

Abstract
Protein quality control in cardiomyocytes is crucial to maintain cellular homeostasis. The accumulation of damaged organelles, such as mitochondria and misfolded proteins in the heart is associated with heart failure. During the process to identify novel mitochondria-specific autophagy (mitophagy) receptors, we found FK506-binding protein 8 (FKBP8), also known as FKBP38, shares similar structural characteristics with a yeast mitophagy receptor, autophagy-related 32 protein. However, knockdown of FKBP8 had no effect on mitophagy in HEK293 cells or H9c2 myocytes. Since the role of FKBP8 in the heart has not been fully elucidated, the aim of this study is to determine the functional role of FKBP8 in the heart. Cardiac-specific FKBP8-deficient (Fkbp8−/−) mice were generated. Fkbp8−/− mice showed no cardiac phenotypes under baseline conditions. The Fkbp8−/− and control wild type littermates (Fkbp8+/+) mice were subjected to pressure overload by means of transverse aortic constriction (TAC). Fkbp8−/− mice showed left ventricular dysfunction and chamber dilatation with lung congestion 1 week after TAC. The number of apoptotic cardiomyocytes was dramatically elevated in TAC-operated Fkbp8−/− hearts, accompanied with an increase in protein levels of cleaved caspase-12 and endoplasmic reticulum (ER) stress markers. Caspase-12 inhibition resulted in the attenuation of hydrogen peroxide-induced apoptotic cell death in FKBP8 knockdown H9c2 myocytes. Immunocytological and immunoprecipitation analyses indicate that FKBP8 is localized to the ER and mitochondria in the isolated cardiomyocytes, interacting with heat shock protein 90. Furthermore, there was accumulation of misfolded protein aggregates in FKBP8 knockdown H9c2 myocytes and electron dense deposits in perinuclear region in TAC-operated Fkbp8−/− hearts. The data suggest that FKBP8 plays a protective role against hemodynamic stress in the heart mediated via inhibition of the accumulation of misfolded proteins and ER-associated apoptosis.
Abbreviations: αMHC-Cre, α-myosin heavy chain promoter-driven Cre recombinase; BCL2L13, Bcl-2-like protein 13; CCCP, mitochondrial uncoupler carbonyl cyanide m-chlorophenylhydrazone; ER, endoplasmic reticulum; FKBP8, FK506-binding protein 8; GRP78, glucose-regulated protein 78 kDa; GRP94, glucose-regulated protein 94 kDa; HSP90, heat shock protein 90; LC3, microtubule associated protein 1 light chain 3; mTOR, mammalian target of rapamycin; PPIase, peptidyl prolyl cis/trans-isomerase; siRNA, small interfering RNA; TAC, transverse aortic constriction; TUNEL, terminal deoxynucleotidyl transferase mediated dUTP nick end-labeling
Keywords: FKBP8, Heart failure, Apoptosis, ER stress, Protein misfolding
Highlights
-
•
This study is the first to identify the in vivo role of FKBP8 in the heart.
-
•
Cardiac-specific FKBP8-deficient mice were generated.
-
•
FKBP8 plays a cardio-protective role in response to pressure overload.
-
•
FKBP8 has anti-apoptotic functions via endoplasmic reticulum-mediated pathway.
-
•
FKBP8 prevents the accumulation of misfolded proteins.
1. Introduction
Heart failure is one of the most common diseases affecting people worldwide and is associated with an increased risk of morbidity and mortality [1]. The accumulation of damaged proteins and organelles in failing hearts may be involved in the pathogenesis of heart disease [2]. As cardiomyocytes are terminally differentiated and long-lived cells, it is crucial to control the quality of proteins and organelles, such as mitochondria, under stress [3]. Chaperone/co-chaperone systems are engaged in protein folding and refolding, failure of which results in cellular toxicity [4]. The accumulation of misfolded or unfolded proteins in the endoplasmic reticulum (ER) lumen, known as ER stress, activates unfolded protein response to maintain ER homeostasis by increasing ER-resident chaperones, inhibiting protein translation and accelerating the degradation of unfolded proteins [5].
The ubiquitin-proteasome system and autophagy lysosomal pathway are important for the degradation of intracellular components [6]. The ubiquitin-proteasome system is involved in degrading misfolded proteins via proteolysis through an enzymatic cascade of ubiquitination, while autophagy is responsible for the bulk turnover of long-lived proteins and organelles. Autophagy includes not only nonselective autophagy, but selective autophagy, which targets specific proteins or organelles. Selective autophagy targeting mitochondria, mitophagy, is believed to be vital to remove damaged mitochondria. In yeast, autophagy-related (Atg) 32 protein is an essential mitophagy receptor, comprising a single transmembrane domain spanning the outer mitochondrial membrane and a WXXI motif, which binds to Atg8 [7]. We hypothesized that a mammalian mitophagy receptor would share the following molecular features with Atg32: mitochondrial localization, WXXL/I motifs, and single membrane-spanning topology. The public protein database was screened for novel mammalian Atg32 functional homologue using the molecular profile of Atg32 as a search tool, identifying Bcl-2-like protein 13 (BCL2L13) that mediates mitophagy and mitochondrial fragmentation [8]. In addition to BCL2L13, FK506-binding protein 8 (FKBP8), also known as FKBP38, satisfied the screening criteria.
FKBP8 is a member of the FK506-binding protein family, which has a conserved peptidyl prolyl cis/trans-isomerase (PPIase) domain [9]. PPIase activity is important for both de novo folding of nascent polypeptide chains and the regulation of activities of mature client proteins. FKBP8 is membrane-anchored via the transmembrane domain and distributed predominantly in mitochondria [10]. FKBP8 mediates multiple functions such as protein folding and trafficking as a co-chaperone of heat shock proteins [11], [12], apoptosis [10], [13], [14], cell size regulation [15] and the mammalian target of rapamycin (mTOR) signaling [16]. Conventional FKBP8-deficient mice die at embryonic day 13.5 or shortly after birth due to prominent malformation of the nervous system including defect in neural tube closure [17], [18]. However, the role of FKBP8 in the heart has not been fully elucidated.
The aim of the present study was to determine the functional role of FKBP8 in the heart. Cardiac-specific FKBP8-deficient mice were generated for this purpose and provided the first evidence that FKBP8 plays a protective role against pressure overload in the heart.
2. Methods
Detailed methods are provided in the Online Supplement.
2.1. Generation of cardiac-specific FKBP8-deficient mice
We generated mice with the Fkbp8flox allele and crossed them with mice expressing α-myosin heavy chain promoter-driven Cre recombinase transgenic mice (αMHC-Cre) to obtain cardiac-specific FKBP8-deficient mice [19]. All procedures were carried out in accordance with the King's College London Ethical Review Process Committee and UK Home Office (Project License No. PPL70/7260 and 70/8889).
2.2. Transverse aortic constriction (TAC) and echocardiography
The 8–11 weeks old mice were subjected to TAC or sham surgeries as previously reported [20]. Echocardiography was conducted with a Vevo 2100 system (Visual Sonics, Toronto, Canada) on conscious mice [21].
2.3. Western blot analysis and mitochondrial fractioning
Western blot analysis from frozen left ventricular tissues or cultured cells was performed as previously described [21]. Mitochondrial fractions were isolated from mouse hearts by differential centrifugation methods [22].
2.4. Histological analysis
O.C.T.-embedded samples were stained with hematoxylin-eosin, Masson's trichrome (Masson's Trichrome Stain Kit, Polysciences, Warrington, PA), wheat germ agglutinin (Alexa Fluor 488 conjugated, Thermo Fisher Scientific, Waltham, MA), or immunohistochemical staining with S100A4 (FSP1) antibody (ab41532, Abcam, Cambridge, UK) followed by avidin peroxidase (VECTASTAIN Elite ABC Kit, Vector Laboratories, Burlingame, CA), DAB peroxidase substrate kit (Vector Laboratories) and counterstaining with hematoxylin. Fibrosis fraction and cross-sectional areas of cardiomyocytes were measured using the NIH ImageJ software (National Institutes of Health, Bethesda, MD) [22].
2.5. Reverse transcription quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the left ventricle using the Trizol reagent (Thermo Fisher Scientific). RT-qPCR was performed to determine mRNA levels for Nppa, Nppb, Myh7, Col3a1, Col1a2. All data were normalized to Actb and expressed as a fold increase of the control group [21]. Primer sequences were described in the Online Supplement.
2.6. Transmission electron microscopy
The heart was perfused with ice cold 0.1 mmol/L PIPES buffer containing 2.5% polyvinylpyrrolidone and 0.1% sodium sitrite, and then fixed with 0.1 mmol/L PIPES buffer containing 2% glutaraldehyde, 2% paraformaldehyde, 0.1% sodium itrite and 2.5% polyvinylpyrrolidone. H9c2 cells were fixed with 2.5% glutaraldehyde in 0.1 mmol/L cacodylate buffer. Samples were processed according to our standard procedures [22].
