

Abstract
The metallo-β-lactamases (MBLs) are a growing threat to the continued efficacy of β-lactam antibiotics. Recently, aspergillomarasmine A (AMA) was identified as an MBL inhibitor, but the mode of inhibition was not fully characterized. Equilibrium dialysis and metal analysis studies revealed that 2 equivalents, eq, of AMA effectively removes 1 eq of Zn(II) from MBLs NDM-1, VIM-2, and IMP-7 when the MBL is at micromolar concentrations. Conversely, 1H NMR studies revealed that 2 eq of AMA remove 2 eq of Co(II) from Co(II)-substituted NDM-1, VIM-2, and IMP-7 when the MBL/AMA are at millimolar concentrations. Our findings reveal that AMA inhibits the MBLs by removal of the active site metal ions required for β-lactam hydrolysis among the most clinically significant MBLs.
Keywords: Aspergillomarasmine A, Metallo-β-lactamase, NDM-1, VIM-2, IMP-7, antibiotic resistance
Graphical Abstract
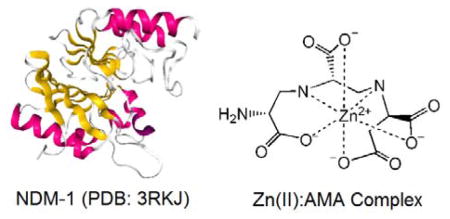
β-Lactams are the most important class of antibiotics used in the fight against infectious disease. Regrettably, their continued efficacy against pathogenic bacteria is threatened by the rapid spread of resistance mechanisms.1–5 Furthermore, due to the tremendous challenge that identifying novel antibiotics poses, the rate at which they have been discovered has slowed substantially in recent decades.6 The most common mechanism of resistance in Gram-negative bacteria is the expression of β-lactamases, which inactivate these antibiotics by hydrolyzing the invariant β-lactam bond.7 Therefore, much effort has been dedicated to the discovery of β-lactamase inhibitors, which are given in combination with an existing β-lactam to restore efficacy of current generation β-lactam antibiotics. This strategy has proven highly successful in the clinic to combat β-lactamases that hydrolyze β-lactams via an active site Ser residue as a catalytic nucleophile.1,8 Four such Ser β-lactamase inhibitors are in current clinical use, and several others are in human clinical trials.9
Metallo-β-lactamases (MBLs) are a class of β-lactamases, which pose a serious threat to public health, and currently, clinical inhibitors for the MBLs are not commercially available.8–10 MBLs have an unusually broad substrate specificity and are capable of inactivating all types of β-lactam antibiotics, with the exception of monobactams, and bind either one or two Zn(II) ions in their active sites.11 The three most concerning clinically observed MBLs are the class B1 enzymes, New Delhi MBL (NDM), Verona Integron-encoded MBL (VIM), and IMiPenemase (IMP) type enzymes.12 These three MBLs, of which there exist many variants, have become especially problematic because of the acquisition of their plasmids by other pathogenic bacteria through horizontal gene transfer.13–16 To further complicate this issue, often times the bla containing plasmids also harbor resistance genes for several other antimicrobials resulting in multidrug-resistant pathogenic strains.15
Recently, a fungal natural product, aspergillomarasmine A (AMA) (Figure 1), was reported to be a potent inhibitor of NDM-1 and VIM-2 (IC50 = 4.0 and 9.6 μM, respectively), and to a lesser extent, IMP-7 and was found to re-sensitize resistant Gram-negative bacteria to carbapenems.17 Inductively-coupled plasma mass spectrometry demonstrated that AMA removed one Zn(II) ion from NDM-1 and that enzyme activity could be restored in vitro by supplementation with excess Zn(II).17 These results were consistent with a Zn(II)-removal mechanism of action; however, a detailed study of the mode of MBL inhibition by AMA was not performed with NDM-1 and other MBLs. Data are still needed on the inhibition mechanism of MBLs other than NDM-1. It is also unclear if AMA acts by metal ion sequestration after the metal ion releases from the active site or through active participation in metal removal with any of the MBLs. Addressing these fundamental questions may help explain the microbiological differences reported between bacterial isolates expressing different MBLs, as well as the apparent selectivity observed for NDM-1 and VIM-2.
Figure 1.
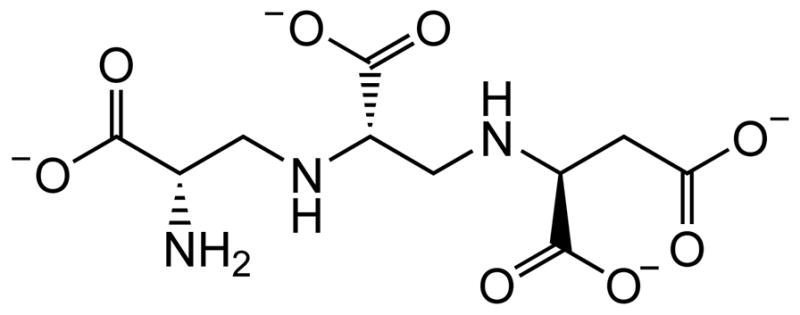
Structure of AMA at pH 7.
Most MBL inhibitors fall into a small subset of classes, one of which are the metal chelators such as ethylenediaminetetraacetic acid (EDTA) and 2,6-dipicolinic acid (DPA).18,19 These chelators are thought to inhibit the catalytic activity of the MBLs via active removal or sequestration of the active site metal ions.20,21 The clinical use of chelator-based inhibitors raises concerns with non-selective metal removal towards other important physiological metalloproteins.22 Another class of MBL inhibitors are the sulfhydryl-containing compounds such as D- and L-captopril.23 L-Captopril is a clinically approved angiotensin-converting enzyme (ACE) inhibitor that is prescribed to treat hypertension. This class of inhibitors has been shown to bind to the MBLs by forming a μ-bridging species between the two active site Zn(II) ions via the thiolate anion, displacing the bridging hydroxide anion native to class B1 MBLs.23,24
Herein, we report a detailed investigation into the mode of inhibition of AMA on three of the most clinically significant types of MBLs: NDM-1, VIM-2, and IMP-7. Using equilibrium dialysis, followed by metal analysis with inductively coupled plasma atomic emission spectroscopy (ICP-AES), the effects of AMA on metal content are shown. We also report a technique to probe the inhibition mechanism using paramagnetic 1H NMR on the diCo(II)-substituted analogs of each enzyme. While diCo(II)-substituted analogs of NDM-1, VIM-2, and IMP-1 were previously described,16,25,26 paramagnetic 1H NMR data of CoCo-VIM-2 and CoCo-IMP-1 are not reported. Here, we describe a new method for Co(II)-substitution of IMP-7, wherein the enzyme has sufficient stability to afford high quality 1H NMR data, providing critical spectroscopic access to this important group of enzymes.
RESULTS AND DISCUSSION
Generation of diCo(II)-substituted MBLs
To elucidate the mode of inhibition of AMA and the MBLs, NDM-1, VIM-2, and IMP-7 were over-expressed and purified. To probe the mechanism of MBL inhibition by AMA, 1H NMR was one technique chosen to probe the two active site metal ions. Because Zn(II) has a full d orbital with no unpaired electrons, very few spectroscopic studies can be performed on the Zn(II)-metalloforms of the MBLs. Therefore, diCo(II)-substituted analogs of NDM-1, VIM-2, and IMP-7 were generated. Co(II)-substitution is a technique that has been employed to probe the structure and function of the MBLs, as well as many other Zn(II)-metalloproteins, and results in catalytic activity that is similar to that of the native state.11,16,25,27–32 Co(II)-substituted analogs of NDM-1, VIM-2, and IMP-1 have been previously reported;16,25,26 however, the IMP-1 analog suffered from poor stability at room temperature and a 1H NMR spectrum could not be attained. Therefore, we chose to explore a different IMP variant (IMP-7) to probe this type of MBL.
Significant challenges arose in generating a Co(II)-substituted analog of IMP-7. The enzyme was over-expressed and purified with a His6-tag as described. It was found that the His-tagged protein exhibited very poor solubility at pH 7.5 with salt concentrations as high as 0.5 M. The enzyme was also particularly prone to precipitation when imidazole, used to elute the protein from the Ni-affinity column, was dialyzed out of solution. This result, likely also coupled with the challenge in quantifying the enzyme with UV-Vis, made assessing catalytic activity and metal analysis with ICP-AES unreliable. To address this, thrombin was used to cleave the His-tag from IMP-7, resulting in a protein with much higher stability and no evidence of precipitation.
