

Summary
Dopamine controls essential brain functions through volume transmission. Different from fast synaptic transmission, where neurotransmitter release and receptor activation are tightly coupled by an active zone, dopamine transmission is widespread and may not necessitate these organized release sites. Here, we determine whether striatal dopamine secretion employs specialized machinery for release. Using superresolution microscopy, we identified co-clustering of the active zone scaffolding proteins bassoon, RIM and ELKS in ~30% of dopamine varicosities. Conditional RIM knockout disrupted this scaffold and, unexpectedly, abolished dopamine release, while ELKS knockout had no effect. Optogenetic experiments revealed that dopamine release was fast and had a high release probability, indicating the presence of protein scaffolds for coupling Ca2+-influx to vesicle fusion. Hence, dopamine secretion is mediated by sparse, mechanistically specialized active zone-like release sites. This architecture supports spatially and temporally precise coding for dopamine and provides molecular machinery for regulation.
In brief
Secretion of dopamine requires specialized release machinery
Introduction
In the brain, two principle modes of chemical transmission support neuronal communication. In fast synaptic transmission, a presynaptic terminal rapidly and precisely signals to a postsynaptic partner. This contrasts with volume transmission, where neuromodulators diffuse to mediate effects in many cells over a large area (Agnati et al., 1995). For fast neurotransmitters, sub-millisecond timing of release is mediated by precisely assembled protein networks called active zones. Active zones are composed of scaffolding proteins that are attached to the presynaptic plasma membrane opposed to postsynaptic receptors. The active zone docks synaptic vesicles, enhances release probability, aligns fusion with postsynaptic neurotransmitter receptors, and mediates presynaptic plasticity (Südhof, 2012). Vertebrate active zones contain bassoon/piccolo, RIM, and ELKS as key scaffolds, Munc13s for vesicle priming, and RIM-BPs to support RIM in the tight coupling of docked vesicles to Ca2+ channels (Acuna et al., 2015, 2016; Altrock et al., 2003; Augustin et al., 1999; Han et al., 2011; Kaeser et al., 2011; Liu et al., 2014; Wang et al., 2016). The nature of volume transmission indicates that precise localization and timing of secretion are not necessary for this signaling mode. Thus, whether volume transmission employs similar active zone machinery for release is an open question.
The midbrain dopamine system is an important neuromodulatory network, with cell bodies residing in the substantia nigra pars compacta (SNc) and the ventral tegmental area (VTA). Axons from these dopamine neurons project to the striatum, where dopamine regulates circuits for movement, motivation and reward (Howe and Dombeck, 2016; Schultz, 1998; Surmeier et al., 2014). In the striatum, dopamine receptors are mostly localized outside of synapses (Cailld et al., 1996; Uchigashima et al., 2016; Yung et al., 1995), many axonal dopamine varicosities are not associated with postsynaptic densities (Descarries et al., 1996), and dopamine receptors are G-protein coupled, whose signaling is more than two orders of magnitude slower than that of ionotropic receptors (Missale et al., 1998). These properties have led to the conclusion that dopamine is a volume transmitter that signals slowly and inaccurately, and suggests that an active zone that warrants speed and accuracy may be unnecessary for dopamine release.
Striatal dopamine release is often subdivided into tonic and phasic modes, which are both triggered by action potentials. Tonic release generates a mild, widespread increase in extracellular dopamine through non-synchronous spontaneous firing of some dopamine neurons, while phasic release results in a fast and transient dopamine increase due to synchronized burst firing (Grace, 2016). Phasic release may support fast and spatially localized dopamine signaling (Floresco et al., 2003; Marcott et al., 2014), and recent studies supported a sub-second time window for dopamine coding (Howe and Dombeck, 2016; Yagishita et al., 2014). Thus, functional studies suggest that at least some dopamine signaling is rapid.
Here, we examined whether there are specialized release sites that support dopamine secretion for fast signaling. We tested for three defining properties of active zones in axonal arbors of midbrain dopamine neurons in the striatum. First, we determined whether dopamine axons contained active zone protein scaffolds. Using superresolution microscopy, we found protein assemblies consisting of bassoon, RIM and ELKS in ~30% of dopamine varicosities. Second, we assessed whether these proteins participated in dopamine release. We generated dopamine neuron-specific knockout mice and found that RIM deletion abolished dopamine release, and the loss of release was accompanied by a disruption of the active zone scaffolds. Remarkably, ELKS was dispensable for striatal dopamine release, suggesting mechanistic specialization of the dopamine release machinery. Third, we tested the functional properties of dopamine release using optogenetics and found that dopamine release was fast and had a very high initial release probability, strongly indicating the necessity of active zone-like sites. Thus, striatal dopamine release is mediated by sparse release hot spots in dopamine axons to generate a precisely localized dopamine signal with rapid kinetics.
Results
Dopamine axons contain small clusters of the active zone protein bassoon
At fast synapses, active zones are identified by the presence of the molecular scaffold bassoon (Altrock et al., 2003; Wang et al., 2016). To investigate whether dopamine neurons contain similar release sites, we tested whether bassoon clusters are present in dopamine axons in the striatum. We labeled dopamine axons with tyrosine hydroxylase (TH) antibodies and co-stained with antibodies against bassoon in sections of the dorsal striatum. Because the striatum contains synapses at a very high density and dopamine axons account only for a small fraction of the striatum, it is not possible to determine whether a given bassoon cluster is within a dopamine axon by diffraction-limited or two-dimensional microscopy. We therefore employed three-dimensional structured illumination superresolution microscopy (3D-SIM, Figures 1, S1, and Movie S1), which enhanced resolution in all three dimensions (Gustafsson et al., 2008). To analyze the 3D-SIM images, we calculated the overlap between bassoon clusters and dopamine axons in 3D reconstructions (Figure 1B). Bassoon clusters for which more than 40% of the volume overlapped with TH-labeling were considered to be within dopamine axons (Figures 1B and S1A). The detected bassoon clusters could reflect bassoon within dopamine axons, or they could be detection artifacts due to the high density of nearby synapses that contain bassoon. To distinguish between these possibilities, we locally shuffled the position of each bassoon cluster within a 1 μm3 cube and calculated how much random overlap this generated with TH-positive axons (Figures 1B–1E). If the association of bassoon and TH was an artifact, bassoon density and size should be similar before and after shuffling. In contrast, we found that in every image, bassoon cluster density within dopamine axons decreased and the cluster size increased upon shuffling (Figures 1D and 1E). This indicates that there are bassoon clusters within dopamine axons, and they are smaller than the ones outside.
Figure 1. Dopamine axons in the dorsal striatum contain bassoon clusters.

(A) Projections of 1 μm thick image stacks of the mouse striatum labeled with TH (magenta) and bassoon (green) antibodies, acquired by confocal (top) or 3D-SIM microscopy (bottom).
(B) Schematic illustrating analyses of 3D-SIM images. The overlap between bassoon and TH was calculated after object recognition in each channel. A bassoon cluster was considered to be within TH when the volume overlap was >40% (Figure S1A). To determine whether this association is different from artificial overlap, each bassoon cluster was locally shuffled within 1 × 1 × 1 μm3 (shuffled bassoon), and the overlap of shuffled bassoon with TH was calculated. 1,000 rounds of shuffling and overlap calculation were averaged.
(C) Representative 3D-SIM images showing distribution of bassoon clusters (green) and TH-labeled dopamine axons (magenta) in dorsal striatal slices (top). Images were obtained by volume rendering (left) of the raw image stack (top, 10 × 10 × 2 μm3, a zoom-in of 5 × 2 × 2 μm3 is shown below), surface rendering of objects (middle), and surface rendering after local shuffling of bassoon objects (right). The bottom rows show bassoon clusters within dopamine axons. For each zoom-in image, a 90° rotation around the x-axis is shown below the standard x-y-z image. For a 3D representation, see Movie S1.
(D, E) Quantification of the density (D) and volume (E) of bassoon clusters within dopamine axons before and after local shuffling. Each circle represents the average result of a region containing 6,000–11,000 bassoon clusters. n = 35 regions/4 mice. For individual overlap bins, see Figure S1A.
(F) Schematic of the striatal synaptosome preparation. See Figures S1B–S1E for assessment of the subcellular fractionation by Western blotting, confocal microscopy and electron microscopy.
(G) Image of bassoon and synaptobrevin-2 staining in TH positive (top) or negative (bottom) striatal synaptosomes. Bassoon and synaptobrevin-2 were imaged by STED microscopy, TH was imaged by confocal microscopy.
(H) Quantification of the bassoon area in TH positive and TH negative synaptosomes. n = 48 synaptosomes/2 mice (TH positive), 1005/2 (TH negative).
Data in H are mean ± SEM. *** p < 0.001, * p < 0.05; paired t test for (D, E), Mann-Whitney rank sum test for (H).
To assess bassoon localization in dopamine axons with an independent method, we prepared synaptosomes from the striatum (Figures 1F, S1B–S1E), labeled them with antibodies against bassoon, TH and the vesicular SNARE protein synaptobrevin-2, and imaged them using stimulated emission depletion (STED) superresolution microscopy (Figure 1G). In TH-positive and in TH-negative synaptosomes, bassoon labeling coincided with synaptobrevin-2 positive vesicles, and the bassoon labeling often aligned with the edge of the synaptobrevin signal. This independently establishes that dopamine axons contain vesicle clusters that are associated with bassoon. The average area of bassoon clusters in TH positive synaptosomes was 24.2 % smaller than that in TH negative synaptosomes (Figure 1H), confirming that bassoon clusters within dopamine axons are smaller than the average striatal bassoon clusters.
Bassoon, RIM and ELKS form active zone-like structures in dopamine axons
We next tested whether dopamine axons contain RIM and ELKS (Figure 2), two important redundant scaffolds for active zone assembly at fast synapses (Wang et al., 2016). While it is desirable to measure absolute cluster densities in dopamine axons, this is not possible through subtraction with the shuffling method. Shuffling only provides an estimate of the maximum influence of artifactual overlap, but not the real false positive rate in the actual analysis. For RIM and ELKS, we were able to address this limitation by conditional gene knockout. We removed RIM (RIM cKODA) or ELKS (ELKS cKODA) specifically from dopamine neurons by crossing mice with floxed alleles for Rims1 and 2 (Kaeser et al., 2011) or Erc1 and 2 (Liu et al., 2014) to DATIRES-Cre mice (Backman et al., 2006), respectively (Figures S2 and S3). We then compared cluster densities within dopamine axons in RIM cKODA and ELKS cKODA mice with corresponding sibling control mice. We found that RIM and ELKS clustered in dopamine axons, and conditional knockout reduced cluster densities progressively with increasing volume overlap with TH (Figures 2A–2D), an effect that was not present after shuffling (Figures S2 and S3). The clusters detected after knockout are a combination of background staining (~25% in immunofluorescent stainings with the antibodies used here, (Wang et al., 2016)) and of presynaptic inputs onto dopamine axons (Threlfell et al., 2012), which may be too close to be reliably distinguished by 3D-SIM. We calculated the absolute density of RIM and ELKS clusters to be 0.31 and 0.22 per μm of axon (Figures 2B, 2D), respectively, after subtracting all signal (>0% volume overlap) in RIM and ELKS cKODA mice from corresponding control mice.
Figure 2. Bassoon, RIM and ELKS co-cluster in dopamine axons.

(A) Representative surface rendered images (5 × 5 × 2 μm3) of RIM clusters within dopamine axons from RIM control and RIM cKODA mice, in which RIM is removed specifically in dopamine neurons by breeding conditional RIM1 and RIM2 knockout mice to DATIRES-Cre mice (Figure S2). RIM control mice were siblings that lack Cre.
(B) Histogram of RIM cluster densities within dopamine axons across 20% bins of overlap. RIM control n = 24 regions/4 mice, RIM cKODA n = 22/4 (p < 0.001 for genotype, p < 0.001 for overlap, and p < 0.01 for interaction; two-way ANOVA, p-values of pairwise post tests indicated in figure). For detailed sample images and data analyses including shuffling, see Figure S2.
(C, D) Same as (A) and (B), except showing representative images (C) and quantification (D) of ELKS clusters in ELKS control and ELKS cKODA mice. ELKS control n = 29/4, ELKS cKODA 27/4 (p < 0.001 for genotype, p < 0.001 for overlap, and p = 0.37 for interaction; two-way ANOVA, p-values of pairwise post tests indicated in figure). For detailed sample images and data analyses including shuffling, see Figure S3.
(E) Confocal images of striatal synaptosomes. Filled arrowheads indicate synaptosomes containing bassoon and TH (insets). Hollow arrowheads indicate particles containing TH but not bassoon.
(F) Quantification of the percentage of synaptosomes that contain RIM, ELKS or DAT in three different types of synaptosomes, defined by specific markers as indicated below the plots. Each circle represents the average result of an area with 1,500–6,000 synaptosomes. n = 18 areas/3 mice for RIM, 12/3 for ELKS and 9/3 for DAT.
(G) Representative STED images of RIM (left) and ELKS (right) in synaptosomes co-labeled with bassoon in TH positive (top) and TH negative (bottom) synaptosomes.