2.7. Mitochondrial enzyme activities
The mitochondrial fraction was freshly prepared from mouse hearts. Mitochondrial electron transport chain complex activities of NADH cytochrome-c oxidoreductase (complex I + III) and succinate cytochrome-c oxidoreductase (complex II + III) were evaluated using spectrophotometric methods [22]. The data were expressed as a relative ratio to the control group.
2.8. Terminal deoxynucleotidyl transferase mediated dUTP nick end-labeling (TUNEL) staining
O.C.T.-embedded heart sections were stained with TUNEL kit (Takara Bio, Otsu, Japan) and anti-actin (α-sarcomeric) antibody (A2172, Sigma-Aldrich, St. Louis, MO) followed by the secondary antibody Texas Red Anti-Mouse IgM (Vector Laboratories). For H9c2 cells, the cells were fixed in 4% paraformaldehyde, stained with the TUNEL kit. Samples were mounted with ProLong Gold Antifade Reagent with DAPI (Thermo Fisher Scientific). The number of TUNEL-positive nuclei and total nuclei was counted, and expressed as the number of TUNEL-positive cardiomyocytes per 105 nuclei for the heart tissue and as the percentage of TUNEL-positive nuclei to total nuclei for H9c2 cells.
2.9. Cell culture and transfection with small interfering RNA (siRNA) and plasmid DNA
H9c2 rat embryonic cardiac myoblasts and HEK293 cells were cultured in Dulbecco's modified Eagle's medium with 10% fetal bovine serum, 100 μg/mL of streptomycin and 100 IU/mL of penicillin at 37 °C in the presence of 5% CO2. For siRNA transfection in H9c2 cells, cells were transfected with scrambled negative control siRNA (4390843, Thermo Fisher Scientific) or FKBP8-specific siRNA (4390771, s66102, Thermo Fisher Scientific) using Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to manufacturer's instructions. For plasmid DNA transfection, HEK293 cells were transfected using Lipofectamine 3000 (Thermo Fisher Scientific) according to manufacturer's protocols.
2.10. Cell viability assay
The numbers of viable cells were analyzed using a Cell-Titer Blue assay (Promega, Fitchburg, WI). The caspase-12 specific inhibitor, Z-ATAD-FMK (BioVision, Milpitas, CA), was incubated prior to hydrogen peroxide (Sigma-Aldrich) stimulation. The data were expressed as a relative ratio to the control.
2.11. Isolation of mouse adult cardiomyocytes and rat neonatal cardiomyocytes and immunofluorescence
Mouse adult cardiomyocytes were isolated using a Langendorff system as previously reported [20]. Rat neonatal cardiomyocytes were isolated from hearts of 1 to 2 day old Sprague-Dawley rats [20]. Immunofluorescence was performed as described in the Online Supplement.
2.12. Construction of DNA plasmid
Constructs of mouse Fkbp8 (NM_001111066.1) and heat shock protein 90 alpha family class A member 1 (Hsp90aa1) (NM_010480.5) were obtained by conventional restriction enzyme-based cloning. Detailed methods were described in the Online Supplement.
2.13. Immunoprecipitation
Samples were prepared from HEK293 cells transfected with plasmid DNA or mouse heart tissue. Detailed methods were described in the Online Supplement.
2.14. Detection for protein aggregates
Protein aggregates were detected with Proteostat Aggresome Detection Kit (Enzo Life Sciences, Farmingdale, NY) according to manufacturer's instruction [23]. H9c2 cells were seeded onto coverslips and transfected with siRNA, and then cells were fixed with 4% paraformaldehyde, permeabilized by Triton X-100, labeled with Proteostat aggresome dye, and mounted by ProLong Gold Antifade Reagent with DAPI. For co-staining with Proteostat dye, cells were incubated with anti-ubiquitin (BML-PW8810-0100, Enzo Life Sciences) or anti-p62 (ab56416, Abcam) antibody followed by Alexa Fluor 488 (A-21200, Thermo Fisher Scientific) or Alexa Fluor 647 (A-31571, Thermo Fisher Scientific).
2.15. Statistical analysis
All data are expressed as the mean ± SEM. Student's t-test was used for the comparison of paired data. One way analysis of variance followed by the Bonferroni's post hoc test was applied for multiple comparisons. All data were analyzed with IBM SPSS Statistics 22.0 (IBM, Armonk, NY). A probability value < 0.05 was considered statistically significant.
3. Results
3.1. FKBP8 is not involved in mitochondrial damage-induced mitophagy
Mouse Fkbp8 encodes 403 amino acids containing a C-terminal single transmembrane domain and sequence alignment revealed two WXXL/I motifs at positions 83–86 and 379–382, which are consensus sequences for the binding sites of microtubule associated protein 1 light chain 3 (LC3), a mammalian homologue of Atg8 (Supplementary Fig. 1A) [24]. Firstly, we tested whether FKBP8 interacts with LC3B mediated through the WXXL/I motifs. A yeast two-hybrid assay using Gal4-fused LC3B and activation domain-fused FKBP8 showed that the cells expressing LC3B and FKBP8 grew on selective plates (Supplementary Fig. 1B). The FKBP8 mutant, which contains W83A I86A W379A L382A amino acid substitution in the WXXL/I motifs, also could grow on the plates. Thus, the interaction between FKBP8 and LC3B was not mediated via the WXXL/I motifs, but seems to be rather non-specific. To estimate the role of FKBP8 in mitophagy, FKBP8 was knocked down in a stable cell line of HEK293 expressing mitochondrial targeted mKeima [8], a coral-derived acid-stable lysosomal proteases-resistant fluorescent protein. Knockdown of endogenous FKBP8 was performed using siRNA (Supplementary Fig. 1C). The knockdown cells were incubated with a mitochondrial uncoupler carbonyl cyanide m-chlorophenylhydrazone (CCCP), which induces mitophagy, and stained with LysoTracker Green before microscopic analysis (Supplementary Fig. 1D). CCCP treatment increased the number of puncta with a high 561/488 nm excitation ratio, reflecting increased lysosomal localization of mKeima. The rainbow-color ratio images were converted into a single-color intensity channel (Lookup table in Supplementary Fig. 1D). The high-ratio mKeima dots were colocalized with LysoTracker-positive dots, confirming lysosomal localization. Knockdown of FKBP8 did not change the number of high-ratio mKeima and LysoTracker double positive dots, suggesting that FKBP8 is not involved in CCCP-induced mitophagy (Supplementary Fig. 1E). Furthermore, GFP-LC3 was transfected in FKBP8 knockdown H9c2 myocytes [25]. The number of mitochondria positive for GFP-LC3-labeled autophagic structures was significantly elevated after CCCP stimulation, but there was no difference between control and FKBP8 knockdown H9c2 myocytes (Supplementary Fig. 1F).
To clarify the role of FKBP8 during cardiac remodeling, the change in FKBP8 expression levels in wild type C57BL/6J mouse hearts after pressure overload was estimated. In our model, mice show cardiac hypertrophy without cardiac dysfunction 1 week after TAC, while they exhibit heart failure phenotypes 4 weeks after the operation. The protein level of FKBP8 in the heart was upregulated at both 1 and 4 weeks after TAC compared to the corresponding sham-operated mice (Supplementary Fig. 2), indicating that FKBP8 may play a pathophysiological role during pressure overload-induced cardiac remodeling.
3.2. Generation and characterization of cardiac-specific FKBP8-deficient mice
Cardiac-specific FKBP8-deficient mice were generated to establish the in vivo role of FKBP8 in the heart. We designed a gene targeting strategy to conditionally inactivate the Fkbp8 gene by inserting loxP sites in intron 2 and 6 (Fig. 1A). The neomycin resistance gene was inserted between exon 6 and the downstream loxP site. The diphtheria toxin A gene was positioned at the 3′ end of the targeting construct for negative selection. Homologous recombinants were identified by PCR and Southern blot analysis (Fig. 1B). The ES cells with Fkbp8-floxed allele were injected into blastocysts to obtain Fkbp8flox/+ mice. The homozygous floxed Fkbp8 mice (Fkbp8flox/flox) appeared normal and were externally indistinguishable from littermates of other genotypes. Fkbp8flox/flox mice were crossed with αMHC-Cre mice [19] to generate Fkbp8flox/flox; αMHC-Cre+ (Fkbp8−/−) mice. Fkbp8flox/flox; αMHC-Cre− littermates were used as controls (Fkbp8+/+). The Fkbp8+/+ and Fkbp8−/− mice were born normally at the expected Mendelian ratio (58 and 55 mice, respectively), and they grew to adulthood and were fertile. Then, the efficiency of Fkbp8 ablation was determined in the heart by immunoblot analysis. The protein level of FKBP8 significantly decreased in Fkbp8−/− hearts by 88% compared to Fkbp8+/+ mice (Fig. 1C). Echocardiographic analysis revealed that ablation of Fkbp8 in cardiomyocytes had no effect on cardiac morphology and function under baseline conditions at 8–10 weeks old (Supplementary Table 1). No differences in any physiological parameters were observed in Fkbp8−/− mice compared to Fkbp8+/+ mice (Supplementary Table 1), indicating that Fkbp8−/− mice showed normal global cardiac structure and function.