Co(II)-substitution was then attempted using the direct addition method (Method A). In this procedure, the apo-enzyme was prepared by stripping the Zn(II) ions from the active site using EDTA. Then, CoCl2 or ZnCl2 were added, and kinetic measurements were taken using saturating amounts of Chromacef (30 μM) (Figure 2). It was found that the apo-enzyme exhibited only ~1.5% of the activity of His-free native Zn(II)-IMP-7, and its incubation with 5 – 100 μM Zn(II) or Co(II) did not restore significant catalytic activity. This result suggests that removal of the Zn(II) ions from the active site with EDTA effectively inactivates the protein and may cause a conformational change, prohibiting the metal ions from re-binding to the active site. This property was not previously observed with the IMP-1 variant, as Co(II)-substitution was achieved using the direct addition method.26 To determine if catalytic activity could be restored, the same inactivated protein preparation was unfolded and refolded in the presence of Zn(II) or Co(II) using Method B (using EDTA or 1,10-phenanthroline), and activity was reexamined (Figure 3). It was found that refolded Zn(II)-IMP-7 had activity near 90% of the untreated form, suggesting that this refolding method was successful in returning the enzyme to its native confirmation with bound Zn(II) ions. The slight reduction in activity is likely a result of the enzyme being exposed to the extensive dialysis steps in Methods A and B. The refolded Co(II)-analog also exhibited catalytic activity, but to a lesser extent than the Zn(II)-metalloform with 30% activity. This result suggests that the Co(II)-analog also adopts a native confirmation, but is not able to hydrolyze Chromacef at the same rate as the native Zn(II)-metalloform.
Figure 2.
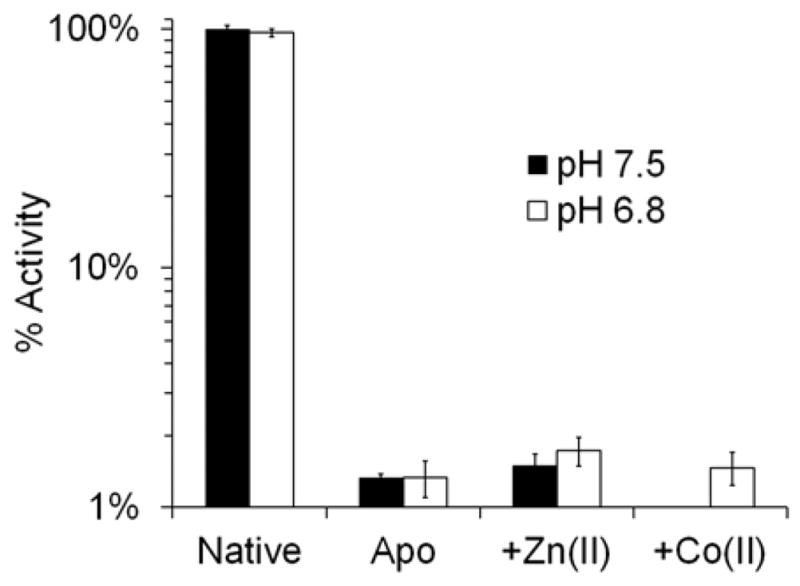
Effects of metal removal and subsequent metal addition on enzymatic activity of IMP-7 at pH 6.8 and 7.5. The native Zn(II)-enzyme was chelated with EDTA using Method A resulting in the apo-enzyme. Native Zn(II)-IMP-7 and apo-IMP-7 (10 nM) were assayed with saturating concentrations of Chromacef (30 μM) in order to measure the rate of catalytic hydrolysis. The apo-enzyme had negligible catalytic activity (1.5 %) and the addition of ZnCl2 or CoCl2 (5 μM) was unsuccessful in restoring activity. The inability to restore catalytic activity suggests that removal of the active site Zn(II) ions with EDTA causes structural changes in the active site that prevents metal ions from binding upon subsequent addition.
Figure 3.
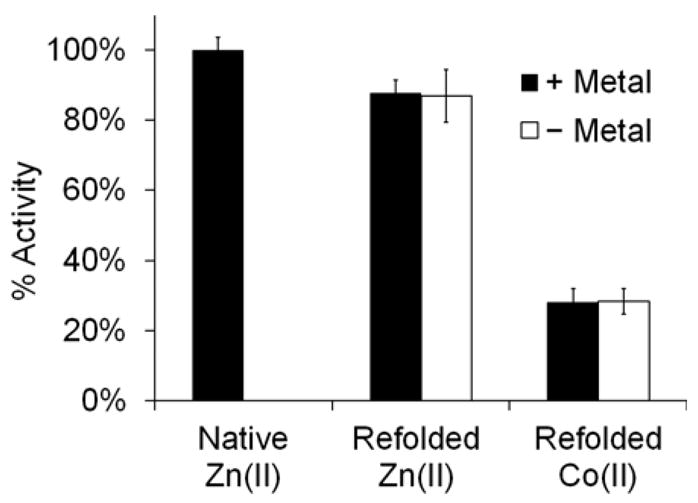
Effects of refolding apo-IMP-7 around Zn(II) or Co(II) ions on catalytic hydrolysis with (5 μM) and without added metal ions in the assay buffer. Zn(II) and Co(II)-metalloforms of IMP-7 were generated using Method B which includes denaturation of the apo-enzyme with 6 M urea and refolding it around Zn(II) or Co(II) in Buffer D. IMP-7 (10 nM) activity was assayed, as in Figure 2, with saturating concentrations of Chromacef (30 μM) and indicates that refolding IMP-7 around Zn(II) results in nearly complete restoration of catalytic activity compared to the native enzyme (90 %). The Co(II)-analog also exhibits catalytic competency, but with a reduced relative activity of 30 %.
Equilibrium dialysis and ICP-AES
Each MBL, in its native Zn(II)-metalloform, was diluted to 50 μM and introduced to an initial dialysis step to remove unbound and loosely-bound metal ions. ICP-AES was then used to measure the metal content of NDM-1, VIM-2, and IMP-7 and was found to be 1.9, 2.0, and 1.8 molar equivalents respectively. Each enzyme was then incubated with varying concentrations of AMA (0, 100, 250, and 500 μM). All samples were then dialyzed for several hours, allowing AMA and zinc ions to reach equilibrium within the total volume of the dialysis buffer. The resulting samples were then diluted (2 μM), and ICP-AES was used to determine metal content. These data show that as the concentration of AMA increased, the zinc content of NDM-1, VIM-2, and IMP-7 decreased (Figure 4). While all three enzymes bind two zinc ions in their active sites, we found that the samples without AMA treatment contained ~1.3–1.4 molar equivalents of zinc. Native mass spectrometry (native MS) was used to probe the metal binding states of each MBL that had not been treated with AMA. Spectra were first collected under denaturing conditions to attain the total mass of the apo-enzymes. Native MS was then used to analyze the equilibrium dialysis samples for the presence of various metal binding states (apo, monoZn(II), diZn(II))(Figure S6–S11). The results suggest that the diZn(II)-metalloform is the predominant species for NDM-1, but that as much as 30% of the sample consists of the monoZn(II)-metalloform. However for the VIM-2 and IMP-7 samples, only the diZn(II)-metalloforms were detected. Apo-enzyme was not detected in any of the native mass spectra obtained for any of the three proteins. Comparing the native MS data to the ICP-AES analysis on the MBLs without AMA treatment, there appears to be some discrepancy. The native MS results suggest that analysis by ICP-AES should have revealed a metal to protein stoichiometry close to 2 molar equivalents of zinc. The reason for this discrepancy is unclear, but variation exists in the literature regarding metal analysis of MBLs. In an experiment very similar to our equilibrium dialysis studies, authors reported a stoichiometry of 2.7 molar equivalents of zinc for NDM-1.17 In another study, authors reported that NDM-1 binds 1.5 molar equivalents of zinc.33 Furthermore, as-isolated, NDM-1 has been reported to bind 1.0 molar equivalents of zinc.16,34
Figure 4.
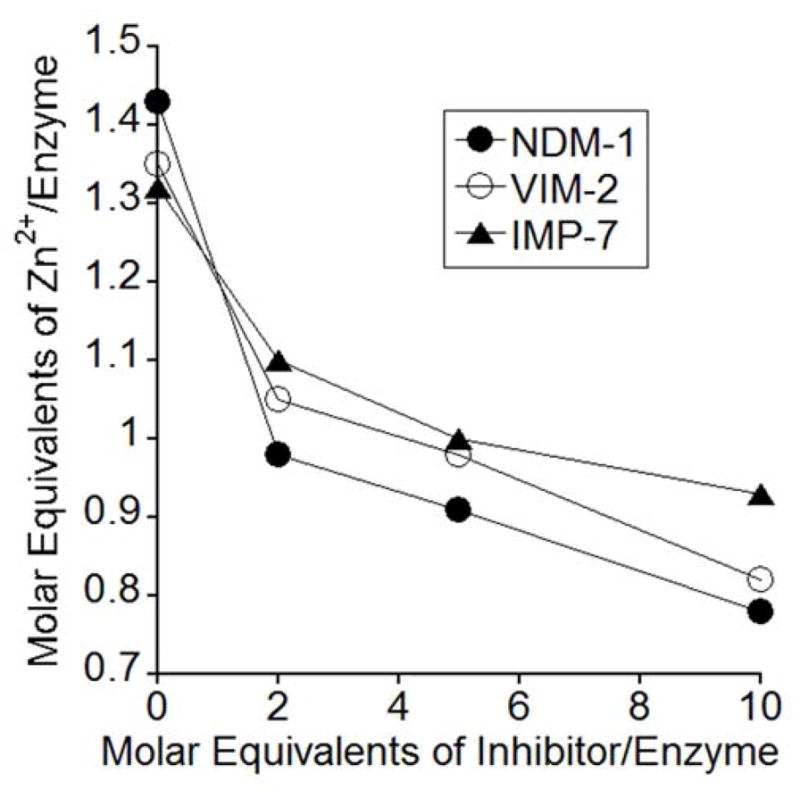
Effects of AMA on Zn(II) content of NDM-1, VIM-2, and IMP-7. Each enzyme (50 μM) was incubated with 0, 100, 250, and 500 μM AMA, dialyzed, then diluted (2 μM). ICP-AES was used to measure metal content of the resulting samples. AMA caused the Zn(II) concentration of all three proteins to decrease, indicating it acts to abstract Zn(II) from the enzyme active site.