All data are mean ± SEM. *** p < 0.001, * p < 0.05, ns, not significant; two-way ANOVA for (B, D) and Kruskal-Wallis analysis of variance with post hoc Dunn’s test for (F).
A defining property of an active zone is the assembly of several scaffolding proteins into a functional release site. The similar densities of RIM and ELKS clusters suggest active zone-like co-clustering in dopamine axons, but a direct test of co-clustering requires three colors in brain slices (two test proteins and TH). Chromatic aberration currently limits enhanced resolution in our 3D-SIM experiments to two colors. We instead performed confocal microscopy on striatal synaptosomes plated at low density (4–12 synaptosomes/100 μm2, Figure 2E). In TH positive synaptosomes, the presence of RIM and ELKS highly correlated with the presence of bassoon (Figure 2F). In contrast, the dopamine transporter (DAT) was associated with TH positive synaptosomes independent of whether they contained bassoon or not (Figures 2E and 2F). Given the extensive direct and indirect protein interactions between RIM, ELKS and bassoon at classical active zones (Südhof, 2012), the presence of these proteins within the same synaptosome strongly suggests co-clustering. We directly tested for co-clustering at the scale of tens of nanometers using STED imaging and found that RIM and ELKS were both tightly associated with bassoon within a small area of TH positive synaptosomes (Figure 2G). These data establish that RIM, ELKS, and bassoon co-cluster to form active zone-like structures in dopamine axons.
RIM is essential for dopamine release
To test whether these active zone-like sites contribute to dopamine release, we employed amperometry in acute brain slices of the dorsal striatum (Figures S4A and S4B). In control slices, electrical stimulation triggered biphasic dopamine release as described before (Threlfell et al., 2012; Zhou et al., 2001). A first component amounting to 20% of the total response is induced by action potential firing of dopamine axons (Figures S4C and S4D). A second, slightly delayed component accounting for 80% of the total response is triggered by cholinergic inputs on dopamine axons, which are co-activated during electrical stimulation (Figures S4C and S4D). At fast synapses, simultaneous removal of RIM1 and RIM2 from active zones decreases release by 50–80% depending on the synapse tested (Acuna et al., 2016; Han et al., 2011; Kaeser et al., 2011). Unexpectedly, dopamine release was essentially abolished in RIM cKODA mice. While release increased with stimulation intensity in RIM control slices, electrical stimuli failed to trigger detectable dopamine release in RIM cKODA slices at any stimulation intensity (Figures 3A and 3B). Short stimulus trains (10 Hz, 10 stimuli), mimicking synchronous burst firing often observed in dopaminergic and cholinergic neurons (Grace and Bunney, 1984; Threlfell et al., 2012), also failed to induce dopamine release in RIM cKODA slices (Figures 3C and 3D). Locally puffing 100 mM KCl for 10 s increased the peak amplitude in RIM control slices four-fold. In RIM cKODA slices, only minimal dopamine release was detected following this strong depolarization (Figures 3E and 3F), establishing that the failure to detect dopamine release in RIM cKODA mice is due to the loss of dopamine release, and not due to low sensitivity of the amperometric electrode (which has an estimated detection threshold of ~50 nM). These experiments establish a central requirement for RIM in dopamine release.
Figure 3. RIM is essential for dopamine release in the dorsal striatum.

(A) Sample traces of dopamine release evoked by electrical stimulation in slices of RIM cKODA and sibling RIM control mice. Dopamine release was measured using constant voltage amperometry, and dopamine concentrations were calculated based on electrode calibration (Figures S4A and S4B). In all electrophysiological experiments, RIM cKODA are mice in which RIM is removed specifically in dopamine neurons, and RIM control mice are siblings of RIM cKODA that have one wild-type allele for each RIM gene.
(B) Quantification of peak amplitudes as shown in (A). RIM control n = 10 slices/3 mice, RIM cKODA 10/3 (p < 0.001 for genotype, stimulation intensity and interaction; two-way ANOVA).
(C) Sample traces (average of 4 sweeps) of dopamine release evoked during 10 Hz electrical stimulation in RIM control and RIM cKODA mice.
(D) Quantification of (C). Amplitudes were normalized to the average first amplitude in RIM control. RIM control n = 9 slices/3 mice, RIM cKODA n = 9/3 (p < 0.001 for genotype, stimulus number and interaction; two-way ANOVA).
(E) Sample traces of dopamine release evoked by a local puff of 100 mM KCl for 10 s in RIM control and RIM cKODA mice.
(F) Quantification of peak amplitude in (E). RIM control n = 9 slices/4 mice, RIM cKODA n = 10/4.
All data are mean ± SEM. *** p < 0.001; two-way ANOVA for (B, D) and Mann-Whitney rank sum test for (F).
ELKS is dispensable for dopamine release
Similar to but somewhat milder than RIM removal, knockout of ELKS1α and ELKS2α at hippocampal synapses decreases release by 40–60% (Held et al., 2016; Liu et al., 2014). Interestingly, when we characterized dopamine release in ELKS cKODA and ELKS control mice, we found that ELKS deletion had no effect on release induced by single electrical stimuli, 10 Hz trains, or local KCl puffs (Figure 4). Thus, ELKS1α and ELKS2α are not needed for dopamine exocytosis. These data establish that the secretory machine for dopamine is different from active zones at fast synapses, where RIM and ELKS participate in release, but neither is required.
Figure 4. ELKS1α and ELKS2α are dispensable for dopamine release in the dorsal striatum.

(A) Sample traces of dopamine release evoked by electrical stimulation in slices of ELKS cKODA and sibling ELKS control mice. In all electrophysiological experiments, ELKS cKODA are mice in which ELKS1α and ELKS2α are removed specifically in dopamine neurons, and ELKS control mice are siblings of ELKS cKODA that have one wild-type allele for each ELKS encoding gene.
(B) Quantification of peak amplitude in (A). ELKS control n = 12 slices/4 mice, ELKS cKODA 12/4 (p = 0.53 for genotype, p < 0.001 for stimulation intensity and p = 0.92 for interaction; two-way ANOVA).
(C) Sample traces (average of 4 sweeps) of dopamine release evoked during 10 Hz electrical stimulation in ELKS control and ELKS cKODA mice.
(D) Quantification of (C). Amplitudes were normalized to the average first amplitude in ELKS control. ELKS control n = 12 slices/4 mice, ELKS cKODA n = 12/4 (p = 0.96 for genotype, p < 0.001 for stimulus number and p = 0.96 for interaction; two-way ANOVA).
(E) Sample traces of dopamine release evoked by a local puff of 100 mM KCl for 10 s in ELKS control and ELKS cKODA mice.
(F) Quantification of peak amplitude in (E). ELKS control n = 13 slices/5 mice, ELKS cKODA n = 15/5.
All data are mean ± SEM. ns, not significant; two-way ANOVA for (B, D) and Mann-Whitney rank sum test for (F).
RIM organizes release sites in dopamine axons
The strong impairment of dopamine release in RIM cKODA mice could be due to direct involvement of RIM in secretion, for example as a required scaffold for the assembly of a functional release site, or it could be due to roles of RIM in dopamine neuron development. We investigated two key upstream processes that could be mistaken for massive impairments in dopamine vesicle exocytosis. We first evaluated whether removal of RIM affected the morphology of striatal dopamine axons and dopamine vesicle clusters labeled by TH and vesicular monoamine transporter 2 (VMAT2), respectively. No significant change in the shape or density of dopamine axons or vesicle clusters was detected (Figures 5A and 5B). This is consistent with the 3D-SIM experiments, where we did not detect obvious defects in dopamine axons of RIM cKODA mice (Figures 2A and S2).
Figure 5. RIM organizes release sites in dopamine neurons.

(A) Representative confocal images of VMAT2 and TH staining of the dorsal striatum in RIM control and RIM cKODA mice.
(B) Quantification of fluorescence intensity, size and density of VMAT2 and TH staining for images shown in (A). Each dot represents the mean of a single animal (average of 6–8 images). RIM cKODA values were normalized to their corresponding RIM control. For VMAT2, RIM control n = 3 mice, RIM cKODA, n = 3. For TH, RIM control n = 4, RIM cKODA n = 4.
(C) Quantification of tissue dopamine levels in the striatum measured by ELISA from mice after intraperitoneal injection of reserpine or DMSO two hours before the tissue harvest. Each dot is an average of two measurements from one animal. For DMSO, RIM control n = 4 mice, RIM cKODA n = 4. For reserpine, RIM control n = 5, RIM cKODA n = 3.
(D) Quantification of in vivo extracellular dopamine levels in the striatum measured by microdialysis in anesthetized mice before and during reverse dialysis of TTX. Dopamine levels are expressed normalized to the average in RIM control before reverse dialysis of TTX. RIM control n = 5 mice, RIM cKODA n = 5 (p < 0.001 for baseline levels, not significant for levels after TTX).
(E) Representative surface rendered images (5 × 5 × 2 μm3) of bassoon clusters within dopamine axons from RIM control and RIM cKODA mice.
(F) Quantification of density and size of bassoon clusters within dopamine axons in RIM control and RIM cKODA mice.
(G) Representative STED images of bassoon within dopamine synaptosomes from RIM control and RIM cKODA mice. Vesicles in dopaminergic neurons were labeled by crossing Cre-dependent synaptophysin-tdTomato mice (SYP-tdTomato) with DATIRES-Cre mice. For analysis of SYP-tdTomato expression in dopamine axons, and for analyses of Munc13 and Munc18 clusters, see Figure S5.
(H) Quantification of the bassoon area in (G). Each circle is the average of an image with 600–2100 synaptosomes. RIM control n = 14 images/3 mice, RIM cKODA n = 18/3.
All data are mean ± SEM. *** p < 0.001, **, p<0.01, ns, not significant; Mann-Whitney rank sum test for (B, F), Kruskal-Wallis analysis of variance with post hoc Dunn’s test for (C, D, H).
We next tested whether dopamine synthesis or vesicular loading was affected by RIM cKODA. We used enzyme-linked immunosorbent assays (ELISA) to measure dopamine levels in striatal homogenates from mice injected intraperitoneally with DMSO or the irreversible VMAT2 inhibitor reserpine, which depletes vesicular dopamine in the striatum (Kuczenski, 1977). No difference in tissue dopamine levels between RIM cKODA and RIM control animals was observed, and reserpine caused similar depletion of tissue dopamine in both genotypes (Figure 5C). This strongly indicates that dopamine is synthesized normally and stored in the striatum in VMAT2 containing vesicles in RIM cKODA mice. We next used in vivo microdialysis to measure extracellular dopamine levels in the dorsal striatum and found that they were strongly decreased in RIM cKODA mice. Remarkably, upon reverse dialysis of tetrodotoxin (TTX) to locally block action potentials, extracellular dopamine levels were identical in RIM cKODA and control mice (Figure 5D). Together with the data presented in Figure 3, these experiments establish that dopamine neuron development, dopamine synthesis and its vesicular loading are not detectably impaired in RIM cKODA mice, but that dopamine release mediated by action potentials is essentially abolished in these mice.
To test whether RIM has essential roles as a release site scaffold in dopamine neurons, we analyzed whether removal of RIM impairs clustering of bassoon. In 3D SIM imaging, RIM cKODA mice had a 26% reduction in bassoon cluster density within dopamine neurons. This could be mediated by the loss of bassoon protein, or by disruption of the active zone cluster resulting in a more wide-spread distribution of bassoon that is not detected in the cluster analysis. Remarkably, the size of the detected clusters increased by 47% (Figures 5E–5F), supporting the latter scenario. To better assess bassoon distribution, we labeled vesicle clusters in dopamine neurons using mouse genetics by Cre-dependent expression of synaptophysin-tdTomato (SYP-tdTomato) in DATIRES-Cre mice (Figures S5A–S5D) and analyzed bassoon cluster size in synaptosomes of these mice by STED microscopy. Bassoon labeling in SYP-tdTomato positive synaptosomes became more widespread and covered much of the area of dopamine vesicle clusters (Figures 5G and 5H). These data suggest that RIM removal results disrupts bassoon clustering, leading to a more widespread distribution of bassoon. In hippocampal neurons, RIM knockout does not disrupt bassoon clustering (Kaeser et al., 2011), but its bassoon scaffolding role is redundant with ELKS in these neurons (Wang et al., 2016). Together with the finding that ELKS knockout has no effect on dopamine release, our data indicate that there is less redundancy at release sites in dopamine axons compared to classical active zones.
One explanation for the loss of neurotransmitter release in RIM cKODA mice could be that proteins required for synaptic vesicle fusion, for example Munc13 or Munc18, are also required for dopamine release and are completely absent from RIM cKODA dopamine neurons. We assessed this possibility by assessing Munc13 and Munc18 localization at dopamine release sites. We found that Munc13 was present in dopamine synaptosomes and was reduced but not absent in RIM cKODA mice (Figures S5E–S5I). Munc18 was unaffected by RIM removal (Figures S5H and S5I). Hence, the loss of dopamine release is not due to complete loss of Munc13 or Munc18. In aggregate, our data indicate that RIM serves as a scaffold that organizes multiple proteins, at least Munc13 and bassoon, into active zone-like sites to support dopamine release.
