

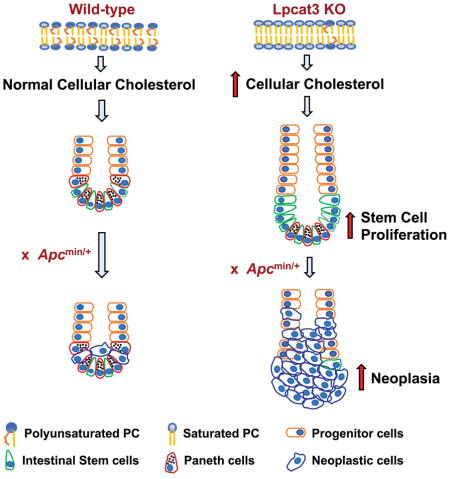
Summary
Adequate availability of cellular building blocks, including lipids, is a prerequisite for cellular proliferation, but excess dietary lipids are linked to increased cancer risk. Despite these connections, specific regulatory relationships between membrane composition, intestinal stem cell (ISC) proliferation and tumorigenesis are unclear. We reveal an unexpected link between membrane phospholipid remodeling and cholesterol biosynthesis and demonstrate that cholesterol itself acts as a mitogen for ISCs. Inhibition of the phospholipid-remodeling enzyme Lpcat3 increases membrane saturation and stimulates cholesterol biosynthesis, thereby driving ISC proliferation. Pharmacologic inhibition of cholesterol synthesis normalizes crypt hyperproliferation in Lpcat3-deficient organoids and mice. Conversely, increasing cellular cholesterol content stimulates crypt organoid growth, and providing excess dietary cholesterol or driving endogenous cholesterol synthesis through SREBP-2 expression promotes ISC proliferation in vivo. Finally, disruption of Lpcat3-dependent phospholipid and cholesterol homeostasis dramatically enhances tumor formation in Apcmin mice. These findings identify a critical dietary-responsive phospholipid-cholesterol axis regulating ISC proliferation and tumorigenesis.
In brief
Tontonoz and colleagues show that the phospholipid remodeling enzyme Lpcat3 regulates intestinal stem cell and progenitor cells by stimulating cholesterol biosynthesis. Furthermore, enhancing cholesterol availability, either by providing it in the diet or through genetic manipulation, promotes tumorigenesis in Apcmin/+ mice.
Introduction
Mammalian cell proliferation requires adequate supply of energy and cellular building blocks, including phospholipids and cholesterol–two major components of cellular membranes (Boroughs and DeBerardinis, 2015; Ito and Suda, 2014). In addition to their roles as biomass components, phospholipids and cholesterol play regulatory roles as signaling molecules that engage specific receptors and transcription factors (Repa and Mangelsdorf, 2000; Simons and Toomre, 2000; Spector and Yorek, 1985). It has also become clear that dynamic changes in phospholipid and cholesterol metabolism can impact membrane composition and alter membrane biophysical properties, thereby influencing a variety of cell processes (Holzer et al., 2011; Spector and Yorek, 1985). In normal tissues, tissue-restricted adult stem cells undergo rapid self-renewal and differentiation to maintain tissue homeostasis. Whether changes in phospholipid or cholesterol metabolism or membrane composition may affect stem cell function is not well understood.
The small intestine is an excellent model system to study adult stem cell biology. Vigorous self-renewal of the intestinal epithelium is essential for optimal gut function. A small number of intestinal stem cells (ISCs) in the crypts continuously divide to produce daughter stem cells and highly proliferative progenitor cells called transit-amplifying (TA) cells (Barker, 2014). The latter migrate from the crypt and differentiate into all of the cell types in the intestinal villi, including absorptive enterocytes, secretory goblet cells, enteroendocrine cells and Paneth cells. Proper balance between ISC self-renewal and differentiation is crucial to preserve the integrity of the intestinal epithelium and maintain its homeostasis (Simons and Clevers, 2011). Dysregulation of intestinal homeostasis is known to cause severe intestinal pathologies, including cancer. ISCs have been shown to be the cells of origin for the intestinal tumors that develop in mice carrying mutations in tumor suppressor gene adenomatous polyposis coli (Apc) (Barker et al., 2009; Schwitalla et al., 2013).
Prior studies have suggested links between phospholipid metabolism and intestinal tumorigenesis, although the underlying mechanisms remain to be fully elucidated. Polymorphisms in the secretory phospholipase A2 (Pla2g2a) gene, which encodes an enzyme catalyzing the deacylation of sn-2 fatty acids during phospholipid remodeling, influences the incidence of intestinal tumors in Apc multiple intestinal neoplasia (Min) mice (Dietrich et al., 1993; MacPhee et al., 1995). Loss of Pla2g2a function increases Apcmin-induced tumor number (Kennedy et al., 1995), whereas overexpression of Pla2g2a reduces tumor multiplicity and size (Cormier et al., 1997). In contrast, deletion of another phospholipase A2 member, cytosolic phospholipase A2 (Pla2g4) has been reported to suppress Apcmin-induced tumorigenesis (Hong et al., 2001). Although it is still unknown how these phospholipases affect tumor initiation and growth, such studies suggest that phospholipid remodeling may be involved in intestinal tumorigenesis. Similarly, cholesterol consumption has long been associated with increased gastrointestinal cancer risk in epidemiological studies (Jarvinen et al., 2001); however, the causal mechanisms have been largely unexplored. Given that ISCs are the cells of origin of intestinal tumors, it is reasonable to speculate that phospholipid remodeling and cholesterol availability may contribute to tumorigenesis by modulating ISC function.
In this study, we investigated whether and how phospholipid and cholesterol metabolism regulate the proliferation of ISC and progenitor cells. We recently identified the phospholipid-remodeling enzyme, lysophosphatidylcholine acyltransferase 3 (Lpcat3), as a critical determinant of membrane phospholipid composition (Rong et al., 2013). Lpcat3 catalyzes the incorporation of polyunsaturated fatty acids at the sn-2 site of lysophospholipids, giving rise to polyunsaturated phospholipids. Loss of Lpcat3 selectively reduces polyunsaturated phosphatidylcholine (PC) in membranes, resulting in decreased membrane fluidity (Rong et al., 2015; Wang et al., 2016). Here we show that Lpcat3 deficiency in intestine enhances ISC proliferation through induction of cholesterol biosynthesis. We demonstrate that cholesterol acts as a mitogen for ISCs and that increasing cellular cholesterol content is sufficient to drive stem cell proliferation both in vivo and ex vivo. Finally, we show that loss of Lpcat3 or activation of SREBP-2 in Apcmin mice markedly promote intestinal tumor formation. These findings reveal a previously unrecognized link between phospholipid remodeling and cholesterol metabolism that modulates intestinal stem cell homeostasis and tumorigenesis.
Results
Loss of Lpcat3 in intestine induces crypt hyperproliferation
In the course of characterizing intestine-specific Lpcat3-deficient mice (Lpcat3F/F, CreVil)(Wang et al., 2016), we noted mucosal hypertrophy in the duodenum and jejunum with profound lengthening of villi and longer small intestines. Since hypertrophy of villi is often observed as a compensatory enlargement of the absorptive surface in response to malabsorption, we conducted further studies to investigate whether this phenotype was a direct or indirect consequence of Lpcat3 deletion in intestinal epithelium. To circumvent the problem of developmental compensation, we crossed Lpcat3-floxed mice with inducible villin-Cre mice (el Marjou et al., 2004) to generate tamoxifen-inducible intestine-specific knockout mice (Lpcat3F/F, CreERT2). At 8 weeks of age, we treated Lpcat3F/F and Lpcat3F/F, CreERT2 mice with tamoxifen to activate Cre recombinase activity. Realtime PCR confirmed >90% reduction in Lpcat3 mRNA in knockout intestines 3 weeks after tamoxifen injection (Figure S1A). Lpcat3 expression remained very low in Lpcat3F/F, CreERT2 mice, even 14 weeks post injection (Figure S1G), indicating that Cre recombinase was also activated in the ISC population (considering the rapid turnover of intestinal epithelium).
Acute Lpcat3 inactivation induced a rapid loss of body weight (up to 10%), but the mice regained body weight after 2~3 weeks (Figure S1B). Similar to constitutive Lpcat3F/Fl, CreVil mice, the length and diameter of Lpcat3F/F, CreERT2 intestines were longer and wider than those of control intestines (Figures S1C and S1D). Although histological analysis did not show multiple branched villi, acute loss of Lpcat3 induced expanded crypt height, indicating hyperproliferation of crypts (Figure 1A). Quantification revealed an approximate 2-fold increase in crypt height and a slight increase in villus length in Lpcat3F/F, CreERT2 mice compared to floxed littermate controls (Figure 1B). There was no change in crypt density (Figure S1E). Similar phenotypes were observed in female mice and male mice even 14 weeks post tamoxifen injection (Figures S1F and S1G-S1I). The fact that these changes appeared after tamoxifen treatment in adult Lpcat3F/F, CreERT2 mice fed a low-fat chow diet suggested that they were unlikely to be secondary to altered lipid absorption.
Figure 1. Lpcat3 deficiency induces hyperproliferation of intestinal crypt and ISCs.

(A) Representative histology of Duodenum and Jejunum from Lpcat3F/F (F/F) and Lpcat3F/F. CreERT2+ (CreERT2) mice 3 weeks after tamoxifen injection.
(B) Quantification of crypt height and villus length in a (~50 crypts and ~20 villi per mouse, 4 mice/group).
