

Abstract
Over five decades of research have yielded a large body of information on how purified proteins attain their native state when refolded in the test tube, starting from a chemically or thermally denatured state. Nevertheless, we still know little about how proteins fold and unfold in their natural biological habitat: the living cell. Indeed, a variety of cellular components, including molecular chaperones, the ribosome, and crowding of the intracellular medium, modulate folding mechanisms in physiologically relevant environments. This review focuses on the current state of knowledge in protein folding in the cell with emphasis on the early stage of a protein’s life, as the nascent polypeptide traverses and emerges from the ribosomal tunnel. Given the vectorial nature of ribosome-assisted translation, the transient degree of chain elongation becomes a relevant variable expected to affect nascent protein foldability, aggregation propensity and extent of interaction with chaperones and the ribosome.
Keywords: ribosome, molecular chaperones, ribosomal exit tunnel, nascent protein, molecular crowding, ribosome-bound nascent chain
PRINCIPLES OF IN VITRO PROTEIN FOLDING
Since Christian Anfinsen’s pioneering article on the relation between protein sequence and structure in 1954 (1) and his formulation of the thermodynamic hypothesis of protein folding in 1962 (35), thousands of articles have been written on how proteins travel through energy landscapes and reach their native state. The large majority of this body of work considers the in vitro refolding mechanisms of pure proteins, starting from a thermally or chemically denatured state diluted into a buffer at physiologically relevant pH. Most experimental and computational studies have so far been carried out on small single-domain proteins. Multidomain proteins are still largely unexplored and have started to receive attention only recently (4).
Over five decades of research on the mechanisms of protein folding in vitro have revealed that there is a wide variability in the way different proteins fold in the test tube (9, 15, 77). Nonetheless, a few important trends of general significance have emerged. The main concepts are worth a summary here because they can be considered the basis for understanding fundamental aspects and mechanistic differences once proteins are allowed to fold and unfold in the complex cellular environment.
First, protein folding does not proceed via a random search (51), and protein energy landscapes are highly funneled (8, 99). The above facts greatly contribute to optimize the efficiency of the conformational search to reach the native state. As a result, a variety of parallel paths are typically present as proteins fold, each generally comprising the formation of numerous transiently populated species, i.e., kinetic intermediates (some experimentally undetectable) separated by energy barriers in the case of rugged landscapes, or progressively evolving conformations undergoing barrierless diffusion toward the native state. The latter scenario typically applies only to very small (<60 residues) proteins. In experimental studies, single-exponential kinetics is often observed. It is important to keep in mind that single-exponential folding is fully compatible with the concept of parallel folding pathways, and it does not necessarily imply a truly two-state folding, which is rarely observed. Indeed, multiple unfolded or partially folded conformations often interconvert faster than the rate-determining steps; hence they do not give rise to distinct kinetic phases (22). In addition, computer simulations suggest that kinetic intermediates are usually present, yet they may be poorly populated and therefore experimentally undetectable (16). Several proteins fold via experimentally detectable folding intermediates, which in some cases are en route to the native state.
Second, individual elements of secondary structure may form very fast (18), as in the case of α-helices (typically <1 μs), but are usually not stable in the absence of long-range tertiary contacts. Therefore, protein folding is generally not a rigorously hierarchical process, and it is extremely rare that high populations of secondary structure (e.g., helices) fold first, followed by collapse and tertiary structure formation. This idea is schematically illustrated in Figure 1 as the class of paths denoted by dashed gray lines, comprising type 1, 2, and 5 species. Studies on isolated polypeptides representing portions of primary structure of entire proteins show that individual helices and sheets are usually unstructured in the absence of surrounding tertiary contacts (23). Investigations on the early stages of protein folding showed that only small populations of secondary structure are detectable before chain collapse [exceptions are some members of the engrailed homeodomain family and protein A (15)]. Furthermore, protein variants containing destabilized versions of highly intrinsically helical regions of the chain are folding competent (12).
Figure 1.
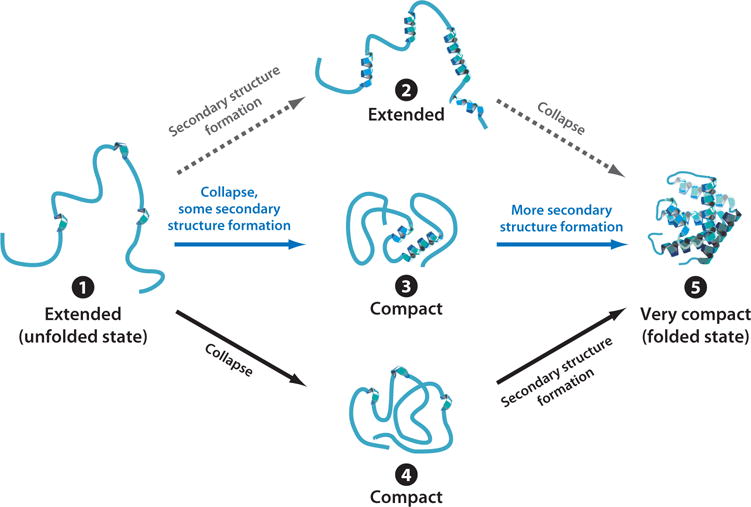
Scheme illustrating limiting in vitro protein folding mechanisms denoted by dashed gray (rarely observed), dark blue, and black arrows. The experiments leading to the formulation of these models are typically performed in purified protein solutions and involve the refolding of unfolded states generated chemically or by temperature jumps. Note that the species other than the unfolded and folded states (denoted 2, 3, and 4) may be either intermediates, transition states, or transient species populated along diffusive downhill routes.
Third, the timescale for protein chain collapse is highly variable (nanoseconds to seconds) and sequence dependent (77). Collapse may (a) occur after most of the secondary structure is formed, as rarely observed experimentally (gray path in Figure 1); (b) be concurrent with most secondary structure formation, as seen in a number of apparently two-state folders (blue path in Figure 1) giving rise to relatively slow collapse with topology-dependent rates; (c) be concurrent with some secondary structure formation followed by slower acquisition of additional secondary structure, as in proteins with detectable folding intermediates such as apomyoglobin (apoMb) (42) (blue path in Figure 1), or (d) precede most secondary structure formation (black path in Figure 1).
The sequence determinants for the above options are not entirely clear yet and represent an outstanding challenge in in vitro protein folding. On the other hand, there are two apparent emerging trends. Collapse is slower when it occurs concomitantly with secondary structure formation, pointing to the kinetic difficulties in assembling secondary and tertiary structure together. In addition, secondary structure formation starting from a collapsed intermediate is also typically slow, pointing to the kinetic challenges in sampling conformational space from collapsed species (especially in large proteins). The above is true even if these species have significant internal dynamics, for instance, in the case of molten globules, and may bear a solvated nonpolar core.
Fourth, the starting species of in vitro folding experiments, the so-called unfolded state, is sometimes far from lacking a structure; therefore, it is not truly unfolded (60, 80). Only expanded highly dynamic unfolded state ensembles follow the three criteria outlined above. Unfolded states bearing significant secondary structure and/or compaction are clearly posed to apply biases to the conformational search, sometimes making it more efficient. The presence of secondary/tertiary structure in proteins under strongly denaturing conditions is particularly interesting in the context of this review, given that the unfolded state populated under physiologically relevant conditions sometimes behaves differently from a self-avoiding Gaussian chain (71).
Fifth, a significant fraction (∼40% in Eukarya) of the proteins expressed in the cell is actually natively unfolded (102). Representatives of this class are known as intrinsically disordered proteins (IDPs) and lack a well-defined independent structure at physiologically relevant pH and ionic strength. IDPs often fold upon interaction with their biological counterparts: Their folding mechanisms, still poorly explored, are beyond the scope of this work.
PROTEIN FOLDING IN THE CELL
In Vitro and In Vivo Protein Folding
Based on the results of pioneering nuclear magnetic resonance (NMR) experiments in live cells, the native structure of medium-size proteins in the intracellular environment is believed to be similar to the one populated in vitro in buffered solution. However, folding mechanisms in the cell are bound to be different from in vitro folding (Figure 2) due to the presence of a different unfolded state (see below); molecular chaperones; the ribosome; a highly crowded medium (200–300 mg ml−1 total protein concentration); cofactors such as heme, NADH, and others; intracellular processes such as posttranslational modifications; and quality-control processes such as protein degradation. In addition, some proteins are also subject to translocation into and out of different cell compartments, secretion, and cotranslational insertion into membranes. The latter processes are neglected in this review, which focuses on the folding of cytosolic soluble proteins.
Figure 2.
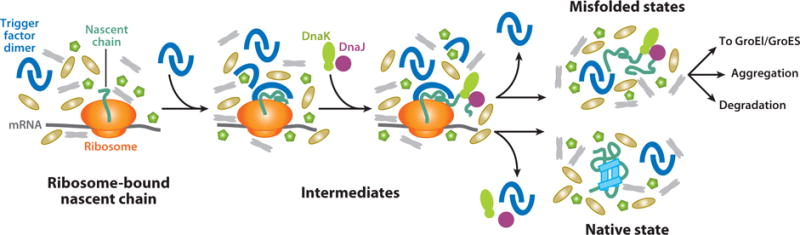
Schematic representation of key aspects of cotranslational protein folding in the crowded milieu of the cellular cytosol.