Properties of dopamine release indicate necessity of an active-zone like release site
We observed a strong depression during short stimulus trains in control slices (Figure 3C) that was not mediated by disynaptic inhibition or autoinhibition (Figures S6A and S6B). Synapses with a high vesicular release probability (p) depress strongly during action potential trains due to depletion of releasable vesicles (Zucker and Regehr, 2002). The depression of dopamine release suggests a high p, but it may in part be due to activation of cholinergic interneurons by electrical stimulation, which strongly enhances dopamine release and potentially increases depletion (Figure S4C) (Threlfell et al., 2012; Zhou et al., 2001). To investigate dopamine release independent of cholinergic innervation, we expressed oChIEF, a fast channelrhodopsin (Lin et al., 2009), selectively in dopamine neurons using Cre-dependent adeno-associated viruses (AAVs, Figures 6A and 6B). A 1 ms flash of blue light applied to the striatal slice in the recording area induced dopamine release that was triggered by a dopamine neuron action potential, established by its sensitivity to TTX but not DHβE (Figures 6C and S6C). The 20–80% rise time of the amperometric current was 1.8 ± 0.2 ms, indicating a millisecond time scale for dopamine secretion. In RIM control mice, dopamine release in the dorsal striatum strongly depressed during a short train (10 Hz, 10 stimuli) and reached a steady state after 3 stimuli (Figures 6D and 6E). The paired-pulse ratio (PPR), inversely correlated with p (Zucker and Regehr, 2002), was 0.41 at 100 ms interstimulus interval (Figure 6F). Release remained depressed for >1s (Figures S6D–S6G), likely reflecting slow replenishment after depletion (Cragg, 2003). Similar results were observed in the ventral striatum (Figures 6G–6I), which receives projections from functionally and molecularly distinct VTA dopamine neurons (Brichta et al., 2015). This indicates shared release properties between different types of dopamine neurons.
Figure 6. Dopamine release has a high release probability that requires RIM.

(A) Schematic showing the experimental setup. DATIRES-Cre mice were transduced with a Cre-dependent AAV expressing oChIEF-tdTomato in the SNc and the VTA at P21–23 and recorded at P41–76.
(B) oChIEF-tdTomato expression and TH immunofluorescence in striatum and midbrain.
(C) Representative traces (average of 4 sweeps) and quantification of dopamine release evoked by light stimulation (1 ms pulse of blue light applied at the recording site) in dorsal striatal slices expressing oChIEF in dopamine axons before (black) and after (magenta) blocking action potentials with TTX. n = 5 slices/4 mice.
(D) Example traces (average of 4 sweeps) of dopamine release evoked by 10 Hz light stimulation in dorsal striatum.
(E) Quantification of (D). Amplitudes are normalized to the average first amplitude in RIM control. RIM control n = 15 slices/5 mice, RIM cKODA n = 13/5 (p < 0.001 for genotype and decay, p < 0.01 for interaction; two-way ANOVA).
(F) Quantification of the paired pulse ratio (PPR) for the first two pulses in (E). RIM control n = 14/5, RIM cKODA n = 9/4. Responses smaller than 15 pA were not included in the PPR analysis.
(G–I) Analyses identical to (D–F) but in the ventral striatum. RIM control n = 10/3, RIM cKODA n = 10/3 for (H, p < 0.001 for genotype and stimulus number, p < 0.01 for interaction; two-way ANOVA). RIM control n = 10/3, RIM cKODA n = 8/3 for (I). For additional analyses of optogenetically induced dopamine release, see Figure S6.
(J) Example traces of extracellular recordings in dorsal striatum from RIM control and RIM cKODA mice under 10 Hz light stimulation. For detailed analyses, see Figure S7.
(K) Quantification of (J) with responses normalized to the initial response. RIM control n = 7/3, RIM cKODA n = 8/3 (p = 0.17 for genotype, p < 0.05 for stimulus number, and p = 0.37 for interaction; two-way ANOVA). The inset bar graph indicates absolute initial amplitude. Each circle represents an average response of 100 stimuli from 1 slice.
(L) Representative traces (average of 4 sweeps) and quantification of dopamine release in dorsal striatum evoked in 2 mM (black) and 4 mM (magenta) [Ca2+]ex by a 1 ms light stimulus. n = 9 slices/4 mice. For PPR, analyses in ventral striatum and comparison of RIM control and RIM cKODA mice, see Figure S6.
All data are mean ± SEM. *** p < 0.001, ** p < 0.01, * p < 0.05, ns, not significant; paired t test for (C, L), two-way ANOVA for (E, H, K) and Mann-Whitney rank sum test for (F, I, K).
The somatic action potential amplitudes of dopamine neurons gradually decreases during burst firing (Grace and Bunney, 1984), which could contribute to the depression of release. We thus measured the firing of dopamine axons extracellularly upon optogenetic activation (Figure S7) and found that action potentials reliably followed 10 Hz stimulation (Figures 6J and 6K), ruling out that the strong depression during train stimulation is due to action potential dampening.
Finally, increasing the extracellular Ca2+ concentration ([Ca2+]ex) from 2 to 4 mM only modestly increased peak dopamine release by 13.7 ± 4.6 % (Figure 6L) and only slightly decreased PPR (Figures S6D–S6G), indicating that enhancing [Ca2+]ex does not strongly increase p. Although we cannot fully exclude other factors contributing to release properties, these experiments strongly indicate that dopamine release has a very high initial p. Fast exocytosis and a high p require release machinery that connects the Ca2+ source, Ca2+ sensors and release ready vesicles (Kaeser et al., 2011; Südhof, 2012). Thus, these data strongly support the necessity of protein scaffolds that form dopamine release sites. Consistent with RIM providing for such a scaffold, RIM cKODA mice had a very strong impairment in optogenetically triggered dopamine release at 2 and 4 mM [Ca2+]ex in the dorsal striatum (Figures 6D–6F and S6) and in the ventral striatum (Figures 6G–6I and S6). These data support that RIM organizes release sites to mediate a high initial p of dopamine release. Importantly, the amplitude of the axonal action potentials was unchanged in RIM cKODA slices, and the action potentials also followed 10 Hz stimulus trains (Figures 6J, 6K and S7), establishing that loss of firing does not account for loss of release in RIM cKODA mice and that the density and excitability of dopamine axons are similar in RIM cKODA and control mice.
Release sites are sparse in dopamine axons
Nerve terminals releasing fast neurotransmitters often contain one or multiple active zones. In contrast, dopamine axons appeared to have a low density of release sites with approximately one active zone-like cluster per 4 μm of axon (Figure 2). This suggests that the active zone density in dopamine axons may be lower than in axons releasing fast neurotransmitters. To test this hypothesis, we assessed the distribution of active zone-like sites relative to vesicle clusters in dopamine synaptosomes. We found that only 26.3% of the dopamine vesicle clusters, defined by the coexistence of TH and the vesicle marker synaptobrevin-2, contained the release site marker bassoon (Figures 7A and 7B). In contrast, 52.6% of TH negative synaptosomes contained bassoon. Similar results were observed when active zones were labeled using ELKS or RIM (Figures 7A–7D). Importantly, approximately 30% TH positive synaptosomes contained RIM independent of whether they were co-labeled with VMAT2 or synaptobrevin-2 (Figures 7C and 7D), making it unlikely that release sites are exclusively associated with varicosities that do not contain dopamine-laden vesicles (Zhang et al., 2015). To test whether many dopamine vesicle clusters lack active zone-like sites in vivo, we labeled vesicles in dopamine neurons by expression of SYP-tdTomato in dopamine neurons (Figures 7E, 7F, S5A–S5D). Only 34% of the labeled vesicle clusters contained bassoon. For comparison, we labeled vesicles in glutamatergic synapses in the striatum, and found that 72% of their vesicle clusters were associated with bassoon. While there are likely some false negatives in this experiment, the quantitative match between the synaptosome experiment and the brain 3D-SIM establish that dopamine varicosities contain active zone-like release sites significantly less frequently than glutamatergic nerve terminals.
Figure 7. Many dopamine varicosities lack active zone-like release sites.

(A) Representative confocal images of sparsely plated striatal synaptosomes stained for synaptobrevin-2, TH and bassoon (top) or ELKS (bottom). Filled arrowheads indicate synaptosomes containing all three markers. Hollow arrowheads indicate synaptosomes containing synaptobrevin-2 and TH but not bassoon or ELKS.
(B) Quantification of (A) showing the percentage of particles that contain bassoon or ELKS. Each circle represents the average result of an area with 1,500–6,000 synaptosomes. n = 18 areas/3 mice for bassoon and 9/3 for ELKS.
(C) Striatal synaptosomes stained for RIM, VMAT2 and TH (n = 14/3); RIM, synaptobrevin-2 and TH (n = 17/3); or RIM, synaptobrevin-2 and VGluT1 (n = 17/3). Filled arrowheads indicate synaptosomes (insets) containing both markers (VMAT2 and TH, synaptobrevin-2 and TH, or synaptobrevin-2 and VGluT1) and RIM. Hollow arrowheads indicate synaptosomes containing the same markers but not RIM.
(D) Quantification of (C) showing the percentage of particles that contain RIM. n = 14 areas/3 mice for VMAT2 and TH positive synaptosomes, 17/3 for synaptobrevin-2 and TH positive synaptosomes and 17/3 for synaptobrevin-2 and VGluT1 positive synaptosomes.
(E) Representative surface rendered 3D-SIM images of bassoon within vesicle clusters in the striatum. Vesicles in dopaminergic neurons or in glutamatergic neurons were labeled by crossing Cre-dependent synaptophysin-tdTomato mice (SYP-tdTomato) with DATIRES-Cre or VGluT1IRES-Cre mice, respectively.
(F) Quantification of (E) showing the percentage of vesicle clusters associated with bassoon. SYP-tdTomato x DATIRES-Cre n = 49 regions/4 mice, SYP-tdTomato x VGluT1IRES-Cre n = 46/3.
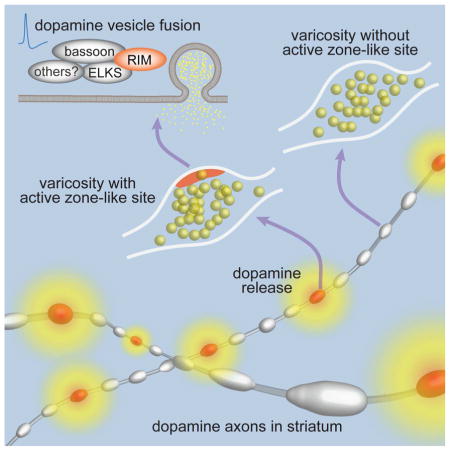
(G) Current model of cellular and molecular architecture for dopamine secretion. Most dopamine varicosities contain clusters of dopamine vesicles and DAT along dopamine axons, but only ~30% of them contain active zone-like release sites composed of the scaffolds bassoon, RIM, ELKS and likely other active zone proteins including Munc13. When action potentials propagate through dopamine axons, dopamine vesicles fuse at these sites with a very high release probability, and fusion requires the presence of RIM. This architecture generates a fast, local rise in dopamine followed by a rapid decay, which may support fast dopamine coding.
All data are mean ± SEM. *** p < 0.001, ** p < 0.01, * p < 0.05, ns, not significant; Kruskal-Wallis analysis of variance with post hoc Dunn’s test for (B, D), Mann-Whitney rank sum test for (F).
Discussion
Here, we find that striatal axons of midbrain dopamine neurons contain active zone-like release sites (Figure 7G) that are mechanistically distinct from active zones at fast synapses. Action potential-triggered dopamine release requires these sites, and only 30% of dopamine vesicle clusters are associated with them, suggesting that many dopamine varicosities may not release dopamine in response to action potentials.
Molecular architecture and mechanisms of dopamine release sites
At active zones of fast synapses, removal of RIM or ELKS impairs release by 40%–80% (Acuna et al., 2016; Han et al., 2011; Held et al., 2016; Kaeser et al., 2011; Liu et al., 2014). In dopamine axons, we observed a much stronger dependence of release on RIM and surprisingly, independence of ELKS. Molecular and mechanistic differences between dopamine release sites and classical active zones are further supported by several additional findings. First, the bassoon cluster size in dopamine axons was smaller than in other striatal synapses. Second, removal of RIM disrupted bassoon clusters in dopamine axons but not in axons of hippocampal neurons (Kaeser et al., 2011). Third, previous findings indicate that, different from fast synapses, dopamine release relies on T- and L-type voltage-gated calcium channels in addition to N- and P/Q-type channels (Brimblecombe et al., 2015). In general, functional heterogeneity between different subtypes of neurons and neurotransmitters is well established, but how the molecular mechanisms of the release machinery account for such diverse properties is largely unexplored. The midbrain dopamine pathway is an ideal system to dissect this question due to the genetic accessibility, the available measurements of dopamine release, and the behavioral and clinical importance of the system. We make a first key step forward by identifying essential components of a mechanistically distinct release site that is specialized for secretion of a neuromodulator.