(C) Representative images and quantification of EdU staining of proliferating cells in crypts (~30 crypts from 3 mice/group). Tamoxifen-injected mice were i.p. injected with EdU (10 mg/kg) 2 h, 24 h and 48 h before sacrificing.
(D–E) Representative images of immunohistochemistry (IHC) staining and quantification of Olfm4-positive and Id1-positive ISCs and progenitor cells in Jejunum from F/F and CreERT2 mice 3 weeks after tamoxifen injection (~100 crypts from 4 mice/group).
Values are means ± SEM. Statistical analysis was performed with Student’s t test. * P < 0.05, ** P < 0.01; *** P<0.001; **** P<0.0001. Scale bars: 20 μm (D, E), 100 μm (A, C).
To further investigate potential intrinsic effects of Lpcat3 activity on crypt proliferation and epithelial turnover, we examined the incorporation and migration of ethynyl-2′-deoxyuridine (EdU)-labelled cells at 2 h, 24 h and 48 h post EdU pulse labelling (Figure 1C). As shown in Figure 1C, there was ~ 3-fold increase in EdU-positive proliferating cells in crypts of Lpcat3F/F, CreERT2 mice and these proliferative cells migrated much higher along the villus than they did in their control counterparts. Specifically, at 24 h EdU-labeled cells migrated to the tip of villi in Lpcat3F/F, CreERT2 intestines, while they reached only 1/3 of the villus height in control intestines.
To assess the impact of Lpcat3 deficiency on ISC frequency and differentiation, we performed immunostaining for several cell markers in tamoxifen-treated control and Lpcat3F/F, CreERT2 mice. Immunohistochemistry analysis for Olfactomedin 4 (Olfm4), a marker of Lgr5+ ISCs (van der Flier et al., 2009), showed a ~53% increase in Olfm4+ cells in Lpcat3F/F, CreERT2 jejunum compared to those in Lpcat3F/F controls (Figures 1D). Similarly, immunostaining for Id1, a marker of both stem cells and progenitor cells (Zhang et al., 2014), revealed a ~2-fold increase in Id1+ cells in Lpcat3-deficient intestines compared to controls (Fig. 1E), suggesting that both ISCs and progenitor cells were hyperproliferative. Increased numbers of Id1+ cells were also observed in Lpcat3F/F, CreERT2 colon (Figure S1J). By contrast, loss of Lpcat3 reduced the frequency of Lysozyme+ Paneth cells and PAS+ goblet cells by 52% and 35%, respectively (Figures S2A and S2B), whereas the frequency of chromogranin A1 (ChgA)+ enteroendocrine cells were not affected (Figure S2C). We did not observe a change in inflammatory cytokine expression in enterocytes of Lpcat3-deficient mice (Figure S2D)
Phospholipid remodeling regulates crypt organoid growth ex vivo
To further investigate the effect of Lpcat3 deficiency on the proliferation of ISCs, we employed an ex vivo culture system to assess the ability of isolated intestinal crypts to form organoids in 3-D culture as described previously (Sato et al., 2009). Consistent with crypt hyperproliferation in vivo, loss of Lpcat3 led to an increase in size, number and complexity (higher number of buds) of organoids (Figures 2A–2D). Thus, loss of Lpcat3 enhanced ISC proliferation both in vivo and ex vivo, demonstrating that the effects were intrinsic to the ISCs.
Figure 2. Lpcat3 deficiency and saturated phosphatidylcholine (PC) promote crypt organoid growth ex vivo.

(A) Representative images of day-7 organoids derived from F/F and CreERT2 crypts.
(B–D) Quantification of the size (B, n=7 mice/group), number (C, n=11 mice/group) and structure (D, n=5 mice/group) of organoids derived from F/F and CreERT2 crypts. Each dot represents the mean of 3–4 wells from one mouse. The organoid structure was scored based on the number of buds: sphere (no bud), organoid 1 (one bud), organoid 2 (two buds) and organoid 3+ (three or more buds).
(E) Representative images of day-5 organoids treated with PBS (control), saturated (16:0/18:0) PC, and polyunsaturated (16:0/20:4) PC (50 μM).
(F–H) Quantification of the structure (F, n=3 mice/group), size (G, n=6 mice/group) and number (H, n=6 mice/group) of organoids treated with PBS and different PC species. Each dot represents the mean of 3–4 wells from one mouse.
(I) Number of secondary organoids per dissociated primary organoid treated with PBS and different PC species (n=5).
Values are means ± SEM. Statistical analysis was performed with Student’s t test (B and C) and two-way ANOVA (G–I). * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001. Scale bars: 200 μm.
To gain insight into how the enzymatic activity of Lpcat3 was linked to ISC proliferation, we performed lipidomic analyses on isolated crypts. Consistent with our previous observations in enterocytes, Lpcat3 deficiency in crypts selectively decreased polyunsaturated (18:2- and 20:4-containing) PC, concomitant with an increase in saturated and monosaturated PC (Figure S3A). Total PC content was not altered by Lpcat3 deficiency (Figure S3B). Arachidonic acid is a substrate for prostaglandin E2 (PGE2) biosynthesis catalyzed by cyclooxygenases (Cox-1 and Cox-2). Prior studies have demonstrated that PGE2 augments ISC function to facilitate regeneration after injury (Cohn et al., 1997; Houchen et al., 2000; Miyoshi et al., 2017). Given that loss of Lpcat3 might lead to an accumulation of AA, we considered the possibility that increased PGE2 production could be involved in Lpcat3 deficiency-induced ISC proliferation. However, the expression of the PGE2 biosynthetic enzymes Cox-1 and Cox-2 was not altered by Lpcat3 deficiency (Figure S3C), and PGE2 levels were decreased rather than increased in Lpcat3-deficient intestines (Figure S3D). Thus, Lpcat3-dependent ISC hyperproliferation was unlikely to be mediated by PGE2.
The above findings suggested that the change in ISC proliferation in Lpcat3F/F, CreERT2 mice was caused by the change in membrane phospholipid composition per se. We therefore assessed the effects of different PC species on crypt organoid growth. We treated crypt cultures with liposomes containing saturated (16:0/18:0) or polyunsaturated (16:0/20:4) PC. Treatment with liposomes containing 16:0/20:4 PC significantly reduced the size, complexity, and colony formation efficiency in organoids derived from Lpcat3-deficient crypts (Figures 2E–2H). Interestingly, compared to treatment with PBS control, saturated 16:0/18:0 PC treatment increased organoid size, without affecting their structure or number.
We next assessed the effect of different PC species on the self-renewal capacity of ISCs by passaging the primary organoids in the presence of PC treatment. As shown in Figure 2I, Lpcat3-deficient primary organoids generated more secondary organoids than WT primary organoids. Moreover, exposure of the cells to 16:0/20:4 PC liposomes reduced secondary organoid formation in Lpcat3-deficient organoids. By contrast, neither saturated nor polyunsaturated PC affected the growth or self-renewal of organoids from WT crypts (Figures 2E–2I). These findings are consistent with the expectation that active Lpcat3 in WT cells continuously remodels phospholipids to maintain their composition. Together, these data directly implicate membrane phospholipid composition as a determinant of ISC proliferation and self-renewal.
Lpcat3 deficiency induces cholesterol biosynthesis
To begin to explore mechanisms underlying crypt hyperproliferation in the absence of Lpcat3, we analyzed the expression of genes involved in several signaling pathways known to promote ISC proliferation, including Wnt, Notch and Yap pathways (Fevr et al., 2007; Gregorieff et al., 2015; VanDussen et al., 2012). Expression of mRNAs encoding mediators in these pathways was not altered in Lpcat3-deficient crypts compared to controls (Figures S4A and S4B). The PPARδ pathway has recently been implicated in regulating ISC proliferation during high-fat diet feeding (Beyaz et al., 2016). We found no change in the expression of PPARδ target genes between Lpcat3-deficient and control crypts (Figure S4C). These data collectively suggested that alternative mechanisms were likely to be driving proliferation in the absence of Lpcat3.
To uncover such mechanisms and to determine the consequence of loss of Lpcat3 activity for intestinal cell gene expression, we profiled gene expression in crypts isolated from WT and Lpcat3-deficient mice. Strikingly, Gene Ontology (GO) analysis revealed strong enrichment for genes involved in sterol biosynthetic processes in the absence of Lpcat3 (Figure 3A). Expression of the majority of the genes in the cholesterol biosynthetic pathway was increased by ~1.5 to ~2.6 fold (Figure 3B). These changes were validated by realtime PCR (Figure 3C). Similarly, expression of several cholesterol biosynthetic genes was also upregulated in Lpcat3 deficient colons (Figure S4D). Western blot analysis revealed a marked increase in nuclear SREBP-2 in Lpcat3-deficient crypts (Figure 3D), suggesting that post-transcriptional activation of SREBP-2 likely drive the enhanced expression of cholesterol biosynthesis. Interestingly, the effect of Lpcat3 deficiency was selective for SREBP-2 compared to SREBP-1 target genes. There was only a slight increase in Srebf1c and Fasn expression, while the expression of other SREBP-1 target genes was not altered (Figure S4E). In agreement with the augmented cholesterol biosynthetic gene expression, cellular free cholesterol content was increased by ~25% in Lpcat3-deficient crypts compared to controls (Figure 3E). To exclude the possibility that augmented cholesterol biosynthesis was secondary to chronic metabolic changes as a result of higher proliferation, we tested the consequences of acute suppression of Lpcat3 expression ex vivo. We isolated crypts from Lpcat3F/F and Lpcat3F/F, CreERT2 mice and induced Cre recombinase activity in the presence of 4-hydroxytamoxifen (4-OHT). 4-OHT treatment in organoids from Lpcat3F/F, CreERT2 crypts increased expression of a battery of cholesterol biosynthetic genes compared to those treated with DMSO control (Figure 3F). As expected, 4-OHT treatment had no effect in organoids from Lpcat3F/F crypts. Consistent with our observations in vivo, Srebf1 target genes were negligibly affected by acute loss of Lpcat3 ex vivo (Figure S4F). These data suggest that cholesterol biosynthesis is directly coupled with phospholipid remodeling in the intestine.