B1 MBLs can exhibit variable affinity for zinc between their Zn1 and Zn2 sites.34–36 The position of a positively-charged residue, specifically Lys125 in the case of NDM-1, near the Zn2 site has been shown to be a contributing factor in reducing the zinc binding affinity at this location.34–36 Therefore, it is likely that the reduced zinc content of the untreated MBLs in the equilibrium dialysis experiments is primarily due to Zn(II) being removed from the Zn2 site. Likewise, when AMA is added to the MBLs, there is an initial drop in zinc content to ~1.0 – 1.1 molar equivalents when 2 molar equivalents of AMA are added. As the concentration of AMA increases, the effect on zinc content becomes less pronounced whereat 10 equivalents of AMA, the final content is 0.8 to 0.9 equivalents.
This equilibrium dialysis experiment was also carried out using EDTA as well as L-captopril (Figure S1). The results are as expected, with L-captopril, a known competitive inhibitor, not showing any evidence of zinc ion removal with the MBLs. Conversely, EDTA, a potent metal chelator with high zinc affinity,37 exhibits a dramatic reduction in zinc content in the same experiment. These data also show that EDTA exhibits more effective metal removal than AMA. Overall, these results indicate that AMA acts by removing or sequestering zinc ions from the active site of each MBL, with a likely preference for the metal ion in the Zn2 site.
The equilibrium dialysis experiment was also performed on the Co(II)-metalloforms of NDM-1, VIM-2, and IMP-7, but the data suggest that the active site Co(II) ions readily become oxidized to Co(III) throughout the dialysis steps required for this experiment.38 Low cobalt concentrations were observed once measured by ICP-AES, and the concentrations did not appear to be affected by AMA concentration. The problem of Co(II) to Co(III) oxidation has been reported for other Co(II)-substituted metalloproteins.39–41 While Co(III) is exchange-inert, having a low ligand-exchange rate,42 it is most stable in an octahedral configuration.43 The tetrahedral Zn1 site of the B1 MBLs may exhibit rapid dissociation upon oxidation to Co(III) as has been reported for a Co(III)-carboxypeptidase analog.40 It is less clear how Co(III) in the Zn2 site would behave as the native Zn(II)-metalloform is 5-coordinate, but in Co(II)-analogs of B1 MBLs, it has been shown that this site can adopt a 6-coordinate arrangement.16,44,45 This information, as well as the low ligand-exchange rate of Co(III), may explain why the equilibrium dialysis experiment was unsuccessful.
Kinetic measurements of post-equilibrium dialysis fractions
To further explore the effects of AMA on the MBLs, kinetic measurements were taken of the post-equilibrium dialysis fractions. The 10 molar equivalent AMA samples were assayed with saturating concentrations of Chromacef (30 μM), and compared to the untreated enzymes, to identify differences in the effect of AMA on the three enzymes, as well as to try to determine if metal removal is selective between the Zn1 and Zn2 sites. Importantly, we note that the samples treated with 10 molar equivalents of AMA are not expected to have significant concentrations of AMA as they were post-dialysis samples. Compared to the untreated samples, the only expected difference is the reduced metal content as observed in Figure 4. The results show that the activity of NDM-1, VIM-2, and IMP-7 drops to 38, 22, and 13%, respectively (Figure 5). Addition of 10 μM ZnCl2 to the treated samples restored some activity with NDM-1 (61%), but had a negligible effect on VIM-2 and IMP-7. The results of the Zn(II) experiment align with our expectations. EDTA-treated NDM-1 can be reactivated by simple addition of Zn(II).16 Conversely, when VIM-2 and IMP-7 are treated with EDTA, reintroduction of metal ions fails to restore activity, suggesting that VIM-2 and IMP-7 partially unfold during the dialysis steps upon metal removal. This characteristic explains why VIM-2 and IMP-7 cannot be Co(II)-substituted using the direct addition method.25 Unlike VIM-2 and IMP-7, NDM-1 does not partially unfold and can be re-activated upon addition of metal. While Chelex-treated buffer was used for all kinetic measurements, the buffers still contained very low Zn(II) concentrations (10–100 nM).46 NDM-1 is likely to bind some of the remaining Zn(II) in the buffer as the kinetic measurements were taken at a very low concentration of enzyme (20 nM).
Figure 5.

Effects of AMA and equilibrium dialysis on enzyme kinetics. After equilibrium dialysis (Figure 4), the NDM-1, VIM-2, and IMP-7 samples, treated with 10 molar equivalents of AMA, were assayed and compared to the untreated enzymes to further probe the mechanism of action of AMA. NDM-1 (20 nM), VIM-2 (20 nM), and IMP-7 (10 nM) were assayed with saturating concentrations of Chromacef (30 μM). The 10 eq samples were run with and without excess Zn(II) (10 μM) to explore its effect on restoring activity. As expected, all samples that had been treated with AMA showed significantly reduced activity. Introduction of Zn(II) to these samples resulted in a significant increase in activity with NDM-1, but had a negligible effect on VIM-2 and IMP-7.
VIM-2 and IMP-1 were previously shown to exhibit positively cooperative metal binding, wherein the binding of metal to the Zn1 site promotes binding of the Zn2 site.25,26 Samples containing 1 molar equivalent of Zn(II) per enzyme were shown to result in ~50 % activity compared to samples containing 2 molar equivalents.25,26 The kinetics of the samples containing 1 molar equivalent of Zn(II) suggest that half of the protein is fully-loaded with 2 metal ions and half exists in the metal-free form. Conversely, NDM-1 exhibits sequential metal binding, wherein metal preferentially binds the Zn1 site before binding the Zn2 site.16 In the mononuclear form, each enzyme is expected to have significantly reduced activity. The diminished kinetic activity of samples treated with 10 molar equivalents of AMA, each containing 0.8 to 0.9 molar equivalents of Zn(II), suggests that AMA removes at least one metal from each enzyme, rather than removing both metal ions from half of the enzyme. Again, due to differences in metal binding affinity between the Zn1 and Zn2 sites, Zn(II) is likely removed from the Zn2 site preferentially and only at high concentrations of inhibitor will the Zn1 site start to become removed or sequestered. These data also indicate that AMA has a lower zinc affinity for Zn(II) than the Zn1 site of the MBLs.
1H NMR Spectroscopy of diCo(II)-Substituted NDM-1, VIM-2, and IMP-7
To further investigate the inhibition mechanism of AMA, we used paramagnetic 1H NMR spectroscopy. The aim was to detect if a long-lived ternary complex exists between AMA, metal ions, and enzyme, or if AMA acted exclusively through active site metal ion sequestration after the metal ion was removed from the active site. Evidence of an intermediate ternary complex would suggest active participation of AMA in metal removal. We also hoped to be able to determine if AMA was abstracting metal from the Zn1 site, Zn2 site, or both. EDTA was used as a model for an inhibitor that scavenges active site metals in a complete and non-selective manner.18,20,21 Alternatively, L-captopril was used as a model for competitive inhibition, as it is known to form a ternary complex between the MBL, Zn(II) ions, and the inhibitor.24,47–52
The 1H NMR spectrum for CoCo-NDM-1 was previously assigned.16 Most notably, the resonances at 110, 82, 73, and 65 ppm were assigned to exchangeable protons from the side chains of four histidine residues in the NDM-1 active site (Figure 6, bottom). The broad signal at 170 ppm was assigned to the β-methylene protons on the Co(II)-bound cysteine in the M2 site. Finally, the sharp resonance at 48 ppm was assigned to the Co(II)-bound aspartate residue in the M2 site. Also important to note are the resonances < 0 ppm, which have been shown to primarily arise from the M2 site. The titration of EDTA into CoCo-NDM-1 resulted in a loss in the Co(II)-bound protein resonances and the appearance of a strong signal at 130 ppm (Figure 6, middle and top), characteristic of the Co(II)-EDTA complex (Figure S2). The enzyme also lost its usual pink color. Once two molar equivalents of EDTA had been reached, none of the Co(II)-bound protein resonances remained, indicating complete removal. Also in the one equivalent spectrum, the loss of the protein signal appears to be uniform and confirms that EDTA does not show selectivity between the two metal binding sites.
Figure 6.

300 MHz 1H NMR spectra of CoCo-NDM-1 titrated with EDTA. (Bottom) CoCo-NDM-1 (1 mM) resting spectrum. (Middle and top) CoCo-NDM-1 (1 mM) titrated with 1 and 2 mM EDTA. Control spectrum of Co(II) and EDTA shown in Figure S2. As EDTA was titrated into NDM-1, resonances arising from the Co(II)-bound protein diminish and are replaced by a sharp resonance at 130 ppm arising from the Co(II)-EDTA complex, indicating EDTA removes and sequesters the active site metal ions of NDM-1.
