ERK1/2 activity and reduce RSK activation (13). HDQ-P1-Luc cells were introduced by IC injection into female NSG mice and treatment began 2 h after injection. The animals were imaged just before treatment to ensure viability and distribution of the cells in vivo. This approach recapitulates the clinical scenario of tumor cells within the circulation, which have been proposed to act as a negative prognostic marker and demonstrate similar therapeutic responsiveness as the metastatic tumor (1). By 24 h both (1b) and trametinib decreased the total in vivo bioluminescence by three-fold (Fig. 5A, B). Moreover, the number of metastatic foci in both the skeleton and the viscera was reduced by drug treatment (Fig. 5C). Treatments were stopped on day 5 and on day 6 the total in vivo bioluminescence was reduced three-fold by drug treatment in comparison to the control (Fig. 5D). To confirm these findings we measured the bioluminescence of the liver and adrenal glands ex vivo and observed a five-fold reduction in metastatic burden in mice treated with either drug (Fig. 5E–H). We conclude that inhibition of RSK or its upstream activator, MEK, decreases metastatic colonization. Moreover, these observations with HDQ-P1 confirm those obtained with MDA-MB-231.
Figure 5. Pharmacological inhibition of metastatic colonization by (1b).

(A) Bioluminescence images of NSG mice injected IC with HDQ-P1-Luc cells at t=1h and 24h after injection. At 2h after injection mice were treated with vehicle, (1b) (40 mg/kg) IP Q12h or trametinib (tram) (2 mg/kg) IP Q24h. Inhibition of RSK or MEK decreases total metastatic burden (B) and the number of metastatic foci in individual organs (t=24h) (C). Bar, mean ± SD (n=4 mice/group, *p<0.05). (D) Inhibition of RSK or MEK decrease total metastatic burden (t=6d). (n=4 mice/group). Representative ex vivo bioluminescence images of livers (E) and adrenal glands (G) (t=6d). Ex vivo analysis confirms that inhibiting RSK or MEK activity decreased the metastatic burden in livers (F) and adrenals (H). (n=4 mice/group).
Inhibition of RSK does not activate AKT
Inhibiting “global regulators” such as MEK results in a number of side effects and their ability to induce an effective clinical response appears limited (12). MEK inhibition can result in activation of AKT (44) and based on these results there are clinical trials underway combining MEK inhibitors with an AKT or PI3K inhibitor (45). Consistent with the literature, we observed that treatment of MDA-MB-231 cells with trametinib enhanced the levels of phosphoSer 473 AKT (pAKT), which is necessary for AKT activity (Fig. 6A). In contrast, activation of AKT was not observed in response to (1b). In comparison to MDA-MB-231, HDQ-P1 have high basal levels of active AKT but consistent with the results observed in MDA-MB-231, (1b) did not increase AKT activity in contrast to trametinib (Fig. 6B). Taken together, these results indicate that RSK inhibition by itself will effectively target the TNBC metastatic process but not have the undesirable side effect of activating AKT.
Figure 6. (1b) does not activate AKT.
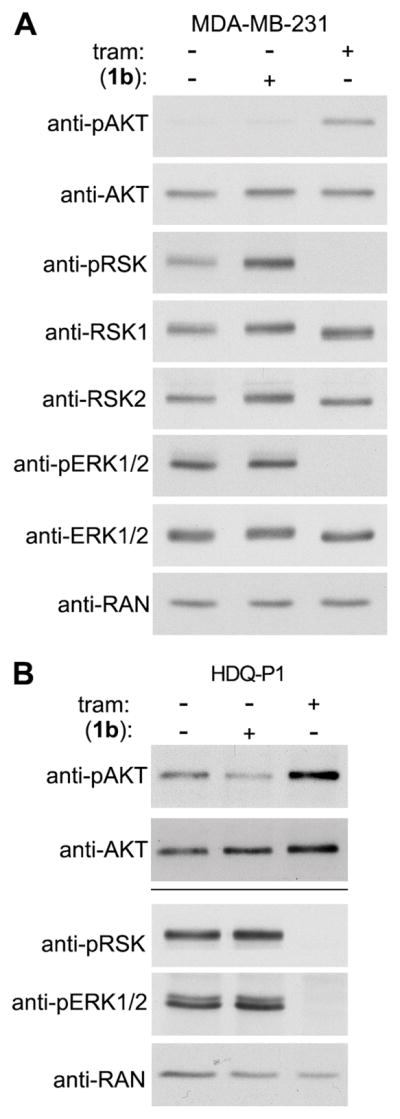
Analysis of lysates from (A) MDA-MB-231 and (B) HDQ-P1 cells treated with vehicle, trametinib (1 μM) or (1b) (25 μM) for 2h. Bar = separate gels.