Figure 3. Loss of Lpcat3 induces cholesterol biosynthetic pathway and increases free cholesterol content in crypts.

(A) Gene ontology (GO) analysis of upregulated genes (≥2 fold) in Lpcat3 deficient crypts identified by gene expression analysis. RNAs from 5 mice/group were pooled and subjected to microarray analysis.
(B) Diagram showing the relative expression of genes involved in cholesterol biosynthesis from the microarray analysis in (A).
(C) Expression of selective genes in cholesterol biosynthetic pathway in F/F and CreERT2 crypts analyzed by realtime PCR (n=11 F/F mice, and 9 CreERT2 mice).
(D) Western blot analysis of nuclear SREBP-2 protein in crypts isolated from F/F and CreERT2 intestines (n=5 mice/group).
(E) Free cholesterol content in crypts isolated from F/F and CreERT2 intestines (n=5 mice/group).
(F) Expression of cholesterol biosynthetic genes in F/F and CreERT2 organoids treated with vehicle (DMSO) or 4-hydroxytamoxifen (4-OHT, 100 nM) (n=7~8).
Values are means ± SEM. Statistical analysis was performed with Student’s t test. * P < 0.05, ** P < 0.01, *** P<0.001.
Inhibition of cholesterol biosynthesis rescues hyperproliferation
To determine whether there might be a functional relationship between cholesterol biosynthesis and crypt hyperproliferation, we first tested if blocking cholesterol biosynthesis could rescue the phenotype. Indeed, suppression of cholesterol biosynthesis by low dose simvastatin (an Hmgcr inhibitor; 1 μM) or Ro 48-8071 (an Lss inhibitor, 1 μM) treatment inhibited the growth of Lpcat3F/F, CreERT2 crypts (Figure 4A). Supplementation of the cultures with mevalonate at levels sufficient to permit protein prenylation but not sterol synthesis (Rowell et al., 1997), did not rescue the inhibition by either Simvastatin or Ro 48-8071. By contrast, administration of cholesterol, but not epicholesterol, fully rescued organoid growth (Figure 4A and S5A), confirming that sterol synthesis is required for crypt proliferation ex vivo.
Figure 4. Inhibition of cholesterol biosynthesis rescues hyperproliferation of Lpcat3 deficient crypts ex vivo and in vivo.

(A) Representative images of Lpcat3 deficient organoids treated with vehicle (DMSO), Simvastatin (Simv, Hmgcr inhibitor, 1 μM), Ro 48-8071 (Ro48, Lss inhibitor, 1 μM), or inhibitors supplemented with mevalonate (Mev, 200 μM) or MβCD-cholesterol (Chol, 50 μM) (n=4 mice/group).
(B) Representative histology and quantification of crypt height in Jejunum of Control and Lpcat3 deficient mice treated with vehicle or Ro48 (~50–100 crypts per mouse, 3 CreERT2 and 2 F/F mice/group). Tamoxifen injected F/F and CreERT2 mice were gavaged with vehicle or Ro48 (15 mg/kg/day) for 8 days before sacrificing.
(C) Representative images and quantification of EdU positive proliferating cells in Jejunum crypts (~50 crypts per mouse, 3 CreERT2 and 2 F/F mice/group). Vehicle or Ro48 treated mice were i.p. injected with EdU (10 mg/kg) 2 h before sacrificing.
(D) Representative images of IHC staining and quantification of Olfm4 positive ISCs in Jejunum of CreERT2 mice treated with vehicle or Ro48 (~50 crypts per mouse, 3 CreERT2 and 2 F/F mice/group).
Values are means ± SEM. Statistical analysis was performed with two-way ANOVA (B–D). * P < 0.05, ** P < 0.01, **** P<0.0001, n.s. not significant. Scale bars: 20 μm (D), 100 μm (B and C), and 200 μm (A).
Next we examined if inhibition of cholesterol biosynthesis could normalize crypt hyperproliferation in vivo. We gavaged tamoxifen-injected Lpcat3F/F and Lpcat3F/F, CreERT2 mice with Ro 48-8071, which has previously been shown to suppress cholesterol biosynthesis in intestine (Chuang et al., 2014). Histological analysis revealed an appreciable decrease in crypt height in Lpcat3F/F, CreERT2 mice treated with Ro 48-8071 compared to those treated with vehicle control (Fig. 4B). Quantification showed a ~15% decrease in crypt height, while villus length was not affected by Ro 48-8071 treatment (Figures 4B and S5B). In contrast to Lpcat3F/F, CreERT2 mice, no overt histological differences were observed in Lpcat3F/F control mice treated with Ro 48-8071 and vehicle. EdU-labeling analysis demonstrated an even more pronounced effect of Ro 48-8071 on ISC and progenitor cell proliferation. There was a ~45% decrease in EdU-positive proliferating cells in Lpcat3-deficient mice treated with Ro 48-8071 compared to those treated with vehicle (Figure 4C). Again, no difference was observed in Lpcat3F/F control mice between Ro 48-8071 and vehicle treatment. More importantly, Ro 48-8071 treatment reduced the number of Olfm4+ and Id1+ ISCs and progenitor cells in Lpcat3F/F, CreERT2 intestines, but had no effect in Lpcat3F/F control intestines (Figures 4D, S5C and S5G). The frequency of Paneth cells and goblet cells was not affected by Ro 48-8071 treament (Figures S5D and S5E). Furthermore, the inhibition of crypt proliferation by Ro 48-8071 was not secondary to increased cell apoptosis, because there was no difference in cleaved caspase-3 immunostaining between groups (Figure S5F). Thus, inhibition of endogenous cholesterol synthesis partially rescued the hyperproliferation observed in the setting of Lpcat3 deficiency, consistent with the hypothesis that excess cholesterol synthesis was driving proliferation in this context. Importantly, however, inhibition of cholesterol synthesis did not impair normal crypt proliferation in control mice, likely due to the availability of sufficient exogenous cholesterol to support the normal rate of proliferation.
Excess cholesterol drives ISC proliferation
We next tested if increasing cellular cholesterol content per se was sufficient to promote crypt proliferation. We cultured crypts isolated from C57BL/6 mice in the presence of cyclodextrin-cholesterol complexes to forcibly load the cells with excess cholesterol (Christian et al., 1997; Klein et al., 1995). Remarkably, cholesterol administration increased the size of organoids without affecting colony formation efficiency (Figure 5A), suggesting that cholesterol acts as a mitogen for ISCs. To test this notion in vivo, we fed C57BL/6 mice with a 1.25% cholesterol diet for 2 weeks, and analyzed ISC and progenitor cell proliferation by EdU labeling (Figure 5B). We found a ~20% increase in EdU-positive proliferating cells in mice fed cholesterol diet compared to those fed chow diet (Figure 5B). Immunostaining showed a corresponding increase in Olfm4+ ISCs (Figure 5C). Furthermore, cholesterol diet feeding resulted in a ~15% increase in free cholesterol content in purified crypts (Figure S6A). Thus, excess exogenous (dietary) cholesterol is a stimulus for ISC proliferation in WT mice.
Figure 5. Excess cholesterol promotes crypt organoid growth ex vivo and ISC proliferation in vivo.

(A) Representative images and quantification of organoid size derived from wild-type mice treated with vehicle control (Ctrl) or MβCD-Cholesterol (50 μM) (n=4 mice). The connected points represent data from organoids derived from the same individual mouse.
(B) Representative images and quantification of EdU positive proliferating cells in Jejunum of chow or high cholesterol diet fed mice. C57BL/6 mice were fed chow or 1.25% cholesterol diet for 2 weeks before sacrificing (n=5, 7 mice/group, ~50 crypts per mouse).
(C) Representative images of IHC staining and quantification of Olfm4-positive ISCs in Jejunum of mice as in B.
(D) Representative images and quantification of EdU-positive proliferating cells in Jejunum of 10 weeks old WT and Srebf2 intestinal transgenic (Srebf2 Tg) mice (~50 crypts per mouse, 5 mice/group).
(E) Representative images of IHC staining and quantification of Olfm4 positive ISCs in Jejunum of WT and Srebf2 transgenic mice (~40 crypts per mouse, 4 mice/group).
(F) Number of secondary organoids per dissociated WT primary organoids treated with MβCD or MβCD-cholesterol and from Srebf2 Tg mice (n=6).
Values are means ± SEM. Statistical analysis was performed with unpaired Student’s t test (C), paired Student’s t test (A) and one-way ANOVA. * P < 0.05, ** P < 0.01, **** P<0.0001, n.s. not significant. Scale bars: 20 μm (C, E), 100 μm (B, D), and 200 μm (A).