A control spectrum of Co(II) with L-captopril showed hyperfine shifted resonances were not present as far out as 500 ppm, indicating a complex is not formed in the absence of protein under the conditions employed (Figure S2). As CoCo-NDM-1 was titrated with L-captopril, the solution turned a bright blue color, which suggests an additional bridging thiol, consistent with previously published crystal structures.24,49,53 The 1H NMR spectra did not show evidence of metal abstraction with concentrations as high as five molar equivalents of L-captopril (Figure 7). Alternatively, a severe change in the spectrum is observed, indicating the formation of a ternary complex in which captopril interacts with both metal binding sites. These data show a significant change in the electronic environment of the active site. Notably the two cysteine β-methylene protons appear to have separated in the spectra containing L-captopril and may account for the resonances at 212 and 188 ppm. This result suggests a change in their dihedral angles. Additionally, the relatively strong intensity and the location of the resonance at 172 ppm may come from the two methylene protons adjacent to the thiol of L-captopril. It is expected that a new resonance at this chemical shift would appear due to the structural similarities between cysteine and the L-captopril thiolate. Another important feature of these spectra is the enhanced resolution and stronger signal intensity of all resonances. This result can be accounted for by enhanced T1 relaxation time, indicating a tighter coupling of the two active site Co(II) ions.27 This phenomenon has also been reported on binuclear Cu(II) complexes. The long relaxation time of Cu(II) in 1H NMR results in very broad, unresolvable resonances. But, in these binuclear complexes a significant reduction in linewidth was observed.54–56
Figure 7.

300 MHz 1H NMR spectra of CoCo-NDM-1 titrated with L-captopril. (Bottom) CoCo- NDM-1 (1 mM) spectrum. (Middle and top) CoCo-NDM-1 (1 mM) titrated with 3 and 5 mM L-captopril. Control spectrum of Co(II) and L-captopril, without protein, showed no hyperfine shifted resonances (Figure S2). Titration of NDM-1 with L-captopril resulted in a severe change in the 1H NMR spectrum, indicating it forms a ternary complex with the protein and active site Co(II) ions. Unlike the 1H NMR spectrum of CoCo-NDM-1 titrated with EDTA, there is no evidence of metal removal or sequestration.
A control spectrum of the Co(II)-AMA complex, without protein, was collected (Figure 8, top). The titration CoCo-NDM-1 with AMA resulted in a stepwise reduction in the signal intensity of Co(II)-bound protein peaks (Figure 8). Once 2 molar equivalents of AMA had been added, the spectrum was identical to the spectrum of the Co(II)-AMA complex, indicating that both Co(II) ions had been removed from the active site of NDM-1. The loss of signal appears to be uniform, with no indication that some resonances decay at a faster rate than others. This result suggests that metal abstraction is indiscriminate between the M1 and M2 sites at millimolar concentrations of enzyme. Moreover, the lack of any additional or shifted resonances at any point in the titration argues against the formation of a tertiary complex. However, it is possible that a short-lived ternary complex exists, but cannot be detected in our 1H NMR experiment which takes several hours. Similarly, titration of CoCo-VIM-2 (Figure 9) and CoCo-IMP-7 (Figure 10) with AMA also resulted in the loss of the Co(II)-bound protein signals and the formation of resonances arising from the Co(II)-AMA complex. In total, the results of the 1H NMR titrations with AMA best fit the model of EDTA. This result suggests that AMA acts via metal ion sequestration with the diCo(II)-substituted MBLs when the enzyme and AMA are at millimolar concentrations.
Figure 8.

200 MHZ 1H NMR spectra of CoCo-NDM-1 titrated with AMA. (Bottom) CoCo-NDM-1 (1 mM) 1H NMR spectrum. (Middle 3 spectra) CoCo-NDM-1 (1 mM) titrated with 0.5, 1, and 2 mM AMA. (Top) Control spectrum of CoCl2 (2 mM) and AMA (4 mM), without NDM-1. As AMA was titrated into NDM-1, resonances arising from the Co(II)-bound protein diminish and are replaced by resonances arising from the Co(II)-AMA complex, indicating AMA removes and sequesters the active site metal ions of NDM-1.
Figure 9.
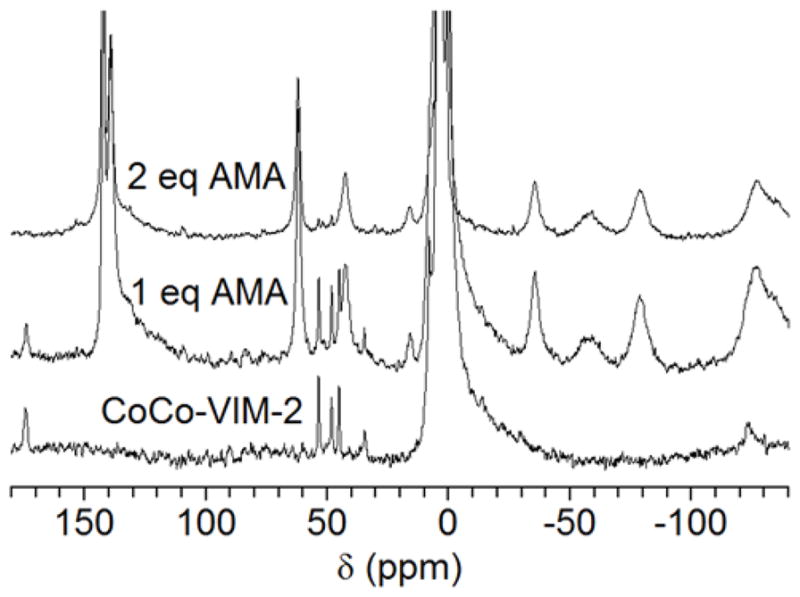
200 MHz 1H NMR spectra of CoCo-VIM-2 titrated with AMA. (Bottom) CoCo-VIM-2 (1 mM) 1H NMR spectrum. (Middle and top) CoCo-VIM-2 (1 mM) titrated with 1 mM and 2 mM AMA. As AMA was titrated into VIM-2, resonances arising from the Co(II)-bound protein diminish and are replaced by resonances arising from the Co(II)-AMA complex, indicating AMA removes and sequesters the active site metal ions of VIM-2.
Figure 10.

200 MHz 1H NMR spectra of CoCo-IMP-7 titrated with AMA. (Bottom) CoCo-IMP-7 (400 μM) 1H NMR spectrum. (Middle and top) CoCo-IMP-7 (400 μM) titrated with 0.4 and 0.8 mM AMA. As IMP-7 was titrated with AMA, resonances arising from the Co(II)-bound protein diminish and are replaced by resonances arising from the Co(II)-AMA complex, indicating AMA removes and sequesters the active site metal ions of IMP-7.
Conclusions
Herein we report a detailed investigation into the inhibition mechanism of AMA with NDM-1, VIM-2, and IMP-7. Within this analysis, we also overcame many challenges in working with IMP-7. Stripping the metal from IMP-7 with EDTA inactivates the enzyme and that activity cannot be restored by reintroducing Zn(II). However, apo-IMP-7 could be reactivated by unfolding the enzyme and refolding it around Zn(II), suggesting that treatment with a chelator causes structural changes to the enzyme that excludes metal ions from binding to the active site. This property may be unique to IMP-7, as IMP-1 was Co(II)-substituted using the direct addition method.26 Using this unfolding/refolding strategy a new method to prepare Co(II)-substituted IMP-7 was identified. Moreover, the resulting CoCo-IMP-7 enzyme exhibited stability high enough to perform paramagnetic 1H NMR spectroscopy. We hope that this Co(II)-substitution method will be useful for future spectroscopic investigations of IMP-type enzymes.