The Unfolded State
Protein folding and unfolding in the cell can occur either during or after protein biosynthesis, i.e., co- or posttranslationally. In both cases, the nature of the unfolded state is poorly understood, yet likely profoundly different from the nonphysiological unfolded state ensemble of in vitro experiments in denaturants. For instance, in the case of full-length proteins in aqueous media at pH 7, the unfolded state is more compact than in the presence of denaturants (101). The effect of molecular crowding on the unfolded state under native conditions has yet to be studied in depth.
Molecular Chaperones
Molecular chaperones are key components of the cellular environment in bacteria, eukarya, and archaea. Their identity and roles have been reviewed elsewhere (17, 30, 37). Chaperones assist protein folding in the cell by preventing protein misfolding and aggregation and possibly also promoting folding. Interestingly, many chaperones in bacteria have overlapping specificities and their roles can sometimes be swapped (32), except for the bacterial GroEL/ES, the lack of which is lethal to the cell.
FOLDING ON THE RIBOSOME: WHAT IS SPECIAL ABOUT IT?
Incomplete Protein Chains from Single-Domain Proteins Do not Generally Assume a Native-Like Conformation
The presence of ribosome-bound incomplete protein chains is one of the unique features of cotranslational events. In 1967, i.e., soon after the discovery that the biosynthesis of most proteins is catalyzed by the ribosome and proceeds vectorially from the N terminus to the C terminus, Phillips (65) formulated the hypothesis that the N-terminal portion of nascent proteins may start folding during translation. Two years later, Taniuchi & Anfinsen (81) responded by showing that cotranslational folding is unlikely for small- and medium-size single-domain proteins because individual purified N-terminal fragments of staphylococcal nuclease (SNase) of increasing length do not achieve any stable fold until their length closely approaches that of the complete protein. Since then, additional experimental model studies on SNase showed that the C-terminally truncated protein can indeed become compact yet partially disordered with only some of its secondary structure if very few residues are removed from its C terminus (29). This finding suggests that the thermodynamic driving force for native-like tertiary structure formation develops during the very latest stages of chain elongation. Analogous studies on chymotrypsin inhibitor 2 and barnase are in agreement with the above ideas (61). Computational studies based on the burial of nonpolar surface as a function of chain elongation further support this concept (49, 50).
Incomplete Protein Chains can be Prone to Aggregation
Chain elongation model studies on purified model polypeptides from the medium-size (17 kDa) all-α-helical protein sperm whale apoMb (13) provide additional support to the idea that the native fold can be achieved only at lengths close to that of the complete primary structure. In addition, this study shows that incomplete N-terminal chains (from 36 to 119 residues of the 153-residue full-length protein), rich in nonpolar residues, exhibit a strong tendency to aggregate and form nonnative β-strands. The above misfolding/aggregation progressively decreases in magnitude as chain length approaches the full-length protein. This model system study highlights a unique feature of incomplete protein chains bearing a high nonpolar content: their tendency to aggregate. Aggregation of incomplete nascent chains is not tolerable in the cellular environment. The ability of the ribosome to keep chains maximally segregated during translation was demonstrated by a recent cryo-electron tomography study of Escherichia coli polysomes (7). Individual ribosome components of the polysome adopt a staggered or pseudohelical mutual arrangement, with nascent chains maximally spaced and pointing toward the cytosol. This 3D arrangement is naturally posed to minimize self-association of nascent proteins. Another investigation showed that the ribosome’s ability to keep chains segregated prevents the aggregation of incomplete chains of the tailspike protein from the Salmonella phage P22 even in the absence of the cotranslationally active trigger factor (TF) chaperone (25). As soon as ribosome release of the tailspike nascent chains is induced, the incomplete-length chains undergo self-association. The intrinsic ability of the ribosome to prevent the aggregation of rhodanese and lysozyme was also shown (33).
The Ribosome and Its Exit Tunnel Provide a Unique Environment for Nascent Chain Conformational Sampling
The tethering of all nascent polypeptides to the ribosome leads to expanding the function of this amazing machine from that of an mRNA-decoding center and catalyst for peptide bond formation to an obligatory scaffold, and possibly interaction counterpart, during the cotranslational conformational sampling of nascent polypeptides and proteins. The last few years have witnessed enormous progress in the elucidation of the archaeal and bacterial ribosome structure, structure-function relations, and assembly (66, 70, 79, 88, 94). The structures of the 50S large subunit and the entire ribosome solved at high resolution by X-ray crystallography (2, 36, 74, 91) provide ideal support to all studies of protein folding in and out of the exit tunnel. Figure 3a shows the high-resolution 3D structure of the E. coli ribosome, including the small and large subunits and the ribosomal proteins. Figure 3b,c provide schematics of a section of both the bacterial and archaeal ribosomes, respectively, highlighting the ribosomal exit tunnel and the proteins that directly face the tunnel’s interior (L4, L22, L24) or are in close proximity to the tunnel (L23 and L29 in bacteria, and L23, L29, and L39e in archaea). Nascent proteins traverse the tunnel from the ribosome active site (i.e., the peptidyl transferase center, which houses the nascent protein C terminus) up until the tunnel’s exit (31, 85). The tunnel is not completely straight and has a bend. Its length spans 80 to 100 Å, depending on where the exit-side end of the tunnel is defined (Figure 3d) (85).
Figure 3.
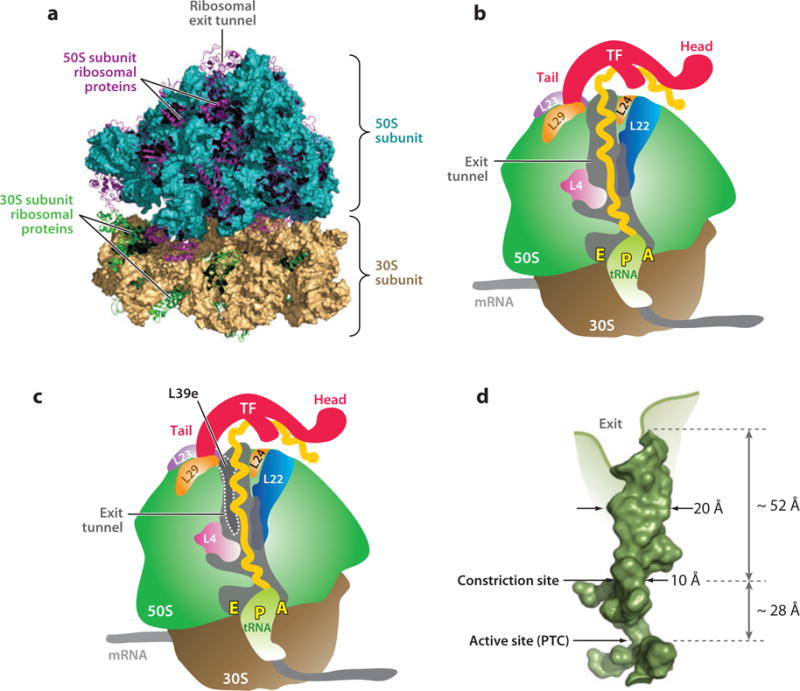
(a) Crystal structure of the Escherichia coli ribosome at 3.5 Å resolution (PDB IDs: 2AVY and 2AW4) (72). The ribosomal RNA is represented as surfaces (23S and 5S RNAs, turquoise; 16S RNA, beige). Ribosomal proteins are shown as ribbons (proteins in 50S subunit, purple; proteins in 30S subunit, green). Schematic representation of a vertical section of the 70S (b) prokaryotic and (c) archaeal ribosomes highlighting the ribosomal proteins facing or near the exit tunnel and the ribosome-associated TF chaperone. A representative hypothetical nascent polypeptide is drawn in yellow. (d) Structure of the ribosomal exit tunnel (PDB file kindly provided by N.R. Voss and P.B. Moore) (85). Abbreviations: PTC, peptidyl transferase center; TF, trigger factor.
Cotranslationally Active Molecular Chaperones Assist the Earliest Stages of a Protein’s Life
The identity of cotranslationally active molecular chaperones varies depending on the kingdom of life and specific organism (30). For instance, in prokaryotes, the ribosome-associated dragon-shaped TF chaperone welcomes a large fraction of all nascent proteins emerging from the ribosomal tunnel, due to its high local concentration (89), and forms an arch above the tunnel (Figure 3b). The resulting constrained environment encompasses sufficient space to host the folding of a small protein domain (3, 28) and serves as a protective shield (38) for nascent chains capable of interacting with it (82). TF latches onto the ribosome via the L23 and L29 proteins (Figure 3b). TF does not exist in eukarya, and it is replaced by a number of other ribosome-associated chaperones (86).
The cotranslationally active chaperone DnaK (i.e., bacterial Hsp70) plays a role complementary to that of TF. The mechanism of action of DnaK and its co-chaperones DnaJ and GrpE has been reviewed (58). More than one Hsp70 are found in eukarya (30).
Finally, the ribosome itself may be far more than a spectator in co- and posttranslational protein folding. For instance, earlier studies by Dasgupta and colleagues (97, 98) and Hardesty (48) showed that ribosomes can promote the folding of denatured proteins. This finding prompted the Hardesty group to suggest that, among its many other activities, the ribosome also plays the role of a chaperone. Future studies hold promise to shed additional light on this interesting proposal.