Fig. 1.
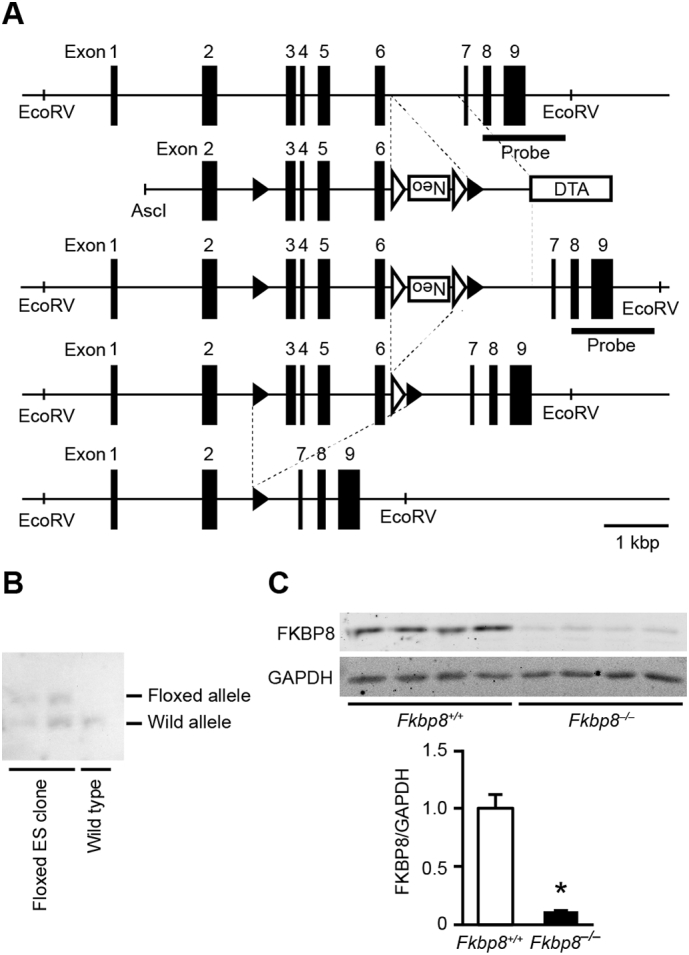
Generation of cardiac-specific FKBP8-deficient mice.
A, Targeting modification of the Fkbp8 gene. Schematic structures of the wild type genomic Fkbp8 sequence, the targeting construct, the targeted allele, the floxed allele after flippase recognition target sites (FRT)-mediated neomycin-resistance gene (Neo) deletion, the deleted allele after Cre-mediated recombination are indicated from top to bottom. The black and white arrowheads indicate loxP and FRT sites, respectively. The targeting construct includes the PGK-Neo cassette flanked by loxP sites and a diphtheria toxin A gene (DTA). The bar labeled as “probe” corresponds to the sequence used for Southern blotting. Scale bar indicates 1 kbp length. B, Genomic analysis of embryonic stem (ES) cells. Genomic DNA extracted from ES cells was digested with EcoRV and analyzed by Southern blotting with the probe. Wild type and floxed allele show 9480 and 11,178 bp, respectively. C, Protein expression levels of FKBP8 in Fkbp8+/+ and Fkbp8−/− hearts. Left ventricular homogenates from Fkbp8+/+ and Fkbp8−/− mice were analyzed by Western blotting with anti-FKBP8 antibody (top). Densitometric analysis is shown (bottom). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the loading control. The average value of FKBP8-to-GAPDH ratio in Fkbp8+/+ was set equal to 1. Values are presented as the mean ± SEM from 4 mice in each group. *P < 0.05 versus Fkbp8+/+ mice.
3.3. FKBP8-deficient mice developed heart failure in response to pressure overload
To examine the role of FKBP8 in the development of cardiac remodeling, mice were subjected to pressure overload by means of TAC [26]. Echocardiographic analysis was performed 1 week after TAC (Fig. 2A and B). The diastolic interventricular septum wall thickness (IVSd) and left ventricular (LV) posterior wall thickness (LVPWd) were elevated in TAC-operated Fkbp8+/+ and Fkbp8−/− mice compared to the corresponding sham-operated mice. While IVSd was not different between TAC-operated Fkbp8+/+ and Fkbp8−/− mice, LVPWd in TAC-operated Fkbp8−/− mice was significantly reduced compared to TAC-operated Fkbp8+/+ mice. The calculated LV mass was elevated by TAC, but not different between TAC-operated Fkbp8+/+ and Fkbp8−/− mice. The end-diastolic and end-systolic LV internal dimensions were significantly elevated and fractional shortening, an index of contractility, was reduced in TAC-operated Fkbp8−/− mice compared to both sham-operated Fkbp8−/− and TAC-operated Fkbp8+/+ mice. Although TAC increased heart weight-to-tibia length ratio in both Fkbp8+/+ and Fkbp8−/− mice, there was no significant difference between the two groups (Fig. 2C). Lung weight-to-tibia length ratio, an index of lung congestion, was significantly elevated in TAC-operated Fkbp8−/− mice compared to both sham-operated Fkbp8−/− and TAC-operated Fkbp8+/+ mice. These data suggest that Fkbp8−/− mice developed congestive heart failure in response to pressure overload.
Fig. 2.

Development of heart failure in response to pressure overload in cardiac-specific FKBP8-deficient mice.
A, Representative images of M-mode echocardiograms from sham- and TAC-operated Fkbp8+/+ and Fkbp8−/− mice. Scale bars, 0.2 s and 5 mm, respectively. B, Echocardiographic parameters. IVSd, diastolic interventricular septum wall thickness; LVPWd, diastolic left ventricular posterior wall thickness; LV mass, left ventricular mass; LVIDd, end-diastolic left ventricular internal dimension; LVIDs, end-systolic left ventricular internal dimension; LVFS, left ventricular fractional shortening. C, Physiological parameters. HW, heart weight; TL, tibia length. Sham-operated Fkbp8+/+ mice (n = 7), TAC-operated Fkbp8+/+ mice (n = 9), sham-operated Fkbp8−/− mice (n = 8), TAC-operated Fkbp8−/− mice (n = 9). The values are expressed as the mean ± SEM. Open and closed bars represent sham- and TAC-operated groups, respectively. *P < 0.05 versus the corresponding sham-operated group. †P < 0.05 versus TAC-operated Fkbp8+/+ mice.
3.4. TAC-operated FKBP8-deficient mice exhibited myocardial remodeling
Hematoxylin-eosin staining showed interstitial cellular infiltration in Fkbp8−/− hearts 1 week after TAC (Fig. 3A). Masson's trichrome staining revealed interstitial fibrosis in both TAC-operated Fkbp8+/+ and Fkbp8−/− hearts, but the extent of fibrosis in Fkbp8−/− mice was greater than in Fkbp8+/+ mice (Fig. 3A, B and Supplementary Fig. 3). Immunohistological examination using anti-fibroblast-specific protein-1 antibody indicated that the infiltrating cells in TAC-operated Fkbp8−/− hearts were mainly fibroblasts (Fig. 3A). Although wheat germ agglutinin staining showed TAC increased the cross-sectional area of cardiomyocytes in both Fkbp8+/+ and Fkbp8−/− mice, there was no significant difference between the two groups (Fig. 3A and C). TAC-operated Fkbp8−/− mice displayed higher mRNA expression of Nppa, which is a biochemical marker for cardiac remodeling, than TAC-operated Fkbp8+/+ mice (Fig. 3D). Although Nppb and Myh7 mRNA expression were elevated by pressure overload in both Fkbp8+/+ and Fkbp8−/− mice, there were no significant differences between the two groups. The mRNA levels of Col3a1 and Col1a2, markers for fibrosis, were significantly higher in TAC-operated Fkbp8−/− mice than those in TAC-operated Fkbp8+/+ mice. These data indicate that deficiency of FKBP8 exacerbated TAC-induced cardiac remodeling including cardiac dysfunction, chamber dilatation and fibrosis, but FKBP8 was not involved in TAC-induced cardiac hypertrophy.
Fig. 3.