Our data indicate that RIM operates as a molecular scaffold for dopamine secretion. Dopamine release is fast and has a high p, which establishes that release must be mediated by a scaffold that tethers primed, dopamine-laden vesicles to the site of Ca2+-influx. Conditional knockout of RIM in dopamine neurons abolishes release and impairs clustering of bassoon and Munc13. Hence, it is likely that RIM mediates dopamine release through autonomous scaffolding of multiple release site proteins. At fast synapses, RIM has three known functions which all show redundancy with other proteins: the scaffolding of active zone proteins (Acuna et al., 2016; Wang et al., 2016), vesicle priming (Deng et al., 2011; Held et al., 2016) and Ca2+ channel tethering (Acuna et al., 2016; Kaeser et al., 2011). Because RIM scaffolding functions are more prominent in dopamine neurons than in other neurons, dopamine release likely relies on a simpler scaffold with less redundancy.
Some midbrain dopamine neurons co-release glutamate from a distinct axonal compartment (Zhang et al., 2015). It is unlikely that the active zone-like structure solely reflects those glutamatergic synapses, because dopamine axons in the dorsal striatum, where all 3D-SIM experiments were performed, release little or no glutamate (Stuber et al., 2010). Furthermore, we labeled dopamine axons and varicosities with TH or VMAT2, markers that may be excluded from glutamate-containing areas (Zhang et al., 2015). Hence, the sparse active zone-like clusters are associated with dopamine containing vesicles and most likely embody their fusion sites. One interesting observation is that some extracellular dopamine remains after RIM knockout or after blocking action potentials (Figure 5D). While we cannot exclude that tissue damage contributes to the leftover dopamine in microdialysis, these data support that action potential-independent modes of dopamine release exist, for example through spontaneous release or through DAT reversal. The RIM independence of this mode suggests that it does not require active zone-like sites, and varicosities that do not contain this structure may also contribute to this form of release.
Implications for dopamine signaling
A prominent model is that burst firing of dopamine neurons generates phasic dopamine release, which codes for events (Grace, 2016). Our findings establish that during burst firing, the active zone-like site generates a fast rise in local dopamine that is rapidly dampened. We demonstrate that the fast depression of release during burst firing is mainly due to the unusually high p, which leads to rapid depletion. Combined with dopamine reuptake, this terminates the dopamine signal within hundreds of milliseconds. This dynamic dopamine signal, controlled by the release machinery we identify here, likely contributes to the sub-second precision by which dopamine modulates locomotion (Howe and Dombeck, 2016) and spine plasticity (Yagishita et al., 2014). Hence dopamine transmission relies on fast, sophisticated release machinery that provides for precise coding ability.
Our data further reveal that stimulation trains result in dopamine release similar to that evoked by single spikes, because only the first action potential efficiently triggers release. This is different from the model that burst firing of dopamine neurons cell-autonomously induces more dopamine release. One explanation is that burst firing is synchronized across many dopamine neurons while tonic firing is not, resulting in rapid accumulation of dopamine. An alternative explanation is that single spikes generated in the axon initial segment fail to propagate in extensive unmyelinated axonal arbors, while burst firing strongly increases the chance of action potentials reaching the varicosities (Matsuda et al., 2009; Pissadaki and Bolam, 2013). Thus, the increase in extracellular dopamine during burst firing may be due to an enhanced rate of spike propagation. Since even low frequency firing in the axon results in depression of dopamine release, dopamine neurons that were previously silent or only fired at very low frequencies are probably more efficient in coding events when recruited.
Recent experiments suggest that only ~20% of dopamine varicosities release dopamine (Pereira et al., 2016), and a similar percentage of varicosities is associated with postsynaptic specializations (Cailld et al., 1996; Descarries et al., 1996; Yung et al., 1995). Remarkably, dopamine may also mediate relatively fast postsynaptic currents (Gantz et al., 2013; Marcott et al., 2014). Furthermore, synaptic contacts of dopamine neurons are composed of dopaminergic presynaptic and GABAergic postsynaptic structures (Uchigashima et al., 2016), consistent with the observation that dopamine neurons synaptically co-release dopamine and GABA (Tritsch et al., 2012). These observations, together with our findings that only ~30% of dopamine varicosities contain active zone-like release sites, raise the possibility that dopamine is exclusively released at synaptic sites in the striatum. However, because dopamine receptors appear to be present mostly outside of synaptic structures (Cailld et al., 1996; Uchigashima et al., 2016; Yung et al., 1995), dopamine would still need to diffuse after release to extrasynaptic sites for signaling. To determine how dopamine codes precisely timed events, future studies will need to assess localization of dopamine receptors relative to its secretory sites.
Active zone-like release sites may be important for regulation and in disease
At fast synapses, active zone protein function and release are regulated during synaptic plasticity, for example through Ca2+ signaling and phosphorylation (Südhof, 2012). Active zone-like release sites in dopamine neurons might be controlled similarly, for example by modification of RIM, to regulate dopamine signaling. Previous studies have found heterogeneity of dopamine release properties across varicosities, including variable p and silent varicosities (Cragg, 2003; Pereira et al., 2016). Variable composition or occurrence of release sites could be a tunable feature that contributes to these functional properties, and dopamine release could be regulated by activating or silencing entire vesicle clusters through adding or removing active zone-like sites. An alternative function of the varicosities void of release sites could be local storage of vesicles or other secretory material. This may be particularly important for midbrain dopamine neurons. With a single axon extending over several meters (Matsuda et al., 2009), the distant soma may not be sufficient for continuous supply of secretory material. This complex cellular architecture may be one explanation for the vulnerability of dopamine neurons, and local storage within varicosities may reflect a cellular strategy to counteract this problem.
Finally, mutations in RIM and other active zone proteins are often associated with brain disease (Fromer et al., 2014; O’Roak et al., 2012). We show that RIM is essential for dopamine secretion. This suggests that mutations in RIM and other proteins in the release machinery may contribute to neurological disease by altering neuromodulation, a point that will have to be better incorporated into the interpretations of roles for secretory proteins in neurological disease.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Pascal S. Kaeser (kaeser@hms.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Knock-in mice with bicistronic Cre recombinase expression from the dopamine transporter (DAT) gene locus (RRID:IMSR_JAX:006660, B6.SJL-Slc6a3tm1.1(cre)Bkmm/J) (Backman et al., 2006) were used for removal of active zone proteins or expression of marker proteins specifically in dopamine neurons and are referred to as DATIRES-Cre mice. For removal of RIM specifically in dopamine neurons (RIM cKODA), previously generated RIM1 (RRID:IMSR_JAX:015832, Rims1 tm3Sud/J) and RIM2 (RRID:IMSR_JAX:015833, Rims2tm1.1Sud/J) double floxed mice (Kaeser et al., 2011) were crossed with DATIRES-Cre mice (Figure S2A). Similarly, dopamine neuron-specific ELKS1α/2α knockouts (ELKS cKODA) were obtained by crossing ELKS1 (RRID:IMSR_JAX:015830, Erc1tm2.1Sud/J) and ELKS2 (RRID:IMSR_JAX:015831, Erc2tm1.2Sud/J) double floxed mice (Liu et al., 2014) with DATIRES-Cre mice (Figure S3A). For labeling vesicles in dopamine neurons, mice with knockin of a CAG promoter-driven loxP-STOP-loxP-synaptophysin-tdTomato cassette (SYP-tdTomato) in the Rosa26 locus (RRID: IMSR_JAX:012570, B6;129S-Gt(ROSA)26Sortm34.1(CAG-Syp/tdTomato)Hze/J) were crossed with DATIRES-Cre mice. For labeling vesicles in glutamate neurons, the SYP-tdTomato mice were crossed with knock-in mice with bicistronic expression of Cre recombinase from the vesicular glutamate transporter 1 gene locus (VGluT1IRES-Cre) (RRID:IMSR_JAX:023527, B6;129S-Slc17a7tm1.1(cre)Hze/J). Mice were group-housed with free access to water and food and experiments were performed at 21–112 days of age in animals of both sexes. All animal experiments were approved by the Harvard University Animal Care and Use Committee.
METHOD DETAILS
3D-SIM imaging and data processing
Male and female mice (postnatal 43–76 days) were deeply anesthetized with 5% isoflurane and perfused transcardially with 30 ml phosphate buffer (PBS) at room temperature. Fixation was performed with perfusion of 50 ml 4% paraformaldehyde (PFA) in PBS at 4 °C. Coronal slices containing striatum (20 μm thick) were cut using a vibratome (Leica, VT1000s) in ice-cold PBS. For RIM and ELKS staining, epitope retrieval was performed by incubating fixed slices for 12–16 hours at 60 °C in retrieval solution containing 10 mM Tris Base, 1 mM EDTA, 0.05% Tween 20, pH 9.0. All slices were blocked in 10% goat serum, permeabilized in 0.25% Triton X-100, stained in primary antibodies for 12 h at 4 °C and secondary antibodies for 2 h at room temperature, and mounted on #1.5 cover glasses (GG-18-1.5-pdl, neuVitro) with H-1000 mounting medium (Vectashield). Primary antibodies used for 3D-SIM staining were: mouse anti-bassoon (1:500, RRID: AB_11181058), mouse anti-RIM1 (1:500, 610907, RRID: AB_10611855), rabbit anti-TH (1:1,000, AB152, RRID: AB_390204), guinea pig anti-TH (1:1,000, RRID: AB_2619897), rabbit anti-ELKS2α (custom made (Held et al., 2016), 1029 bleed 5, affinity purified). Secondary antibodies tagged with Alexa 488 or Alexa 568 were used for detection. 3D-SIM data were collected on a DeltaVision OMX V4 Blaze system (GE Healthcare) equipped with a 60x/1.42 N.A. oil immersion objective and separate Edge 5.5 sCMOS cameras (PCO) for each channel. Z-stacks were acquired with a z-step of 125 nm and with 15 raw images per plane (five phases, three angles). Spherical aberration was minimized using immersion oil matching. Super-resolution images were computationally reconstructed from the raw datasets with a channel-specific measured optical transfer function and a Wiener filter constant of 0.002–0.003 in softWoRx 6.1.3 (GE Healthcare). Channel misregistration was measured using a control slide and multi-channel datasets were registered using the image registration function in softWoRx. 3–5 regions of interest (ROIs) were imaged from the dorsal striatum in each slice with imaging volumes of 40 × 40 × 4 μm3. For image analysis, ROIs ranging from 35 × 33 × 2.5 to 39 × 39 × 3.4 μm3 were manually selected in each image stack. A custom Matlab program was produced to analyze the 3D data sets. Binary images in each layer of the stack were generated, with the threshold determined by automatic two-dimensional segmentation (Otsu algorithm). Objects in each channel were recognized by the size of the cluster formed by contiguous voxels. Size filters for object recognition were as follows: TH-positive axons, 0.04–20 μm3; vesicle clusters, 0.04–1.2 μm3; active zone markers, 0.003–0.04 μm3. The size of objects in individual channels and the overlap between objects from different channels were calculated. Histograms showing the number of clusters in 20% overlap bins are shown in Figures S1 and 2. An object was considered to be within the other when at least 40% of its voxels colocalized with the voxels from the other object in Figures 1, 7, S2 and S3. Shuffled objects were generated by randomly relocating each of the objects in one channel locally within 1 × 1 × 1 μm3 after object recognition (Figure 1B). The overlap between shuffled objects and objects from the other channel were then quantified. The effect of shuffling was evaluated by averaging 1,000 rounds of shuffling and overlap quantification. To estimate the length of dopamine axons, image stacks of tyrosine hydroxylase (TH) staining were first blurred by three-dimensional Gaussian filtering with a sigma value of 1. A three-dimensional skeletonization was then performed using homotopic thinning algorithm and the total length of the skeleton was calculated. The absolute active zone protein cluster density along dopamine axons was estimated by calculating the differences in the cluster densities between cKODA mice and control mice using an overlap threshold of >0% to include all clusters in close proximity to dopamine axons. In conditional genetic 3D-SIM experiments (Figures 2 and 5), cKODA mice were mice with homozygote floxed alleles for RIM1 and RIM2, or ELKS1 and ELKS2, and a heterozygote DATIRES-Cre allele. Control mice were siblings of the corresponding cKODA mice that had homozygote floxed alleles (either for RIM or ELKS, respectively) but were negative for the DATIRES-Cre knockin mutation. All data analyses were automated and were done identical for all conditions. All volume and surface rendering of the representative images for figures were performed in Imaris (BITPLANE).
Production and injection of AAV viruses
Adeno-associated viruses (AAV)-DJ encoding a double-floxed inverted oChIEF-tdTomato expressed under a human synapsin 1 promoter was produced in HEK293T cells. 72 h after transfection with Ca2+ phosphate, cells were collected, lysed by three freeze-thaw cycles and the AAVs were purified after harvesting of the cell lysate using iodixanol gradient ultracentrifugation. Genomic titers were measured by quantitative PCR, and infectious titers were estimated by transducing cultured hippocampal neurons with serial dilutions of the AAV. AAVs were injected unilaterally into the substantia nigra pars compacta (SNc) or ventral tegmental area (VTA) of DATIRES-Cre mice containing conditional floxed alleles for RIM. At postnatal days 21–23, mice were anesthetized using 5% isoflurane and mounted on a stereotaxic frame. 1.5–2 % isoflurane was used to maintain a stable anesthesia during the surgery. After exposing the skull, a small hole was drilled, and the AAV (in DMEM, 1012–1013 genome copies/ml) was injected through a microinjector at a rate of 0.1 μl/min for a total volume of 0.5–1 μl. Injection coordinates were 0.8 mm anterior, 1.3 mm lateral of Lambda and 4.4 mm below pia for SNc, and 0.6 mm anterior, 0.7 mm lateral from lambda and 4.8 mm below pia for VTA. After recovery from the surgery, the mice were returned to their home cage for at least 19 days prior to recording. Stereotaxic procedures were performed under protocols approved by the Harvard University Animal Care and Use Committee.