The equilibrium dialysis results demonstrated that Zn(II) content decreases with increasing concentrations of AMA for each of the three MBLs tested. The initial drop to ~1 molar equivalent of zinc is consistent with results recently published with NDM-1 and AMA,.17 Previous studies on other MBLs, including NDM-1,34 have suggested that zinc binding to the Zn1 site is tighter than zinc binding to the Zn2 site.29,36,57 Therefore, we hypothesize that AMA preferentially removes zinc from the Zn2 site and removes the metal ions from the Zn1 site at higher AMA concentrations. The absence of any significant differences in this experiment between the three enzymes suggests that AMA acts by metal removal with each MBL. In the Co(II)-substituted MBL 1H NMR experiments, the same trend was observed. In each case, resonances from the Co(II)-bound protein decreased with increasing concentrations of AMA. These experiments also support a metal removal mechanism. Due to the absence of any additional or shifted resonances throughout the titrations, we could not find evidence to support active participation of AMA in metal removal. Though it is possible a short-lived complex exists that cannot be captured in our 1H NMR experiments, our data suggest that AMA acts primarily through metal ion sequestration. However, in contrast with the equilibrium dialysis results, the 1H NMR results show complete and indiscriminate abstraction of metal from both the M1 and M2 sites. These results may be explained by differences in the affinity for the MBLs between Co(II) and Zn(II) or by the concentrations of enzymes/AMA in these experiments as compared to their respective Kd values. To address the issue of whether AMA binds tighter to zinc than cobalt, we used ITC to measure the dissociation constant (Kd) between AMA and zinc and cobalt (Table 1 and Figure S3, S4, and S5). Its clear that AMA does not preferentially bind zinc over cobalt. Efforts to evaluate the Kd values of zinc and cobalt binding to metal-free NDM-1 were unsuccessful due to protein precipitation during the titrations (data not shown). We also attempted to use ITC to determine the Kd values for AMA binding to the Zn(II)- and Co(II)-analogs of NDM-1. The resulting thermograms suggested multiple equilibria and reliable Kd values could not be determined (data not shown). Therefore, we cannot definitively explain why AMA appears to preferentially remove zinc from the Zn2 site in the equilibrium dialysis studies while AMA indiscriminately removes cobalt in the 1H NMR studies. It is likely that this behavior can be explained by the differing concentration of enzymes in the two experiments: the Kd for the zinc AMA complex is similar to the concentration of the enzyme in the equilibrium dialysis studies while the Kd for cobalt AMA complex is 3 to 4 orders of magnitude lower than the concentration of the enzyme in the 1H NMR studies.
Table 1.
Thermodynamics of AMA/metal interaction
| Interaction | Kd (nM) | ΔH (kJ mol−1) | ΔS (J mol−1 K−1) | ΔG (kJ mol−1) |
|---|---|---|---|---|
| Zn(II) / AMA | 200 ± 60 | 2.7 ± 0.2 | 140 ± 3 | −38.2 ± 0.7 |
| Co(II) / AMA | 250 ± 40 | 1.4 ± 0.1 | 130 ± 2 | −37.7 ± 0.4 |
Previously published data showed that, when combined with meropenem, AMA is more effective at killing bacterial isolates expressing NDM and VIM-type MBLs than it is on those expressing IMP-type enzymes.17 This result suggests that AMA exhibits greater inhibitory activity on NDM and VIM MBLs. However, we did not detect any differences between the three enzymes, and significant inhibition was seen for all three MBLs. Furthermore, some controversy exists within the literature on mononuclear IMP enzymes. It has been shown that when IMP-1 is exposed to several different chelators, it results in a mononuclear Zn(II)-IMP-1, which was detected using MS-ESI.19 It was also found that this mononuclear form of the enzyme still exhibited significant catalytic activity. These data are in alignment with the observation that AMA is more active on bacterial isolates expressing NDM and VIM. Treatment of IMP-1 with AMA at micromolar enzyme/AMA concentrations could result in the mononuclear Zn(II)-enzyme, which still may have catalytic activity, thus allowing for the survival of bacterial isolates expressing IMP. Taken together, these data suggest that in vivo IMP-1 may be catalytically competent in the mononuclear form. However, other studies do not support this finding.58,59 Several labs showed that mutagenesis of essential metal binding residues results in the mononuclear enzyme and that they are severely deficient of activity. The D120A mutant crystal structure shows that it binds only a metal ion to the Zn1 site, and that it is not catalytically competent.58 Additionally, H116A, H118A, H196A, C221A, and C221S mutants (BBL nomenclature)60 of IMP-1 also show diminished activity while binding only ~1 molar equivalent of metal.59 These data are in alignment with our kinetic measurements. As such, it is unclear why differences were observed between bacterial isolates expressing IMP-type enzymes when treated with AMA compared to NDM and VIM-type enzymes. Further research would be necessary to address this question.
In total, we found that AMA inhibits NDM-1, VIM-2, and IMP-7 via a metal ion sequestration mechanism. Furthermore, we found that the inhibition mechanism was conserved between each of the three enzymes throughout our experiments. It is our hope that these data will be useful for further research on AMA as it moves forward as a lead compound in the development of clinically useful inhibitors of the MBLs. The introduction of substituents that increase the specificity of AMA towards the MBL active sites might prove to be an effective strategy of using AMA as a scaffold for novel MBLs inhibitors.
METHODS
Materials
The His-IMP-7 expression vector and AMA were provided by Dr. Gerard Wright (McMaster University, ON, Canada). E. coli BL21(DE3) competent cells were purchased from Lucigen Corporation (Middleton, WI). Miller Luria-Bertani (LB) growth medium, dialysis tubing, Slide-A-Lyzer Cassettes, HisPur Ni-NTA columns, as well as all buffers used were purchased from ThermoFisher Scientific (Waltham, MA). Isopropyl β-D-thiogalactoside (IPTG) was purchased from Gold Biotechnology (St. Louis, MO). L-Captopril was purchased from Acros Organics (Geel, Belgium). Amicon Ultra-4 centrifugal units and YM-10 membranes were purchased from EMD Millipore (Darmstadt, Germany). Kinetic assays were conducted with the substrate, Chromacef, from Gladius Pharmaceuticals, Inc. (Montreal, QC, Canada). Q-Sepharose anion-exchange resin, Sephacryl S-200 gel filtration resin, and HiTrap Benzamidine FF columns were purchased from GE Healthcare (Marlborough, MA). All buffers were made with Nanopure water from a Barnstead filtration system. To remove metal contamination from all indicated buffering solutions, Bio-Rad Chelex-100 resin was used (Hercules, CA). For all 1H NMR spectroscopic studies, 5 mm D2O matched Advanced NMR Shigemi Microtubes with an 8 mm bottom were used, purchased from Shigemi Inc. (Allison Park, PA).
Over-expression and purification of NDM-1
BL21(DE3) E. coli cells containing the pET26b-ndm-1 plasmid, which expresses NDM-1 with a Δ36 N-terminal truncation, were used to inoculate a 50 mL LB starter culture containing kanamycin (25 μg/mL).16 The culture was allowed to grow overnight at 37 °C in a shaker. The starter culture (10 mL) was then used to inoculate 4 × 1 L LB containing kanamycin (25 μg/mL). These flasks were incubated at 37 °C in a shaker until cultures reached an optical density (OD600) of 0.6 to 0.8. The incubator was then set to 22 °C, and the cultures were allowed to cool at room temperature for 30 min. ZnCl2 (100 μM) and IPTG (0.5 mM) were then added to the cultures, and the cultures were incubated for 18 h at 22 °C. The cells were then harvested using centrifugation (10,800 × g) for 10 min. The pellet was resuspended with 25 mL 50 mM HEPES, pH 7.5, containing 500 mM NaCl (Buffer B). Lysis was performed with three passes through a French Press. The lysate was centrifuged for 30 min (32,600 × g) at 4 °C. The supernatant was then dialyzed (10 kDa MWCO) versus 2 L of 50 mM HEPES, pH 7.5, (Buffer A) overnight. An FPLC was used to equilibrate a Q-Sepharose column (1.5 cm × 28 cm with a 28 mL bed volume) with buffer A using a flowrate of 2.0 mL/min for 1.5 h. The lysate was then loaded onto the column and eluted with a linear NaCl gradient from 0–500 mM (Buffer A and B). Fractions containing NDM-1 were identified with SDS-PAGE. These fractions were pooled and concentrated to 1–2 mL in an Amicon ultracentrifugation concentrating unit with a YM-10 membrane. The resulting protein mixture was then loaded onto a Sephacryl S-200 gel filtration column (1.5 cm × 40 cm with a 60 mL bed volume) and run with 50 mM HEPES, pH 7.5, containing 150 mM NaCl (Buffer C). SDS-PAGE was used to identify fractions containing pure protein.
Over-expression and purification of VIM-2 and IMP-7
VIM-2 was over-expressed and purified as previously described,25 except using Buffer A and B for ion exchange chromatography, and Buffer C for gel filtration. IMP-7 was over-expressed using the pET-24a-his-imp-7 plasmid17 using the same methods described for expressing NDM-1 up until purification of the lysate. The lysate supernatant was incubated with 3 mL of HisPur Ni-NTA resin for 1 h on a rocker at 4 °C. The resin was poured into a gravity-flow column and washed with Buffer C containing 25 mM imidazole to remove unbound and loosely-bound protein. The purified protein was eluted using Buffer C containing 250 mM imidazole. SDS-PAGE was used to confirm that the elution product contained pure IMP-7. Thrombin (2 mg) was added to the purified protein, which was then incubated on a rocker at 4 °C for 18 h to cleave the His-tag. In order to remove the thrombin, the protein solution was then passed through a 1 mL GE Healthcare HiTrap Benzamidine FF column. The protein solution was then dialyzed (10 kDa MWCO) versus 1 L Buffer C for 18 h to remove the His-tag and imidazole.