The Kinetics of TF Binding/Unbinding is Coordinated with Chain Elongation Rates and Nascent Protein Folding
The lifetime of TF-ribosome complexes is much longer (multiple seconds) (55, 68) than the average lifetime of nascent chain-TF complexes (≥ms) in the absence of the ribosome (56). However, the latter lifetime can be modulated by the extent of nascent chain interaction with TF; i.e., it can increase significantly with the size of the nascent protein’s nonpolar-region-binding TF and with ribosome-induced proximity. Furthermore, the presence of the ribosome enhances the association rates between the nascent protein chain and TF. Upon measuring the apparent association and dissociation rates of ribosome-nascent chain complexes with fluorescently labeled TF, Rutkowska et al. (68) proposed a kinetic model for the interplay of chaperone binding/release, cotranslational chain elongation, and protein folding, as shown in Figure 4. This interesting scheme shows how fast association and release of nascent chain to TF (within the ribosomal complex) is compatible with polypeptide chain elongation. However, when the emerged nonpolar region is sufficiently large to slow down release of TF from the nascent chain, the TF chaperone may stay bound to the nascent chain even if released from the ribosome. Additional kinetic studies will certainly clarify how the above events are coordinated with co-and post-translational folding, so that nascent and newly synthesized proteins have a kinetic (and thermodynamic) opportunity to sample conformational space during translation and upon release from the ribosome.
Figure 4.

Model for the dynamic interaction of the trigger factor (TF) chaperone with ribosomes. The symbol t1/2 denotes the half-life for the dissociation of the TF-ribosome binary complex or the apparent half-life for the dissociation of the TF-ribosome-nascent chain ternary complex. (a,b) The apparent association rate constant of TF (green) to ribosomes increases when a peptide chain emerges from the ribosomal exit tunnel. (c) Some longer nascent chains can increase the half-life t1/2 for complex dissociation up to ~53 s. (d) The association rate of TF for ribosomes eventually decreases when a large nascent polypeptide is exposed to the ribosomal surface. TF may remain associated with some nascent chains even after dissociation of TF from its ribosome-binding site. Nonpolar stretches serving as TF binding sites are in blue. Adapted from Reference 68.
Kinetic Considerations on Cotranslational Protein Folding
The best way to study folding at the exit tunnel is undoubtedly to watch the development of nascent protein structure and dynamics concurrently with translation. As shown in the next section, following up on this opportunity is especially desirable for large proteins, given that their translation rates approach intrinsic folding rates (62) and that it is likely that codon usage and ribosomal pausing are posed to affect the actual mechanism of folding.
Indeed, studying the cotranslational folding of fairly small single-domain proteins would also be extremely useful to verify that translation rates are slower than conformational sampling on the ribosome. However, to the best of our knowledge no such studies have been performed, although there are excellent prospects for progress in this area in the near future.
Cotranslational protein folding studies need to preserve the natural translation rates (so that they can be compared with folding rates) and are therefore best performed in vivo. However, working in an in vivo environment is challenging due to (a) the difficulties in selectively detecting folding in the complex cellular environment, and (b) the inability to synchronize translation given the stochastic nature of the process. Biological approaches pioneered by A. Helenius and F.U. Hartl have solved challenge a by monitoring protein activity cotranslationally, and challenge b by pulse-chase experiments often performed in bulk spheroplasts.
WHAT A DIFFERENCE TRANSLATION MAKES
Biosynthesis Rates Affect the Extent of Cotranslational Folding in Multidomain Proteins and Can Be Ad Hoc Modulated
Protein synthesis proceeds at variable rates in different environments and organisms (see Reference 93 and table 1 in Reference 13). For instance, translation rates are faster in vivo than under cell-free conditions. In addition, translation proceeds faster in prokarya (15–20 amino acids s−1) than in eukarya (3–4 amino acids s−1), leading to an average timescale for the production of a small-/medium-size protein of ∼10 s and ∼65 s in prokarya and eukarya, respectively. These fairly long timescales are similar to chaperone binding/unbinding times and longer than the folding/unfolding timescales of small proteins. The above suggests that, in vivo, nascent chains encoding small proteins may have sufficient time to adopt preferred conformations as they are synthesized. On the other hand, very large proteins take multiple seconds to fold and may or may not attain stable conformations cotranslationally. Accordingly, the absence (62) or presence (63) of in vivo cotranslational folding in E. coli seems to be highly protein and codon dependent. Rare codon clusters (14), sometimes localized at interdomain junctions in large proteins, are emerging as important sites for an orchestrated pausing. This pausing is responsible for facilitating cotranslational domain folding before synthesis of the following domain is initiated (92). A proper balance between translation rates and co- and posttranslational folding is important for the production of active ribosome-released multidomain proteins. For instance, mutant ribosomes displaying slower translation than wild-type E. coli ribosomes enhance the production of active multidomain proteins (of eukaryotic origin) in bacteria (76). A detailed review of this topic was recently published by Zhang & Ignatova (93).
Preparation and Analysis of RNCs for Model Studies on the Conformation of Nascent Proteins at Equilibrium
The highest resolution information on protein folding at the exit tunnel has so far been achieved via studies on purified arrested ribosomes bearing nascent proteins, sometimes labeled with fluorophores or NMR-active tags. These studies implicitly assume that nascent protein chains have the opportunity to conformationally equilibrate faster than the rate of translation. This assumption is likely acceptable in many cases, especially for small proteins. However, in general, caution should be exercised and it is desirable to assess the validity of this approximation in each case.
The most common methods to prepare ribosome-bound nascent chains (RNCs) for model studies at equilibrium are outlined in Figure 5a. An exhaustive overview of these methodologies is beyond the scope of this review. Therefore, we simply provide general guidelines here.
Figure 5.
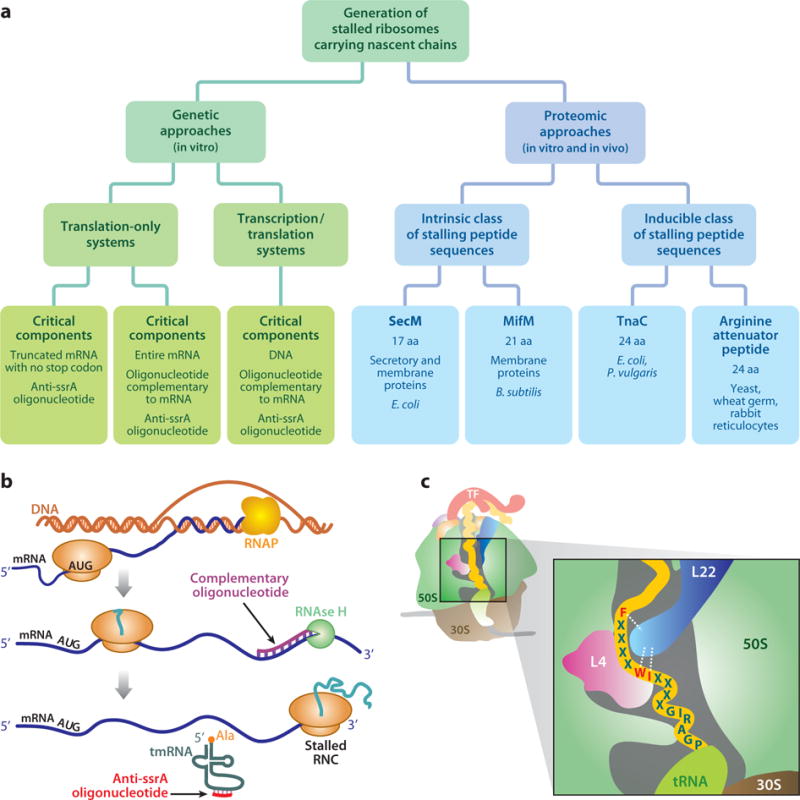
(a) Overview of currently available methods to generate RNCs of well-defined chain length. Step-by-step procedures based on (b) in vitro (cell-free) coupled transcription-translation and (c) SecM stalling. For simplicity, cotranslationally active chaperones are omitted. The 17-residue SecM peptide-stalling sequence (FXXXXWIXXXXGIRAGP) is shown inside the ribosomal tunnel. The underlined amino acids (in red) experience critical interactions with the ribosomal tunnel (white dashed lines) with L22. Abbreviations: RNAP, RNA polymerase; RNC, ribosome-bound nascent chain; X, any residue.
RNCs can be prepared via genetic approaches exploiting addition or in situ production of truncated mRNAs. These methodologies have been employed in vitro, either in cell-free systems (e.g., from E. coli, wheat germ, or rabbit reticulocyte) (20, 96, 103) or via reconstituted systems containing all the necessary components for translation (e.g., PURE, protein synthesis using recombinant elements, based on prokaryotic components) (64, 75). As an example, a procedure for the generation of RNCs in cell-free systems via coupled transcription/translation is shown in Figure 5b (20, 96). Reconstituted in vitro expression systems have recently emerged as a convenient option because of their complete lack of nucleases, proteases, tmRNA, and other undesired components.
Alternatively, stalled ribosomes can be generated via protein-based approaches by controlling translation arrest via special gene products, i.e., short amino acid sequences (typically ∼15–25 residues) that interact strongly with specific portions of the ribosomal tunnel and cause translation to stop (Figure 5a). The most popular of these approaches is based on generating very stable RNCs via the 17-residue SecM arrest sequence (26, 67, 69), as shown in Figure 5c. The SecM approach is particularly convenient when RNCs are generated in vivo, given the affordability of the method and its high yields.
On the other hand, the SecM sequence is by far not the only available method to generate arrested ribosomes by protein-based approaches. Other strategies, based on both intrinsic and inducible classes of ribosome-stalling sequences (e.g., MifM, TnaC, and the Arg attenuator peptide), have been reviewed recently (41) (Figure 5a).