Discussion
The importance of RSK in regulating metastasis in vivo has not been thoroughly investigated. Kang et al (46) reported that silencing RSK2 decreased metastatic colonization to the lymph nodes using a human head and neck squamous cell carcinoma line. They further followed up on these observations using the RSK CTKD inhibitor, FMK-MEA, which resulted in a modest decrease in metastatic tumor burden from 97% to 79% (47). In a screen Lara et al (48) identified that loss of RSK1 increased motility in lung cancer lines but in contrast Zhou et al (49) found that inhibition of RSK activity was associated with decreased motility in lung cancer lines. It is possible that the discrepancy between these studies results from the ability of RSK1 to act as a scaffold and regulate other signaling pathways. RSK has also been proposed as a drug target for TNBC based on observations that it decreased the levels of the surface marker CD44, which is reported to be associated with cancer stem cells (19). We demonstrated using genetic and pharmacological approaches in vitro and in vivo that reducing RSK1/2 activity or RSK1 or RSK2 levels inhibits multiple steps within the metastatic program. Furthermore, we confirmed that activated RSK is present in the majority of TNBCs. Our results strongly suggest that RSK is a viable drug target for TNBC metastasis.
We also report the generation and validation of a novel SL0101 analogue, C6″-n-propyl cyclitol SL0101 (1b) that is specific for RSK1/2. The specificity of the inhibitor for RSK1/2 is demonstrated by our observations that silencing RSK1/2 eliminates responsiveness to (1b) in MDA-MB-231 proliferation and survival assays. In in vitro assays using multiple TNBC cell lines the new analogue inhibits the major steps involved in the metastatic process, which include motility, proliferation and survival in a non-adherent environment. Additionally, (1b) was as effective at inhibiting metastatic colonization in vivo as the FDA-approved MEK inhibitor, trametinib. Activation of the AKT pathway is proposed as a mechanism to account for the lack of efficacy of selumetinib, a MEK inhibitor, in combination with the anti-estrogen, fulvestrant, in a phase II clinical trial (50). We propose that inhibitors of RSK will offer greater flexibility in designing combination cancer therapies than MEK inhibitors, as there will be no positive feedback loop that results in activation of AKT.
Supplementary Material
Acknowledgments
Financial Support:
Susan G. Komen #IIR12223770 (D.A. Lannigan), NIH GM088839 (G.A. O’Doherty.) and NSF CHE-0749451 (G.A. O’Doherty)
Kinome illustrations reproduced courtesy of Cell Signaling Technology, Inc (www.cellsignal.com). We thank the Univ. Virginia Biorepository and Tissue Procurement Facility for human breast samples and P. Brulet and R. Kemler, Developmental Studies Hybridoma Bank, through the NICHD, Dept. Biology, Univ. Iowa for the cytokeratin Endo-A (K8) monoclonal.
Footnotes
No conflicts.