Preparation of Co(II)-substituted analogs
The Co(II)-analog of NDM-1 was generated using direct addition of Co(II) to the metal free enzyme, as previously described (Method A).16 VIM-2 and IMP-7 were Co(II)-substituted by removing the active site metal ions with EDTA or 1,10-phenanthroline, and then unfolding the protein and refolding it around Co(II) (Method B). VIM-2 or IMP-7 (200 – 400 μM) were dialyzed (10 kDa MWCO) versus the following 1 L buffers for ~8 h per step at 4 °C: 50 mM HEPES, pH 6.8, containing 150 mM NaCl (Buffer D) and 2 mM 1,10-phenanthroline, Buffer D containing 2 mM 1,10-phenanthroline, Buffer D containing 2 mM 1,10-phenanthroline, Buffer D, 6 M urea, Buffer D containing 300 μM CoCl2, and 1 mM TCEP, Buffer D containing 1 mM TCEP, and Buffer D containing 1 mM TCEP. Any precipitate was removed by centrifugation (18,000 × g). Each of the buffers were treated with Chelex resin, and all of the glassware used was rinsed with 20% HNO3 to remove contaminating metal ions.
Equilibrium dialysis and metal analysis using ICP-AES
Each enzyme was diluted to 50 μM in Buffer C (final volume of 4 mL), and to remove unbound and loosely bound metal ions from the enzymes, subsequently dialyzed (10 kDa MWCO) for 4 h at 4 °C versus 1 L of Chelex-treated Buffer C. To 1 mL aliquots of the samples, 0, 100, 250, and 500 μM AMA were incubated for 30 min on ice. The subsequent mixtures, including the samples without AMA, were then dialyzed versus 1 L Chelex-treated 20 mM HEPES, pH 6.8, for 4 h at 4 °C in 3 mL Slide-A-Lyzer 10 kDa MWCO cassettes. The reduced buffer concentration and absence of NaCl were necessary for accurate metal analysis in following steps. After dialysis, the enzymes were diluted to 2 μM in Nanopure water. Five calibration samples were prepared in 2 mM HEPES (to match the final HEPES concentration of the enzyme samples) using Nanopure water at 0, 1, 2, 4, and 8 μM each containing Zn(II), Co(II), Fe(II), Cu(II), and Ni(II). These samples were used to generate a calibration curve with a PerkinElmer Optima 7300V inductively coupled plasma spectrometer with atomic emission spectroscopy (ICP-AES). The emission lines detected were 202.548, 228.616, 238.196, 327.394, and 231.604 nm, respectively. The enzyme mixtures treated with various concentrations of AMA were then measured with ICP-AES. The resulting Zn(II) concentrations were then divided by the enzyme concentrations to give the molar equivalents of Zn(II) per enzyme.
Kinetic measurements of IMP-7
ZnZn-IMP-7 and CoCo-IMP-7 were prepared using both Method A and Method B for kinetic analyses. Chromacef (30 μM) and IMP-7 (10 nM) were added to a 96-well plate in Chelex-treated Buffer C or Buffer D with a final volume of 200 μL. CoCl2 or ZnCl2 (5 – 100 μM) was added where indicated. After the enzyme was added to the 96-well plate, measurements began immediately in order to capture initial velocities. All kinetic measurements were collected using a Biotek Synergy HT plate reader set to record the A442 every 7 seconds for a total duration of 63 seconds at room temperature. Each measurement was recorded in triplicate. The slope of each line was used as the reaction rate and native diZn(II)-IMP-7 was standardized to 100% activity. All of the other reaction rates were divided by the reaction rate of native diZn(II)-IMP-7 in order to determine the relative % activity. The error bars indicate the standard deviation between the triplicate measurements.
Kinetic measurements of post-equilibrium dialysis fractions
After equilibrium dialysis, NDM-1, VIM-2, and IMP-7 samples, treated with 10 molar equivalents of AMA, were used for kinetic measurements compared to the untreated enzymes. NDM-1 and VIM-2 were diluted to a final concentration of 20 nM, and IMP-7 was diluted to 10 nM in order to achieve linearity for all three enzymes. The kinetic measurements were collected as described above using saturating concentrations of Chromacef (30 μM) and Chelex-treated Buffer C. ZnCl2 (10 μM) was added where indicated to explore the effect of excess Zn(II) on activity restoration.
1H NMR sample preparation and data collection
The Co(II)-analogs of NDM-1 and VIM-2 were concentrated to ~1 mM in an Amicon Ultra-4 centrifugal unit with an Ultracel-10 membrane. CoCo-IMP-7 was not stable at 1 mM and only 400 μM could be reached. Each NMR sample was buffered with Buffer D. To a D2O-matched 5 mm Shigemi Advanced NMR Microtube, 400 μL of enzyme with 10% D2O was added. Excess Co(II) (3 mM) was required to collect an NMR spectrum of VIM-2. The AMA stock was dissolved in water at high concentration (25 mM) so it could be titrated into the NMR samples in small aliquots. A 100 mM EDTA stock was prepared in water, and a 100 mM L-captopril stock dissolved in DMSO. Spectra were collected at 292 K on Bruker DPX200 (QNP probe) or Bruker DPX300 (BBI probe) 1H NMR spectrometers operating at frequencies of 200.13 MHz and 300.16 MHz, respectively. Data collection was performed using a frequency-switching method in which a long, low power 270 ms presaturation pulse was centered on the water signal, then a high power 3 μs pulse was focused on 90 ppm.31 The presaturation pulse allowed for suppression of the water signal and enhancement of the paramagnetic hyperfine shifted resonances. Spectra collected on the 200 MHz 1H NMR consisted of 200,000 transients (~12 h of signal averaging) of 4k data points over a 400 ppm spectral window. Due to greater signal to noise, data collected on the 300 MHz 1H NMR was reduced to 30,000 transients (~3 h signal averaging) and 16k data points over a 333 ppm spectral window. All data processing was performed using TopSpin 3.5pl5.
Isothermal titration calorimetry of AMA with Zn(II) and Co(II)
The ITC thermograms were obtained on a MicroCal Auto-ITC200 instrument (Malvern). AMA, CoCl2, and ZnCl2 were dissolved in HEPES buffer (20 mM, pH 6.8) and all the solutions were degassed for 10 min in a sonication bath. For performing Zn(II)/AMA binding experiment, 0.50 mM ZnCl2 was titrated into 0.05 mM AMA, while for Co(II)/AMA binding experiment, 1.00 mM CoCl2 was titrated into 0.10 mM AMA. The syringe content was titrated over 26 aliquots of 1.5 μL (with the exception of the first injection which was 0.5 μL) by 120 s intervals. The reference power was set at 2.0 μcal/sec and the experiments were performed at 25 °C. The peaks were integrated and processed using Origin 7.0 software and the errors of the thermodynamic parameters were estimated by Monte Carlo simulation of the standard errors of three experiments.
Native mass spectrometry of the MBLs
All experiments were performed on a Thermo Fisher Scientific Orbitrap Elite mass spectrometer (San Jose, CA) modified to perform ultraviolet photodissociation (UVPD) experiments in the HCD cell using a 193 nm ArF excimer laser (Coherent ExciStar XS), as described previously.61 Protein solutions at 10 μM were loaded into Au-coated borosilicate emitters in 50% (v/v) acetonitrile or 20 mM ammonium acetate (pH 6.8) and ionized offline using a nano-ESI source. UV photoactivation of the denatured proteins was achieved using one pulse applied at an energy of 2 mJ/pulse. MS1 and MS/MS spectra (60–500 scans) were acquired at 240K resolving power (at m/z 400). Processing of MS1 and MS/MS spectra was performed using Xtract and ProSight 3.0 to identify fragment ions.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Health (NIH Grant GM111926 and R01GM121714), Miami University, the National Science Foundation (Grant CHM1509285), and the Robert A. Welch Foundation (F-1572 and F-1155). Research reported in this publication was supported in part by the National Institutes of Health (NIH), through the National Institute of Allergy and Infectious Diseases (R01AI100560, R01AI063517, and R01AI072219 to RAB). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. This study was also supported in part by funds and/or facilities provided by the Cleveland Department of Veterans Affairs, the Veterans Affairs Merit Review Program (Award 1I01BX001974 to RAB, and the Geriatric Research Education and Clinical Center VISN 10 (RAB). Financial support was also provided by Utrecht University and The Netherlands Organization for Scientific Research. The authors thank Seth M. Cohen of the University of California at San Diego for helpful discussions.
ABBREVIATIONS
- MBL
metallo-β-lactamase
- AMA
aspergillomarasmine A
- NDM
New Delhi metallo-β-lactamase
- VIM
Verona integron-encoded metallo-β-lactamase
- IMP
imipenemase
- EDTA
ethylenediaminetetraacetic acid
- DPA
2,6-dipicolinic acid
- ITC
isothermal titration calorimetry
- Native MS
native mass spectrometry
- IPTG
isopropyl β-D-thiogalactoside
- ICP-AES
inductively coupled plasma atomic emission spectroscopy
Footnotes
Notes
The authors declare no competing financial interest.