To address whether increased endogenous cholesterol synthesis was a stimulus for ISC proliferation we analyzed intestinal-specific Srebf2 transgenic mice (Ma et al., 2014). These mice have enhanced cholesterol biosynthesis and increased cellular cholesterol levels specifically in enterocytes. Interestingly, similar to Lpcat3F/F, CreERT2 mice, Villin-Srebf2 transgenic mice also have longer intestines, consistent with the hypothesis that augmented cholesterol biosynthesis may promote intestinal crypt growth. In further support of this idea, EdU labeling revealed a ~60% increase in EdU-positive cells in Villin-Srebf2 transgenic mice compared to littermate control mice (Figure 5D). Moreover, immunostaining showed ~50% more Olfm4+ and Id1+ ISCs and progenitor cells in Villin-Srebf2 transgenic mice compared to controls (Figure 5E and S6B). Passaging of primary organoids demonstrated a ~40% and 2-fold increase in secondary organoid formation in MβCD-cholesterol treated and Srebf2 Tg organoids, respectively (Figure 5F). Thus, increased endogenous cholesterol biosynthesis and cholesterol content is sufficient to drive ISC and progenitor cell proliferation and ISC self-renewal.
Loss of Lpcat3 dramatically enhances tumor formation
Given that ISCs have been demonstrated to be cells of origin for intestinal tumors, we hypothesized that Lpcat3 might impact intestinal tumorigenesis. To test this idea, we crossed Lpcat3F/F, CreERT2 mice with Apcmin mice to generate Lpcat3F/F, CreERT2, Apcmin and Lpcat3F/F, Apcmin control mice. At 6 weeks of age, we i.p. injected Lpcat3F/F, CreERT2, Apcmin and Lpcat3F/F, Apcmin mice with tamoxifen to activate Cre recombinase activity. Similar to Lpcat3F/F, CreERT2 mice, Lpcat3F/F, CreERT2, Apcmin mice exhibited a slight loss of body weight within the first 2 weeks and regained body weight at 3 weeks post tamoxifen injection (Figure 6A). Surprisingly, however, their body weight dropped precipitously thereafter, and ~80% of Lpcat3F/F, CreERT2, Apcmin mice died 4–5 weeks after Lpcat3 inactivation (Figures 6A and 6B). In contrast, most of the Lpcat3F/F, Apcmin mice survived until 12 weeks post tamoxifen injection. Hematocrit analysis revealed a marked reduction in red blood cell mass in Lpcat3F/F, CreERT2, Apcmin mice (Figures 6C and 6D), which likely contributed to their moribundity. Since intestinal tumors cause bleeding and anemia in Apcmin mice, this finding suggested a worsening of the intestinal pathology in the absence of Lpcat3. Indeed, gross examination and histological analysis showed that Lpcat3F/F, CreERT2, Apcmin intestines were overwhelmed with tumors, whereas Lpcat3F/F, Apcmin control intestines developed only sporadic tumors at this age (Figures 6F and 6G). Quantification of tumor numbers showed a >10-fold increase in Duodenum and Jejunum, and ~3-fold increase in Ileum and colon of Lpcat3F/F, CreERT2, Apcmin compared to Lpcat3F/F, Apcmin mice (Figure 6E).
Figure 6. Lpcat3 deficiency dramatically enhances tumor formation in the intestine.

(A) Body weight change in tamoxifen-injected Lpcat3F/F, CreERT2, Apcmin/+ and Lpcat3F/F, Apcmin/= mice (n=8, 9 mice/group).
(B) Survival curve of tamoxifen-injected Lpcat3F/F, CreERT2, Apcmin/+ and Lpcat3F/F, Apcmin/+ mice (n=9, 11 mice/group).
(C) Representative pictures of centrifuged blood from Lpcat3F/F, CreERT2, Apcmin/+ and Lpcat3F/F, Apcmin/+ mice 5 weeks post tamoxifen injection.
(D) Hematocrit analysis of tamoxifen-injected mice as in C (n=4, 5 mice/group).
(E) Quantification of tumor numbers in tamoxifen-injected mice as in C.
(F) Representative gross images of duodenum and jejunum from tamoxifen-injected mice as in C.
(G) Representative images of H&E-stained histological sections (“Swiss roll” preparation) of duodenum and jejunum from mice as in C.
(H) Expression of selective genes in cholesterol biosynthetic pathway in Lpcat3F/F, CreERT2, Apcmin/+ and Lpcat3F/F, Apcmin/+ intestines analyzed by realtime RT-PCR (n=6, 7 mice/group).
Values are means ± SEM. Statistical analysis was performed with Student’s t test (A, C, E and H) and Log-rank (Mantel-Cox) test (B). * P < 0.05, *** P<0.001, **** P<0.0001, n.s. not significant. Scale bars: 400 μm (G).
Loss of Lpcat3on an Apc+/+ background did not cause a change in inflammatory cytokine expression (Figure S2D) or marked infiltration of F4/80+ or Ly6G+ inflammatory cells (Figure S7A, B), suggesting that inflammation was unlikely to be the driver of tumor formation in the absence of Lpcat3. Although the expression of several cytokines was increased in Lpcat3F/F, CreERT2, Apcmin/+ mice compared to Apcmin/+ mice (Figure S7C), we speculate that this may be secondary to the massive tumor burden in Lpcat3F/F, CreERT2, Apcmin/+ mice. We did not observe obvious infiltration of inflammatory cells by Ly6G and F4/80 staining (Figure S7A, B). Similar to Lpcat3F/F, CreERT2 mice on the Apc+/+ background, the expression of cholesterol biosynthetic genes was increased in small intestine in Lpcat3F/F, CreERT2, Apcmin/+ mice compared to Lpcat3F/F, Apcmin/+ mice (Figure 6H). Interestingly, some of these genes were also upregulated in Apcmin/+ mice compared to WT mice (Figure S7D). These data suggested that Lpcat3 deficiency might enhance tumor growth by inducing cholesterol biosynthesis.
To directly test if increased cholesterol biosynthesis promotes intestinal tumorigenesis, we crossed Srebf2 Tg mice with Apcmin/+ mice to generate Srebf2 Tg, Apcmin/+ mice. Strikingly, Srebf2 Tg, Apcmin/+ mice developed more tumors than Apcmin/+ control mice at 12-week of age (Figure 7A and 7B) and had reduced hematocrits consistent with their elevated tumor burden (Figure S7E). Finally, we asked if blocking endogenous cholesterol biosynthesis would reduce tumor growth in Lpcat3F/F, CreERT2, Apcmin/+ mice. As shown in Figure 7C and 7D, Ro 48-8071 treatment decreased tumor numbers compared to vehicle treatment, implicating enhanced cholesterol synthesis as a major contributor to tumorigenesis in Lpcat3-deficient mice.
Figure 7. Cholesterol availability is a key determinant of tumor formation in Apcmin/+ mice.

(A) Representative gross images of duodenum and jejunum from Srebf2 Tg, Apcmin/+ and Apcmin/+ mice.
(B) Quantification of tumor numbers in mice as in A (n=6, 7 mice/group).
(C) Representative gross images of duodenum and jejunum from vehicle and Ro48 treated Lpcat3F/F, CreERT2, Apcmin/+ mice.
(D) Quantification of tumor numbers in mice as in C (n=6 mice/group).
Values are means ± SEM. Statistical analysis was performed with Student’s t test (B, D). * P < 0.05, ** P<0.01, *** P<0.001, **** P<0.0001, n.s. not significant.
Discussion
The fatty acyl chain composition of phospholipids determines membrane biophysical properties and influences various cellular processes. Previous studies have identified Lpcat3, a phospholipid-remodeling enzyme, as a critical determinant of polyunsaturated phospholipid abundance and membrane fluidity (Rong et al., 2013; Rong et al., 2015; Wang et al., 2016). Here we have shown that Lpcat3 also unexpectedly impacts ISC proliferation through control of cholesterol biosynthesis. We showed that increasing cellular cholesterol content is sufficient to drive ISC proliferation both ex vivo and in vivo, suggesting that cholesterol acts as a mitogen for ISCs. Finally we showed that Lpcat3 activity is important for suppressing Apcmin-induced tumor formation. These findings outline a previously unrecognized link between phospholipid remodeling and cholesterol biosynthesis that regulate ISC homeostasis and tumorigenesis.
Lands’ cycle, in which Lpcat3 participates, involves the hydrolysis of sn-2 fatty acids by phospholipase A2 (PLA2) and their re-acylation by lysophospholipid acyltransferases. A recent study has shown that overexpression of secreted phospholipase A2 (sPLA2, encoded by Pla2g2a) inhibits ISC function by suppressing Wnt signaling (Schewe et al., 2016). sPLA2 is highly expressed in Paneth cells and appears to act as a stem cell niche factor that indirectly affects ISC proliferation. However, whether the enzymatic activity of sPLA2 and its role in phospholipid remodeling is relevant for ISC function remains unknown. In the future it would be interest to determine if the composition of membrane phospholipids–the substrates of Pla2g2a–is altered in the intestines of mice deficient in this enzyme. We have shown that changes in phospholipid composition per se modulate ISC function. Individual phospholipid species differentially affect crypt organoid growth in culture, confirming that crypt hyperproliferation in the absence of Lpcat3 is a consequence of altered membrane composition. The fact that loss of Lpcat3 did not alter Wnt signaling nor increase the frequency of Paneth cells suggests that alterations in the stem cell niche may not contribute to increased proliferation in Lpcat3-deficient intestines. Rather, our data support a cell-autonomous effect due to altered phospholipid remodeling and increased cholesterol biosynthesis in ISCs and progenitor cells.