Electrophysiological recordings
Male and female mice (postnatal 41–103 days) were deeply anesthetized by isoflurane and decapitated. Sagittal brain slices containing the striatum (250 μm thick) were cut using a vibratome (Leica, VT1200s) in ice-cold cutting solution containing (in mM): 75 NaCl, 2.5 KCl, 7.5 MgSO4, 75 Sucrose, 1 NaH2PO4, 12 Glucose, 26.2 NaHCO3, 1 Myo-inositol, 3 Pyruvic acid, 1 Ascorbic acid. After cutting, slices were incubated at room temperature in incubation solution containing (in mM): 126 NaCl, 2.5 KCl, 2 CaCl2, 1.3 MgSO4, 1 NaH2PO4, 12 Glucose, 26.2 NaHCO3, 1 Myo-inositol, 3 Pyruvic acid, 1 Ascorbic acid for at least 1h before use. Recordings were performed at 30–32 °C, and slices were placed in a recording chamber continuously perfused with artificial cerebrospinal fluid (ACSF) containing (in mM): 126 NaCl, 2.5 KCl, 2 CaCl2, 1.3 MgSO4, 1 NaH2PO4, 12 Glucose, 26.2 NaHCO3 at 2.5–3 ml/min (heated to 30–32 °C). All solutions were constantly bubbled with 95% O2 and 5% CO2. All recordings were completed within 4 hours of slicing. In conditional genetic experiments for electrophysiology (Figures 3, 4, S6 and S7), cKODA mice were mice with homozygote floxed alleles for RIM1 and RIM2, or ELKS1 and ELKS2, and a heterozygote DATIRES-Cre allele. Control mice were siblings of the corresponding cKODA mice that had heterozygote floxed alleles (either for RIM or ELKS, respectively) and carried a heterozygote DATIRES-Cre knockin mutation to control for potentially confounding effects on dopamine measurements by a tendency towards slightly lower DAT levels in these mice (Backman et al., 2006). Thus, the control mice are heterozygote for the targeted genes in these experiments. Because RIM control, ELKS control and ELKS cDKODA mice have indistinguishable dopamine release (Figures 3 and 4), we conclude that heterozygosity for RIM did not strongly impair dopamine release.
Amperometric recordings were performed using carbon-fiber microelectrodes (CFE, 7 μm in diameter, 50–100 μm in length) that were fabricated in the laboratory using commercially available carbon fiber filament (Goodfellow). The CFE was calibrated immediately before use by puffing freshly made dopamine solutions in ACSF (0, 1, 5, 10, 20 μM, Figure S4A). The recording electrode was held at a constant voltage of 600 mV and placed approximately 50 μm below the slice surface in the dorsal striatum. Signals were sampled at 10 KHz and low-pass filtered at 400 Hz. Dopamine release was evoked by electrical or optogenetic stimulation every 2 min.
Electrical stimulation was applied through a unipolar glass pipette (tip diameter 3–5 μm) filled with ACSF. The electrode was placed approximately 20 μm below the slice surface in the dorsal striatum and 100–150 μm away from the CFE (Figure S4B). The frequency, duration and magnitude of the stimulus were controlled through a linear stimulus isolator (A395, World Precision Instruments). A biphasic wave (0.25 ms in each phase) was applied to evoke responses.
Potassium chloride stimulation was applied by puffing a buffer containing (in mM) 50 NaCl, 100 KCl, 2 CaCl2, 1.3 MgSO4, 12 Glucose, 10 HEPES, pH 7.3 at the recording site through a 200 μm wide tube for 10 s at 9 μl/s controlled by a microinjector pump. Only one stimulus was applied per slice.
Optogenetic stimulation was limited to dopaminergic axons in the striatum by AAV mediated, Cre-dependent expression of oChIEF-tdTomato in dopamine neurons. For the activation of oChIEF, brief pulses of light (470 nm; 1 ms duration; 7.5 mW/mm2) were delivered at the recording site by a light-emitting diode. In the PPR analysis, responses smaller than 15 pA were excluded.
Extracellular recordings were performed with a glass pipette (2–3 μm in tip diameter) filled with ACSF and placed approximately 20 μm below the slice surface in an area of the dorsal striatum with even expression of oChIEF-tdTomato (Figure S7A). Brief pulses of blue light (470 nm; 1 ms duration; 7.5 mW/mm2) were delivered directly at the recording site at 1 Hz for 200–1,200 times. Train stimulation was applied every 10s. 100 sweeps were averaged for quantification (Figure S7B).
ELISA to measure tissue dopamine levels
Male and female mice (postnatal days 66–112) were injected intraperitoneally with DMSO (0.1 ml) or reserpine (5 mg/kg, 0.1 ml, dissolved in DMSO). 2 hours after injection, forebrains containing dorsal and ventral striatum were harvested and homogenized with 30 strokes with a glass Teflon homogenizer in buffer containing (in mM): 25 HEPES, 320 sucrose, 4 EDTA and 1x protease inhibitor cocktail (P8340, Sigma-Aldrich) pH 7.4. 5 μl of buffer was added for each mg of brain tissue. The homogenate was incubated for 1 hour at 4 °C in 1% Triton X-100 on a rotator and centrifuged at 25,000 g for 45 min at 4 °C. 50 μl of the supernatant was used for the Enzyme-linked immunosorbent assay (ELISA). The ELISA assay was performed according to the manufacturer’s protocol using a Dopamine ELISA kit (Rocky Mountain Diagnostics, BA E-5300). Fractions were measured 2 times each at 1:5 and 1:25 dilutions. The average of the two 1:25 measurements were used for further analyses because the measured values for this dilution were all within the linear range of the standard curve. Dopamine concentrations were calculated based on a standard curve that was obtained by using defined dopamine concentrations (0, 0.5, 1.5, 5, 20, 80 ng/ml). Dopamine levels are expressed as ng/mg tissue (Figure 5C). RIM control and RIM cKODA mice were as described in electrophysiological recordings.
In vivo microdialysis to measure dopamine
Male and female mice (postnatal 61–75 days) were anesthetized using 5% isoflurane and mounted on a stereotaxic frame. 1% isoflurane was used to maintain stable anesthesia during microdialysis. Upon surgical exposure of the skull, a small hole was drilled with a hand drill, and a microdialysis probe (6 kDa cut-off, CMA 11, Harvard Apparatus) was inserted through the hole into the dorsal striatum (coordinates: 1.0 mm anterior, 2.0 mm lateral of bregma, and 3.3 mm below pia). The probe was continuously perfused with ACSF containing (in mM): 155 NaCl, 2.5 KCl, 1.2 CaCl2, 1.2 MgCl2, and 5 glucose at a speed of 1 μl/min. Samples were collected every 15 min and measured using an HPLC connected to an electrochemical detector (Eicom). TTX (10 μM dissolved in ACSF) was reversely dialyzed into the brain when the dopamine level was stable for at least 1 hr. RIM control and RIM cKODA mice were as described in electrophysiological recordings.
Confocal imaging in brain sections
Immunofluorescent labeling of striatal slices was performed as described for 3D-SIM imaging. Primary antibodies used were: mouse anti-bassoon (1:500, RRID: AB_11181058), guinea pig anti-TH (1:1,000, RRID: AB_2619897), rabbit anti-VMAT2 (1:500, RRID: AB_10013884). Single sections (Figures 5A and 5B) or z stacks (Figure 1A) were taken on an Olympus FV1000 laser scanning microscope. RIM control and RIM cKODA mice were as described in electrophysiological recordings. Images were taken from 6–8 regions (200 × 200 μm2 each) from each animal. ROIs containing the test protein were generated by automatic segmentation in ImageJ (Otsu algorithm), and the average intensity, size and density were then calculated.
Striatal synaptosome preparation
Coronal brain slices containing striatum were cut (1 mm thick) using a vibratome (Leica, VT1200s) as described in the electrophysiological recordings. The dorsal striatum was dissected out from 3–4 slices and homogenized by 12 strokes with a glass Teflon homogenizer in 1 ml homogenizing buffer containing (in mM) 4 HEPES, 320 sucrose, and 1x protease inhibitor cocktail, pH 7.4. After the addition of 1 ml homogenizing buffer, the homogenate was centrifuged at 1,000 g for 10 min at 4 °C. The supernatant (S1) was taken and centrifuged at 12,500 g for 15 min at 4 °C. The supernatant (S2) was removed and the pellet (P2) was re-homogenized in 1 ml homogenizing buffer with 6 slow strokes in a glass Teflon homogenizer. After the addition of 1 ml homogenizing buffer, the P2 homogenate was added to the top of a sucrose gradient made of 5 ml 1.2 M sucrose and 5 ml 0.8 M sucrose, and was centrifuged at 69,150 g for 70 min at 4 °C. The synaptosome layer (1–1.5 ml) was collected from the interface of the two sucrose layers (Figure S1C).
Analyses of striatal synaptosomes
For confocal microscopy, synaptosomes were diluted 10–40 times in homogenizing buffer and spun down (4,000 g, 10 min) on #1.5 coverglasses coated with poly-d-lysine. 4% PFA in PBS was added for 20 min to fix the synaptosomes. The synaptosomes were blocked in 3% bovine serum albumin, permeabilized in 0.1% Triton X-100, stained in primary antibodies for 12 h at 4 °C and secondary antibodies for 2 h at room temperature. Primary antibodies used were: mouse anti-bassoon (1:500, RRID: AB_11181058), mouse anti-RIM1 (1:500, RRID: AB_10611855), rabbit anti-VMAT2 (1:500, RRID: AB_10013884), rabbit anti-TH (1:1,000, RRID: AB_390204), rabbit anti-synaptobrevin-2 (1:1,000, RRID: AB_887810), rabbit anti-DAT (1:100, RRID: AB_90808), rabbit anti-ELKS2α (custom made (Held et al., 2016), 1029 bleed 5), guinea pig anti-TH (1:1,000, RRID: AB_2619897), guinea pig anti-bassoon (1:1,000, RRID: AB_2290619), guinea pig anti-VGluT1 (1:1,000, RRID: AB_887878), rabbit anti-Munc13-1 (1:1000, RRID: AB_887733), rabbit anti-Munc18 (1:1000, SySy 116003). Secondary antibodies were tagged with Alexa 488, Alexa 568 or Alexa 633. A single optical section (200 × 200 μm2) was taken for each area. Image analysis was carried out using ImageJ. Briefly, background was subtracted from all channels using the “rolling ball” ImageJ plugin with a radius of 1 μm. ROIs were first defined by bassoon, TH, VMAT2 or synaptobrevin-2 by automatic segmentation and a size filter of 0.2–1 μm2. The average intensity of signals in other channels within individual ROIs was then measured. An ROI was considered to contain the target protein when the average fluorescence intensity of that particular protein within the ROI was more than 3 times the average intensity of all pixels in the image, and was considered as not containing the target protein when the intensity of the target protein within the ROI was below the average intensity of all pixels.
For STED microscopy, samples were prepared as described for confocal microscopy, but different secondary antibodies were used. The secondary antibodies were tagged with Oregon 488 (for STED imaging), Alexa 555 (for STED imaging) or Alexa 633 (for confocal imaging). The glass coverslips were mounted on slides in ProLong Diamond Antifade mounting medium (ThermoFisher). Images were acquired on a Leica SP8 STED 3X microscope using a 100x oil immersion objective (NA: 1.4) in a specific order to reduce bleaching (633 nm, 555 nm and 488 nm). Fluorophores were excited at corresponding wavelengths. Depletion lasers for STED imaging were at 40% power for Alexa 555 (STED laser: 660 nm) and at 50% power for Oregon 488 (STED laser: 592 nm). For quantification, ROIs containing bassoon were generated by automatic segmentation in ImageJ, and the areas of the ROIs that overlapped with synaptobrevin-2 with or without TH were calculated.
For electron microscopy (EM), the synaptosome layer was spun down at 16,000 g for 15 min at 4 °C. The pellet was washed with 1 ml of synaptosome homogenizing buffer, and again centrifuged. The supernatant was removed and the pellet was fixed in 2% PFA, 2.5% glutaraldehyde in 0.1M cacodylate buffer pH 7.4 with 2 mM CaCl2 on ice for 1 hour. The synaptosome pellet was then washed three times with 0.1 M cacodylate buffer and processed immediately using standard EM embedding and cutting protocols as described (Wang et al., 2016). Images were taken with a transmission electron microscope (JEOL 1200 EX at 80 kV accelerating voltage).