Histological and biochemical characterization of Fkbp8−/− mice after TAC.
A, Hematoxylin-eosin-stained (HE), Masson's trichrome-stained, immunostained with anti-fibroblast-specific protein-1 (FSP1) antibody and wheat germ agglutinin-stained (WGA) sections of hearts from Fkbp8+/+ and Fkbp8−/− mice. Scale bar, 50 μm. B, Quantitative analysis of fibrosis fraction in Masson's trichrome-stained sections. The values are expressed as the mean ± SEM (n = 3). C, Cross-sectional area of cardiomyocytes in WGA-stained sections. Data are presented as the mean ± SEM (n = 3). D, The mRNA expression of Nppa, Nppb, Myh7, Col3a1 and Col1a2. Actb was used as the loading control. The average value for sham-operated Fkbp8+/+ mice was set equal to 1. The values represent the mean ± SEM (n = 5–7). Open and closed bars represent sham- and TAC-operated groups, respectively. *P < 0.05 versus the corresponding sham-operated group. †P < 0.05 versus TAC-operated Fkbp8+/+ mice.
3.5. Deficiency of FKBP8 had no effect on mitochondrial morphology, enzymatic function, mitophagy or mTOR signaling
Since FKBP8 is reported to be expressed in outer mitochondrial membrane [10], mitochondrial morphology and function were examined to explore the mechanisms underlying the cardiac phenotypes observed in TAC-operated Fkbp8−/− mice. Ultrastructural analysis demonstrated that sarcomere structures were preserved in Fkbp8−/− hearts 1 week after TAC and there was no apparent difference in mitochondrial morphology and intra-mitochondrial structures among groups (Fig. 4A). Mitochondrial functions estimated by the enzymatic activity of complex I + III and II + III were not different among groups (Fig. 4B). Likewise, the protein expression levels of mitochondrial proteins such as succinate dehydrogenase complex flavoprotein subunit A (SDHA), cytochrome c oxidase subunit IV (COX IV), and voltage-dependent anion channel (VDAC) were not different among groups (Fig. 4C). For the assessment of mitophagy in the heart, mitochondrial DNA content which can be degraded by mitophagy [27] and the level of LC3B-II in mitochondrial fraction which can reflect mitochondrial autophagosome formation [28] were examined, and there were no differences among groups (Supplementary Fig. 4A and 4B). Although it has been recently reported that FKBP8 recruits lipidated LC3A to mitochondria [29], the level of LC3A-II in mitochondrial fraction was not different among groups (Supplementary Fig. 4C). Thus, these results suggest that mitochondrial morphological change, function, and mitophagy were not associated with cardiac dysfunction observed in TAC-operated Fkbp8−/− mice. It has been reported that FKBP8 is an endogenous inhibitor of mTOR [16]. Although phosphorylation levels of both S6 and eukaryotic initiation factor 4E-binding protein 1 (4E-BP1), downstream signaling molecules of mTORC1, were significantly elevated by TAC, there were no differences between TAC-operated Fkbp8+/+ and Fkbp8−/− hearts (Fig. 4D), indicating that mTOR signaling is not involved in cardiac phenotypes observed in Fkbp8−/− hearts after hemodynamic stress.
Fig. 4.

Mitochondrial morphology and function, and mTOR signaling in TAC-operated Fkbp8−/− mice.
A, Electron microscopic analysis. Intra-mitochondrial structures are shown in the insets. Scale bars represent 2 μm in the main panels and 500 nm in the insets. B, The activities of mitochondrial complex I + III and complex II + III in mitochondrial fraction isolated from sham- and TAC-operated hearts. The average value for sham-operated Fkbp8+/+ mice was set to 1. Values are expressed as the mean ± SEM (n = 3–4). C, Western blot analysis of mitochondrial proteins. Heart extracts were immunoblotted with the indicated antibodies. The panels show densitometric analysis. GAPDH was used as the loading control. The average value for sham-operated Fkbp8+/+ mice was set equal to 1. Data are presented as the mean ± SEM (n = 3). D, Western blot analysis on S6 and 4E–BP1. p-S6 and t-S6 indicate phosphorylated and total S6, respectively. A higher concentration of 4E–BP1 antibody was used (high) to visualize the α-form of 4E-BP1. p-S6 to t-S6 ratios and γ-form of 4E–BP1 to total 4E-BP1 (t-4E-BP1) ratios are shown in the graphs. The average value for sham-operated Fkbp8+/+ mice was set to 1. Data are presented as the mean ± SEM (n = 3). Open and closed bars represent sham- and TAC-operated groups, respectively. *P < 0.05 versus the corresponding sham-operated group.
3.6. Cardiomyocyte apoptosis increased in FKBP8-deficient hearts after TAC
Apoptotic cardiomyocyte death plays an important role in the development and progression of heart failure [30]. Cardiomyocyte apoptosis was evaluated in hearts 1 week after TAC by means of TUNEL assay and immunoblot analysis using anti-caspase antibodies. The numbers of TUNEL-positive cardiomyocytes in sham-operated Fkbp8+/+ and sham-operated Fkbp8−/− mice were similar (Fig. 5A). However, TAC-operated Fkbp8−/− mice exhibited more TUNEL-positive cardiomyocytes than TAC-operated Fkbp8+/+ mice. The expression level of cleaved caspase-3 was significantly higher in TAC-operated Fkbp8−/− hearts than in TAC-operated Fkbp8+/+ hearts (Fig. 5B). When the activation levels of the initiator caspases upstream of caspase-3 were evaluated, the expression of cleaved caspase-8 and caspase-9, which are involved in the death receptor (extrinsic) and mitochondrial (intrinsic) pathways, respectively, was not detectable in all groups. The expression levels of cleaved caspase-12, which is specifically associated with ER stress-mediated apoptosis, were significantly elevated in TAC-operated Fkbp8−/− hearts compared to both sham-operated Fkbp8−/− and TAC-operated Fkbp8+/+ hearts. Although the protein expression level of Bax or Bcl-2 in isolated mitochondrial fraction was significantly elevated in TAC-operated Fkbp8−/− hearts compared to sham-operated Fkbp8−/− hearts, there was no significant difference in Bax or Bcl-2 level between TAC-operated Fkbp8+/+ and Fkbp8−/− hearts (Fig. 5C). The ratios of Bax to Bcl-2 protein were not different among the four groups. To support the involvement of ER stress-associated cell death in the genesis of cardiac dysfunction in Fkbp8−/− mice, the expression levels of ER stress markers such as glucose-regulated protein 94 kDa (GRP94), glucose-regulated protein 78 kDa (GRP78) and protein disulfide-isomerase (PDI) were assessed. These ER stress markers were significantly elevated by TAC in both Fkbp8+/+ and Fkbp8−/− mice, but these protein levels in TAC-operated Fkbp8−/− hearts were higher than in TAC-operated Fkbp8+/+ hearts (Fig. 5D). Furthermore, phosphorylation level of protein kinase R–like endoplasmic reticulum kinase (PERK) and protein expression level of activating transcription factor 4 (ATF4) were significantly upregulated in TAC-operated Fkbp8−/− hearts (Supplementary Fig. 5).
Fig. 5.

Increased ER stress-mediated apoptosis in TAC-operated Fkbp8−/− mice.