Equilibrium dialysis data assessing Zn(II) content of NDM-1 and VIM-2 as effected by EDTA and L-captopril (Figure S1), and 200 MHz 1H NMR control spectra of Co(II) with EDTA, L-captopril, and AMA (Figure S2). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Drawz SM, Bonomo RA. Three decades of β-lactamase inhibitors. Clin Microbiol Rev. 2010;23(1):160–201. doi: 10.1128/CMR.00037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blair JMA, Webber MA, Baylay AJ, Ogbolu DO, Piddock LJV. Molecular mechanisms of antibiotic resistance. Nat Rev Microbiol. 2015;13(1):42–51. doi: 10.1038/nrmicro3380. [DOI] [PubMed] [Google Scholar]
- 3.Li X-Z, Plésiat P, Nikaido H. The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin Microbiol Rev. 2015;28(2):337–418. doi: 10.1128/CMR.00117-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Acker H, Van Dijck P, Coenye T. Molecular mechanisms of antimicrobial tolerance and resistance in bacterial and fungal biofilms. Trends Microbiol. 2014;22(6):326–333. doi: 10.1016/j.tim.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 5.Karaiskos I, Giamarellou H. Multidrug-resistant and extensively drug-resistant Gram-negative pathogens: current and emerging therapeutic approaches. Expert Opin Pharmacother. 2014;15(10):1–20. doi: 10.1517/14656566.2014.914172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown ED, Wright GD. Antibacterial drug discovery in the resistance era. Nature. 2016;529(7586):336–343. doi: 10.1038/nature17042. [DOI] [PubMed] [Google Scholar]
- 7.Meini MR, Llarrull LI, Vila AJ. Overcoming differences: The catalytic mechanism of metallo-β-lactamases. FEBS Lett. 2015;589(22):3419–3432. doi: 10.1016/j.febslet.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Papp-Wallace KM, Bonomo RA. New β-Lactamase Inhibitors in the Clinic. Infect Dis Clin North Am. 2016;30(2):441–464. doi: 10.1016/j.idc.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wright GD. Antibiotic Adjuvants: Rescuing Antibiotics from Resistance. Trends Microbiol. 2016;24(11):862–871. doi: 10.1016/j.tim.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 10.Bush K. Bench-to-bedside review: The role of β-lactamases in antibiotic-resistant Gram-negative infections. Crit care. 2010;14(3):224. doi: 10.1186/cc8892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crowder MW, Spencer J, Vila AJ. Metallo-β-lactamases: Novel weaponry for antibiotic resistance in bacteria. Acc Chem Res. 2006;39(10):721–728. doi: 10.1021/ar0400241. [DOI] [PubMed] [Google Scholar]
- 12.Mojica MF, Bonomo RA, Fast W. B1-Metallo-β-Lactamases: Where Do We Stand? Curr Drug Targets. 2016;17(9):1029–1050. doi: 10.2174/1389450116666151001105622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toney JH, Hammond GG, Fitzgerald PMD, Sharma N, Balkovec JM, Rouen GP, Olson SH, Hammond ML, Greenlee ML, Gao YD. Succinic Acids as Potent Inhibitors of Plasmid-borne IMP-1 Metallo-β-lactamase. J Biol Chem. 2001;276(34):31913–31918. doi: 10.1074/jbc.M104742200. [DOI] [PubMed] [Google Scholar]
- 14.Docquier JD, Lamotte-Brasseur J, Galleni M, Amicosante G, Frère JM, Rossolini GM. On functional and structural heterogeneity of VIM-type metallo-β-lactamases. J Antimicrob Chemother. 2003;51(2):257–266. doi: 10.1093/jac/dkg067. [DOI] [PubMed] [Google Scholar]
- 15.Yong D, Toleman MA, Giske CG, Cho HS, Sundman K, Lee K, Walsh TR. Characterization of a new metallo-β-lactamase gene, bla NDM-1, and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India. Antimicrob Agents Chemother. 2009;53(12):5046–5054. doi: 10.1128/AAC.00774-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang H, Aitha M, Marts AR, Hetrick A, Bennett B, Crowder MW, Tierney DL. Spectroscopic and mechanistic studies of heterodimetallic forms of metallo-β-lactamase NDM-1. J Am Chem Soc. 2014;136(20):7273–7285. doi: 10.1021/ja410376s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.King AM, Reid-Yu SA, Wang W, King DT, De Pascale G, Strynadka NC, Walsh TR, Coombes BK, Wright GD. Aspergillomarasmine A overcomes metallo-β-lactamase antibiotic resistance. Nature. 2014;510(7506):503–506. doi: 10.1038/nature13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bush K, Jacoby GA. Updated functional classification of β-lactamases. Antimicrob Agents Chemother. 2010;54(3):969–976. doi: 10.1128/AAC.01009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Siemann S, Brewer D, Clarke AJ, Dmitrienko GI, Lajoie G, Viswanatha T. IMP-1 metallo-β-lactamase: Effect of chelators and assessment of metal requirement by electrospray mass spectrometry. Biochim Biophys Acta - Gen Subj. 2002;1571(3):190–200. doi: 10.1016/S0304-4165(02)00258-1. [DOI] [PubMed] [Google Scholar]
- 20.Walsh TR, Toleman MA, Poirel L, Nordmann P. Metallo-β-lactamases: The quiet before the storm? Clin Microbiol Rev. 2005;18(2):306–325. doi: 10.1128/CMR.18.2.306-325.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nordmann P, Dortet L, Poirel L. Carbapenem resistance in Enterobacteriaceae: Here is the storm! Trends Mol Med. 2012;18(5):263–272. doi: 10.1016/j.molmed.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 22.von Nussbaum F, Schiffer G. Aspergillomarasmine A, an inhibitor of bacterial metallo-β-lactamases conferring blaNDM and blaVIM resistance. Angew Chem Int Ed Engl. 2014;53(44):11696–11698. doi: 10.1002/anie.201407921. [DOI] [PubMed] [Google Scholar]
- 23.Fast W, Sutton LD. Metallo-β-lactamase: Inhibitors and reporter substrates. Biochim Biophys Acta - Proteins Proteomics. 2013;1834(8):1648–1659. doi: 10.1016/j.bbapap.2013.04.024. [DOI] [PubMed] [Google Scholar]
- 24.Brem J, Van Berkel SS, Zollman D, Lee SY, Gileadi O, McHugh PJ, Walsh TR, McDonough MA, Schofield CJ. Structural basis of metallo-β-lactamase inhibition by captopril stereoisomers. Antimicrob Agents Chemother. 2016;60(1):142–150. doi: 10.1128/AAC.01335-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aitha M, Marts AR, Bergstrom A, Moller AJ, Moritz L, Tumer L, Nix JC, Bonomo RA, Page RC, Tierney DL, et al. Biochemical, Mechanistic, and Spectroscopic Characterization of Metallo-β-lactamase VIM-2. Biochemistry. 2014;53(46):7321–7331. doi: 10.1021/bi500916y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Griffin DH, Richmond TK, Sanchez C, Moller AJ, Breece RM, Tierney DL, Bennett B, Crowder MW. Structural and kinetic studies on metallo-β-lactamase IMP-1. Biochemistry. 2011;50(42):9125–9134. doi: 10.1021/bi200839h. [DOI] [PubMed] [Google Scholar]
- 27.Bertini I, Banci L, Luchinat C. Proton magnetic resonance of paramagnetic metalloproteins. Methods Enzymol. 1989;177:246–263. doi: 10.1016/0076-6879(89)77014-2. [DOI] [PubMed] [Google Scholar]
- 28.Banci L, Dugad LB, La Mar GN, Keating Ka, Luchinat C, Pierattelli R. 1H nuclear magnetic resonance investigation of cobalt(II) substituted carbonic anhydrase. Biophys J. 1992;63(August):530–543. doi: 10.1016/S0006-3495(92)81607-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Orellano EG, Girardini JE, Cricco JA, Ceccarelli EA, Vila AJ. Spectroscopic characterization of a binuclear metal site in Bacillus cereus β-lactamase II. Biochemistry. 1998;37(28):10173–10180. doi: 10.1021/bi980309j. [DOI] [PubMed] [Google Scholar]
- 30.Crawford PA, Yang KW, Sharma N, Bennett B, Crowder MW. Spectroscopic studies on cobalt(II)-substituted metallo-β-lactamase ImiS from Aeromonas veronii bv. sobria. Biochemistry. 2005;44(13):5168–5176. doi: 10.1021/bi047463s. [DOI] [PubMed] [Google Scholar]
- 31.Riley EA, Petros AK, Smith KA, Gibney BR, Tierney DL. Frequency-switching inversion-recovery for severely hyperfine-shifted NMR: Evidence of asymmetric electron relaxation in high-spin Co(II) Inorg Chem. 2006;45(25):10016–10018. doi: 10.1021/ic061207h. [DOI] [PubMed] [Google Scholar]