For Western blotting, the striatal homogenate, S1, S2, P1 and P2 fractions were diluted 6-fold with homogenizing buffer to equal the final dilution of the sucrose gradient and synaptosome fractions. The samples were boiled in SDS loading buffer, run on SDS-PAGE gels, and standard western blotting procedures were followed as described (Wang et al., 2016). Primary antibodies used for blotting were as follows: mouse anti-synaptophysin 1 (1:1,000, RRID: AB_887824), rabbit anti-synaptobrevin-2 (1:1,000, RRID: AB_887810), rabbit anti-SNAP25 (1:4,000, RRID: AB_887790), rabbit anti-RIM (1:500, RRID: AB_2617051), rabbit anti-panELKS (1:1,000, custom made P224, a gift from Dr. T.C. Südhof), and mouse anti-β-actin (1:2,000, RRID: AB_476692). Enhanced chemiluminescence was used for detection of HRP-coupled secondary antibodies.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are expressed as mean ± SEM. Statistical analyses were performed in Prism (GraphPad). Paired two-tailed t test was used in Figures 1D, 1E, 6C, 6L, S4D and S7C. Mann-Whitney rank sum test was used in Figures 1H, 3F, 4F, 5B, 5F, 6F, 6I, 6K, 7F, S2D, S3D, S5B, S5I and S7C. Kruskal-Wallis analysis of variance with post hoc Dunn’s test was used in Figures 2F, 5C, 5H, 7B, 7D, S5F, S6E and S6G. Two-way ANOVA followed by post hoc Bonferroni test was used in Figures 2B, 2D, 3B, 3D, 4B, 4D, 6E, 6H, 6K, S1A, S2C, S3C, S6B and S7E. No statistical method was used to determine sample size. Significance was set as * p < 0.05, ** p < 0.01, and *** p < 0.001 for all data. For genotype comparisons by electrophysiology, the experimenter was blinded for data acquisition and analyses for all experiments except for Figures 6, S6 and S7. For genotype comparisons by microscopy, the experimenter was blinded for all data analyses.
DATA AND SOFTWARE AVAILABILITY
All data, codes for analyses, materials and animals are available from the corresponding author upon request. Code for 3D-SIM analyses has been deposited to GitHub.
Supplementary Material
(A) Histogram of actual and locally shuffled bassoon cluster densities within dopamine axons across 20% bins of overlap. n = 35 regions/4 mice. Each region contains 6000–11,000 bassoon clusters. Rectangle area indicates bassoon clusters that have more than 40% of volume overlapped with TH and are considered to be within dopamine axons. The 40%–100% range was selected based on the observations that proteins expressed specifically in dopamine axons overlapped with TH mainly within this range (Figure S5D), and that the density of RIM and ELKS clusters within dopamine axons dropped rapidly in this range when removed genetically (Figures 2B and 2D).
(B) Schematic of striatal synaptosome preparation.
(C) Western blotting of the fractions obtained during a synaptosome preparation. The synaptosome layer is enriched with vesicle markers (synaptophysin-1 and synaptobrevin-2) and active zone markers (RIM and ELKS).
(D) Representative images of bassoon and synaptobrevin-2 staining in a synaptosome preparation taken by confocal (top) and STED (bottom) microscopy.
(E) Representative image of electron microscopic analysis of the synaptosome layer reveals enrichment of membrane enclosed structures filled with synaptic vesicles.
Data in A are mean ± SEM.
(A) Schematic of the conditional RIM knockout in dopamine neurons (RIM cKODA). Exons 6 (E6) or 26 (E26) were flanked by loxP sites in the conditional RIM1 and RIM2 knockout mice, respectively, to generate double conditional RIM1/2 knockout mice (Kaeser et al., 2011). When crossed with DATIRES-Cre mice (Backman et al., 2006), Cre recombinase removed RIM1 and RIM2 proteins in dopamine neurons.
(B) Representative images showing RIM and TH labeled dopamine axons, and RIM within dopamine axons in the dorsal striatum of RIM control and RIM cKODA mice. The volume and surface rendered images were obtained from 10 × 10 × 2 μm3 image stacks. For each zoom-in image, a 90° rotation around the x-axis is shown below the standard x-y-z image.
(C) Histogram of locally shuffled RIM cluster densities within dopamine axons across 20% bins of overlap. After shuffling of RIM control and RIM cKODA clusters, no difference in cluster density is detected. RIM control n = 24 regions/4 mice, RIM cKODA n = 22/4 (p = 0.83 for genotype, p < 0.001 for overlap, and p = 0.63 for interaction; two-way ANOVA).
(D) Quantification of actual and shuffled RIM clusters with >40% volume overlap with dopamine axons from RIM control and RIM cKODA mice. RIM cluster density is decreased by 49% in RIM cKODA axons. n as in C.
All data are mean ± SEM. *** p < 0.001, ns, not significant; two-way ANOVA for (C), Mann-Whitney rank sum test for (D).
(A) Schematic of the conditional ELKS knockout in dopamine neurons. Exons 2 (E2) and 3 (E3) were flanked by loxP sites in the conditional ELKS1α knockout mice, and exon 3 (E3) in ELKS2α knockout mice to generate double conditional ELKS1α/2α knockout mice (Liu et al., 2014). When crossed with DATIRES-Cre mice (Backman et al., 2006), Cre recombinase removed ELKS1α and ELKS2α proteins in dopamine neurons.
(B) Representative images showing ELKS and TH labeled dopamine axons, and ELKS within dopamine axons in the dorsal striatum of ELKS control and ELKS cKODA mice. The volume and surface rendered images are obtained from 10 × 10 × 2 μm3 image stacks. For each zoom-in image, a 90° rotation around the x-axis is shown below the standard x-y-z image.
(C) Histogram of locally shuffled ELKS cluster densities within dopamine axons across 20% bins of overlap. After shuffling of ELKS control and ELKS cKODA clusters, no difference in cluster density is detected. ELKS control n = 29 regions/4 mice, ELKS cKODA n = 27/4 (p = 0.33 for genotype, p < 0.001 for overlap, and p = 0.99 for interaction; two-way ANOVA).
(D) Quantification of actual and shuffled ELKS clusters with >40% volume overlap with dopamine axons from ELKS control and ELKS cKODA mice. ELKS cluster density is decreased by 45% in ELKS cKODA axons. n as in C.
All data are mean ± SEM. *** p < 0.001; two-way ANOVA for (C), Mann-Whitney rank sum test for (D).
(A) Setup and example traces of the calibration of carbon fiber electrodes (CFE). The CFE was held at 600 mV. Pipettes filled with various concentrations of dopamine (0, 1, 5, 10, 20 μM) were placed one-by-one close to the tip of the CFE, and dopamine was puffed continuously onto the CFE for 5 s.
(B) Quantification of the experiment shown in (A). The current amplitudes were plotted against the dopamine concentration, and the standard curve was generated by linear regression. The CFE exhibits no signs of desensitization during the puff, and the current amplitude has a linear relationship with the dopamine concentration.
(C) Setup of the amperometric recording and sample traces of electrically evoked dopamine release before (black) and after (magenta) the application of nicotinic acetylcholine receptor antagonist DHβE (1 μM) in sagittal striatal slices of wild type mice. Electrical stimulation evoked two phases of dopamine release (arrow indicating the first phase), with the second phase inhibited by DhβE.
(D) Quantification of the experiment shown in (C). n = 6 slices/3 mice.
All data are mean ± SEM. *** p < 0.001; paired t test.
(A) Representative confocal images of sparsely plated striatal synaptosomes expressing synaptophysin-tdTomato (SYP-tdTomato) in dopamine axons stained for synaptobrevin-2 and TH. The filled arrowheads indicate SYP-tdTomato positive synaptosomes (inset 1) containing synaptobrevin-2 and TH. The hollow arrowhead indicates a particle (inset 2) containing TH but not synaptobrevin-2. SYP-tdTomato is restricted to synaptobrevin-2 positive synaptosomes, but is not present in TH positive structures without vesicle marker synaptobevin-2.
(B) Quantification of (A), showing the percentage of SYP-tdTomato positive synaptosomes in TH positive particles that do or do not contain synaptobrevin-2. These data establish that SYP-tdTomato reliably marks vesicles in dopamine terminals. n = 12 areas/3 mice.
(C) Representative images of SYP-tdTomato (green) and TH staining (magenta) in dorsal striatum acquired using 3D-SIM. The example images are volume- or surface-rendered assemblies of a 10 × 10 × 2 μm3 image stack.
(D) Histogram of density of vesicle clusters along dopamine axons in 20% overlap bins. 76.3 ± 1.7 % of SYP-tdTomato clusters have >40% overlap within dopamine axons (rectangle area, n = 28 regions/3 mice).
(E) Confocal images of striatal synaptosomes stained for Munc13, bassoon and TH. Filled arrowheads indicate synaptosomes containing bassoon and TH (insets). Hollow arrowheads indicate particles containing TH but not bassoon.
(F) Quantifications of (E) showing that Munc13 is colocalized with bassoon. n = 10 areas/3 mice.
(G) Representative STED images of Munc13 and bassoon in TH positive synaptosomes showing that Munc13 and bassoon are tightly associated within a small cluster.
(H) Representative confocal images of synaptosomes from RIM control and RIM cKODA mice stained for RIM, Munc13 and Munc18. Dopamine vesicle clusters were labeled by genetic expression of SYP-tdTomato.
(I) Quantifications of (H). Removal of RIM caused 76% decrease in RIM positive dopamine synaptosomes with the remaining signal likely being antibody background (Wang et al., 2016), 26% decrease in Munc13 positive dopamine synaptosomes and no significant change in Munc18 positive dopamine synaptosomes. For RIM and Munc18 quantification, n =15 areas/3 mice in RIM control and RIM cKODA mice. For Munc13 quantification n = 15/3 in RIM control and 18/3 in RIM cKODA mice.
All data are mean ± SEM. *** p < 0.001, **, p<0.01, ns, not significant; Mann-Whitney rank sum test for (B) and (I), Kruskal-Wallis analysis of variance with post hoc Dunn’s test for (F).
(A) Example traces (average of 4 sweeps) of dopamine release during a 10 Hz electrical stimulation train in wild type mice before (black traces) and after the application of a mixture of 5 μM NBQX to block AMPA and kainate receptors, 50 μM D-APV to block NMDA receptors, 5 μM Gabazine to block GABAA receptors, 0.3 μM S-Sulpiride to block D2 receptors, and 0.1 μM CGP55845 to block GABAB receptors (magenta traces, cocktail).
(B) Summary plot of the experiment shown in (A) with responses normalized to the initial response before adding the drug cocktail. Because no significant difference was seen after adding the inhibitor cocktail, we conclude that the fast decay of release is not caused by disynaptic inhibition, or feedback inhibition mediated by D2 or GABAB receptor activation. n = 6 slices/3 mice (p = 0.76 for drug application, p < 0.001 for stimulus number, and p = 0.93 for interaction; two-way ANOVA).
(C) Example traces (average of 4 sweeps) and quantification of light-evoked dopamine release in wild type mice before (black) and after (magenta) the application of nicotinic receptor blocker DhβE (1 μM). DhβE did not reduce optogenetically induced dopamine release. n = 6/2.
(D) Example traces of dopamine transients evoked by paired light stimulation (1 s interval, 1 ms duration, blue bars) at 2 and 4 mM extracellular Ca2+ ([Ca2+]ex) in dorsal striatal slices of RIM control mice and RIM cKODA mice.
(E) Quantification of peak dopamine transient and PPR of (D). RIM control n = 14/5 at 2 mM [Ca2+]ex and 9/4 at 4 mM [Ca2+]ex, RIM cKODA mice n = 13/5 at 2 mM [Ca2+]ex and 9/4 at 4 mM [Ca2+]ex. Removal of RIM caused a dramatic decrease in dopamine release in the dorsal striatum, and significantly increased the paired-pulse ratio (PPR) at both [Ca2+]ex. Responses smaller than 15 pA were not included in PPR analysis.
(F, G) Same as (D, E), but showing results obtained from ventral striatum. RIM control n = 10/3 at 2 mM [Ca2+]ex and 7/3 at 4 mM [Ca2+]ex for amplitude and PPR, RIM cKODA mice n = 9/3 at 2 mM [Ca2+]ex and 7/3 at 4 mM [Ca2+]ex. Removal of RIM caused a dramatic decrease in dopamine release in the ventral striatum, and significantly increased the paired-pulse ratio (PPR) at both [Ca2+]ex.
All data are mean ± SEM. *** p < 0.001, ns, not significant; two-way ANOVA for (B), paired t test for (C), and Kruskal-Wallis analysis of variance with post hoc Dunn’s test for (E, G).
(A) Images of sagittal slices expressing oChiEF-tdTomato from RIM control and RIM cKODA mice. Extracellular recordings were performed by placing a glass pipette on the slice in an area with even expression of oChiEF-tdTomato (square). Light stimulation was applied directly to the recording site.
(B) Examples of extracellular field recordings, with a single sweep in gray, and the average of 100 sweeps in magenta. Blue bar indicates light stimulation.
(C) Representative traces of extracellular recordings (left) before (black) and after (magenta) application of 1 μM TTX. Similar inhibition of the peak amplitude (right) was observed after adding TTX (which abolishes neurotransmitter release, Figure 6C) in RIM control (n = 7 slices/3 mice) and RIM cKODA mice (n = 7/3). Data shown were normalized to the amplitude before TTX application (the absolute amplitude is shown in Figure 6K). The field potentials consist of a TTX sensitive dopamine axon fiber volley and an oChIEF component that generated a voltage sag by passive depolarization caused by activation of oChIEF. These experiments establish that the loss of dopamine release upon knockout of RIM is not caused by strongly impaired action potential initiation or propagation, or large reductions in fiber number or excitability.