A, Triple staining of mouse hearts with TUNEL (green), anti-α-sarcomeric actin antibody (red) and DAPI (blue). White arrows indicate TUNEL-positive nuclei. The graph shows quantitative analysis on the number of TUNEL-positive cardiomyocytes. Data are expressed as the mean ± SEM (n = 3). Scale bar, 50 μm. B, Immunoblot analysis for caspases by indicated antibodies. The graphs show densitometric analysis for cleaved caspase-3 (n = 6) and cleaved caspase-12 (n = 4). Data are normalized by GAPDH. The average value for sham-operated Fkbp8+/+ mice was set to 1. Data are presented as the mean ± SEM. C, Immunoblot analysis for Bax and Bcl-2 in mitochondrial fraction prepared from heart homogenates. Densitometric analysis is shown in the graphs. Data are normalized by SDHA and presented as the mean ± SEM (n = 3). D, Immunoblot analysis of ER stress markers. The graphs show densitometric analysis. The average value in sham-operated Fkbp8+/+ mice was set to 1. Data are presented as the mean ± SEM (n = 3). Open and closed bars represent sham- and TAC-operated groups, respectively. *P < 0.05 versus the corresponding sham-operated group. †P < 0.05 versus TAC-operated Fkbp8+/+ mice. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.7. Caspase-12 inhibition effectively attenuated H2O2-induced cardiomyocyte apoptosis in FKBP8-deficient H9c2 cells
ER stress and oxidative stress are closely linked and reactive oxygen species (ROS) can cause protein misfolding in the ER, leading to ER stress [31]. Although pressure overload significantly promoted ROS generation in the heart assessed by heme oxygenase 1 (HO-1) expression, there was no difference between TAC-operated Fkbp8+/+ and Fkbp8−/− hearts (Supplemental Fig. 6). To investigate the role of caspase-12 in FKBP8-related apoptotic cell death, the effect of caspase-12 inhibition on hydrogen peroxide (H2O2)-induced cell death in FKBP8 knockdown H9c2 myocytes was examined. The FKBP8 siRNA (siFKBP8) significantly decreased the endogenous protein levels of FKBP8 compared to control siRNA (siCTRL) (Fig. 6A). We incubated FKBP8 knockdown cells with 50 μmol/L of H2O2 for 6 h to evaluate cell viability. There was no difference in cell viability between cells transfected with siCTRL and siFKBP8 without H2O2 stimulation (Fig. 6B). Although H2O2 did not induce cell death in siCTRL-transfected cells in our experimental conditions, the cell viability was significantly reduced in H2O2-treated FKBP8 knockdown myocytes, indicating that FKBP8 is involved in H2O2-induced cell death. Z-ATAD-FMK, which is a caspase-12 inhibitor, improved cell viability in H2O2-treated FKBP8 knockdown H9c2 myocytes. H2O2 increased the number of TUNEL-positive nuclei in FKBP8 knockdown myocytes, while Z-ATAD-FMK treatment attenuated the increase in apoptotic cell number (Fig. 6C). Furthermore, the levels of cleaved caspase-3 and caspase-12 in Z-ATAD-FMK- and H2O2-treated FKBP8 knockdown cells were lower than in vehicle- and H2O2-treated FKBP8 knockdown cells (Fig. 6D). However, Z-ATAD-FMK had no effect on H2O2-induced upregulation of GRP94 and GRP78 in FKBP8 knockdown myocytes. These data indicate caspase-12 is involved in H2O2-induced apoptotic cell death in FKBP8 knockdown myocytes and exists downstream of ER stress.
Fig. 6.

Effects of caspase-12 inhibition on H2O2-induced cell death in H9c2 cells.
A, Immunoblots of FKBP8 in knockdown H9c2 cells. H9c2 cells were transfected with FKBP8-specific siRNA (siFKBP8) or non-targeting control siRNA (siCTRL) for 96 h. The graph shows densitometric analysis. The value of FKBP8-to-GAPDH ratio in siCTRL was set equal to 1. Data are presented as the mean ± SEM (n = 3). *P < 0.05 versus siCTRL. B, Transfected cells were incubated with Z-ATAD-FMK (3 μmol/L) or DMSO for 2 h before H2O2 administration (50 μmol/L). Six h after H2O2 stimulation, cellular viability was determined by Cell-Titer Blue assay. The value in the cells transfected with siCTRL and incubated without H2O2 nor Z-ATAD-FMK was set to 1. Values represent the mean ± SEM from 3 independent experiments with triplicates. *P < 0.05 versus all other groups. C, Z-ATAD-FMK inhibited apoptotic cell death caused by H2O2 in FKBP8 knockdown H9c2 cells. Representative images of double staining of TUNEL (green) and DAPI (blue). White arrows indicate TUNEL positive nuclei. The graph shows quantitative analysis of TUNEL positive nuclei. Values are presented as the mean ± SEM (n = 5). *P < 0.05 versus all other groups. †P < 0.05 versus all other groups except the group transfected with siCTRL and incubated with H2O2. Scale bar, 50 μm. D, Western blot analysis of caspase-12, caspase-3 and KDEL. The graphs show densitometric analysis. Data are normalized to GAPDH and presented as the mean ± SEM from 4 to 6 independent experiments. *P < 0.05 versus all other groups. †P < 0.05 versus all other groups except the siCTRL-transfected group followed by H2O2 without Z-ATAD-FMK. ‡P < 0.05 versus the groups without H2O2. §P < 0.05 versus all other groups except the siFKBP8-transfected group followed by H2O2 with Z-ATAD-FMK. || P < 0.05 versus all other groups except the siFKBP8-tranfected group followed by H2O2 without Z-ATAD-FMK. Open and closed bars represent siCTRL- and siFKBP8-transfected groups, respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.8. FKBP8 was localized at the ER in the isolated cardiomyocytes and bound to heat shock protein 90 (HSP90)
We then examined subcellular localization of FKBP8 in the cardiomyocytes. Confocal microscopic analysis using anti-KDEL and anti-Tomm20 antibodies demonstrated that endogenous FKBP8 was localized to both ER and mitochondria in adult cardiomyocytes isolated from Fkbp8+/+ mouse heart (Fig. 7A and B). Triple labeling of endogenous FKBP8 and KDEL in combination with MitoTracker was performed in cultured rat neonatal cardiomyocytes. We focused on areas where the ER is enriched (Supplementary Fig. 7A) or tubular structure of mitochondria is evident (Supplementary Fig. 7B). The results provided further evidence of localization of FKBP8 in both ER and mitochondria. To confirm ER localization of FKBP8, rat neonatal cardiomyocytes were transfected with tdTomato-ER-3 containing KDEL sequence to target it to the ER. The expression of tdTomato showed a clear mesh pattern in the peripheral area of the cells, which is characteristic of ER structure, and was colocalized with endogenous FKBP8 staining (Fig. 7C).
Fig. 7.

Subcellular localization of FKBP8 in cardiomyocytes and its interaction with heat shock protein 90 (HSP90).
A, Endogenous FKBP8 in the ER in mouse adult cardiomyocytes. Isolated adult cardiomyocytes from mouse hearts were stained with anti-FKBP8 (green) and anti-KDEL (red) antibodies. Images in boxed areas at higher magnification are shown in right panels. Scale bars, 10 μm and 5 μm, respectively. B, Endogenous FKBP8 in mitochondria in mouse adult cardiomyocytes. The cells were stained with anti-FKBP8 (green) and anti-Tomm20 (red) antibodies. Images in boxed areas at higher magnification are shown in right panels. Scale bars, 10 μm and 5 μm, respectively. C, Endogenous FKBP8 in rat neonatal cardiomyocytes. Cultured rat neonatal cardiomyocytes transfected with tdTomato-ER-3 (red) were stained with MitoTracker (blue), then fixed, and stained with anti-FKBP8 antibody (green). Boxed areas are highlighted in the lower panels. Scale bars are 10 μm and 1 μm. D, Coimmunoprecipitation of FKBP8 and HSP90. Forty-eight h after transfection of HEK293 cells with HA-FKBP8 and Flag-HSP90, cell lysates were immunoprecipitated with anti-HA or anti-Flag antibody in the presence of 1 mmol/L CaCl2 or EGTA. Immunoprecipitates (IP) were analyzed by immunoblotting (IB). E, Coimmunoprecipitation of FKBP8 and HSP90 in mouse heart tissue. Heart homogenates were immunoprecipitated with anti-FKBP8 antibody and subjected to immunoblotting. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
To investigate how FKBP8 is involved in ER quality control, we examined whether FKBP8 interacted with chaperon proteins such as heat shock proteins. It has been reported that FKBP8 interacted with HSP90, which is a major molecular chaperone to assist protein folding [4], [12], [32]. The coimmunoprecipitation assay demonstrated the interaction of FKBP8 with HSP90 in HEK293 cells in the absence or presence of Ca2 + (Fig. 7D). Using protein lysates from mouse hearts, endogenous FKBP8 was shown to interact with HSP90 (Fig. 7E). Taken together, these data suggest that FKBP8 might act as a co-chaperone coordinating with HSP90 at the ER.
3.9. Deficiency of FKBP8 resulted in the accumulation of misfolded proteins in cardiomyocytes
ER stress is the consequence of the accumulation of misfolded proteins. The aggregation of misfolded proteins was visualized using Proteostat aggresome detection dye, a molecular rotor fluorescent dye which specifically detects misfolded protein aggregates [23], [33]. Knockdown of FKBP8 in H9c2 cells induced the accumulation of Proteostat-positive aggregates in the perinuclear region as well as in cultured rat neonatal cardiomyocytes (Fig. 8A and Supplementary Fig. 8A). Next, ultrastructure of misfolded protein aggregates was characterized. There were aggresome-like electron dense deposits with membranous contents and vacuoles in FKBP8 knockdown H9c2 cells (Fig. 8B). We examined the effect of H2O2 stimulation on misfolded protein aggregates. We observed the cells 1.5 h after H2O2 administration, when cell viability was maintained (Fig. 8C). H2O2 did not increase the percentage of Proteostat-positive cells in FKBP8 knockdown or control cells. The p62 facilitates degradation of ubiquitinated protein aggregates by autophagy [34]. We examined whether the Proteostat-positive structures in FKBP8 knockdown H9c2 cells were ubiquitinated and/or contained p62 using anti-ubiquitin or anti-p62 antibody and found that the Proteostat-positive structures were not costained with ubiquitin nor p62 (Fig. 8D and E).