References
- 1.Banys-Paluchowski M, Krawczyk N, Meier-Stiegen F, Fehm T. Circulating tumor cells in breast cancer-current status and perspectives. Critical reviews in oncology/hematology. 2016;97:22–9. doi: 10.1016/j.critrevonc.2015.10.010. [DOI] [PubMed] [Google Scholar]
- 2.Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13:4429–34. doi: 10.1158/1078-0432.CCR-06-3045. [DOI] [PubMed] [Google Scholar]
- 3.Badve S, Dabbs DJ, Schnitt SJ, Baehner FL, Decker T, Eusebi V, et al. Basal-like and triple-negative breast cancers: a critical review with an emphasis on the implications for pathologists and oncologists. Mod Pathol. 2011;24:157–67. doi: 10.1038/modpathol.2010.200. [DOI] [PubMed] [Google Scholar]
- 4.Craig DW, O’Shaughnessy JA, Kiefer JA, Aldrich J, Sinari S, Moses TM, et al. Genome and transcriptome sequencing in prospective metastatic triple-negative breast cancer uncovers therapeutic vulnerabilities. Mol Cancer Ther. 2013;12:104–16. doi: 10.1158/1535-7163.MCT-12-0781. [DOI] [PubMed] [Google Scholar]
- 5.Infante JR, Papadopoulos KP, Bendell JC, Patnaik A, Burris HA, 3rd, Rasco D, et al. A phase 1b study of trametinib, an oral Mitogen-activated protein kinase kinase (MEK) inhibitor, in combination with gemcitabine in advanced solid tumours. Eur J Cancer. 2013;49:2077–85. doi: 10.1016/j.ejca.2013.03.020. [DOI] [PubMed] [Google Scholar]
- 6.Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell. 2012;149:307–21. doi: 10.1016/j.cell.2012.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giltnane JM, Balko JM. Rationale for targeting the Ras/MAPK pathway in triple-negative breast cancer. Discov Med. 2014;17:275–83. [PubMed] [Google Scholar]
- 8.Network TCGA. Comprehensive molecular portraits of human breast tumors. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cossu-Rocca P, Orru S, Muroni MR, Sanges F, Sotgiu G, Ena S, et al. Analysis of PIK3CA Mutations and Activation Pathways in Triple Negative Breast Cancer. PLoS One. 2015;10:e0141763. doi: 10.1371/journal.pone.0141763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoeflich KP, O’Brien C, Boyd Z, Cavet G, Guerrero S, Jung K, et al. In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin Cancer Res. 2009;15:4649–64. doi: 10.1158/1078-0432.CCR-09-0317. [DOI] [PubMed] [Google Scholar]
- 11.Jing J, Greshock J, Holbrook JD, Gilmartin A, Zhang X, McNeil E, et al. Comprehensive predictive biomarker analysis for MEK inhibitor GSK1120212. Mol Cancer Ther. 2012;11:720–9. doi: 10.1158/1535-7163.MCT-11-0505. [DOI] [PubMed] [Google Scholar]
- 12.Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nature reviews Drug discovery. 2014;13:928–42. doi: 10.1038/nrd4281. [DOI] [PubMed] [Google Scholar]
- 13.Eisinger-Mathason TS, Andrade J, Lannigan DA. RSK in tumorigenesis: connections to steroid signaling. Steroids. 2010;75:191–202. doi: 10.1016/j.steroids.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doehn U, Hauge C, Frank SR, Jensen CJ, Duda K, Nielsen JV, et al. RSK is a principal effector of the RAS-ERK pathway for eliciting a coordinate promotile/invasive gene program and phenotype in epithelial cells. Mol Cell. 2009;35:511–22. doi: 10.1016/j.molcel.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Larrea MD, Hong F, Wander SA, da Silva TG, Helfman D, Lannigan D, et al. RSK1 drives p27Kip1 phosphorylation at T198 to promote RhoA inhibition and increase cell motility. Proc Natl Acad Sci U S A. 2009;106:9268–73. doi: 10.1073/pnas.0805057106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vial D, McKeown-Longo PJ. Epidermal growth factor (EGF) regulates alpha5beta1 integrin activation state in human cancer cell lines through the p90RSK-dependent phosphorylation of filamin A. J Biol Chem. 2012;287:40371–80. doi: 10.1074/jbc.M112.389577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gawecka JE, Young-Robbins SS, Sulzmaier FJ, Caliva MJ, Heikkila MM, Matter ML, et al. RSK2 protein suppresses integrin activation and fibronectin matrix assembly and promotes cell migration. J Biol Chem. 2012;287:43424–37. doi: 10.1074/jbc.M112.423046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smolen GA, Zhang J, Zubrowski MJ, Edelman EJ, Luo B, Yu M, et al. A genome-wide RNAi screen identifies multiple RSK-dependent regulators of cell migration. Genes Dev. 2010;24:2654–65. doi: 10.1101/gad.1989110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stratford AL, Reipas K, Hu K, Fotovati A, Brough R, Frankum J, et al. Targeting p90 ribosomal S6 kinase eliminates tumor-initiating cells by inactivating Y-box binding protein-1 in triple-negative breast cancers. Stem cells. 2012;30:1338–48. doi: 10.1002/stem.1128. [DOI] [PubMed] [Google Scholar]