The mechanisms that link Lpcat3-dependent phospholipid remodeling to cholesterol biosynthesis in the intestine remain to be elucidated. Our data suggest that loss of Lcapt3 leads to changes in phospholipid composition that affect the activation of nuclear SREBP-2, perhaps by promoting processing. Recent studies have shown that ER phospholipid composition in liver is a determinant of SREBP-1 processing (Rong et al., 2017). In contrast these observations in liver, we observe a selective effect on SREBP-2 pathway activity in the intestine. Thus, the effects of phospholipid metabolism on SREBP-1 and SREBP-2 are likely to be complex and cell-type specific. Several signaling pathways have been shown to regulate SREBP activity in cancer cells, thereby linking cell proliferation with lipid biosynthesis, including the PI3K/AKT/mTOR and p53 pathways (Du et al., 2006; Freed-Pastor et al., 2012; Porstmann et al., 2008). Interestingly, prior studies have suggested that membrane phospholipid composition can modulate the activity of cellular signaling pathways, including the AKT pathway (Koeberle et al., 2013). It will be interesting to investigate if any of these signaling pathways may be involved in the effects of Lpcat3 on cholesterol biosynthesis.
The most surprising finding of this study was the mitogenic effect of cholesterol itself on ISC proliferation. Recent studies have suggested that ISC self-renewal may be linked to the availability of dietary nutrients. For example, a triglyceride-rich diet has been shown to activate the PPARδ signaling pathway to enhance ISC and progenitor cell stemness and tumorigenesis (Beyaz et al., 2016), and calorie restriction facilitates ISC proliferation through mTORC1 and Sirt1 in Paneth cells and ISCs (Igarashi and Guarente, 2016; Yilmaz et al., 2012). Such studies suggest that organismal nutritional status and metabolism are coupled with ISC function. Our study substantiated this concept by showing that cholesterol availability modulates ISC proliferation. We showed that pharmacological inhibition of cholesterol biosynthesis dramatically suppressed crypt growth in vivo and ex vivo. On the other hand, increasing cholesterol content via dietary feeding or genetic activation of cholesterol biosynthesis enhanced ISC self-renewal.
It is to be expected that reducing the availability of cholesterol, an essential component of cell membranes, would impede crypt growth. On the contrary, the observation that simply providing more cholesterol to the cell drives proliferation is unexpected. Our data suggesting that the rate of endogenous cholesterol synthesis in ISCs is a limiting factor for proliferation under physiological conditions. At the same time, inhibition of cholesterol synthesis did not impair normal crypt proliferation in control mice, likely due to the availability of sufficient exogenous cholesterol to support the normal rate of proliferation. The details of how excess cholesterol stimulates ISC proliferation remain to be determined. In a previous study, Hartman et al. demonstrated that dietary cholesterol regulates follicle stem cells proliferation in Drosophila by modulating the release of Hedgehog (Hh) protein (Hartman et al., 2013). Cholesterol homeostasis has also been linked to hematopoietic stem cell proliferation through regulating IL-23/G-CSF production (Westerterp et al., 2012; Yvan-Charvet et al., 2010). Interestingly, the Hedgehog pathway has been implicated in regulating ISC proliferation (Crosnier et al., 2006). Therefore, it will be interesting to investigate if excess cholesterol modulates the Hedgehog pathway or other signal transduction pathways in intestine. Another possibility is that cholesterol may regulate ISC function through conversion to a metabolite, such as 27-hydroxycholesterol, which has been shown to influence proliferation in cancer cells (Nelson et al., 2013). Further investigation will be needed to discriminate between these and alternative mechanisms.
ISCs have been demonstrated to be cells of origin for intestinal tumors. Our observations that Lpcat3 deficiency and increased cholesterol biosynthesis dramatically enhance both ISC proliferation and Apcmin-induced tumor formation are consistent with this notion. Cholesterol metabolism has long been linked with the risk of gastrointestinal cancer (Broitman, 1986; Jacobs et al., 2012; Mamtani et al., 2016). Although it is still under debate whether the use of statins reduces the risk of intestinal cancer, it is widely accepted that dietary cholesterol increases the risk. However, the underlying mechanisms are still not clear. We showed here that high cholesterol diet feeding increased cellular cholesterol levels in crypts and that cellular cholesterol content regulates the proliferation of ISCs. Thus, it is logical to hypothesize that dietary cholesterol presumably may promote tumorigenesis by modulating ISC homeostasis.
In conclusion, our findings highlight a link between phospholipid remodeling and cholesterol biosynthesis that regulates ISC function and tumorigenesis. Future studies will explore whether manipulating these metabolic axes could be used as a strategy for therapeutic intervention in gastrointestinal diseases.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-Olfm4 | Cell Signaling | Cat# 39141S; RRID: AB_2650511 |
| Rabbit polyclonal anti-Lysozyme | Dako | Cat# A0099; RRID: AB_2341231 |
| Rabbit polyclonal anti-Chromogranin A | ImmunoStar | Cat# 20086, RRID: AB_572226 |
| Rabbit polyclonal anti-cleaved caspase 3 | Cell Signaling | Cat# 9661, RRID: AB_2341188 |
| Rabbit monoclonal anti-Id1 | BioCheck | Cat# BCH-1/37-2, RRID:AB_2713996 |
| Rat monoclonal anti-Ly-6G | BioLegend | Cat# 127601, RRID:AB_1089179 |
| Rat monoclonal anti-F4/80 | BioLegend | Cat# 123101, RRID:AB_893504 |
| Rabbit monoclonal anti-SREBP2 | EMD Millipore | Cat#: MABS1988 |
| F(ab)2-Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Thermo Fisher Scientific | Cat# A-21430 RRID:AB_2535851 |
| R.T.U. Biotinylated Goat Anti-Rat IgG Antibody | Vector laboratories | BP-9400 |
| DAKO EnVision+ Single Reagents (HRP. Rabbit) | Agilent | K400311-2 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tamoxifen | Sigma-Aldrich | T5648 |
| Matrigel | Fisher | CB-40230C |
| JAG-1 | Anaspec. | AS-61298 |
| EGF Recombinant Mouse Protein | Thermofisher | PMG8041 |
| GlutaMAX™ Supplement | Thermofisher | 35050-061 |
| Noggin | Peprotech | 250-38 |
| R-spondin | Thermo Fisher Scientific | 4645-RS-025 |
| Y-27632 | Sigma-Aldrich | Y0503-1MG |
| N-Acetyl-L cysteine | Sigma-Aldrich | A7250-5g |
| B27 | Thermo Fisher Scientific | 12587-010 |
| N2 | Thermo Fisher Scientific | 17502-048 |
| 4-hydroxytamoxifen | Sigma | H7904 |
| Simvastatin | EMD Biosciences | 567021-5MG |
| Ro 48-8071 | Cayman Chemical | 10006415 |
| Methyl-β-cyclodextrin | Cyclodextrin Technologies Inc | TRMB-P |
| Cholesterol | Sigma | C8667-1G |
| Mevalonolactone | Sigma | M4667-1G |
| IntestiCult™ Organoid Growth Medium (Mouse) | Stemcell technologies | 6005 |
| Critical Commercial Assays | ||
| Prostaglandin E2 Parameter Assay Kit | Fisher | KGE004B |
| Amplex® Red Cholesterol Assay Kit | Life Technologies | A12216 |
| Deposited Data | ||
| Microarray data | This paper | GSE98516 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Lpcat3 Floxed mice | Rong et al., 2015 | N/A |
| Mouse: VillinCre-ERT2: Tg(Vil-cre/ERT2)23Syr | Generated by Sylvie Robine (Institut Curie) and provided by Nicholas Davidson (Washington University) | N/A |
| Mouse: C57BL/6J-ApcMin/J | The Jackson Laboratory | Stock No: 002020 |
| Mouse: Srebf2 intestinal transgenic mice | Ma et al., 2014 | N/A |
| Oligonucleotides | ||
| See table S1 | ||
| Software and Algorithms | ||
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/scientificsoftware/prism/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Other | ||
| ProLong® Diamond Antifade Mountant with DAPI | Thermo Fisher | P36966 |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Peter Tontonoz (ptontonoz@mednet.ucla.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Lpcat3fl/fl mice were generated as described (Rong et al., 2015; Wang et al., 2016). Lpcat3fl/fl mice were crossed with Villin-CreERT2 mice to generate Lpcat3 tamoxifen inducible intestine-specific knockout (Lpcat3f/fl,CreERT2 or CreERT2) mice. Lpcat3f/fl,CreERT2 mice were crossed with Apcmin mice to generate Lpcat3fl/fl, CreERT2, Apcmin and Lpcat3fl/fl, Apcmin control mice. Cre activity was induced by intraperitoneal injection with tamoxifen suspended in corn oil (1 mg/day for 5 days). Srebf2 intestinal transgenic (Srebf2 Tg) mice were described before (Ma et al., 2014). Srebf2 Tg mice were crossed with Apcmin/+ mice to generate Srebf2 Tg, Apcmin/+ mice. C57BL/6 wild-type were obtained from The Jackson Laboratory. All mice were housed under pathogen-free conditions in a temperature controlled room with a 12-hr light/dark cycle. All experiments were performed with male mice unless otherwise stated. Mice were fed a chow diet or a 1.25% cholesterol diet (Research Diets #C12826). Small intestines were excised and cut into three segments with length ratios of 1:3:2 (corresponding to duodenum, jejunum and ileum). Intestine tissues were frozen in liquid nitrogen and stored at −80°C, or fixed in 10% formalin, or frozen in OCT for cryosectioning. Tissue histology was performed in the UCLA Translational Pathology Core Laboratory. Animal experiments were conducted in accordance with the UCLA Animal Research Committee.