Fig. 8.

Knockdown or knockout of FKBP8 results in the accumulation of misfolded protein in cardiomyocytes.
A, Detection of misfolded protein aggregates in H9c2 cells. Ninety-six h after transfection with siCTRL or siFKBP8, the cells were stained with Proteostat fluorescent dye (red) and DAPI (blue), and subjected to confocal microscopic analysis. Boxed areas at higher magnification (high) are shown in right panels. Scale bars are 100 μm and 10 μm, respectively. B, Electron micrographs of H9c2 cells transfected with siCTRL or siFKBP8. Boxed areas at higher magnification are shown in right panels. Scale bars are 10 μm and 2 μm, respectively. C, The effect of H2O2 on protein aggregates. H9c2 cells transfected with siCTRL or siFKBP8 were fixed 1.5 h after H2O2 stimulation and stained with Proteostat dye (red) and DAPI (blue). The percentage of Proteostat-positive cells to total nuclei is shown in the graph (n = 3). More than 50 cells were counted in each group. *P < 0.05 versus the corresponding siCTRL group. Scale bar, 50 μm. D, Detection of ubiquitinated protein and p62 in Proteostat-positive structures. Ninety-six h after transfection with siCTRL or siFKBP8, the cells were stained with Proteostat fluorescent dye (red), followed by anti-ubiquitin (green) or anti-p62 (green) antibody, and mounted with DAPI (blue). Scale bar, 50 μm. E, Higher magnified images of triple staining from boxed areas in Fig. 8D. Upper and lower panels show ubiquitin and p62 staining in FKBP8 knockdown cells, respectively. Scale bar, 5 μm. F, Ultrastructural analysis of the perinuclear region in sham- and TAC-operated Fkbp8+/+ and Fkbp8−/− mice. Scale bar, 1 μm. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Since the ubiquitin-proteasome system and autophagy lysosomal pathway are important for the degradation of misfolded proteins [2], proteasome and autophagy activity were investigated in FKBP8 knockdown H9c2 cells (Supplementary Fig. 8B and 8C). Knockdown of FKBP8 in H9c2 cells had no effect on proteasome activity nor the protein level of LC3B-II. Furthermore, Tat-Beclin 1, an autophagy-inducible peptide [35], increased the level of LC3B-II (Supplementary Fig. 8D), but had no effect on the accumulation of Proteostat-positive structures in FKBP8 knockdown H9c2 cells (Supplementary Fig. 8E). Moreover, Tat-Beclin 1 did not affect accumulation of Proteostat-positive aggregates or cellular viability in H2O2-treated FKBP8 knockdown H9c2 cells (Supplementary Fig. 8F and G).
Protein aggregates in pressure overloaded hearts are observed in the perinuclear area as electron dense deposits [36]. Ultrastructural analysis showed that TAC-operated Fkbp8−/− hearts displayed disorganization in the perinuclear region with high electron dense structures and membranous structures (Fig. 8F). Taken together, these findings suggest that deficiency of FKBP8 resulted in the accumulation of misfolded protein during hemodynamic stress, leading to cardiac dysfunction.
4. Discussion
The purpose of this study was to elucidate the role of FKBP8 in the heart and it was the first to identify the in vivo role of FKBP8 in the heart using a loss-of-function mouse model. We showed that cardiac-specific FKBP8-deficient mice had no effect on cardiac structure or function under basal conditions, indicating that FKBP8 is not essential for cardiac development or postnatal growth. It is possible that other molecular pathways could compensate the loss of FKBP8 function. In stressed hearts, FKBP8 has a cardio-protective role in response to pressure overload by preventing misfolded proteins and resultant ER-associated apoptosis.
Although Atg32 is essential for mitophagy in yeast, no mammalian homologue has been identified. We hypothesized that a mammalian mitophagy receptor would share certain molecular features with Atg32 and identified BCL2L13 and FKBP8 as possible candidates. BCL2L13 mediates mitochondrial fragmentation and mitophagy in HEK293 cells [8]. However, knockdown of FKBP8 had no effect on CCCP-induced mitophagy in HEK293 cells or H9c2 myocytes. Furthermore, the FKBP8 mutant containing amino acid substitutions in the WXXL/I motifs was able to bind LC3B. These data suggest that FKBP8 is not involved in mitophagy. Recently, Bhujabal et al. reported that FKBP8 is a novel mitophagy receptor which recruits LC3A and identified the FEVL motif located in the N-terminal site of FKBP8 as a binding site for LC3A [29]. They demonstrated that co-overexpression of FKBP8 and LC3A increased the percentage of the cells containing acidified mitochondria, while overexpression of FKBP8 alone did not activate mitophagy. They also showed FKBP8 recruited lipidated LC3A to mitochondria upon CCCP treatment when the LC3A was exogenously overexpressed in Hela and HEK293 cells, but our results indicate that endogenous LC3A-II level in mitochondrial fraction was not altered in TAC-operated Fkbp8−/− hearts. Although we cannot fully explain this discrepancy regarding the role of FKBP8 in mitophagy, overexpression of FKBP8 with LC3A might non-specifically induce mitophagy.
In this study, ablation of Fkbp8 induced dilated cardiomyopathy and massive cardiomyocyte apoptosis with the activation of caspase-12 and increase in ER stress markers in response to pressure overload. Thus, FKBP8 is indispensable for cardiac adaptation against hemodynamic stress. Caspase-12 is localized in the ER and involved in ER stress-induced apoptosis [5]. ER stress results from the accumulation of excess proteins and misfolded proteins in the ER lumen. When the misfolded protein load exceeds the capacity, ER-initiated cell death is induced via the caspase-12 signaling pathway. There is debate concerning the subcellular localization of FKBP8, with some reports that FKBP8 is localized predominantly to the outer mitochondrial membrane through its C-terminal membrane anchor in HeLa cells [10], while others have showed that FKBP8 is localized to the ER in HEK293 cells and both ER and mitochondria in SH-SY5Y cells [12], [37]. Our results showed that FKBP8 is highly expressed in the ER as well as in mitochondria under basal conditions in cardiomyocytes. It has been reported that FKBP8, along with Bcl-2, translocates from mitochondria to the ER during CCCP-induced mitophagy in mouse embryonic fibroblasts [38]. Mitophagy is known to be transiently upregulated in response to pressure overload in mouse hearts [27]. We attempted to detect TAC-induced changes in subcellular distribution of FKBP8, but due to high background signal in heart sections using commercially available anti-FKBP8 antibodies, it was not possible to prove the translocation of FKBP8 from mitochondria to the ER upon pressure overload. However, TAC did not change mitochondrial protein levels in Fkbp8+/+ hearts and the levels were not different between TAC-operated Fkbp8+/+ and Fkbp8−/− hearts. In addition, mitochondrial Bcl-2 levels were similar between sham- and TAC-operated Fkbp8+/+ hearts and those in TAC-operated Fkbp8−/− hearts were not different compared with TAC-operated Fkbp8+/+ hearts. Although the extent of mitophagy in pressure overloaded hearts may not be sufficient to induce global degradation of mitochondrial protein content, translocation of FKBP8 with Bcl-2 from mitochondria to the ER during mitophagy was not evident in our study. The distribution of FKBP8 in the cell might be cell-type specific or depend on cellular conditions under stress.
It has been reported that FKBP8 is involved in apoptosis. FKBP8 binds to Bcl-2 and Bcl-xL, recruiting them to mitochondria and inhibiting mitochondria-initiated apoptosis in HeLa cells [10]. FKBP8 translocation to the ER prevents unwanted apoptosis during mitophagy induced by CCCP [38]. Conversely, in neuronal SH-SY5Y cells, association between Bcl-2 and Ca2 +/calmodulin/FKBP8 complex participates in the promotion of apoptosis [37]. The in vitro and in vivo studies presented here clearly showed the anti-apoptotic function of FKBP8 in cardiomyocytes. The function of FKBP8 in apoptosis is perhaps cell-type specific due to various Bcl-2 interaction partners and their expression pattern [37]. In this study, we detected an increase in protein levels of cleaved caspase-12, but not that of cleaved caspase-8 or 9 in TAC-operated Fkbp8−/− hearts. Furthermore, the Bax/Bcl-2 ratio in the mitochondrial fraction was not different between TAC-operated Fkbp8+/+ and Fkbp8−/− hearts. In addition, the inhibition of caspase-12 attenuated H2O2-induced cell death in H9c2 cells. Taken together, these data indicate that FKBP8 is involved in caspase-12-mediated apoptosis, but not in the mitochondrial apoptotic pathway. Apoptotic death of cardiomyocytes plays a pivotal role in the progression of cardiac remodeling [30]. Considering the very high level of apoptosis in TAC-operated Fkbp8−/− hearts, the cardiomyopathic phenotypes observed in TAC-operated Fkbp8−/− mice would be due to the upregulation of caspase-12-mediated apoptosis.