Generation of RNCs at equilibrium enabled analysis of the structure and dynamics of nascent proteins at a level of detail presently unattainable by in vivo studies. This analysis has allowed addressing several questions regarding the stepwise generation of 3D protein structures in nature. Figure 6 provides a global overview of the RNC structural and dynamic aspects that have been addressed thus far. This figure also links these specific folding-related aspects to the technique used to gain the desired information. As shown in Figure 6, a large variety of spectroscopy- and microscopy-based and biological techniques have been used synergistically. Perhaps even more importantly, the diagram shows that the same structural/dynamic question can often be addressed by both biological and spectroscopy/microscopy biophysical approaches. This synergism has been particularly valuable in the field of RNC folding, given the challenging features of RNC for direct biophysical analysis (e.g., large internal dynamics, conformational heterogeneity, and large size of the RNC complexes). Despite the potential of the biophysical methods to provide higher resolution insights, biological approaches taking advantage of properties such as protein activity and antibody response are often efficient and highly informative.
Figure 6.
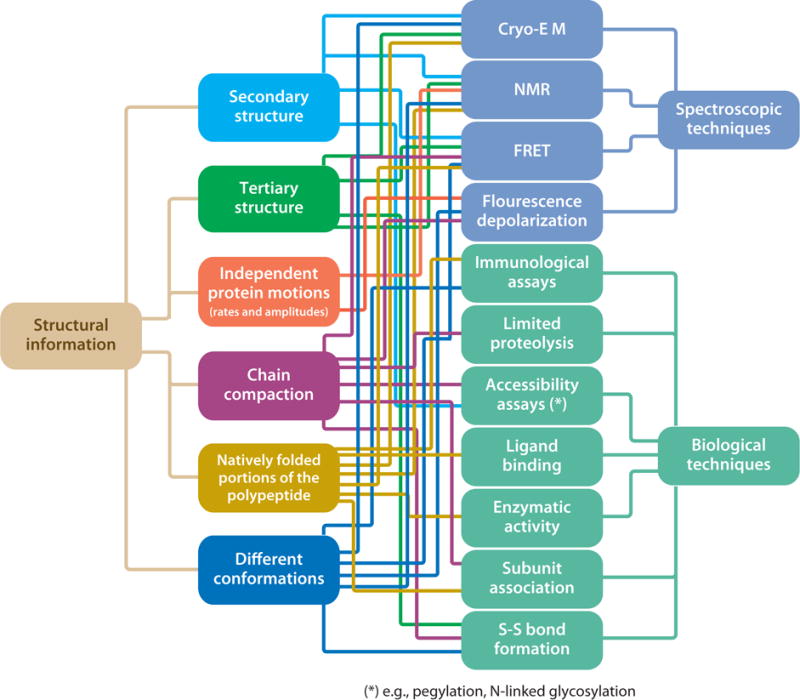
Relationship between specific RNC structural features and biological or spectroscopic techniques employed to elucidate them. Abbreviations: cryo-EM, cryo-electron microscopy; FRET, Förster resonance energy transfer; NMR, nuclear magnetic resonance; RNC, ribosome-bound nascent chain.
Investigations on Nascent Polypeptides Inside the Ribosomal Tunnel
The ribosome exit tunnel (Figure 3d) has a width that ranges from 10 Å (constriction site) to 20 Å (widest region). These dimensions are incompatible with tertiary structure formation within the tunnel’s interior. Major tunnel dynamics, presumably accompanied by extensive ribosome rearrangements, would be required for the tunnel to host even a simple tertiary fold such as a helical hairpin. Therefore, although it is recognized that the ribosome is a dynamic entity, it is likely that no tertiary structure, and only secondary structure, can be populated in nascent chains inside the ribosomal tunnel (85). This argument is supported by the finding of a highly spatially confined environment inside the tunnel, revealed by recent investigations showing that the N terminus of nascent polypeptides buried in the tunnel experiences narrow local motions (21).
The lower limit of the tunnel length is 80 Å, as proposed by Voss et al. (85) for the archaeal ribosome. Given this value, a fully α-helical polypeptide would bury approximately 53 residues (assuming an effective length of 1.5 Å per residue for the α-helix), and a fully extended polypeptide would bury approximately 23 residues (assuming an effective length of 3.5 Å per residue for an extended chain). These geometrical considerations prompt the question of whether any specific secondary structure is supported by the tunnel. Pioneering experiments by Malkin & Rich in 1967 (57), using in vivo pulse-chase techniques followed by cell lysis and proteolysis, showed that approximately 30 to 35 residues of nascent globin are protected from proteolysis in eukaryotic polysomes, implying that those residues are buried inside the ribosomal exit tunnel. These results are consistent with later investigations (reviewed in Reference 47) showing that there are 30 to 40 protected residues in nascent proteins. The above finding supports the presence of a partially helical conformation inside the tunnel. Computational studies by Ziv et al. (95) showed that a helical conformation can be entropically favored in a cylinder that models the ribosomal tunnel’s dimensions. This result suggests that even a Teflon-like noninteracting tunnel (2) may be capable of inducing helical structure, especially in the case of nascent polypeptides whose coil state is highly disordered. Some recent high-resolution experiments further clarify this matter.
Förster resonance energy transfer (FRET) investigations on peptide sequences from a soluble secretory protein and a membrane protein showed that inside the eukaryotic tunnel the former is less helical than the latter (43). As the polypeptide chain elongates, helices can persist beyond the tunnel if they are stable in that environment. For instance, a peptide sequence from an integral membrane protein stays helical outside the tunnel as it is inserted into the membrane. However, the same sequence loses its helicity if the ribosomal surface faces bulk solution and is not bound to the membrane. Moreover, peptide sequences from soluble proteins have negligible helicity both inside and outside the tunnel (43). In summary, the ribosomal tunnel is capable of inducing helicity in nascent polypeptides, and this phenomenon is highly sequence dependent.
Additional investigations from Deutsch and coworkers (52–54) support the above conclusion by exploiting ingenuous accessibility assays, which enabled the detection of distinct tunnel zones characterized by different (highly negative) electrostatic potential. The authors also showed that some of these tunnel regions promote polypeptide chain compaction, suggestive of helix formation (46, 83, 84).
Recent cryo-electron microscopy (cryo-EM) work by Beckman and coworkers (5, 6) provides to date the highest-resolution insights on nascent secondary structure within the exit tunnel. As shown in Figure 7, the authors detected helical structure for sequences with high helical propensity in distinct regions of the tunnel (5). However, sequences with lower intrinsic helical propensity are disordered (5, 73). This work effectively complements and supports the findings by Johnson and Deutsch (46, 52, 84, 90, 103).
Figure 7.

Cryo-EM maps of different peptidyl tRNAs inside the eukaryotic ribosome’s P-site and exit tunnel. (a) 80S–helix 1 RNC, (b) 80S–DPAP RNC, (c) 80S–helix2 RNC, and (d) enlarged view of transparent density of panel a with fitted ribbon model for tRNA and nascent chain. (e, f) Enlarged view of panel c with alternative models for helix 2 nascent chain. Red arrows indicate corresponding region (residues 97–108) modeled as helical (e) or extended (f). (g) Schematic cross-section of 80S–helix 1 RNC representing helix formation within the exit tunnel. Abbreviations: cryo-EM, cyro-electron microscopy; RNC, ribosome-bound nascent chain; DPAP, dipeptidylaminopeptidase; PTC, peptidyl transferase center. Adapted by permission from Macmillan Publishers Ltd: Nature Structural & Molecular Biology (Reference 5), copyright (2010).
Whether the secondary structure formation in distinct regions of the tunnel results from specific polypeptide-tunnel interactions or whether it is driven (or at least contributed by) by entropic effects is still unclear and in need of further investigation. In the specific case of the SecM and TnaC ribosome-stalling sequences, convincing evidence for tunnel-polypeptide interactions was presented (73, 90).
Investigations on Nascent Proteins Emerging from the Ribosomal Tunnel
Our understanding of polypeptide conformation and dynamics as nascent proteins emerges from the ribosomal tunnel is not as advanced as our knowledge on nascent peptide structure inside the tunnel. The many experimental challenges presented by out-of-tunnel RNCs include the high conformational heterogeneity of the nascent chain and the variable effects introduced by cotranslationally active chaperones. Nevertheless, considerable progress has been made, and recent technical advances hold promise for additional exciting future progress. Comprehensive reviews of earlier work are available (24, 27, 47). Here, we focus on recent findings. In short, several examples of independent nascent structure and dynamics were discovered in RNCs emerging from the tunnel, defying the earlier proposal (81) that proteins acquire an independent conformation only after departing from the ribosome.
Investigations by Merz et al. (59) based on chemical cross-linking showed that nascent proteins with a significant nonpolar content emerging out the tunnel have a tendency to interact with the TF chaperone via its elongated binding surface (Figure 8). It is plausible that, while interacting with TF, the nascent protein also binds/unbinds TF and, possibly, other chaperones as it gets elongated, therefore maintaining its ability to sample conformational space while transiently non-TF-bound.
Figure 8.