METHOD DETAILS
Crypt isolation and organoid culture
Crypts were isolated as described previously (Jabaji et al., 2013; Sato et al., 2009). Briefly, the proximal ~2/3 of small intestine was harvested and flushed with ice-cold phosphate buffered saline (PBS). The intestine was cut into ~5 cm fragments and washed with ice-cold PBS. The fragments were then incubated in 2.5 mM EDTA with gentle shaking at 4°C for 30 min.
The supernatant was removed, fragments were washed with 10 mL of cold PBS, and the sample was vortexed for 30 s with 3 s pulses. The fragments were allowed to settle, and the supernatant was set aside on ice as fraction 1. This process was repeated twice. The fractions were settled down on ice for ~10 min and supernatant was filtered through 70 μm pore cell strainers (BD Biosciences, Bedford, MA). The filtrate was centrifuged at 100 g for 5 min, and the pellet was resuspended in 1 mL of basic medium, consisting of advanced Dulbecco’s modified Eagle medium (DMEM)/Ham’s F12 (Thermo Fisher Scientific, #12634-010) with 2 mM GlutaMAX (Thermo Fisher Scientific, #35050-061), 10 mM HEPES (Thermo Fisher Scientific, #15630080).
Isolated crypts were counted and embedded in Matrigel (Thermo Fisher Scientific, #CB-40230C) at 10 crypts/μl. 150~200 crypts were plated per well of a 48-well plate and cultured in a crypt culture medium, DMEM/F12 (Thermo Fisher Scientific, #12634-010) supplemented with 2 mM GlutaMAX (Thermo Fisher Scientific, #35050-061), 10 mM HEPES (Thermo Fisher Scientific, #15630080), 1× N2 (Thermo Fisher Scientific, #17502-048), 1× B27 (Thermo Fisher Scientific, #12587-010), 1 mM N-Acetyl-L cysteine (Sigma-Aldrich, #A7250-5g), 50 ng/ml EGF (Thermo Fisher Scientific, #PMG8041), 100 ng/ml Noggin (Peprotech, #250-38), 500 ng/ml R-spondin (Thermo Fisher Scientific, #4645-RS-025), 1 μM Jagged-1 (Anaspec, #61298), and 20 ng/ml Y-27632 (Sigma-Aldrich; #Y0503-1MG) (Figure 2 and 4) or IntestiCult™ Organoid Growth Medium (Mouse) (StemCell Technology, #06005) (Figure 5 and S5A). The medium was changed every other day. The results in Figure 2 and 5 are not directly comparable due to the fact that growth media from different commercial sources was used in the two experiments.
Secondary organoid assay
The primary organoids were mechanically disassociated using a p200 pipette and pipetting 50–100 times. The cells were centrifuged, resuspended in Matrigel and replated as described above. Secondary organoids were quantified at day 4.
Organoid measurement
The growth of organoids was evaluated as described with modifications (Lindemans et al., 2015). For organoid size evaluation, the surface area of organoid horizontal cross sections was measured. Organoid perimeters for area measurements were defined manually using ImageJ software. Four to five (2.5x) or ~8–10 (5x) non-overlapping images of all organoids in each well were acquired using a Zeiss Axio Observer Z1 inverted microscope. After 4–5 days in culture, total organoid numbers per well were counted under light microscopy to evaluate colony formation efficiency. The organoid structure was scored based on the number of buds under light microscopy: sphere (no budding), organoid 1 (one bud), organoid 2 (two buds) and organoid 3+ (three or more buds).
Organoid treatment
Organoids were treated as described in the figure legends at the following concentrations: 100 nM 4-hydroxytamoxifen (4-OHT) (Sigma-Aldrich; #H7904), 50 μM 16:0/18:0 (Avanti Polar Lipids, #850456) or 16:0/20:4 PC (Avanti Polar Lipids, #850459) liposome, 1 μM Simvastatin (EMD Biosciences, #567021-5MG), 1 μM Ro 48-8071 (Cayman Chemical, #10006415), 200 μM mevalonic acid, 50 μM cholesterol (Sigma-Aldrich, #C8667) solubilized in methyl-β-cyclodextrin (MβCD) (Cyclodextrin Technologies Inc., #TRMB-P). MβCD-cholesterol complexes were prepared as described (Brown et al., 2002; Ito et al., 2015).
EdU labeling and staining
Mice were i.p. injected with EdU (10 mg/kg) and sacrificed at 2 h, 24 h and 48 h post injection. Intestine tissues were frozen in OCT and cryosectioned (10 μm). EdU staining was conducted using Click-iT™ EdU imaging kit (Thermofisher cat# C10337) according to the manufacturer’s protocol. Briefly, slides containing mounted frozen sections were allowed to thaw to room temperature, and then fixed with 4% paraformaldehyde in phosphate buffer saline (PBS) for 15 min. After washing twice with 3% bovine serum albumin (BSA) in PBS, the sections were permeabilized with 0.5% Triton X-100 in PBS for 20 min. The sections were again washed twice with 3% BSA in PBS and then incubated with a Click-iT™ reaction cocktail (Thermo Fisher Scientific, #C10337) containing Click-iT™ reaction buffer, CuSO4, Alexa Fluor® 488 Azide, and reaction buffer additive for 30 min while protected from light. The sections were washed once more with 3% BSA in PBS. For subsequent DNA staining, sections were washed once with PBS and then incubated with 5ug/mL Hoechst 33342 for 30 min. The slides were then washed twice with PBS and mounted with Vectashield (Vector Laboratories Inc).
Immunohistochemistry and immunofluorescence
Fragments of jejunum were fixed overnight in 10% neutral-buffered formalin at room temperature, embedded in paraffin and sectioned. Sections were deparaffinized and subjected to antigen retrieval with 10 mM sodium citrate (pH 6.0) in a sub-boiling water bath for 20 minutes. Slides were then incubated with the primary antibodies overnight at 4°C unless stated otherwise. The primary antibodies used were rabbit anti-Olfm4 (1:500; Cell Signaling, #39141), rabbit anti-Lysozyme (1:400; Dako, #A0099), rabbit anti-Id1 (1:400; BioCheck, # BCH-1/37-2), rabbit anti-chromogranin A (1:500; Immunostar, #20086, incubated at room temperature for 45 min), rat anti-Ly6G (1:100; BioLegend, #127601 ), rat anti-F4/80 (1:100; BioLegend, #123101), and rabbit anti-cleaved caspase 3 (1:400; Cell Signaling #9661). For IHC staining, slides were further incubated with Envision+ System-HRP Labelled Polymer Anti-Rabbit (Agilent, # K400311-2) or R.T.U. Biotinylated Goat Anti-Rat IgG Antibody (Vector laboratories, # BP-9400) at room temperature for 1 h and developed with ImmPACT DAB Peroxidase (HRP) Substrate (Vector, #SK4105). For immunofluorescence staining, slides were further incubated with Alexa-555 conjugated F(ab′)2-goat anti-rabbit IgG (H+L) cross-adsorbed secondary antibody (Thermo Fisher Scientific, #A21430). Microscopic images were obtained by a Zeiss Axioskop 2 plus fluorescent microscope, and positive cells were quantified by ImageJ software.
Phosphatidylcholine liposome preparation
16:0; 18:0 PC and 16:0; 20:4 PC dissolved in chloroform were evaporated under argon stream to form a thin lipid film. Residual chloroform was thoroughly removed on a vacuum pump. The lipid film was resuspended in saline to 0.5 mM and incubated in 55 °C water bath with occasional vortex. Small unilamellar vesicles (SUV) were obtained with exclusion methods by passing PC suspension through a polycarbonate filter with 30 nm pore size for 10 times.
Gene expression
Total RNA was isolated from tissues or organoids with Trizol (Invitrogen). cDNA was synthesized, and gene expression was quantified by real-time PCR with SYBR Green (Diagenode) and an ABI 7900. Gene expression levels were normalized to 36B4 or Actin. For microarray analysis, RNA from isolated crypts was pooled from n = 5 biological replicates and processed in the UCLA Microarray Core Facility with Gene-Chip Mouse Gene 430.2 Arrays (Affymetrix). Data analysis was performed with GenespringGX (Agilent).
Cell fractionation and western blot analysis
Cell fractionation of isolated crypts was performed following previous publication (Wang et al., 1994). Briefly, crypts were suspended in 0.5 ml buffer C (10 mM HEPES-KOH, pH 7.6, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol, 1 mM EDTA, 1 mM EGTA) supplemented with protease inhibitors, and disrupted by passage ten times through a 22-gauge needle, and centrifuged at 1000 g for 10 min. The crude nuclear pellet was extracted with 100 μl buffer D (20 mM HEPES-KOH, pH 7.6, 25% glycerol, 0.5 M NaCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA) supplemented with protease inhibitors and centrifuged at 12,000 rpm for 30 min. The supernatant was nuclear extract. Twenty micrograms of total nuclear proteins were loaded on 4–12% gel and blotted with SREBP-2 antibody (EMD Millipore, #MABS1988).
Free cholesterol content measurement
Total lipids were extracted from crypts as described by Bligh and Dyer (Bligh and Dyer, 1959). The crypt suspension in PBS was extracted by the addition of 3.75 ml of chloroform/methanol (1:2, v/v) and vortexed for 2 min. Chloroform (1.25 ml) was added and the suspension vortexed for additional 30 s. Then, 1.25 ml of 1.5 M NaCl was added to the mixture and vortexed for another 30 s. The mixture was centrifuged (500 g, 10 min) to separate the organic from the aqueous phase. The chloroform (lower) layer was collected for analysis. Free cholesterol content was measured with Amplex® Red Cholesterol Assay kit (Thermo Fisher scientific, #A12216).