Protein misfolding is linked to the pathogenesis of many diseases including heart failure and neurodegenerative diseases such as Alzheimer's disease, Parkinson's disease and Huntington's disease [3]. The protein aggregates, which are transported on the microtubules and accumulate in the perinuclear region, have been termed aggresomes [39]. Reports of ubiquitin localization to aggresomes are inconsistent. The p62 facilitates degradation of ubiquitinated protein aggregates by autophagy [34]. Desmin-related cardiomyopathy is a well-studied disease, where misfolded proteins are implicated in cardiac dysfunction [2]. The αB-crystallin (CryAB) is a small chaperone of desmin protein. The transgenic mice expressing mutant CryAB (R120G) in the heart displayed the accumulation of protein aggregates containing desmin and dilated cardiomyopathy [40]. Overexpression of CryAB (R120G) in rat neonatal cardiomyocytes resulted in ubiquitin-positive aggresome formation in the perinuclear region [41]. Activation of autophagy attenuated the development of cardiomyopathy in the CryAB (R120G) transgenic mice [42]. In wild type mice, pressure overload promotes development of ubiquitinated aggresome-like structures in the perinuclear region with upregulation of autophagy in the heart [36]. Thus, the continuous presence and chronic accumulation of misfolded proteins in cardiomyocytes can lead to the development of heart failure [2]. Intracellular protein aggregation is a proximal trigger of cardiomyocyte autophagy [31]. In this study, we observed the accumulation of Proteostat fluorescent dye-positive misfolded protein aggregates in FKBP8 knockdown H9c2 myocytes and electron dense deposits in the perinuclear region in the cells and TAC-operated Fkbp8−/− hearts. Proteostat-positive aggregates in FKBP8 knockdown H9c2 cells were not ubiquitinated nor p62-positive. Furthermore, autophagy was not upregulated and the activation of autophagy had no effect on the misfolded protein accumulation in FKBP8 knockdown cells. Thus, it is unlikely that autophagy is involved in the degradation of FKBP8-mediated protein aggregates. Knockdown of FKBP8 induced Proteostat dye-positive protein aggregates without affecting ER stress or cell death, whereas H2O2 treatment in FKBP8 knockdown H9c2 cells accelerated ER stress and cell death without increasing the number of the cells with Proteostat dye-positive protein aggregates. Therefore, protein aggregates themselves in FKBP8 knockdown H9c2 myocytes were not toxic to the cells. However, H2O2 administration resulted in apoptotic cell death, suggesting that FKBP8 knockdown H9c2 cells with Proteostat-positive structures were more susceptible to H2O2 stimuli. H2O2-induced accumulation of misfolded proteins which are not detected by Proteostat dye resulted in ER stress and subsequent caspase-12-mediated apoptosis in FKBP8 knockdown cells. In in vivo, there was no difference in cardiac function or ultrastructure of cardiomyocytes between sham-operated Fkbp8+/+ and Fkbp8−/− hearts, suggesting that compensatory mechanisms prevented the accumulation of misfolded proteins in Fkbp8−/− mice. However, upon pressure overload, Fkbp8−/− mice cannot overcome misfolded protein load, leading to ER stress-induced apoptosis and development of dilated cardiomyopathy, indicating that FKBP8 plays an essential role in the prevention of misfolded protein accumulation and resultant apoptosis in pressure overloaded hearts. It is likely that upregulation of FKBP8 in the heart after TAC is an adaptive mechanism against stress. The precise molecular mechanisms how protein aggregates affect myocardial damage remain to be elucidated.
Chaperones and co-chaperones are essential for protein quality control mechanisms to facilitate protein folding and refolding [4]. Protein misfolding can take place in the ER as well as outside the ER in the cell [32]. Members of the HSP90 family are the most abundant chaperones and highly expressed in cardiomyocytes [4]. We found that FKBP8 binds to HSP90 in cardiomyocytes. FKBP8 is localized to the ER membrane, but almost entirely exposed to the cytosol and contains tetratricopeptide repeat domain (Supplementary Fig. 1A) [9], [10], which is known to interact with HSP90 [43]. FKBP8 may be involved in protein-folding machinery interacting with HSP90 at the ER for protein and ER quality control to prevent accumulation of misfolded proteins. Further study will be required to investigate how the interaction of ER protein FKBP8 with cytosolic protein HSP90 coordinates as chaperone machinery in the cardiomyocytes. FKBP8 is reported to promote the maturation of HERG channel and cystic fibrosis transmembrane conductance regulator (CFTR) [12], [44]. However, we could not detect differences in the protein levels of mature HERG or CFTR between TAC-operated Fkbp8+/+ and Fkbp8−/− hearts (Supplementary Fig. 9A and 9B). The substrates of FKBP8 or the components of accumulated protein aggregates in FKBP8-deficient cardiomyocytes remain to be elucidated. In addition, the mechanism to degrade the FKBP8-related protein aggregates requires further investigation.
In summary, deficiency of FKBP8 induced the development of dilated cardiomyopathy. This is presumably due to increased apoptotic cardiomyocyte death and the accumulation of misfolded proteins. It has been reported that FKBP8 plays various roles in cellular and molecular regulation, so it is possible that the function of FKBP8 might be cell- or stress-type specific. In cardiomyocytes, FKBP8-mediated protein quality control is critical to maintain cellular homeostasis against stress. Our findings provide the first evidence that FKBP8 has protective effects in the heart and upregulation of FKBP8 could be a potential therapeutic target for heart failure patients.
Sources of funding
This work was supported by the British Heart Foundation (CH/11/3/29051, RG/11/12/29052 and RG/16/15/32294) to K.O., Fondation Leducq (15CVD04) to K.O. and JSPS KAKENHI Grant Numbers 15H04822 to K.O. and 15K09140 to O.Y.
Disclosures
None.
Acknowledgments
Acknowledgments
We thank Prof. Noboru Mizushima, University of Tokyo, for providing pEGFP-LC3, and Ms. Brodie Quine and Mr. Mohsin Arain for their technical assistance.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.yjmcc.2017.11.004.