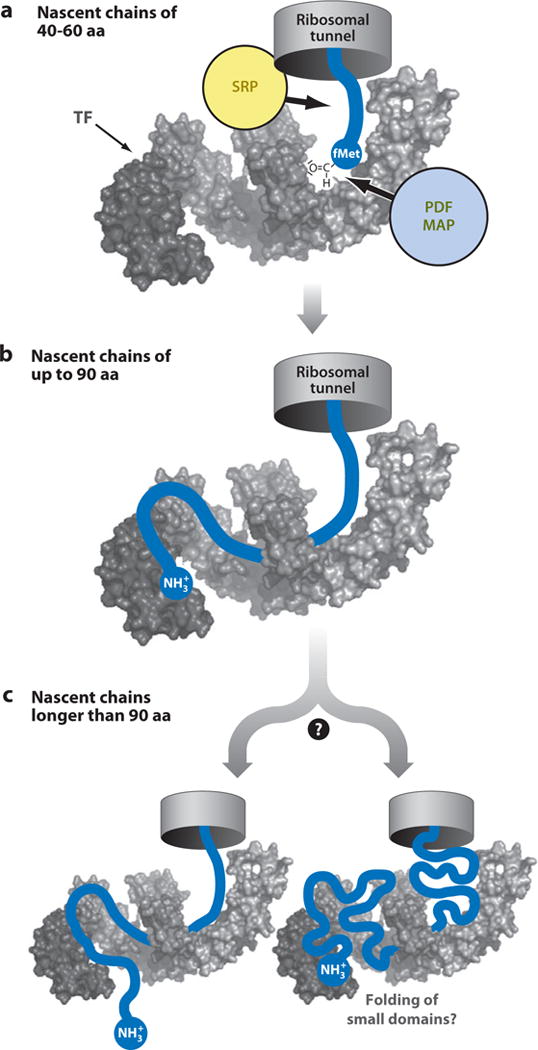
Mode for TF chaperone binding to nascent polypeptides based on cross-linking experiments by Merz et al. (59). TF directs the nascent chains through its interior in a sequence- and length-dependent manner. Interactions with TF are (a) moderate for nascent chains 40 to 60 residues long, and (b) considerable for nascent chains up to 90 residues, where the nascent chain’s N terminus reaches up to the TF PPIase domain (head). (c) Upon further elongation, the nascent chain may leave TF or it may accumulate in the interior of the TF chaperone. Abbreviations: PDF, protein deformylase; MAP, methionine aminopeptidase; SRP, signal recognition particle; TF, trigger factor. Adapted by permission from Macmillan Publishers Ltd: EMBO Journal (Reference 59), copyright (2008).
Cryo-EM images of polypeptides emerging from the ribosomal tunnel (34, 59) provided somewhat moderate structural detail. On the other hand, these studies were important to establish the possibility of tertiary structure formation outside the exit tunnel, in small single domain proteins. Additional evidence on 3D structure development comes from nascent chains from the ion channels, where tertiary structure was detected close to the ribosomal tunnel exit, via accessibility experiments based on side chain pegylation (45, 46). These results are supported by recent computational investigations (100).
Analysis of fluorescence depolarization decays of RNCs and ribosome-released fluorophore-labeled apoMb in the frequency domain (87) enabled Ellis et al. (20) to study the dynamics of nascent apoMb’s N terminus on the subnanosecond timescale and follow the formation of an independent protein domain on the nanosecond timescale, as shown in Figure 9a,b. ApoMb RNCs acquire independent dynamics, indicative of compact or semicompact species, only when a significant portion of the sequence emerges from the ribosomal tunnel. The rotational correlation time reporting on the protein’s nanosecond local motions increases significantly upon nascent protein release from the ribosome, showing that the structure of the full-length RNC differs from that of the ribosome-released native apoMb. RNCs encoding the natively unfolded protein PIR (phosphorylated insulin receptor interaction region) experience no motions on the nanosecond timescale, suggesting that PIR does not fold on the ribosome. The spatial amplitude of the nascent chain local motions is very narrow inside and, surprisingly, even outside the ribosomal tunnel (Figure 9c,d) (21). This is true even when RNCs are depleted of bound chaperones (TF and Hsp70). This result suggests that both the tunnel and the outer surface of the ribosome exert a severe local confinement on nascent apoMb and PIR.
Figure 9.
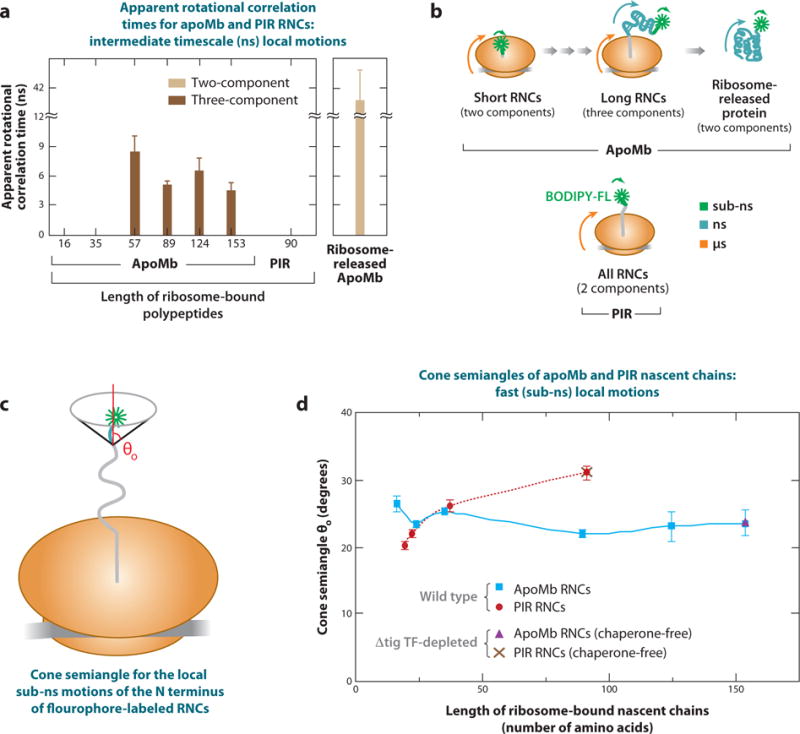
(a) Frequency domain dynamic fluorescence depolarization of ribosome-bound apoMb and PIR nascent chains generated in an Escherichia coli cell-free system. Data are shown only for the nanosecond local motions that reveal the presence of a small compact or semicompact species. (b) Scheme highlighting the motions associated with each fluorescence phase with each associated component of the motion. (c) Scheme illustrating the spatial amplitude of the subnanosecond local motion of the N terminus of the fluorophore-labeled RNC. The symbol θo represents the cone semiangle (in red) assessed in panel d. (d) Amplitude of the fast (subnanosecond) motions experienced by the N termini of nascent apoMb and natively unfolded PIR nascent polypeptides of increasing length under different conditions. Data were collected for samples prepared from either wild-type or Δtig TF-depleted cell strains. Panels a and b adapted with permission from References 20 and 21, respectively. Copyright 2008 and 2009, respectively, American Chemical Society and John Wiley and Sons. Abbreviations: apoMb, apomyoglobin; PIR, phosphorylated insulin receptor interaction region; RNC, ribosome-bound nascent chain; TF, trigger factor chaperone.
The limits of NMR spectroscopy have been pushed by recent studies on RNCs at atomic resolution (10, 11, 19, 39, 40). These investigations revealed that nascent single-domain proteins are not fully structured before they have entirely emerged from the ribosomal tunnel, consistent with the expectation that the C-terminal portion of the chain plays an important role in folding (49, 50).
Taken together, the above findings suggest that relatively small, full-length single-domain nascent proteins may adopt compact conformations outside the ribosomal tunnel. However, the nascent chains whose buried C-terminal residues are not available for folding may retain a considerable degree of disorder. Additional future studies are needed to provide more extensive evidence for these emerging trends.
The influence of the ribosome on protein folding is striking particularly for very large proteins unable to fold in vitro in the absence or presence of molecular chaperones. For such systems (e.g., the trimeric phage P2 tailspike protein), cotranslational folding is an irreplaceable requirement to attain the folded state and exploit biological activity (25). This important concept is illustrated in Figure 10.
Figure 10.
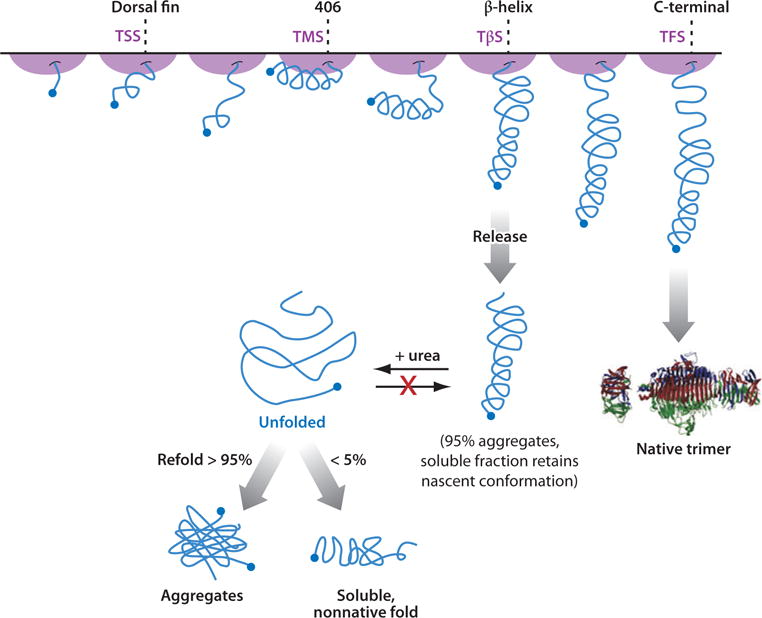
Model for the cotranslational folding of P22 tailspike nascent protein chains. Abbreviations: TSS, tailspike short stalled nascent chain; TMS, tailspike mid-length stalled nascent chain; TβS, tailspike stalled nascent chain with the entire β-helix exposed; TFS, tailspike full stalled nascent chain. Reprinted from the Journal of Molecular Biology, Vol. 383, Evans MS, Sander IM, Clark PL. “Cotranslational folding promotes beta-helix formation and avoids aggregation in vivo” pp. 683–92, Copyright (2008), with permission from Elsevier.
