

Abstract
Background
Most clinical trials comparing treatments evaluate the separate effects on each of several efficacy and toxicity outcomes. However, population-averaged summary measures of treatment differences may not accurately reflect individual responses to treatment, and drawing conclusions about which treatment is “best” is straightforward if one treatment is superior across all outcomes, but challenging when this is not the case.
Methods
We created a study outcome based on expert opinion, which captures the risk/benefit profile of response to a treatment. Treatments were compared using this ordered outcome with standard statistical techniques. To illustrate the approach we used as an example a study designed to evaluate initial antiretroviral therapy (ART) in HIV-1 infected infants, in which results were contradictory across the study’s primary and secondary efficacy and toxicity outcomes. The proposed risk/benefit outcome was evaluated retrospectively in each participant.
Results
In the IMPAACT P1060 study, one treatment regimen (LPV/r-based ART) was superior to the other (NVP-based ART) in reducing viral load (primary outcome), but inferior for immunologic and growth outcomes (important secondary outcomes in resource-limited settings). Treatment comparisons using the risk/benefit outcome indicated that the LPV/r-based ART regimen had a higher proportion of participants with the best overall response to treatment. Comparisons focusing on individual-level responses for the secondary outcomes also favored LPV/r-based ART, results that differed from the original population-averaged analyses’ ones.
Conclusions
Designing studies prospectively using risk/benefit outcomes focusing on an individual’s responses to treatment, more closely matches the needs of clinicians making decisions about how best to treat patients in clinical settings.
Keywords: Clinical Studies, Multiple Outcomes, Risk/Benefit Assessment, HIV/AIDS
Introduction
In clinical trials evaluating treatments, the standard approach is to collect several clinical/biomarker outcomes for each participant, capturing disease status as well as treatment toxicities and tolerability. Differences between treatments are then evaluated using population-averaged treatment differences for each outcome separately. This allows evaluation of the potential risks and benefits of each treatment, but may not provide the right kind of information to help guide medical practitioners making a treatment selection decision for a future patient, especially when there are contradictory findings across outcomes. For example, one treatment may be highly efficacious in treating the disease, but if it can only be tolerated by a small proportion of patients, a better-tolerated treatment with somewhat less efficacy may actually be the “best” treatment to prescribe. Therefore, for a clinical study it seems preferable to use an endpoint which encompasses all relevant outcomes collectively at the patient-level. Such an endpoint would quantify disease burden and treatment toxicity over time, better reflecting the management of individual patients in clinical practice. We refer to this endpoint as the overall patient-level risk/benefit outcome.
To illustrate this approach, we used data from the P1060 clinical trial conducted by the International Maternal Pediatric Adolescent AIDS Clinical Trials (IMPAACT) Group to assess initial antiretroviral therapy (ART) in HIV-1-infected infants and children in resource-limited settings (1, 2). The primary study endpoint focused on efficacy in reducing viral load and showed superiority of the lopinavir/ritonavir (LPV/r)-based regimen versus the nevirapine (NVP)-based regimen. For two secondary outcomes (immunology and growth related) however, the NVP-based regimen was significantly better in adjusted analyses (3). This discrepancy had also been observed in the NEVEREST study (4). These findings raised questions about which of these two regimens might be “best” for use in resource-limited settings, as LPV/r is more challenging to use (it is more expensive, needs refrigeration and is not palatable) and weight gain is a particularly important outcome in resource-limited settings, since poor weight gain is a major risk factor for other morbidities. The contradictory findings left lingering questions about which ART regimen is “best” for use in resource-limited settings.
As a possible solution to this issue, a patient-level outcome might be constructed that integrates the various efficacy and safety outcomes and then treatments can be compared with respect to that outcome. To illustrate how such a risk/benefit outcome can be developed and how it can provide new insight to interpreting results in a clinical trial, four HIV-1 clinicians developed a single ordered, categorical risk/benefit outcome measuring an individual’s disease status and response to initiating ART after one year of treatment. The risk/benefit outcome included components for viral load, immunology, growth, adverse events, ability to stay on study treatment and hospitalizations. We used data from the P1060 trial as a working example of how treatment comparisons based on the risk/benefit outcome could shed new light on the original study findings and illustrate the usefulness of this kind of outcome retrospectively in a clinical trial. We hope the example will motivate researchers to consider using risk/benefit outcomes focusing on individual patient responses prospectively in future clinical trials in other disease areas than HIV-1/AIDS as well.