(D) Example traces (average of 100 sweeps) of extracellular recordings from RIM control and RIM cKODA mice at 20, 40 and 80 Hz light stimulation (1 ms duration, blue bars).
(E) Quantification of (D). Responses were normalized to the initial response. Action potential firing followed in both groups at 10 Hz (Figure 6K), but failed to follow reliably in RIM control and RIM cKODA mice when the frequency exceeded 20 Hz. The pronounced depression of the amplitude with increasing stimulation frequency is most likely due to an increasing number of fibers that fail to follow the light stimulation. All experiments characterizing dopamine release were done at frequencies of 10 Hz or lower, at which action potential firing reliably follows light stimulation in both groups. RIM control n = 6/3, RIM cKODA n = 8/3.
All data are mean ± SEM, *** p < 0.001, ns, not significant; paired t test and Mann-Whitney rank sum test.
Rotating 3D view of bassoon (green) and TH-labeled dopamine axons (magenta) from a 3D-SIM experiment in a dorsal striatal slice (10 × 10 × 2 μm3, same data as image in Figure 1C) showing volume rendered raw images (1–10 s), surface rendered bassoon clusters and dopamine axons (10–17 s), and bassoon clusters within dopamine axons (17–22 s).
Highlights.
Striatal dopamine axons contain active zone-like sites with bassoon, RIM and ELKS.
RIM is essential for scaffolding and dopamine exocytosis, but ELKS is dispensable.
These mechanistically specialized sites support a high initial release probability.
Only ~30% of dopamine varicosities contain active zone-like sites.
Acknowledgments
We thank L. Bickford for technical support, Dr. W. Regehr, Dr. B. Sabatini, and members of the Kaeser laboratory for comments on the manuscript, Dr. T. Südhof for antibodies, Dr. B. Lehnert and Dr. D. Ginty for providing VGluT1IRES-cre and SYP-tdTomato mice, and Dr. B. Sabatini for providing DATIRES-cre mice. This work was supported by grants from the NIH (R01NS083898 and R01NS103484 to PSK), the Harvard Brain Initiative, the Goldenson Foundation, the Lefler Foundation (to PSK), and by Alice and Joseph E. Brooks, Fix and Gordon postdoctoral fellowships (to CL). We also acknowledge the Neurobiology Imaging Facility (supported by a P30 Core Center Grant NS072030), the Electron Microscopy Facility at Harvard Medical School and the Cellular Imaging Core at Boston Children’s Hospital (BCH IDDRC, 1U54HD090255), and Dr. J. Waters and Dr. T. Lambert from the Cell Biology Microscopy Facility.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Author Contributions
Conceptualization, C.L. and P.S.K.; Methodology, C.L., L.K., S.S. and P.S.K.; Software, C.L.; Formal Analysis, C.L., L.K. and P.S.K.; Investigation, C.L., L.K., S.S. and J.W.; Writing-Original Draft, C.L. and P.S.K.; Supervision, P.S.K.; Funding Acquisition P.S.K. and C.L.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acuna C, Liu X, Gonzalez A, Südhof TC. RIM-BPs Mediate Tight Coupling of Action Potentials to Ca2+-Triggered Neurotransmitter Release. Neuron. 2015;87:1234–1247. doi: 10.1016/j.neuron.2015.08.027. [DOI] [PubMed] [Google Scholar]
- Acuna C, Liu X, Südhof TC. How to Make an Active Zone: Unexpected Universal Functional Redundancy between RIMs and RIM-BPs. Neuron. 2016;91:792–807. doi: 10.1016/j.neuron.2016.07.042. [DOI] [PubMed] [Google Scholar]
- Agnati LF, Zoli M, Strömberg I, Fuxe K. Intercellular communication in the brain: Wiring versus volume transmission. Neuroscience. 1995;69:711–726. doi: 10.1016/0306-4522(95)00308-6. [DOI] [PubMed] [Google Scholar]
- Altrock WD, tom Dieck S, Sokolov M, Meyer AC, Sigler A, Brakebusch C, Fässler R, Richter K, Boeckers TM, Potschka H, et al. Functional inactivation of a fraction of excitatory synapses in mice deficient for the active zone protein bassoon. Neuron. 2003;37:787–800. doi: 10.1016/s0896-6273(03)00088-6. [DOI] [PubMed] [Google Scholar]
- Augustin I, Rosenmund C, Sudhof TC, Brose N. Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature. 1999;400:457–461. doi: 10.1038/22768. [DOI] [PubMed] [Google Scholar]
- Backman CM, Malik N, Zhang Y, Shan L, Grinberg A, Hoffer BJ, Westphal H, Tomac AC. Characterization of a mouse strain expressing Cre recombinase from the 3′ untranslated region of the dopamine transporter locus. Genesis. 2006;44:383–390. doi: 10.1002/dvg.20228. [DOI] [PubMed] [Google Scholar]
- Brichta L, Shin W, Jackson-Lewis V, Blesa J, Yap E-L, Walker Z, Zhang J, Roussarie J-P, Alvarez MJ, Califano A, et al. Identification of neurodegenerative factors using translatome–regulatory network analysis. Nat Neurosci. 2015:18. doi: 10.1038/nn.4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brimblecombe KR, Gracie CJ, Platt NJ, Cragg SJ. Gating of dopamine transmission by calcium and axonal N-, Q-, T- and L-type voltage-gated calcium channels differs between striatal domains. J Physiol. 2015;593:929–946. doi: 10.1113/jphysiol.2014.285890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cailld I, Dumartin B, Bloch B. Ultrastructural localization of D 1 dopamine receptor immunoreactivity in rat striatonigral neurons and its relation with dopaminergic innervation. Brain Res. 1996;730:17–31. doi: 10.1016/0006-8993(96)00424-6. [DOI] [PubMed] [Google Scholar]
- Cragg SJ. Variable dopamine release probability and short-term plasticity between functional domains of the primate striatum. J Neurosci. 2003;23:4378–4385. doi: 10.1523/JNEUROSCI.23-10-04378.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Kaeser PS, Xu W, Sudhof TC. RIM Proteins Activate Vesicle Priming by Reversing Autoinhibitory Homodimerization of Munc13. Neuron. 2011;69:317–331. doi: 10.1016/j.neuron.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descarries L, Watkins KC, Garcia S, Bosler O, Doucet G. Dual character, asynaptic and synaptic, of the dopamine innervation in adult rat neostriatum: A quantitative autoradiographic and immunocytochemical analysis. J Comp Neurol. 1996;375:167–186. doi: 10.1002/(SICI)1096-9861(19961111)375:2<167::AID-CNE1>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Floresco SB, West AR, Ash B, Moore H, Grace AA. Afferent modulation of dopamine neuron firing differentially regulates tonic and phasic dopamine transmission. Nat Neurosci. 2003;6:968–973. doi: 10.1038/nn1103. [DOI] [PubMed] [Google Scholar]
- Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, Georgieva L, Rees E, Palta P, Ruderfer DM, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506:179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantz SC, Bunzow JR, Williams JT. Spontaneous inhibitory synaptic currents mediated by a g protein-coupled receptor. Neuron. 2013;78:807–812. doi: 10.1016/j.neuron.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace AA. Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nat Rev Neurosci. 2016;17:524–532. doi: 10.1038/nrn.2016.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. The Control of Firing Pattern in nigral dopamine Neurons: Burst Firing. J Neurosci. 1984;4:2877–2890. doi: 10.1523/JNEUROSCI.04-11-02877.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson MGL, Shao L, Carlton PM, Wang CJR, Golubovskaya IN, Cande WZ, Agard DA, Sedat JW. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys J. 2008;94:4957–4970. doi: 10.1529/biophysj.107.120345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Kaeser PS, Südhof TC, Schneggenburger R. RIM Determines Ca(2+) Channel Density and Vesicle Docking at the Presynaptic Active Zone. Neuron. 2011;69:304–316. doi: 10.1016/j.neuron.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Held RG, Liu C, Kaeser PS. ELKS controls the pool of readily releasable vesicles at excitatory synapses through its N-terminal coiled-coil domains. Elife. 2016:5. doi: 10.7554/eLife.14862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe MW, Dombeck DA. Rapid signalling in distinct dopaminergic axons during locomotion and reward. Nature. 2016;535:505–510. doi: 10.1038/nature18942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser PS, Deng L, Wang Y, Dulubova I, Liu X, Rizo J, Südhof TC, Sudhof TC. RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ-domain interaction. Cell. 2011;144:282–295. doi: 10.1016/j.cell.2010.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczenski R. Differential effects of reserpine and tetrabenazine on rat striatal synaptosomal dopamine biosynthesis and synaptosomal dopamine pools. J Pharmacol Exp Ther. 1977;201:357–367. [PubMed] [Google Scholar]
- Lin JY, Lin MZ, Steinbach P, Tsien RY. Characterization of engineered channelrhodopsin variants with improved properties and kinetics. Biophys J. 2009;96:1803–1814. doi: 10.1016/j.bpj.2008.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Bickford LS, Held RG, Nyitrai H, Sudhof TC, Kaeser PS. The Active Zone Protein Family ELKS Supports Ca2+ Influx at Nerve Terminals of Inhibitory Hippocampal Neurons. J Neurosci. 2014;34:12289–12303. doi: 10.1523/JNEUROSCI.0999-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcott PF, Mamaligas AA, Ford CP. Phasic Dopamine Release Drives Rapid Activation of Striatal D2-Receptors. Neuron. 2014;84:164–176. doi: 10.1016/j.neuron.2014.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda W, Furuta T, Nakamura KC, Hioki H, Fujiyama F, Arai R, Kaneko T. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J Neurosci. 2009;29:444–453. doi: 10.1523/JNEUROSCI.4029-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missale C, Nash SRS, Robinson SW, Jaber M, Caron MG. Dopamine receptors: from structure to function. Physiol Rev. 1998;78:189–225. doi: 10.1152/physrev.1998.78.1.189. [DOI] [PubMed] [Google Scholar]
- O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira DB, Schmitz Y, Mészáros J, Merchant P, Hu G, Li S, Henke A, Lizardi-Ortiz JE, Karpowicz RJ, Morgenstern TJ, et al. Fluorescent false neurotransmitter reveals functionally silent dopamine vesicle clusters in the striatum. Nat Neurosci. 2016;19:578–586. doi: 10.1038/nn.4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pissadaki EK, Bolam JP. The energy cost of action potential propagation in dopamine neurons: clues to susceptibility in Parkinson’s disease. Front Comput Neurosci. 2013;7:1–17. doi: 10.3389/fncom.2013.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz W. Predictive Reward Signal of Dopamine Neurons. J Neurophysiol. 1998;80:1–27. doi: 10.1152/jn.1998.80.1.1. [DOI] [PubMed] [Google Scholar]
- Stuber GD, Hnasko TS, Britt JP, Edwards RH, Bonci A. Dopaminergic terminals in the nucleus accumbens but not the dorsal striatum corelease glutamate. J Neurosci. 2010;30:8229–8233. doi: 10.1523/JNEUROSCI.1754-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. The Presynaptic Active Zone. Neuron. 2012;75:11–25. doi: 10.1016/j.neuron.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Graves SM, Shen W. Dopaminergic modulation of striatal networks in health and Parkinson’s disease. Curr Opin Neurobiol. 2014;29:109–117. doi: 10.1016/j.conb.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threlfell S, Lalic T, Platt NJ, Jennings Ka, Deisseroth K, Cragg SJ. Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron. 2012;75:58–64. doi: 10.1016/j.neuron.2012.04.038. [DOI] [PubMed] [Google Scholar]
- Tritsch NX, Ding JB, Sabatini BL. Dopaminergic neurons inhibit striatal output through non-canonical release of GABA. Nature. 2012;490:262–266. doi: 10.1038/nature11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchigashima M, Ohtsuka T, Kobayashi K, Watanabe M. Dopamine synapse is a neuroligin-2-mediated contact between dopaminergic presynaptic and GABAergic postsynaptic structures. Proc Natl Acad Sci U S A. 2016;113:201514074. doi: 10.1073/pnas.1514074113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SSH, Held RG, Wong MY, Liu C, Karakhanyan A, Kaeser PS. Fusion Competent Synaptic Vesicles Persist upon Active Zone Disruption and Loss of Vesicle Docking. Neuron. 2016;91:777–791. doi: 10.1016/j.neuron.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagishita S, Hayashi-Takagi A, Ellis-Davies GCR, Urakubo H, Ishii S, Kasai H. A critical time window for dopamine actions on the structural plasticity of dendritic spines. Science. 2014;345:1616–1620. doi: 10.1126/science.1255514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung KK, Bolam JP, Smith AD, Hersch SM, Ciliax BJ, Levey AI. Immunocytochemical localization of D1 and D2 dopamine receptors in the basal ganglia of the rat: light and electron microscopy. Neuroscience. 1995;65:709–730. doi: 10.1016/0306-4522(94)00536-e. [DOI] [PubMed] [Google Scholar]
- Zhang S, Qi J, Li X, Wang HL, Britt JP, Hoffman AF, Bonci A, Lupica CR, Morales M. Dopaminergic and glutamatergic microdomains in a subset of rodent mesoaccumbens axons. Nat Neurosci. 2015;18:386–392. doi: 10.1038/nn.3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FM, Liang Y, Dani JA. Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat Neurosci. 2001;4:1224–1229. doi: 10.1038/nn769. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Histogram of actual and locally shuffled bassoon cluster densities within dopamine axons across 20% bins of overlap. n = 35 regions/4 mice. Each region contains 6000–11,000 bassoon clusters. Rectangle area indicates bassoon clusters that have more than 40% of volume overlapped with TH and are considered to be within dopamine axons. The 40%–100% range was selected based on the observations that proteins expressed specifically in dopamine axons overlapped with TH mainly within this range (Figure S5D), and that the density of RIM and ELKS clusters within dopamine axons dropped rapidly in this range when removed genetically (Figures 2B and 2D).