Phospholipid analyses
Isolated crypts were snap frozen in liquid nitrogen. Crypts were homogenized on ice in phosphate buffered saline. Homogenates were subsequently subjected to a modified Bligh-Dyer lipid extraction (Bligh and Dyer, 1959) in the presence of lipid class internal standards including eicosanoic acid, 1-0-heptadecanoyl-sn-glycero-3-phosphocholine, 1,2-dieicosanoyl-sn-glycero-3-phosphocholine, cholesteryl heptadecanoate, and 1,2-ditetradecanoyl-sn-glycero-3-phosphoethanolamine (Demarco et al., 2013). Lipid extracts were diluted in methanol/chloroform (4/1, vol/vol) and molecular species were quantified using electrospray ionization mass spectrometry on a triple quadrupole instrument (Themo Fisher Quantum Ultra) employing shotgun lipidomics methodologies (Han and Gross, 2005). LysoPC molecular species were quantified as sodiated adducts in the positive ion mode using neutral loss scanning for 59.1 amu (collision energy = −28 eV). PC molecular species were quantified as chlorinated adducts in the negative ion mode using neutral loss scanning for 50 amu (collision energy = 24 eV). Individual molecular species were quantified by comparing the ion intensities of the individual molecular species to that of the lipid class internal standard with additional corrections for type I and type II 13C isotope effects (Han and Gross, 2005). Each molecular species indicated in data tables was confirmed by concomitant analyses of samples using product ion scanning for individual fatty acid constituents, including palmitoleic, palmitate, linoleate, oleate, stearate, arachidonate and docosohexadecanoate (product ion scans in m/z of 253.2, 255.2, 279.3, 281.3, 283.3, 303.4 and 327.4, respectively, at a collision energy + 35 eV).
Prostaglandin E2 (PGE2) measurement
Small intestines were harvested and snap frozen in liquid nitrogen. Frozen samples were homogenized in 500 μl of cold PBS containing indomethacin (10 μg/ml) using a tissue homogenizer. The suspension was sonicated in an ice water containing bath sonicator for 20 cycles of 20 seconds of sonication with 20 seconds of cooling, followed by centrifugation for 10 minutes at 12,000 rpm. PGE2 levels in the supernatant was measured using a PGE2 ELISA Kit (Thermo Fisher Scientific, #KGE004B).
Statistical analysis
For all studies, results from quantitative experiments were expressed as means ± SEM and were analyzed using Prism (GraphPad) computer software. Where appropriate, significance was calculated by Student’s t test, one- or two-way ANOVA with Tukey’s or Sidak’s multiple comparison test.
Supplementary Material
Highlights.
Lpcat3 and phospholipid remodeling regulate intestinal stem cell proliferation.
Loss of Lpcat3 enhances cholesterol biosynthesis.
Excess cholesterol increases intestinal stem cell proliferation.
Lpcat3 deficiency and increased cholesterol synthesis promote tumorigenesis.
Acknowledgments
This work was supported by NIH grants HL030568 and DK063491.
Footnotes
Author Contributions
B.W., X.R., E.N.D.P. performed experiments. B.W., X.R., D.A.F., A.M.F. and P.T. designed experiments and interpreted data. J.W., M.G.M., A.M.F., W.A.A. provided essential reagents and interpreted data. B.W., D.A.F. and P.T. wrote the paper.
Declaration of Interests
A.M.F. is a principal and an officer in Bruin Pharma.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barker N. Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nat Rev Mol Cell Biol. 2014;15:19–33. doi: 10.1038/nrm3721. [DOI] [PubMed] [Google Scholar]
- Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ, Clevers H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608–611. doi: 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- Beyaz S, Mana MD, Roper J, Kedrin D, Saadatpour A, Hong SJ, Bauer-Rowe KE, Xifaras ME, Akkad A, Arias E, et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature. 2016;531:53–58. doi: 10.1038/nature17173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol. 2015;17:351–359. doi: 10.1038/ncb3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broitman SA. Dietary cholesterol, serum cholesterol, and colon cancer: a review. Adv Exp Med Biol. 1986;206:137–152. doi: 10.1007/978-1-4613-1835-4_13. [DOI] [PubMed] [Google Scholar]
- Brown AJ, Sun L, Feramisco JD, Brown MS, Goldstein JL. Cholesterol addition to ER membranes alters conformation of SCAP, the SREBP escort protein that regulates cholesterol metabolism. Mol Cell. 2002;10:237–245. doi: 10.1016/s1097-2765(02)00591-9. [DOI] [PubMed] [Google Scholar]
- Christian AE, Haynes MP, Phillips MC, Rothblat GH. Use of cyclodextrins for manipulating cellular cholesterol content. J Lipid Res. 1997;38:2264–2272. [PubMed] [Google Scholar]
- Chuang JC, Valasek MA, Lopez AM, Posey KS, Repa JJ, Turley SD. Sustained and selective suppression of intestinal cholesterol synthesis by Ro 48-8071, an inhibitor of 2,3-oxidosqualene:lanosterol cyclase, in the BALB/c mouse. Biochem Pharmacol. 2014;88:351–363. doi: 10.1016/j.bcp.2014.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn SM, Schloemann S, Tessner T, Seibert K, Stenson WF. Crypt stem cell survival in the mouse intestinal epithelium is regulated by prostaglandins synthesized through cyclooxygenase-1. J Clin Invest. 1997;99:1367–1379. doi: 10.1172/JCI119296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormier RT, Hong KH, Halberg RB, Hawkins TL, Richardson P, Mulherkar R, Dove WF, Lander ES. Secretory phospholipase Pla2g2a confers resistance to intestinal tumorigenesis. Nat Genet. 1997;17:88–91. doi: 10.1038/ng0997-88. [DOI] [PubMed] [Google Scholar]
- Crosnier C, Stamataki D, Lewis J. Organizing cell renewal in the intestine: stem cells, signals and combinatorial control. Nat Rev Genet. 2006;7:349–359. doi: 10.1038/nrg1840. [DOI] [PubMed] [Google Scholar]
- Demarco VG, Ford DA, Henriksen EJ, Aroor AR, Johnson MS, Habibi J, Ma L, Yang M, Albert CJ, Lally JW, et al. Obesity-related alterations in cardiac lipid profile and nondipping blood pressure pattern during transition to diastolic dysfunction in male db/db mice. Endocrinology. 2013;154:159–171. doi: 10.1210/en.2012-1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich WF, Lander ES, Smith JS, Moser AR, Gould KA, Luongo C, Borenstein N, Dove W. Genetic identification of Mom-1, a major modifier locus affecting Min-induced intestinal neoplasia in the mouse. Cell. 1993;75:631–639. doi: 10.1016/0092-8674(93)90484-8. [DOI] [PubMed] [Google Scholar]
- Du X, Kristiana I, Wong J, Brown AJ. Involvement of Akt in ER-to-Golgi transport of SCAP/SREBP: a link between a key cell proliferative pathway and membrane synthesis. Mol Biol Cell. 2006;17:2735–2745. doi: 10.1091/mbc.E05-11-1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el Marjou F, Janssen KP, Chang BH, Li M, Hindie V, Chan L, Louvard D, Chambon P, Metzger D, Robine S. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39:186–193. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- Fevr T, Robine S, Louvard D, Huelsken J. Wnt/beta-catenin is essential for intestinal homeostasis and maintenance of intestinal stem cells. Mol Cell Biol. 2007;27:7551–7559. doi: 10.1128/MCB.01034-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, Barsotti A, Chicas A, Li W, Polotskaia A, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. 2012;148:244–258. doi: 10.1016/j.cell.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorieff A, Liu Y, Inanlou MR, Khomchuk Y, Wrana JL. Yap-dependent reprogramming of Lgr5(+) stem cells drives intestinal regeneration and cancer. Nature. 2015;526:715–718. doi: 10.1038/nature15382. [DOI] [PubMed] [Google Scholar]
- Han X, Gross RW. Shotgun lipidomics: electrospray ionization mass spectrometric analysis and quantitation of cellular lipidomes directly from crude extracts of biological samples. Mass Spectrom Rev. 2005;24:367–412. doi: 10.1002/mas.20023. [DOI] [PubMed] [Google Scholar]
- Hartman TR, Strochlic TI, Ji Y, Zinshteyn D, O’Reilly AM. Diet controls Drosophila follicle stem cell proliferation via Hedgehog sequestration and release. J Cell Biol. 2013;201:741–757. doi: 10.1083/jcb.201212094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer RG, Park EJ, Li N, Tran H, Chen M, Choi C, Solinas G, Karin M. Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell. 2011;147:173–184. doi: 10.1016/j.cell.2011.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong KH, Bonventre JC, O’Leary E, Bonventre JV, Lander ES. Deletion of cytosolic phospholipase A(2) suppresses Apc(Min)-induced tumorigenesis. Proc Natl Acad Sci U S A. 2001;98:3935–3939. doi: 10.1073/pnas.051635898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houchen CW, Stenson WF, Cohn SM. Disruption of cyclooxygenase-1 gene results in an impaired response to radiation injury. Am J Physiol Gastrointest Liver Physiol. 2000;279:G858–865. doi: 10.1152/ajpgi.2000.279.5.G858. [DOI] [PubMed] [Google Scholar]