- 32.Otting G. Protein NMR using paramagnetic ions. Annu Rev Biophys. 2010;39:387–405. doi: 10.1146/annurev.biophys.093008.131321. [DOI] [PubMed] [Google Scholar]
- 33.King D, Strynadka N. Crystal structure of New Delhi metallo-β-lactamase reveals molecular basis for antibiotic resistance. Protein Sci. 2011;20(9):1484–1491. doi: 10.1002/pro.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomas PW, Zheng M, Wu S, Guo H, Liu D, Xu D, Fast W. Characterization of purified New Delhi metallo-β-lactamase-1. Biochemistry. 2011;50(46):10102–10113. doi: 10.1021/bi201449r. [DOI] [PubMed] [Google Scholar]
- 35.Bebrone C. Metallo-β-lactamases (classification, activity, genetic organization, structure, zinc coordination) and their superfamily. Biochem Pharmacol. 2007;74(12):1686–1701. doi: 10.1016/j.bcp.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 36.Wommer S, Rival S, Heinz U, Galleni M, Frère JM, Franceschini N, Amicosante G, Rasmussen B, Bauer R, Adolph HW. Substrate-activated zinc binding of metallo-β-lactamases: Physiological importance of the mononuclear enzymes. J Biol Chem. 2002;277(27):24142–24147. doi: 10.1074/jbc.M202467200. [DOI] [PubMed] [Google Scholar]
- 37.Krezel A, Maret W. The biological inorganic chemistry of zinc ions. Arch Biochem Biophys. 2016;611:3–19. doi: 10.1016/j.abb.2016.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.White WI, Legg JI. Exchange-inert metal-ions as probes of enzyme structure-fuction-relationships - cobalt(III), cobalt(II), and zinc(II) azophenol complexes as models for enzyme azotyrosine complexes. J Am Chem Soc. 1975;97(14):3937–3941. doi: 10.1021/ja00847a012. [DOI] [PubMed] [Google Scholar]
- 39.Crowder MW, Yang KW, Carenbauer AL, Periyannan G, Seifert ME, Rude NE, Walsh TR. The problem of a solvent exposable disulfide when preparing Co(II)-substituted metallo-β-lactamase L1 from Stenotrophomonas maltophilia. J Biol Inorg Chem. 2001;6(1):91–99. doi: 10.1007/s007750000173. [DOI] [PubMed] [Google Scholar]
- 40.Vanwart HE, Vallee BL. Enzymatically inactive, exchange-inert Co(III)-Carboxypeptidase-A - Role of inner sphere coordination in peptide and ester catalysis. Biochemistry. 1978;17(16):3385–3394. doi: 10.1021/bi00609a032. [DOI] [PubMed] [Google Scholar]
- 41.Hu Z, Periyannan GR, Crowder MW. Folding strategy to prepare Co(II)-substituted metallo-β-lactamase L1. Anal Biochem. 2008;378(2):177–183. doi: 10.1016/j.ab.2008.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shinar H, Navon G. Kinetic and Magnetic Properties of Cobalt(III) Ion in the Active Site of Carbonic Anhydrase. Eur J Biochem. 1979;93(2):313–322. doi: 10.1111/j.1432-1033.1979.tb12825.x. [DOI] [PubMed] [Google Scholar]
- 43.Vanwart HE. Introduction of exchange-inert metal-ions into enzymes. Methods Enzymol. 1988;158:95–110. doi: 10.1016/0076-6879(88)58050-3. [DOI] [PubMed] [Google Scholar]
- 44.Periyannan GR, Costello AL, Tierney DL, Yang KW, Bennett B, Crowder MW. Sequential binding of cobalt(II) to metallo-β-lactamase CcrA. Biochemistry. 2006;45(4):1313–1320. doi: 10.1021/bi051105n. [DOI] [PubMed] [Google Scholar]
- 45.Bennett B. EPR of Cobalt-Substituted Zinc Enzymes. In: Hanson G, Berliner L, editors. Metals in Biology: Applications of High-Resolution EPR to Metalloenzymes. Springer New York; New York, NY: 2010. pp. 345–370. [Google Scholar]
- 46.Gunasekera TS, Herre AH, Crowder MW. Absence of ZnuABC-mediated zinc uptake affects virulence-associated phenotypes of uropathogenic Escherichia coli CFT073 under Zn(II)-depleted conditions. FEMS Microbiol Lett. 2009;300(1):36–41. doi: 10.1111/j.1574-6968.2009.01762.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klingler FM, Wichelhaus TA, Frank D, Cuesta-Bernal J, El-Delik J, Müller HF, Sjuts H, Göttig S, Koenigs A, Pos KM, et al. Approved drugs containing thiols as inhibitors of metallo-β-lactamases: Strategy to combat multidrug-resistant bacteria. J Med Chem. 2015;58(8):3626–3630. doi: 10.1021/jm501844d. [DOI] [PubMed] [Google Scholar]
- 48.Zheng M, Xu D. New Delhi metallo-β-lactamase I: Substrate binding and catalytic mechanism. J Phys Chem B. 2013;117(39):11596–11607. doi: 10.1021/jp4065906. [DOI] [PubMed] [Google Scholar]
- 49.King DT, Worrall LJ, Gruninger R, Strynadka NCJ. New delhi metallo-β-lactamase: Structural insights into β-lactam recognition and inhibition. J Am Chem Soc. 2012;134(28):11362–11365. doi: 10.1021/ja303579d. [DOI] [PubMed] [Google Scholar]
- 50.Vella P, Hussein WM, Leung EWW, Clayton D, Ollis DL, Mitić N, Schenk G, McGeary RP. The identification of new metallo-β-lactamase inhibitor leads from fragment-based screening. Bioorganic Med Chem Lett. 2011;21(11):3282–3285. doi: 10.1016/j.bmcl.2011.04.027. [DOI] [PubMed] [Google Scholar]
- 51.Liénard BMR, Garau G, Horsfall L, Karsisiotis AI, Damblon C, Lassaux P, Papamicael C, Roberts GCK, Galleni M, Dideberg O, et al. Structural basis for the broad-spectrum inhibition of metallo-β-lactamases by thiols. Org Biomol Chem. 2008;6(13):2282–2294. doi: 10.1039/b802311e. [DOI] [PubMed] [Google Scholar]
- 52.Heinz U, Bauer R, Wommer S, Meyer-Klaucke W, Papamichaels C, Bateson J, Adolph HW. Coordination geometries of metal ions in D- or L-captopril-inhibited metallo-β-lactamases. J Biol Chem. 2003;278(23):20659–20666. doi: 10.1074/jbc.M212581200. [DOI] [PubMed] [Google Scholar]
- 53.Bertini I, Luchinat C. High Spin Cobalt(II) as a Probe for the Investigation of Metalloproteins. Adv Inorg Biochem. 1984;6:71–111. [PubMed] [Google Scholar]
- 54.Brink JM, Rose RA, Holz RC. Characterization of the Structural and Electronic Properties of Spin-Coupled Dinuclear Copper(II) Centers by Proton NMR Spectroscopy. Inorg Chem. 1996;35(10):2878–2885. doi: 10.1021/ic951472v. [DOI] [Google Scholar]
- 55.Holz RC, Bennett B, Chen G, Ming LJ. Proton NMR Spectroscopy as a Probe of Dinuclear Copper(II) Active Sites in Metalloproteins. Characterization of the Hyperactive Copper(II)-Substituted Aminopeptidase from Aeromonas proteolytica. J Am Chem Soc. 1998;120:6329–6335. doi: 10.1021/ic00099a002. [DOI] [Google Scholar]
- 56.García-Giménez JL, Alzuet G, González-Álvarez M, Castiñeiras A, Liu-González M, Borrás J. A dinuclear copper(II) complex with adeninate bridge ligands and prominent DNA cleavage activity. Structural and spectroscopic characterization and magnetic properties. Inorg Chem. 2007;46(17):7178–7188. doi: 10.1021/ic700751j. [DOI] [PubMed] [Google Scholar]
- 57.Paul-Soto R, Bauer R, Frere JM, Galleni M, Meyer-Klaucke W, Nolting H, Rossolini GM, de Seny D, Hernandez-Valladares M, Zeppezauer M, et al. Mono- and binuclear Zn(II)-β-lactamase. Role of the conserved cysteine in the catalytic mechanism. J Biol Chem. 1999;274(19):13242–13249. doi: 10.1074/jbc.274.19.13242. [DOI] [PubMed] [Google Scholar]
- 58.Yamaguchi Y, Kuroki T, Yasuzawa H, Higashi T, Jin W, Kawanami A, Yamagata Y, Arakawa Y, Goto M, Kurosaki H. Probing the role of Asp-120(81) of metallo-β-lactamase (IMP-1) by site-directed mutagenesis, kinetic studies, and X-ray crystallography. J Biol Chem. 2005;280(21):20824–20832. doi: 10.1074/jbc.M414314200. [DOI] [PubMed] [Google Scholar]
- 59.Haruta S, Yamaguchi H, Yamamoto ET, Eriguchi Y, Nukaga M, O’Hara K, Sawai T. Functional analysis of the active site of a metallo-β-lactamase proliferating in Japan. Antimicrob Agents Chemother. 2000;44(9):2304–2309. doi: 10.1128/AAC.44.9.2304-2309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bush K. The ABCD’s of β-lactamase nomenclature. J Infect Chemother. 2013;19(4):549–559. doi: 10.1007/s10156-013-0640-7. [DOI] [PubMed] [Google Scholar]
- 61.Shaw JB, Li W, Holden DD, Zhang Y, Griep-Raming J, Fellers RT, Early BP, Thomas PM, Kelleher NL, Brodbelt JS. Complete protein characterization using top-down mass spectrometry and ultraviolet photodissociation. J Am Chem Soc. 2013;135(34):12646–12651. doi: 10.1021/ja4029654. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.