(B) Schematic of striatal synaptosome preparation.
(C) Western blotting of the fractions obtained during a synaptosome preparation. The synaptosome layer is enriched with vesicle markers (synaptophysin-1 and synaptobrevin-2) and active zone markers (RIM and ELKS).
(D) Representative images of bassoon and synaptobrevin-2 staining in a synaptosome preparation taken by confocal (top) and STED (bottom) microscopy.
(E) Representative image of electron microscopic analysis of the synaptosome layer reveals enrichment of membrane enclosed structures filled with synaptic vesicles.
Data in A are mean ± SEM.
(A) Schematic of the conditional RIM knockout in dopamine neurons (RIM cKODA). Exons 6 (E6) or 26 (E26) were flanked by loxP sites in the conditional RIM1 and RIM2 knockout mice, respectively, to generate double conditional RIM1/2 knockout mice (Kaeser et al., 2011). When crossed with DATIRES-Cre mice (Backman et al., 2006), Cre recombinase removed RIM1 and RIM2 proteins in dopamine neurons.
(B) Representative images showing RIM and TH labeled dopamine axons, and RIM within dopamine axons in the dorsal striatum of RIM control and RIM cKODA mice. The volume and surface rendered images were obtained from 10 × 10 × 2 μm3 image stacks. For each zoom-in image, a 90° rotation around the x-axis is shown below the standard x-y-z image.
(C) Histogram of locally shuffled RIM cluster densities within dopamine axons across 20% bins of overlap. After shuffling of RIM control and RIM cKODA clusters, no difference in cluster density is detected. RIM control n = 24 regions/4 mice, RIM cKODA n = 22/4 (p = 0.83 for genotype, p < 0.001 for overlap, and p = 0.63 for interaction; two-way ANOVA).
(D) Quantification of actual and shuffled RIM clusters with >40% volume overlap with dopamine axons from RIM control and RIM cKODA mice. RIM cluster density is decreased by 49% in RIM cKODA axons. n as in C.
All data are mean ± SEM. *** p < 0.001, ns, not significant; two-way ANOVA for (C), Mann-Whitney rank sum test for (D).
(A) Schematic of the conditional ELKS knockout in dopamine neurons. Exons 2 (E2) and 3 (E3) were flanked by loxP sites in the conditional ELKS1α knockout mice, and exon 3 (E3) in ELKS2α knockout mice to generate double conditional ELKS1α/2α knockout mice (Liu et al., 2014). When crossed with DATIRES-Cre mice (Backman et al., 2006), Cre recombinase removed ELKS1α and ELKS2α proteins in dopamine neurons.
(B) Representative images showing ELKS and TH labeled dopamine axons, and ELKS within dopamine axons in the dorsal striatum of ELKS control and ELKS cKODA mice. The volume and surface rendered images are obtained from 10 × 10 × 2 μm3 image stacks. For each zoom-in image, a 90° rotation around the x-axis is shown below the standard x-y-z image.
(C) Histogram of locally shuffled ELKS cluster densities within dopamine axons across 20% bins of overlap. After shuffling of ELKS control and ELKS cKODA clusters, no difference in cluster density is detected. ELKS control n = 29 regions/4 mice, ELKS cKODA n = 27/4 (p = 0.33 for genotype, p < 0.001 for overlap, and p = 0.99 for interaction; two-way ANOVA).
(D) Quantification of actual and shuffled ELKS clusters with >40% volume overlap with dopamine axons from ELKS control and ELKS cKODA mice. ELKS cluster density is decreased by 45% in ELKS cKODA axons. n as in C.
All data are mean ± SEM. *** p < 0.001; two-way ANOVA for (C), Mann-Whitney rank sum test for (D).
(A) Setup and example traces of the calibration of carbon fiber electrodes (CFE). The CFE was held at 600 mV. Pipettes filled with various concentrations of dopamine (0, 1, 5, 10, 20 μM) were placed one-by-one close to the tip of the CFE, and dopamine was puffed continuously onto the CFE for 5 s.
(B) Quantification of the experiment shown in (A). The current amplitudes were plotted against the dopamine concentration, and the standard curve was generated by linear regression. The CFE exhibits no signs of desensitization during the puff, and the current amplitude has a linear relationship with the dopamine concentration.
(C) Setup of the amperometric recording and sample traces of electrically evoked dopamine release before (black) and after (magenta) the application of nicotinic acetylcholine receptor antagonist DHβE (1 μM) in sagittal striatal slices of wild type mice. Electrical stimulation evoked two phases of dopamine release (arrow indicating the first phase), with the second phase inhibited by DhβE.
(D) Quantification of the experiment shown in (C). n = 6 slices/3 mice.
All data are mean ± SEM. *** p < 0.001; paired t test.
(A) Representative confocal images of sparsely plated striatal synaptosomes expressing synaptophysin-tdTomato (SYP-tdTomato) in dopamine axons stained for synaptobrevin-2 and TH. The filled arrowheads indicate SYP-tdTomato positive synaptosomes (inset 1) containing synaptobrevin-2 and TH. The hollow arrowhead indicates a particle (inset 2) containing TH but not synaptobrevin-2. SYP-tdTomato is restricted to synaptobrevin-2 positive synaptosomes, but is not present in TH positive structures without vesicle marker synaptobevin-2.
(B) Quantification of (A), showing the percentage of SYP-tdTomato positive synaptosomes in TH positive particles that do or do not contain synaptobrevin-2. These data establish that SYP-tdTomato reliably marks vesicles in dopamine terminals. n = 12 areas/3 mice.
(C) Representative images of SYP-tdTomato (green) and TH staining (magenta) in dorsal striatum acquired using 3D-SIM. The example images are volume- or surface-rendered assemblies of a 10 × 10 × 2 μm3 image stack.
(D) Histogram of density of vesicle clusters along dopamine axons in 20% overlap bins. 76.3 ± 1.7 % of SYP-tdTomato clusters have >40% overlap within dopamine axons (rectangle area, n = 28 regions/3 mice).
(E) Confocal images of striatal synaptosomes stained for Munc13, bassoon and TH. Filled arrowheads indicate synaptosomes containing bassoon and TH (insets). Hollow arrowheads indicate particles containing TH but not bassoon.
(F) Quantifications of (E) showing that Munc13 is colocalized with bassoon. n = 10 areas/3 mice.
(G) Representative STED images of Munc13 and bassoon in TH positive synaptosomes showing that Munc13 and bassoon are tightly associated within a small cluster.
(H) Representative confocal images of synaptosomes from RIM control and RIM cKODA mice stained for RIM, Munc13 and Munc18. Dopamine vesicle clusters were labeled by genetic expression of SYP-tdTomato.
(I) Quantifications of (H). Removal of RIM caused 76% decrease in RIM positive dopamine synaptosomes with the remaining signal likely being antibody background (Wang et al., 2016), 26% decrease in Munc13 positive dopamine synaptosomes and no significant change in Munc18 positive dopamine synaptosomes. For RIM and Munc18 quantification, n =15 areas/3 mice in RIM control and RIM cKODA mice. For Munc13 quantification n = 15/3 in RIM control and 18/3 in RIM cKODA mice.
All data are mean ± SEM. *** p < 0.001, **, p<0.01, ns, not significant; Mann-Whitney rank sum test for (B) and (I), Kruskal-Wallis analysis of variance with post hoc Dunn’s test for (F).
(A) Example traces (average of 4 sweeps) of dopamine release during a 10 Hz electrical stimulation train in wild type mice before (black traces) and after the application of a mixture of 5 μM NBQX to block AMPA and kainate receptors, 50 μM D-APV to block NMDA receptors, 5 μM Gabazine to block GABAA receptors, 0.3 μM S-Sulpiride to block D2 receptors, and 0.1 μM CGP55845 to block GABAB receptors (magenta traces, cocktail).
(B) Summary plot of the experiment shown in (A) with responses normalized to the initial response before adding the drug cocktail. Because no significant difference was seen after adding the inhibitor cocktail, we conclude that the fast decay of release is not caused by disynaptic inhibition, or feedback inhibition mediated by D2 or GABAB receptor activation. n = 6 slices/3 mice (p = 0.76 for drug application, p < 0.001 for stimulus number, and p = 0.93 for interaction; two-way ANOVA).
(C) Example traces (average of 4 sweeps) and quantification of light-evoked dopamine release in wild type mice before (black) and after (magenta) the application of nicotinic receptor blocker DhβE (1 μM). DhβE did not reduce optogenetically induced dopamine release. n = 6/2.
(D) Example traces of dopamine transients evoked by paired light stimulation (1 s interval, 1 ms duration, blue bars) at 2 and 4 mM extracellular Ca2+ ([Ca2+]ex) in dorsal striatal slices of RIM control mice and RIM cKODA mice.
(E) Quantification of peak dopamine transient and PPR of (D). RIM control n = 14/5 at 2 mM [Ca2+]ex and 9/4 at 4 mM [Ca2+]ex, RIM cKODA mice n = 13/5 at 2 mM [Ca2+]ex and 9/4 at 4 mM [Ca2+]ex. Removal of RIM caused a dramatic decrease in dopamine release in the dorsal striatum, and significantly increased the paired-pulse ratio (PPR) at both [Ca2+]ex. Responses smaller than 15 pA were not included in PPR analysis.
(F, G) Same as (D, E), but showing results obtained from ventral striatum. RIM control n = 10/3 at 2 mM [Ca2+]ex and 7/3 at 4 mM [Ca2+]ex for amplitude and PPR, RIM cKODA mice n = 9/3 at 2 mM [Ca2+]ex and 7/3 at 4 mM [Ca2+]ex. Removal of RIM caused a dramatic decrease in dopamine release in the ventral striatum, and significantly increased the paired-pulse ratio (PPR) at both [Ca2+]ex.
All data are mean ± SEM. *** p < 0.001, ns, not significant; two-way ANOVA for (B), paired t test for (C), and Kruskal-Wallis analysis of variance with post hoc Dunn’s test for (E, G).
(A) Images of sagittal slices expressing oChiEF-tdTomato from RIM control and RIM cKODA mice. Extracellular recordings were performed by placing a glass pipette on the slice in an area with even expression of oChiEF-tdTomato (square). Light stimulation was applied directly to the recording site.
(B) Examples of extracellular field recordings, with a single sweep in gray, and the average of 100 sweeps in magenta. Blue bar indicates light stimulation.
(C) Representative traces of extracellular recordings (left) before (black) and after (magenta) application of 1 μM TTX. Similar inhibition of the peak amplitude (right) was observed after adding TTX (which abolishes neurotransmitter release, Figure 6C) in RIM control (n = 7 slices/3 mice) and RIM cKODA mice (n = 7/3). Data shown were normalized to the amplitude before TTX application (the absolute amplitude is shown in Figure 6K). The field potentials consist of a TTX sensitive dopamine axon fiber volley and an oChIEF component that generated a voltage sag by passive depolarization caused by activation of oChIEF. These experiments establish that the loss of dopamine release upon knockout of RIM is not caused by strongly impaired action potential initiation or propagation, or large reductions in fiber number or excitability.
(D) Example traces (average of 100 sweeps) of extracellular recordings from RIM control and RIM cKODA mice at 20, 40 and 80 Hz light stimulation (1 ms duration, blue bars).
(E) Quantification of (D). Responses were normalized to the initial response. Action potential firing followed in both groups at 10 Hz (Figure 6K), but failed to follow reliably in RIM control and RIM cKODA mice when the frequency exceeded 20 Hz. The pronounced depression of the amplitude with increasing stimulation frequency is most likely due to an increasing number of fibers that fail to follow the light stimulation. All experiments characterizing dopamine release were done at frequencies of 10 Hz or lower, at which action potential firing reliably follows light stimulation in both groups. RIM control n = 6/3, RIM cKODA n = 8/3.
All data are mean ± SEM, *** p < 0.001, ns, not significant; paired t test and Mann-Whitney rank sum test.
Rotating 3D view of bassoon (green) and TH-labeled dopamine axons (magenta) from a 3D-SIM experiment in a dorsal striatal slice (10 × 10 × 2 μm3, same data as image in Figure 1C) showing volume rendered raw images (1–10 s), surface rendered bassoon clusters and dopamine axons (10–17 s), and bassoon clusters within dopamine axons (17–22 s).