Appendix A. Supplementary data
Supplementary material
References
- 1.Yancy C.W., Jessup M., Bozkurt B., Butler J., Casey D.E., Jr., Drazner M.H. 2013 ACCF/AHA guideline for the management of heart failure: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2013;128:1810–1852. doi: 10.1161/CIR.0b013e31829e8807. [DOI] [PubMed] [Google Scholar]
- 2.McLendon P.M., Robbins J. Proteotoxicity and cardiac dysfunction. Circ. Res. 2015;116:1863–1882. doi: 10.1161/CIRCRESAHA.116.305372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Willis M.S., Patterson C. Proteotoxicity and cardiac dysfunction—Alzheimer's disease of the heart? N. Engl. J. Med. 2013;368:455–464. doi: 10.1056/NEJMra1106180. [DOI] [PubMed] [Google Scholar]
- 4.Willis M.S., Patterson C. Hold me tight: role of the heat shock protein family of chaperones in cardiac disease. Circulation. 2010;122:1740–1751. doi: 10.1161/CIRCULATIONAHA.110.942250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Minamino T., Kitakaze M. ER stress in cardiovascular disease. J. Mol. Cell. Cardiol. 2010;48:1105–1110. doi: 10.1016/j.yjmcc.2009.10.026. [DOI] [PubMed] [Google Scholar]
- 6.Mizushima N., Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 7.Okamoto K., Kondo-Okamoto N., Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev. Cell. 2009;17:87–97. doi: 10.1016/j.devcel.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 8.Murakawa T., Yamaguchi O., Hashimoto A., Hikoso S., Takeda T., Oka T. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat. Commun. 2015;6:7527. doi: 10.1038/ncomms8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lam E., Martin M., Wiederrecht G. Isolation of a cDNA encoding a novel human FK506-binding protein homolog containing leucine zipper and tetratricopeptide repeat motifs. Gene. 1995;160:297–302. doi: 10.1016/0378-1119(95)00216-s. [DOI] [PubMed] [Google Scholar]
- 10.Shirane M., Nakayama K.I. Inherent calcineurin inhibitor FKBP38 targets Bcl-2 to mitochondria and inhibits apoptosis. Nat. Cell Biol. 2003;5:28–37. doi: 10.1038/ncb894. [DOI] [PubMed] [Google Scholar]
- 11.Wang X., Venable J., LaPointe P., Hutt D.M., Koulov A.V., Coppinger J. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell. 2006;127:803–815. doi: 10.1016/j.cell.2006.09.043. [DOI] [PubMed] [Google Scholar]
- 12.Walker V.E., Atanasiu R., Lam H., Shrier A. Co-chaperone FKBP38 promotes HERG trafficking. J. Biol. Chem. 2007;282:23509–23516. doi: 10.1074/jbc.M701006200. [DOI] [PubMed] [Google Scholar]
- 13.Choi B.H., Feng L., Yoon H.S. FKBP38 protects Bcl-2 from caspase-dependent degradation. J. Biol. Chem. 2010;285:9770–9779. doi: 10.1074/jbc.M109.032466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haupt K., Jahreis G., Linnert M., Maestre-Martinez M., Malesevic M., Pechstein A. The FKBP38 catalytic domain binds to Bcl-2 via a charge-sensitive loop. J. Biol. Chem. 2012;287:19665–19673. doi: 10.1074/jbc.M111.317214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosner M., Hofer K., Kubista M., Hengstschlager M. Cell size regulation by the human TSC tumor suppressor proteins depends on PI3K and FKBP38. Oncogene. 2003;22:4786–4798. doi: 10.1038/sj.onc.1206776. [DOI] [PubMed] [Google Scholar]
- 16.Bai X., Ma D., Liu A., Shen X., Wang Q.J., Liu Y. Rheb activates mTOR by antagonizing its endogenous inhibitor, FKBP38. Science. 2007;318:977–980. doi: 10.1126/science.1147379. [DOI] [PubMed] [Google Scholar]
- 17.Bulgakov O.V., Eggenschwiler J.T., Hong D.H., Anderson K.V., Li T. FKBP8 is a negative regulator of mouse sonic hedgehog signaling in neural tissues. Development. 2004;131:2149–2159. doi: 10.1242/dev.01122. [DOI] [PubMed] [Google Scholar]
- 18.Shirane M., Ogawa M., Motoyama J., Nakayama K.I. Regulation of apoptosis and neurite extension by FKBP38 is required for neural tube formation in the mouse. Genes Cells. 2008;13:635–651. doi: 10.1111/j.1365-2443.2008.01194.x. [DOI] [PubMed] [Google Scholar]
- 19.Nishida K., Yamaguchi O., Hirotani S., Hikoso S., Higuchi Y., Watanabe T. p38α mitogen-activated protein kinase plays a critical role in cardiomyocyte survival but not in cardiac hypertrophic growth in response to pressure overload. Mol. Cell. Biol. 2004;24:10611–10620. doi: 10.1128/MCB.24.24.10611-10620.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakai A., Yamaguchi O., Takeda T., Higuchi Y., Hikoso S., Taniike M. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 21.Omiya S., Omori Y., Taneike M., Protti A., Yamaguchi O., Akira S. Toll-like receptor 9 prevents cardiac rupture after myocardial infarction in mice independently of inflammation. Am. J. Physiol. Heart Circ. Physiol. 2016;311:H1485–H1497. doi: 10.1152/ajpheart.00481.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taneike M., Nishida K., Omiya S., Zarrinpashneh E., Misaka T., Kitazume-Taneike R. mTOR hyperactivation by ablation of tuberous sclerosis complex 2 in the mouse heart induces cardiac dysfunction with the increased number of small mitochondria mediated through the down-regulation of autophagy. PLoS One. 2016;11 doi: 10.1371/journal.pone.0152628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shen D., Coleman J., Chan E., Nicholson T.P., Dai L., Sheppard P.W. Novel cell- and tissue-based assays for detecting misfolded and aggregated protein accumulation within aggresomes and inclusion bodies. Cell Biochem. Biophys. 2011;60:173–185. doi: 10.1007/s12013-010-9138-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Noda N.N., Kumeta H., Nakatogawa H., Satoo K., Adachi W., Ishii J. Structural basis of target recognition by Atg8/LC3 during selective autophagy. Genes Cells. 2008;13:1211–1218. doi: 10.1111/j.1365-2443.2008.01238.x. [DOI] [PubMed] [Google Scholar]
- 25.Kim I., Lemasters J.J. Mitochondrial degradation by autophagy (mitophagy) in GFP-LC3 transgenic hepatocytes during nutrient deprivation. Am. J. Physiol. Heart Circ. Physiol. 2011;300:C308–C317. doi: 10.1152/ajpcell.00056.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oka T., Hikoso S., Yamaguchi O., Taneike M., Takeda T., Tamai T. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485:251–255. doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shirakabe A., Zhai P., Ikeda Y., Saito T., Maejima Y., Hsu C.-P. Drp1-dependent mitochondrial autophagy plays a protective role against pressure overload–induced mitochondrial dysfunction and heart failure. Circulation. 2016;133:1249–1263. doi: 10.1161/CIRCULATIONAHA.115.020502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ebrahimi-Fakhari D., Saffari A., Wahlster L., DiNardo A., Turner D., Lewis T.L., Jr. Impaired mitochondrial dynamics and mitophagy in neuronal models of tuberous sclerosis complex. Cell Rep. 2016;17:1053–1070. doi: 10.1016/j.celrep.2016.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhujabal Z., Birgisdottir A.B., Sjottem E., Brenne H.B., Overvatn A., Habisov S. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 2017;18:947–961. doi: 10.15252/embr.201643147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wencker D., Chandra M., Nguyen K., Miao W., Garantziotis S., Factor S.M. A mechanistic role for cardiac myocyte apoptosis in heart failure. J. Clin. Invest. 2003;111:1497–1504. doi: 10.1172/JCI17664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malhotra J.D., Miao H., Zhang K., Wolfson A., Pennathur S., Pipe S.W. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. U. S. A. 2008;105:18525–18530. doi: 10.1073/pnas.0809677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buchberger A., Bukau B., Sommer T. Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol. Cell. 2010;40:238–252. doi: 10.1016/j.molcel.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 33.Maejima Y., Kyoi S., Zhai P., Liu T., Li H., Ivessa A. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat. Med. 2013;19:1478–1488. doi: 10.1038/nm.3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pankiv S., Clausen T.H., Lamark T., Brech A., Bruun J.A., Outzen H. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 35.Shoji-Kawata S., Sumpter R., Leveno M., Campbell G.R., Zou Z., Kinch L. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494:201–206. doi: 10.1038/nature11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tannous P., Zhu H., Nemchenko A., Berry J.M., Johnstone J.L., Shelton J.M. Intracellular protein aggregation is a proximal trigger of cardiomyocyte autophagy. Circulation. 2008;117:3070–3078. doi: 10.1161/CIRCULATIONAHA.107.763870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Edlich F., Weiwad M., Erdmann F., Fanghanel J., Jarczowski F., Rahfeld J.U. Bcl-2 regulator FKBP38 is activated by Ca2 +/calmodulin. EMBO J. 2005;24:2688–2699. doi: 10.1038/sj.emboj.7600739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saita S., Shirane M., Nakayama K.I. Selective escape of proteins from the mitochondria during mitophagy. Nat. Commun. 2013;4:1410. doi: 10.1038/ncomms2400. [DOI] [PubMed] [Google Scholar]
- 39.Kopito R.R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–530. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- 40.Wang X., Osinska H., Klevitsky R., Gerdes A.M., Nieman M., Lorenz J. Expression of R120G-alphaB-crystallin causes aberrant desmin and alphaB-crystallin aggregation and cardiomyopathy in mice. Circ. Res. 2001;89:84–91. doi: 10.1161/hh1301.092688. [DOI] [PubMed] [Google Scholar]
- 41.Sanbe A., Osinska H., Saffitz J.E., Glabe C.G., Kayed R., Maloyan A. Desmin-related cardiomyopathy in transgenic mice: a cardiac amyloidosis. Proc. Natl. Acad. Sci. U. S. A. 2004;101:10132–10136. doi: 10.1073/pnas.0401900101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bhuiyan M.S., Pattison J.S., Osinska H., James J., Gulick J., McLendon P.M. Enhanced autophagy ameliorates cardiac proteinopathy. J. Clin. Invest. 2013;123:5284–5297. doi: 10.1172/JCI70877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scheufler C., Brinker A., Bourenkov G., Pegoraro S., Moroder L., Bartunik H. Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell. 2000;101:199–210. doi: 10.1016/S0092-8674(00)80830-2. [DOI] [PubMed] [Google Scholar]
- 44.Banasavadi-Siddegowda Y.K., Mai J., Fan Y., Bhattacharya S., Giovannucci D.R., Sanchez E.R. FKBP38 peptidylprolyl isomerase promotes the folding of cystic fibrosis transmembrane conductance regulator in the endoplasmic reticulum. J. Biol. Chem. 2011;286:43071–43080. doi: 10.1074/jbc.M111.269993. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material