- Igarashi M, Guarente L. mTORC1 and SIRT1 Cooperate to Foster Expansion of Gut Adult Stem Cells during Calorie Restriction. Cell. 2016;166:436–450. doi: 10.1016/j.cell.2016.05.044. [DOI] [PubMed] [Google Scholar]
- Ito A, Hong C, Rong X, Zhu X, Tarling EJ, Hedde PN, Gratton E, Parks J, Tontonoz P. LXRs link metabolism to inflammation through Abca1-dependent regulation of membrane composition and TLR signaling. Elife. 2015;4:e08009. doi: 10.7554/eLife.08009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Suda T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat Rev Mol Cell Biol. 2014;15:243–256. doi: 10.1038/nrm3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabaji Z, Sears CM, Brinkley GJ, Lei NY, Joshi VS, Wang J, Lewis M, Stelzner M, Martin MG, Dunn JC. Use of collagen gel as an alternative extracellular matrix for the in vitro and in vivo growth of murine small intestinal epithelium. Tissue Eng Part C Methods. 2013;19:961–969. doi: 10.1089/ten.tec.2012.0710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs RJ, Voorneveld PW, Kodach LL, Hardwick JC. Cholesterol metabolism and colorectal cancers. Curr Opin Pharmacol. 2012;12:690–695. doi: 10.1016/j.coph.2012.07.010. [DOI] [PubMed] [Google Scholar]
- Jarvinen R, Knekt P, Hakulinen T, Rissanen H, Heliovaara M. Dietary fat, cholesterol and colorectal cancer in a prospective study. Br J Cancer. 2001;85:357–361. doi: 10.1054/bjoc.2001.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy BP, Payette P, Mudgett J, Vadas P, Pruzanski W, Kwan M, Tang C, Rancourt DE, Cromlish WA. A natural disruption of the secretory group II phospholipase A2 gene in inbred mouse strains. J Biol Chem. 1995;270:22378–22385. doi: 10.1074/jbc.270.38.22378. [DOI] [PubMed] [Google Scholar]
- Klein U, Gimpl G, Fahrenholz F. Alteration of the myometrial plasma membrane cholesterol content with beta-cyclodextrin modulates the binding affinity of the oxytocin receptor. Biochemistry. 1995;34:13784–13793. doi: 10.1021/bi00042a009. [DOI] [PubMed] [Google Scholar]
- Koeberle A, Shindou H, Koeberle SC, Laufer SA, Shimizu T, Werz O. Arachidonoylphosphatidylcholine oscillates during the cell cycle and counteracts proliferation by suppressing Akt membrane binding. Proc Natl Acad Sci U S A. 2013;110:2546–2551. doi: 10.1073/pnas.1216182110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindemans CA, Calafiore M, Mertelsmann AM, O’Connor MH, Dudakov JA, Jenq RR, Velardi E, Young LF, Smith OM, Lawrence G, et al. Interleukin-22 promotes intestinal-stem-cellmediated epithelial regeneration. Nature. 2015;528:560–564. doi: 10.1038/nature16460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma K, Malhotra P, Soni V, Hedroug O, Annaba F, Dudeja A, Shen L, Turner JR, Khramtsova EA, Saksena S, et al. Overactivation of intestinal SREBP2 in mice increases serum cholesterol. PLoS One. 2014;9:e84221. doi: 10.1371/journal.pone.0084221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPhee M, Chepenik KP, Liddell RA, Nelson KK, Siracusa LD, Buchberg AM. The secretory phospholipase A2 gene is a candidate for the Mom1 locus, a major modifier of ApcMininduced intestinal neoplasia. Cell. 1995;81:957–966. doi: 10.1016/0092-8674(95)90015-2. [DOI] [PubMed] [Google Scholar]
- Mamtani R, Lewis JD, Scott FI, Ahmad T, Goldberg DS, Datta J, Yang YX, Boursi B. Disentangling the Association between Statins, Cholesterol, and Colorectal Cancer: A Nested Case-Control Study. PLoS Med. 2016;13:e1002007. doi: 10.1371/journal.pmed.1002007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi H, VanDussen KL, Malvin NP, Ryu SH, Wang Y, Sonnek NM, Lai CW, Stappenbeck TS. Prostaglandin E2 promotes intestinal repair through an adaptive cellular response of the epithelium. EMBO J. 2017;36:5–24. doi: 10.15252/embj.201694660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson ER, Wardell SE, Jasper JS, Park S, Suchindran S, Howe MK, Carver NJ, Pillai RV, Sullivan PM, Sondhi V, et al. 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science. 2013;342:1094–1098. doi: 10.1126/science.1241908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, Griffiths JR, Chung YL, Schulze A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008;8:224–236. doi: 10.1016/j.cmet.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repa JJ, Mangelsdorf DJ. The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu Rev Cell Dev Biol. 2000;16:459–481. doi: 10.1146/annurev.cellbio.16.1.459. [DOI] [PubMed] [Google Scholar]
- Rong X, Albert CJ, Hong C, Duerr MA, Chamberlain BT, Tarling EJ, Ito A, Gao J, Wang B, Edwards PA, et al. LXRs regulate ER stress and inflammation through dynamic modulation of membrane phospholipid composition. Cell Metab. 2013;18:685–697. doi: 10.1016/j.cmet.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong X, Wang B, Dunham MM, Hedde PN, Wong JS, Gratton E, Young SG, Ford DA, Tontonoz P. Lpcat3-dependent production of arachidonoyl phospholipids is a key determinant of triglyceride secretion. Elife. 2015:4. doi: 10.7554/eLife.06557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong X, Wang B, Palladino EN, de Aguiar Vallim TQ, Ford DA, Tontonoz P. ER phospholipid composition modulates lipogenesis during feeding and in obesity. J Clin Invest. 2017;127:3640–3651. doi: 10.1172/JCI93616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowell CA, Kowalczyk JJ, Lewis MD, Garcia AM. Direct demonstration of geranylgeranylation and farnesylation of Ki-Ras in vivo. J Biol Chem. 1997;272:14093–14097. doi: 10.1074/jbc.272.22.14093. [DOI] [PubMed] [Google Scholar]
- Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, Clevers H. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- Schewe M, Franken PF, Sacchetti A, Schmitt M, Joosten R, Bottcher R, van Royen ME, Jeammet L, Payre C, Scott PM, et al. Secreted Phospholipases A2 Are Intestinal Stem Cell Niche Factors with Distinct Roles in Homeostasis, Inflammation, and Cancer. Cell Stem Cell. 2016;19:38–51. doi: 10.1016/j.stem.2016.05.023. [DOI] [PubMed] [Google Scholar]
- Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Goktuna SI, Ziegler PK, Canli O, Heijmans J, Huels DJ, Moreaux G, et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell. 2013;152:25–38. doi: 10.1016/j.cell.2012.12.012. [DOI] [PubMed] [Google Scholar]
- Simons BD, Clevers H. Strategies for homeostatic stem cell self-renewal in adult tissues. Cell. 2011;145:851–862. doi: 10.1016/j.cell.2011.05.033. [DOI] [PubMed] [Google Scholar]
- Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- Spector AA, Yorek MA. Membrane lipid composition and cellular function. J Lipid Res. 1985;26:1015–1035. [PubMed] [Google Scholar]
- van der Flier LG, Haegebarth A, Stange DE, van de Wetering M, Clevers H. OLFM4 is a robust marker for stem cells in human intestine and marks a subset of colorectal cancer cells. Gastroenterology. 2009;137:15–17. doi: 10.1053/j.gastro.2009.05.035. [DOI] [PubMed] [Google Scholar]
- VanDussen KL, Carulli AJ, Keeley TM, Patel SR, Puthoff BJ, Magness ST, Tran IT, Maillard I, Siebel C, Kolterud A, et al. Notch signaling modulates proliferation and differentiation of intestinal crypt base columnar stem cells. Development. 2012;139:488–497. doi: 10.1242/dev.070763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Rong X, Duerr MA, Hermanson DJ, Hedde PN, Wong JS, Vallim TQ, Cravatt BF, Gratton E, Ford DA, Tontonoz P. Intestinal Phospholipid Remodeling Is Required for Dietary-Lipid Uptake and Survival on a High-Fat Diet. Cell Metab. 2016;23:492–504. doi: 10.1016/j.cmet.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Sato R, Brown MS, Hua X, Goldstein JL. SREBP-1, a membrane-bound transcription factor released by sterol-regulated proteolysis. Cell. 1994;77:53–62. doi: 10.1016/0092-8674(94)90234-8. [DOI] [PubMed] [Google Scholar]
- Westerterp M, Gourion-Arsiquaud S, Murphy AJ, Shih A, Cremers S, Levine RL, Tall AR, Yvan-Charvet L. Regulation of hematopoietic stem and progenitor cell mobilization by cholesterol efflux pathways. Cell Stem Cell. 2012;11:195–206. doi: 10.1016/j.stem.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz OH, Katajisto P, Lamming DW, Gultekin Y, Bauer-Rowe KE, Sengupta S, Birsoy K, Dursun A, Yilmaz VO, Selig M, et al. mTORC1 in the Paneth cell niche couples intestinal stem-cell function to calorie intake. Nature. 2012;486:490–495. doi: 10.1038/nature11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvan-Charvet L, Pagler T, Gautier EL, Avagyan S, Siry RL, Han S, Welch CL, Wang N, Randolph GJ, Snoeck HW, Tall AR. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science. 2010;328:1689–1693. doi: 10.1126/science.1189731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Yantiss RK, Nam HS, Chin Y, Zhou XK, Scherl EJ, Bosworth BP, Subbaramaiah K, Dannenberg AJ, Benezra R. ID1 is a functional marker for intestinal stem and progenitor cells required for normal response to injury. Stem Cell Reports. 2014;3:716–724. doi: 10.1016/j.stemcr.2014.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.