SUMMARY POINTS.
Chain compaction preceding or concurrent with secondary structure formation is a dominant class of mechanisms for the in vitro folding of small- and medium-size proteins, starting from largely unstructured unfolded ensembles.
The unfolded state of full-length proteins is believed to be rather compact in aqueous solution and physiological pH. Hence, secondary structure formation from compact states may be an important motif in posttranslational protein folding in the cell. Landscapes corresponding to this process may be rather rugged.
Incomplete N-terminal protein fragments (lacking the C terminus) often lack much of the native structure and may aggregate in aqueous solution and physiological pH, in the absence of the ribosome and molecular chaperones.
What do incomplete protein chains look like before translation is complete? The answer to this question is still largely unknown but great progress has been made over the past few years. There is a lot of activity in this exciting area.
The ribosomal tunnel is narrow and it provides an extremely spatially constrained environment for nascent polypeptides. The tunnel is capable of inducing helical structure, even in nascent polypeptides (derived from soluble proteins) that lack independent structure in solution. However, this process is highly sequence dependent.
The ribosomal tunnel consists of zones that differ in chemical potential and may promote secondary structure formation to a different degree.
Folding-competent proteins emerging from the ribosomal exit tunnel can assume a compact or semicompact conformation. Small single-domain proteins experience variations in their chain dynamics (and possibly folding) as they are released from the ribosome.
Very large proteins such as P22 tailspike are incapable of reaching their native state unless they are allowed to fold vectorially on the ribosome.
Acknowledgments
We are grateful to all the past and present members of the Cavagnero group for their invaluable contributions. We thank Bernd Bukau, Anna Rutkowska, Elke Deuerling, Frieder Merz, Patricia Clark, Roland Beckmann, and Daniel Wilson for providing figures. The protein folding research in the Cavagnero group was funded by the National Science Foundation (grants MCB-0951209 and MCB-0544182), the National Institutes of Health (grants R21AI079656, R01GM068535, and R21GM071012), the Research Corporation Research Innovation Award, and the Shaw and Vilas Associates Awards.
Glossary
- apoMb
apomyoglobin
- Molten globule
a highly dynamic nonnative compact state lacking a considerable fraction of a protein’s secondary structure
- Transition state ensemble
a collection of conformations that lie at the maximum of energy barriers in protein folding energy landscapes
- IDP
intrinsically disordered protein
- NMR
nuclear magnetic resonance
- SNase
staphylococcal nuclease
- TF
trigger factor
- Spheroplast
a bacterium that has been deprived of the cell wall
- RNC
ribosome-bound nascent chain
- Ribosome exit tunnel
a narrow (10–20 Å) tunnel in the interior of the ribosome large subunit that nascent proteins need to traverse as they are being synthesized, before reaching the ribosome’s surface
- FRET
Förster resonance energy transfer
- cryo-EM
cryo-electron microscopy
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Anfinsen CB, Redfield RR, Choate WI, Page J, Carroll WR. Studies on the gross structure, cross-linkages, and terminal sequences in ribonuclease. J Biol Chem. 1954;207:201–10. [PubMed] [Google Scholar]
- 2.Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 angstrom resolution. Science. 2000;289:905–20. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 3.Baram D, Pyetan E, Sittner A, Auerbach-Nevo T, Bashan A, Yonath A. Structure of trigger factor binding domain in biologically homologous complex with eubacterial ribosome reveals its chaperone action. Proc Natl Acad Sci USA. 2005;102:12017–22. doi: 10.1073/pnas.0505581102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Batey S, Nickson AA, Clarke J. Studying the folding of multidomain proteins. HFSP J. 2008;2:365–77. doi: 10.2976/1.2991513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhushan S, Gartmann M, Halic M, Armache J-P, Jarasch A, et al. Alpha-helical nascent polypeptide chains visualized within distinct regions of the ribosomal exit tunnel. Nat Struct Mol Biol. 2010;17:313–17. doi: 10.1038/nsmb.1756. [DOI] [PubMed] [Google Scholar]
- 6.Bhushan S, Meyer H, Starosta AL, Becker T, Mielke T, et al. Structural basis for translational stalling by human cytomegalovirus and fungal arginine attenuator peptide. Mol Cell. 2010;40:138–46. doi: 10.1016/j.molcel.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 7.Brandt F, Etchells SA, Ortiz JO, Elcock AH, Hartl FU, Baumeister W. The native 3D organization of bacterial polysomes. Cell. 2009;136:261–71. doi: 10.1016/j.cell.2008.11.016. [DOI] [PubMed] [Google Scholar]
- 8.Bryngelson JD, Onuchic JN, Socci ND, Wolynes PG. Funnels, pathways, and the energy landscape of protein folding: a synthesis. Proteins Struct Funct Bioinform. 1995;21:167–95. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 9.Brockwell DJ, Smith DA, Radford SE. Protein folding mechanisms: new methods and emerging ideas. Curr Opin Struct Biol. 2000;10:16–25. doi: 10.1016/s0959-440x(99)00043-3. [DOI] [PubMed] [Google Scholar]
- 10.Cabrita LD, Dobson CM, Christodoulou J. Early nascent chain folding events on the ribosome. Isr J Chem USA. 2010;50:99–108. [Google Scholar]
- 11.Cabrita LD, Hsu STD, Launay H, Dobson CM, Christodoulou J. Probing ribosome-nascent chain complexes produced in vivo by NMR spectroscopy. Proc Natl Acad Sci USA. 2009;106:22239–44. doi: 10.1073/pnas.0903750106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cavagnero S, Dyson HJ, Wright PE. Effect of H helix destabilizing mutations on the kinetic and equilibrium folding of apomyoglobin. J Mol Biol. 1999;285:269–82. doi: 10.1006/jmbi.1998.2273. [DOI] [PubMed] [Google Scholar]
- 13.Chow CC, Chow C, Raghunathan V, Huppert TJ, Kimball EB, Cavagnero S. Chain length dependence of apomyoglobin folding: structural evolution from misfolded sheets to native helices. Biochemistry. 2003;42:7090–99. doi: 10.1021/bi0273056. [DOI] [PubMed] [Google Scholar]
- 14.Clarke TF, 4th, Clark PL. Rare codons cluster. PLOS One. 2008;3:e3412. doi: 10.1371/journal.pone.0003412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daggett V, Fersht AR. Is there a unifying mechanism for protein folding? Trends Biochem Sci. 2003;28:18–25. doi: 10.1016/s0968-0004(02)00012-9. [DOI] [PubMed] [Google Scholar]
- 16.Daggett V, Fersht A. The present view of the mechanism of protein folding. Nat Rev Mol Cell Biol. 2003;4:497–502. doi: 10.1038/nrm1126. [DOI] [PubMed] [Google Scholar]
- 17.Deuerling E, Bukau B. Chaperone-assisted folding of newly synthesized proteins in the cytosol. Crit Rev Biochem Mol Biol. 2004;39:261–77. doi: 10.1080/10409230490892496. [DOI] [PubMed] [Google Scholar]
- 18.Eaton WA, Munoz V, Thompson PA, Henry ER, Hofrichter J. Kinetics and dynamics of loops, alpha-helices, beta-hairpins, and fast-folding proteins. Acc Chem Res. 1998;31:745–53. [Google Scholar]
- 19.Eichmann C, Preissler S, Riek R, Deuerling E. Cotranslational structure acquisition of nascent polypeptides monitored by NMR spectroscopy. Proc Natl Acad Sci USA. 2010;107:9111–16. doi: 10.1073/pnas.0914300107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ellis JP, Bakke CK, Kirchdoerfer RN, Jungbauer LM, Cavagnero S. Chain dynamics of nascent polypeptides emerging from the ribosome. ACS Chem Biol. 2008;3:555–66. doi: 10.1021/cb800059u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ellis JP, Culviner PH, Cavagnero S. Confined dynamics of a ribosome-bound nascent globin: Cone angle analysis of fluorescence depolarization decays in the presence of two local motions. Protein Sci. 2009;18:2003–15. doi: 10.1002/pro.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ellison PA, Cavagnero S. Role of unfolded state heterogeneity and en-route ruggedness in protein folding kinetics. Protein Sci. 2006;15:564–82. doi: 10.1110/ps.051758206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Epand RM, Scheraga A. Influence of long-range interactions on structure of myoglobin. Biochemistry. 1968;7:2864–72. doi: 10.1021/bi00848a024. [DOI] [PubMed] [Google Scholar]