- 20.Smith JA, Poteet-Smith CE, Xu Y, Errington TM, Hecht SM, Lannigan DA. Identification of the first specific inhibitor of p90 ribosomal S6 kinase (RSK) reveals an unexpected role for RSK in cancer cell proliferation. Cancer Res. 2005;65:1027–34. [PubMed] [Google Scholar]
- 21.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Utepbergenov D, Derewenda U, Olekhnovich N, Szukalska G, Banerjee B, Hilinski MK, et al. Insights into the Inhibition of the p90 Ribosomal S6 Kinase (RSK) by the Flavonol Glycoside SL0101 from the 1.5 A Crystal Structure of the N-Terminal Domain of RSK2 with Bound Inhibitor. Biochemistry. 2012;51:6499–510. doi: 10.1021/bi300620c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sapkota GP, Kieloch A, Lizcano JM, Lain S, Arthur JS, Williams MR, et al. Phosphorylation of the protein kinase mutated in Peutz-Jeghers cancer syndrome, LKB1/STK11, at Ser431 by p90(RSK) and cAMP-dependent protein kinase, but not its farnesylation at Cys(433), is essential for LKB1 to suppress cell vrowth. J Biol Chem. 2001;276:19469–82. doi: 10.1074/jbc.M009953200. [DOI] [PubMed] [Google Scholar]
- 24.Kirrane TM, Boyer SJ, Burke J, Guo X, Snow RJ, Soleymanzadeh L, et al. Indole RSK inhibitors. Part 2: optimization of cell potency and kinase selectivity. Bioorg Med Chem Lett. 2012;22:738–42. doi: 10.1016/j.bmcl.2011.10.029. [DOI] [PubMed] [Google Scholar]
- 25.Edgar AJ, Trost M, Watts C, Zaru R. A combination of SILAC and nucleotide acyl phosphate labelling reveals unexpected targets of the Rsk inhibitor BI-D1870. Bioscience reports. 2014;34:e00091. doi: 10.1042/BSR20130094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fryer RM, Muthukumarana A, Chen RR, Smith JD, Mazurek SN, Harrington KE, et al. Mitigation of off-target adrenergic binding and effects on cardiovascular function in the discovery of novel ribosomal S6 kinase 2 inhibitors. J Pharmacol Exp Ther. 2012;340:492–500. doi: 10.1124/jpet.111.189365. [DOI] [PubMed] [Google Scholar]
- 27.Aronchik I, Appleton BA, Basham SE, Crawford K, Del Rosario M, Doyle LV, et al. Novel potent and selective inhibitors of p90 ribosomal S6 kinase reveal the heterogeneity of RSK function in MAPK-driven cancers. Mol Cancer Res. 2014;12:803–12. doi: 10.1158/1541-7786.MCR-13-0595. [DOI] [PubMed] [Google Scholar]
- 28.Jain R, Mathur M, Lan J, Costales A, Atallah G, Ramurthy S, et al. Discovery of Potent and Selective RSK Inhibitors as Biological Probes. J Med Chem. 2015 doi: 10.1021/acs.jmedchem.5b00450. [DOI] [PubMed] [Google Scholar]
- 29.Cohen MS, Hadjivassiliou H, Taunton J. A clickable inhibitor reveals context-dependent autoactivation of p90 RSK. Nature chemical biology. 2007;3:156–60. doi: 10.1038/nchembio859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cohen MS, Zhang C, Shokat KM, Taunton J. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science. 2005;308:1318–21. doi: 10.1126/science1108367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Serafimova IM, Pufall MA, Krishnan S, Duda K, Cohen MS, Maglathlin RL, et al. Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nature chemical biology. 2012;8:471–6. doi: 10.1038/nchembio.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mrozowski RM, Vemula R, Wu B, Zhang Q, Schroeder BR, Hilinski MK, et al. Improving the affinity of SL0101 for RSK using structure-based design. ACS Med Chem Lett. 2012;4:175–9. doi: 10.1021/ml300298v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li M, Li Y, Mrozowski RM, Sandusky ZM, Shan M, Song X, et al. Synthesis and Structure-Activity Relationship Study of 5a-Carbasugar Analogues of SL0101. ACS Med Chem Lett. 2015;6:95–9. doi: 10.1021/ml5004525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X, Li W, Williams M, Terada N, Alessi DR, Proud CG. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J. 2001;20:4370–9. doi: 10.1093/emboj/20.16.4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clark DE, Poteet-Smith CE, Smith JA, Lannigan DA. Rsk2 allosterically activates estrogen receptor alpha by docking to the hormone-binding domain. EMBO J. 2001;20:3484–94. doi: 10.1093/emboj/20.13.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roux PP, Shahbazian D, Vu H, Holz MK, Cohen MS, Taunton J, et al. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J Biol Chem. 2007;282:14056–64. doi: 10.1074/jbc.M700906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eisinger-Mathason TS, Andrade J, Groehler AL, Clark DE, Muratore-Schroeder TL, Pasic L, et al. Codependent functions of RSK2 and the apoptosis-promoting factor TIA-1 in stress granule assembly and cell survival. Mol Cell. 2008;31:722–36. doi: 10.1016/j.molcel.2008.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ries CH, Hoves S, Cannarile MA, Ruttinger D. CSF-1/CSF-1R targeting agents in clinical development for cancer therapy. Current opinion in pharmacology. 2015;23:45–51. doi: 10.1016/j.coph.2015.05.008. [DOI] [PubMed] [Google Scholar]
- 39.Ammirati M, Bagley SW, Bhattacharya SK, Buckbinder L, Carlo AA, Conrad R, et al. Discovery of an in Vivo Tool to Establish Proof-of-Concept for MAP4K4-Based Antidiabetic Treatment. ACS Med Chem Lett. 2015;6:1128–33. doi: 10.1021/acsmedchemlett.5b00215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750–67. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lehmann BD, Pietenpol JA. Identification and use of biomarkers in treatment strategies for triple-negative breast cancer subtypes. J Pathol. 2014;232:142–50. doi: 10.1002/path.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pantel K, Speicher MR. The biology of circulating tumor cells. Oncogene. 2015 doi: 10.1038/onc.2015.192. [DOI] [PubMed] [Google Scholar]
- 43.Neuzillet C, Tijeras-Raballand A, de Mestier L, Cros J, Faivre S, Raymond E. MEK in cancer and cancer therapy. Pharmacol Ther. 2014;141:160–71. doi: 10.1016/j.pharmthera.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 44.Turke AB, Song Y, Costa C, Cook R, Arteaga CL, Asara JM, et al. MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Res. 2012;72:3228–37. doi: 10.1158/0008-5472.CAN-11-3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jokinen E, Koivunen JP. MEK and PI3K inhibition in solid tumors: rationale and evidence to date. Ther Adv Med Oncol. 2015;7:170–80. doi: 10.1177/1758834015571111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kang S, Elf S, Lythgoe K, Hitosugi T, Taunton J, Zhou W, et al. p90 ribosomal S6 kinase 2 promotes invasion and metastasis of human head and neck squamous cell carcinoma cells. J Clin Invest. 2010;120:1165–77. doi: 10.1172/JCI40582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li D, Jin L, Alesi GN, Kim YM, Fan J, Seo JH, et al. The prometastatic ribosomal S6 kinase 2-cAMP response element-binding protein (RSK2-CREB) signaling pathway up-regulates the actin-binding protein fascin-1 to promote tumor metastasis. J Biol Chem. 2013;288:32528–38. doi: 10.1074/jbc.M113.500561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lara R, Mauri FA, Taylor H, Derua R, Shia A, Gray C, et al. An siRNA screen identifies RSK1 as a key modulator of lung cancer metastasis. Oncogene. 2011;30:3513–21. doi: 10.1038/onc.2011.61. [DOI] [PubMed] [Google Scholar]
- 49.Zhou Y, Yamada N, Tanaka T, Hori T, Yokoyama S, Hayakawa Y, et al. Crucial roles of RSK in cell motility by catalysing serine phosphorylation of EphA2. Nat Commun. 2015;6:7679. doi: 10.1038/ncomms8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zaman K, Winterhalder R, Mamot C, Hasler-Strub U, Rochlitz C, Mueller A, et al. Fulvestrant with or without selumetinib, a MEK 1/2 inhibitor, in breast cancer progressing after aromatase inhibitor therapy: A multicentre randomised placebo-controlled double-blind phase II trial, SAKK 21/08. Eur J Cancer. 2015;51:1212–20. doi: 10.1016/j.ejca.2015.03.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.