- 24.Evans MS, Clark TF, Clark PL. Conformations of co-translational folding intermediates. Protein Pept Lett. 2005;12:189–95. doi: 10.2174/0929866053005908. [DOI] [PubMed] [Google Scholar]
- 25.Evans MS, Sander IM, Clark PL. Cotranslational folding promotes beta-helix formation and avoids aggregation in vivo. J Mol Biol. 2008;383:683–92. doi: 10.1016/j.jmb.2008.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Evans MS, Ugrinov KG, Frese M-A, Clark PL. Homogeneous stalled ribosome nascent chain complexes produced in vivo or in vitro. Nat Methods. 2005;2:757–62. doi: 10.1038/nmeth790. [DOI] [PubMed] [Google Scholar]
- 27.Fedorov AN, Baldwin TO. Cotranslational protein folding. J Biol Chem. 1997;272:32715–18. doi: 10.1074/jbc.272.52.32715. [DOI] [PubMed] [Google Scholar]
- 28.Ferbitz L, Maier T, Patzelt H, Bukau B, Deuerling E, Ban N. Trigger factor in complex with the ribosome forms a molecular cradle for nascent proteins. Nature. 2004;431:590–96. doi: 10.1038/nature02899. [DOI] [PubMed] [Google Scholar]
- 29.Flanagan JM, Kataoka M, Shortle D, Engelman DM. Truncated staphylococcal nuclease is compact but disordered. Proc Natl Acad Sci USA. 1992;89:748–52. doi: 10.1073/pnas.89.2.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frydman J. Folding of newly translated proteins in vivo: the role of molecular chaperones. Annu Rev Biochem. 2001;70:603–47. doi: 10.1146/annurev.biochem.70.1.603. [DOI] [PubMed] [Google Scholar]
- 31.Fulle S, Gohlke H. Statics of the ribosomal exit tunnel: implications for cotranslational peptide folding, elongation regulation, and antibiotics binding. J Mol Biol. 2009;387:502–17. doi: 10.1016/j.jmb.2009.01.037. [DOI] [PubMed] [Google Scholar]
- 32.Genevaux P, Keppel F, Schwager F, Langendijk-Genevaux PS, Hartl FU, Georgopoulos C. In vivo analysis of the overlapping functions of DnaK and trigger factor. EMBO Rep. 2004;5:195–200. doi: 10.1038/sj.embor.7400067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghosh N, Hazra K, Sarkar SN. Ribosome facilitates refolding of rhodanese and lysozyme by suppressing aggregation. Prog Biophys Mol Biol. 1996;65:85. [Google Scholar]
- 34.Gilbert RJC, Fucini P, Connell S, Fuller SD, Nierhaus KH, et al. Three-dimensional structures of translating ribosomes by cryo-EM. Mol Cell. 2004;14:57–66. doi: 10.1016/s1097-2765(04)00163-7. [DOI] [PubMed] [Google Scholar]
- 35.Haber E, Anfinsen CB. Side-chain interactions governing the pairing of half-cystine residues in ribonuclease. J Biol Chem. 1962;237:1839–44. [PubMed] [Google Scholar]
- 36.Harms J, Schluenzen F, Zarivach R, Bashan A, Gat S, et al. High-resolution structure of the large ribosomal subunit from a mesophilic eubacterium. Cell. 2001;107:679–88. doi: 10.1016/s0092-8674(01)00546-3. [DOI] [PubMed] [Google Scholar]
- 37.Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol. 2009;16:574–81. doi: 10.1038/nsmb.1591. [DOI] [PubMed] [Google Scholar]
- 38.Hoffmann A, Merz F, Rutkowska A, Zachmann-Brand B, Deuerling E, Bukau B. Trigger factor forms a protective shield for nascent polypeptides at the ribosome. J Biol Chem. 2006;281:6539–45. doi: 10.1074/jbc.M512345200. [DOI] [PubMed] [Google Scholar]
- 39.Hsu ST, Cabrita LD, Fucini P, Christodoulou J, Dobson CM. Probing side-chain dynamics of a ribosome-bound nascent chain using methyl NMR spectroscopy. J Am Chem Soc. 2009;131:8366–67. doi: 10.1021/ja902778n. [DOI] [PubMed] [Google Scholar]
- 40.Hsu ST, Fucini P, Cabrita LD, Launay H, Dobson CM, Christodoulou J. Structure and dynamics of a ribosome-bound nascent chain by NMR spectroscopy. Proc Natl Acad Sci USA. 2007;104:16516–21. doi: 10.1073/pnas.0704664104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ito K, Chiba S, Pogliano K. Divergent stalling sequences sense and control cellular physiology. Biochem Biophys Res Commun. 2010;393:1–5. doi: 10.1016/j.bbrc.2010.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jennings PA, Wright PE. Formation of a molten globule intermediate early in the kinetic folding pathway of apomyoglobin. Science. 1993;262:892–96. doi: 10.1126/science.8235610. [DOI] [PubMed] [Google Scholar]
- 43.Johnson AE. Functional ramifications of FRET-detected nascent chain folding far inside the membrane-bound ribosome. Biochem Soc Trans. 2004;32:668–72. doi: 10.1042/BST0320668. [DOI] [PubMed] [Google Scholar]
- 44.Katzen F, Chang G, Kudlicki W. The past, present and future of cell-free protein synthesis. Trends Biotechnol. 2005;23:150–56. doi: 10.1016/j.tibtech.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 45.Kosolapov A, Deutsch C. Tertiary interactions within the ribosomal exit tunnel. Nat Struct Mol Biol. 2009;16:405–11. doi: 10.1038/nsmb.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kosolapov A, Tu L, Wang J, Deutsch C. Structure acquisition of the T1 domain of Kv1.3 during biogenesis. Neuron. 2004;44:295–307. doi: 10.1016/j.neuron.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 47.Kramer G, Ramachandiran V, Hardesty B. Cotranslational folding—omnia mea mecum porto? Int J Biochem Cell Biol. 2001;33:541–53. doi: 10.1016/s1357-2725(01)00044-9. [DOI] [PubMed] [Google Scholar]
- 48.Kudlicki W, Coffman A, Kramer G, Hardesty B. Ribosomes and ribosomal RNA as chaperones for folding of proteins. Fold Des. 1997;2:101–8. doi: 10.1016/S1359-0278(97)00014-X. [DOI] [PubMed] [Google Scholar]
- 49.Kurt N, Cavagnero S. The burial of solvent-accessible surface area is a predictor of polypeptide folding and misfolding as a function of chain elongation. J Am Chem Soc. 2005;127:15690–91. doi: 10.1021/ja0560682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kurt N, Mounce BC, Ellison PA, Cavagnero S. Residue-specific contact order and contact breadth in single-domain proteins: implications for folding as a function of chain elongation. Biotechnol Progr. 2008;24:570–75. doi: 10.1021/bp070475v. [DOI] [PubMed] [Google Scholar]
- 51.Levinthal C. Are there pathways for protein folding? J Chim Phys Phys-Chim Biol. 1968;65:44–45. [Google Scholar]
- 52.Lu J, Deutsch C. Folding zones inside the ribosomal exit tunnel. Nat Struct Mol Biol. 2005;12:1123–29. doi: 10.1038/nsmb1021. [DOI] [PubMed] [Google Scholar]
- 53.Lu J, Deutsch C. Electrostatics in the ribosomal tunnel modulate chain elongation rates. J Mol Biol. 2008;384:73–86. doi: 10.1016/j.jmb.2008.08.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lu J, Kobertz WR, Deutsch C. Mapping the electrostatic potential within the ribosomal exit tunnel. J Mol Biol. 2007;371:1378–91. doi: 10.1016/j.jmb.2007.06.038. [DOI] [PubMed] [Google Scholar]
- 55.Maier R, Eckert B, Scholz C, Lilie H, Schmid FX. Interaction of trigger factor with the ribosome. J Mol Biol. 2003;326:585–92. doi: 10.1016/s0022-2836(02)01427-4. [DOI] [PubMed] [Google Scholar]
- 56.Maier R, Scholz C, Schmid FX. Dynamic association of trigger factor with protein substrates. J Mol Biol. 2001;314:1181–90. doi: 10.1006/jmbi.2000.5192. [DOI] [PubMed] [Google Scholar]
- 57.Malkin LI, Rich A. Partial resistance of nascent polypeptide chains to proteolytic digestion due to ribosomal shielding. J Mol Biol. 1967;26:329–46. doi: 10.1016/0022-2836(67)90301-4. [DOI] [PubMed] [Google Scholar]
- 58.Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62:670–84. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Merz F, Boehringer D, Schaffitzel C, Preissler S, Hoffmann A, et al. Molecular mechanism and structure of trigger factor bound to the translating ribosome. EMBO J. 2008;27:1622–32. doi: 10.1038/emboj.2008.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Millett IS, Doniach S, Plaxco KW. Toward a taxonomy of the denatured state: small angle scattering studies of unfolded proteins. Adv Protein Chem. 2002;62:241–62. doi: 10.1016/s0065-3233(02)62009-1. [DOI] [PubMed] [Google Scholar]
- 61.Neira JL, Fersht AR. Exploring the folding funnel of a polypeptide chain by biophysical studies on protein fragments. J Mol Biol. 1999;285:1309–33. doi: 10.1006/jmbi.1998.2249. [DOI] [PubMed] [Google Scholar]
- 62.Netzer WJ, Hartl FU. Recombination of protein domains facilitated by co-translational folding in eukaryotes. Nature. 1997;388:343–49. doi: 10.1038/41024. [DOI] [PubMed] [Google Scholar]
- 63.Nicola AV, Chen W, Helenius A. Co-translational folding of an alphavirus capsid protein in the cytosol of living cells. Nat Cell Biol. 1999;1:341–45. doi: 10.1038/14032. [DOI] [PubMed] [Google Scholar]
- 64.Ohashi H, Kanamori T, Shimizu Y, Ueda T. A highly controllable reconstituted cell-free system—a breakthrough in protein synthesis research. Curr Pharm Biotechnol. 2010;11:267–71. doi: 10.2174/138920110791111889. [DOI] [PubMed] [Google Scholar]
- 65.Phillips DC. The hen egg-white lysozyme molecule. Proc Natl Acad Sci USA. 1967;57:483–95. doi: 10.1098/rspb.1967.0034. [DOI] [PubMed] [Google Scholar]
- 66.Ramakrishnan V. What we have learned from ribosome structures. Biochem Soc Trans. 2008;036:567–74. doi: 10.1042/BST0360567. [DOI] [PubMed] [Google Scholar]
- 67.Rutkowska A, Beerbaum M, Rajagopalan N, Fiaux J, Schmieder P, et al. Large-scale purification of ribosome-nascent chain complexes for biochemical and structural studies. FEBS Lett. 2009;583:2407–13. doi: 10.1016/j.febslet.2009.06.041. [DOI] [PubMed] [Google Scholar]
- 68.Rutkowska A, Mayer MP, Hoffmann A, Merz F, Zachmann-Brand B, et al. Dynamics of trigger factor interaction with translating ribosomes. J Biol Chem. 2008;283:4124–32. doi: 10.1074/jbc.M708294200. [DOI] [PubMed] [Google Scholar]
- 69.Schaffitzel C, Ban N. Generation of ribosome nascent chain complexes for structural and functional studies. J Struct Biol. 2007;159:302–10. doi: 10.1016/S1047-8477(07)00167-0. [DOI] [PubMed] [Google Scholar]
- 70.Schmeing TM, Ramakrishnan V. What recent ribosome structures have revealed about the mechanism of translation. Nature. 2009;461:1234–42. doi: 10.1038/nature08403. [DOI] [PubMed] [Google Scholar]
- 71.Schuler B, Eaton WA. Protein folding studied by single-molecule FRET. Curr Opin Struct Biol. 2008;18:16–26. doi: 10.1016/j.sbi.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schuwirth BS, Borovinskaya MA, Hau CW, Zhang W, Vila-Sanjurjo A, et al. Structures of the bacterial ribosome at 3.5 Å resolution. Science. 2005;310:827–34. doi: 10.1126/science.1117230. [DOI] [PubMed] [Google Scholar]
- 73.Seidelt B, Innis CA, Wilson DN, Gartmann M, Armache J-P, et al. Structural insight into nascent polypeptide chain-mediated translational stalling. Science. 2009;326:1412–15. doi: 10.1126/science.1177662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Selmer M, Dunham CM, Murphy FV, 4th, Weixlbaumer A, Petry S, et al. Structure of the 70S ribosome complexed with mRNA and tRNA. Science. 2006;313:1935–42. doi: 10.1126/science.1131127. [DOI] [PubMed] [Google Scholar]
- 75.Shimizu Y, Kanamori T, Ueda T. Protein synthesis by pure translation systems. Methods. 2005;36:299–304. doi: 10.1016/j.ymeth.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 76.Siller E, DeZwaan DC, Anderson JF, Freeman BC, Barral JM. Slowing bacterial translation speed enhances eukaryotic protein folding efficiency. J Mol Biol. 2010;396:1310–18. doi: 10.1016/j.jmb.2009.12.042. [DOI] [PubMed] [Google Scholar]
- 77.Sinha KK, Udgaonkar JB. Early events in protein folding. Curr Sci. 2009;96:1053–70. [Google Scholar]
- 78.Deleted in proof
- 79.Steitz TA. A structural understanding of the dynamic ribosome machine. Nat Rev Mol Cell Biol. 2008;9:242–53. doi: 10.1038/nrm2352. [DOI] [PubMed] [Google Scholar]
- 80.Tang YF, Rigotti DJ, Fairman R, Raleigh DP. Peptide models provide evidence for significant structure in the denatured state of a rapidly folding protein: the villin headpiece subdomain. Biochemistry. 2004;43:3264–72. doi: 10.1021/bi035652p. [DOI] [PubMed] [Google Scholar]
- 81.Taniuchi H, Anfinsen CB. An experimental approach to the study of the folding of staphylococcal nuclease. J Biol Chem. 1969;244:3864–75. [PubMed] [Google Scholar]
- 82.Tomic S, Johnson AE, Hartl FU, Etchells SA. Exploring the capacity of trigger factor to function as a shield for ribosome bound polypeptide chains. FEBS Lett. 2006;580:72–76. doi: 10.1016/j.febslet.2005.11.050. [DOI] [PubMed] [Google Scholar]
- 83.Tu L, Wang J, Deutsch C. Biogenesis of the T1 S1 linker of voltage-gated K+ channels. Biochemistry. 2007;46:8075–84. doi: 10.1021/bi700319f. [DOI] [PubMed] [Google Scholar]
- 84.Tu LW, Deutsch C. A folding zone in the ribosomal exit tunnel for Kv1.3 helix formation. J Mol Biol. 2010;396:1346–60. doi: 10.1016/j.jmb.2009.12.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Voss NR, Gerstein M, Steitz TA, Moore PB. The geometry of the ribosomal polypeptide exit tunnel. J Mol Biol. 2006;360:893–906. doi: 10.1016/j.jmb.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 86.Wegrzyn RD, Deuerling E. Molecular guardians for newborn proteins: ribosome-associated chaperones and their role in protein folding. Cell Mol Life Sci. 2005;62:2727–38. doi: 10.1007/s00018-005-5292-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Weinreis SA, Ellis JP, Cavagnero S. Dynamic fluorescence depolarization: a powerful tool to explore protein folding on the ribosome. Methods. 2010;52:57–73. doi: 10.1016/j.ymeth.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Williamson JR. Biophysical studies of bacterial ribosome assembly. Curr Opin Struct Biol. 2008;18:299–304. doi: 10.1016/j.sbi.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Witt SN. Tethering creates unusual kinetics for ribosome-associated chaperones with nascent chains. Protein Pept Lett. 2009;16:631–34. doi: 10.2174/092986609788490195. [DOI] [PubMed] [Google Scholar]
- 90.Woolhead CA, Johnson AE, Bernstein HD. Translation arrest requires two-way communication between a nascent polypeptide and the ribosome. Mol Cell. 2006;22:587–98. doi: 10.1016/j.molcel.2006.05.021. [DOI] [PubMed] [Google Scholar]
- 91.Yusupov MM, Yusupova GZ, Albion B, Lieberman K, Earnest TN, et al. Crystal structure of the ribosome at 5.5 angstrom resolution. Science. 2001;292:883–96. doi: 10.1126/science.1060089. [DOI] [PubMed] [Google Scholar]
- 92.Zhang G, Hubalewska M, Ignatova Z. Transient ribosomal attenuation coordinates protein synthesis and co-translational folding. Nat Struct Mol Biol. 2009;16:274–80. doi: 10.1038/nsmb.1554. [DOI] [PubMed] [Google Scholar]
- 93.Zhang G, Ignatova Z. Folding at the birth of the nascent chain: coordinating translation with co-translational folding. Curr Opin Struct Biol. 2010;21:1–7. doi: 10.1016/j.sbi.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 94.Zimmerman E, Yonath A. Biological implications of the ribosome’s stunning stereochemistry. ChemBioChem. 2009;10:63–72. doi: 10.1002/cbic.200800554. [DOI] [PubMed] [Google Scholar]
- 95.Ziv G, Haran G, Thirumalai D. Ribosome exit tunnel can entropically stabilize alpha-helices. Proc Natl Acad Sci USA. 2005;102:18956–61. doi: 10.1073/pnas.0508234102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Behrmann M, Koch HG, Hengelage T, Wieseler B, Hoffschulte HK, Muller M. Requirements for the translocation of elongation-arrested, ribosome-associated OmpA across the plasma membrane of Escherichia coli. J Biol Chem. 1998;273:13898–904. doi: 10.1074/jbc.273.22.13898. [DOI] [PubMed] [Google Scholar]
- 97.Chattopadhyay S, Das B, Dasgupta C. Reactivation of denatured proteins by 23S ribosomal RNA: Role of domain V. Proc Natl Acad Sci USA. 1996;93:8284–87. doi: 10.1073/pnas.93.16.8284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Das B, Chattopadhyay S, Das Gupta C. Reactivation of denatured fungal glucose 6-phosphate dehydrogenase and alkaline phosphatase with ribosome. Biochem Biophys Res Commun. 1992;183:774–80. doi: 10.1016/0006-291x(92)90550-5. [DOI] [PubMed] [Google Scholar]
- 99.Dill KA, Bromberg S, Yue K, Fiebig KM, Yee DP, Thomas PD, Chan HS. Principles of protein folding: a perspective from simple exact models. Protein Sci. 1995;4:561–602. doi: 10.1002/pro.5560040401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.O’Brien EP, Christodoulou J, Vendruscolo M, Dobson CM. New scenarios of protein folding can occur on the ribosome. J Am Chem Soc. 2011;133:513–26. doi: 10.1021/ja107863z. [DOI] [PubMed] [Google Scholar]
- 101.Schuler B, Lipman EA, Eaton WA. Probing the free-energy surface for protein folding with single-molecule fluorescence spectroscopy. Nature. 2002;419:743–47. doi: 10.1038/nature01060. [DOI] [PubMed] [Google Scholar]
- 102.Uversky VN, Dunker AK. Understanding protein non-folding. Biochim Biophys Acta. 2010;1804:1231–64. doi: 10.1016/j.bbapap.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Woolhead CA, McCormick PJ, Johnson AE. Nascent membrane and secretory proteins differ in FRET-detected folding far inside the ribosome and in their exposure to ribosomal proteins. Cell. 2004;116:725–36. doi: 10.1016/s0092-8674(04)00169-2. [DOI] [PubMed] [Google Scholar]