Methods
Creating a risk/benefit outcome
The specific components of a risk/benefit outcome relevant to real-world clinical practice and for use in a clinical trial will depend on the disease, evaluation time point and setting. Levels of patient response may be a combination of status at a particular time point and of events occurring between study entry and that time point. Ideally, the outcome would be defined by a panel of experts independent from a specific protocol and before any data are collected.
To illustrate a possible approach in HIV-1, a team of four pediatric HIV-1 clinicians was convened (co-authors PP, AV, MA, LB) to develop an outcome with ordered levels of response, which comprehensively summarized a child’s risk/benefit response to ART after 48 weeks in a resource-constrained setting. All the clinicians leaned towards developing an outcome with more than three levels. They felt having only three levels would obscure information and result in HIV-1 RNA driving the results, which would provide no extra insight into patient response. Each clinician independently developed a patient-level overall outcome measure based on her/his clinical experience with HIV-1 infected infants and children initiating ART. The clinicians and four statisticians (co-authors KA, JL, MH, LJW) then had several extensive discussions, eventually reaching a consensus outcome with four ordinal categories: “responder”, “partial responder”, “poor responder” and “non-responder”. However, there wasa debate on the relative importance of the different components of the outcome, e.g. one clinician gave a lower rank to the weight-for-age z-scores versus the HIV-1 RNA, while another gave it a higher rank. Therefore, final agreement was a compromise for all clinicians and all components were equally weighted.
A participant would be classified into an overall responder category using vital status and five component measures: 1) HIV-1 RNA, 2) adverse events and changes to the ART regimen, 3) hospitalizations (as a measure of clinically-significant morbidity), 4) weight-for-age z-score and 5) CD4%. Each component took into account the status of the child after 48 weeks and how their status had changed since starting ART, capturing both cross-sectional and longitudinal information, and mimicking how a clinician would assess a child in the clinic outside the context of a clinical trial. The full consensus definition is provided in Table 1.
Table 1.
Consensus categorization of each of the six outcome measures used to construct the overall risk/benefit outcome
| OUTCOME | ||||
|---|---|---|---|---|
| Responder | Partial Responder | Poor Responder | Non-Responder | |
| Vital Status | Alive at week 48 | Alive at week 48 | Alive at week 48 | Died before week 48 |
| HIV-1 RNA (copies/mL) | ≤ 400 at week 48 AND no blips after first measurement < 400 | ≤ 400 at week 48 AND single value < 1000 after first measurement < 400 | ≤ 400 at week 48 AND single value (1000 ≤ blip < 4000) after first measurement < 400 | > 400 at week 48;OR ≤ 400 at week 48 but single value ≥ 4000 after first measurement <400;OR ≤ 400 at week 48 and ≥400 at multiple times after first measurement < 400 |
| Toxicities1: Grade 3 or 4 signs and symptoms, laboratories (AEs) and associated ARV changes | No grade 3 or 4 AEs AND no change to randomized regimen due to AEs | Grade 3 or 4 AEs AND no dose modification or temporary interruption to randomized regimen due to an AE | AE of any grade that leads to a dose modification or temporary interruption of randomized regimen | AE of any grade resulting in permanent discontinuation of randomized regimen |
| Hospitalizations | None | 1 hospitalization with discharge on same day as, or day after, day of admission | 1 hospitalization for > 1 day | > 1 hospitalization |
| Weight-for-age z-score2 | z-score ≥ −1 at week 48 AND(≤ 0.5 decline from baseline or increased from baseline) | −2 ≤ z-score < −1 at week 48 AND(≤ 0.5 decline from baseline or increase from baseline) | z-score < −2 at week 48 AND > 0.5 increase from baseline | > 0.5 decrease from baseline (irrespective of z-score at week 48);OR z-score < − 2 at week 48 AND (≤ 0.5 decline from baseline or < 0.5 increase from baseline) |
| CD4% | ≥ 25% at week 48 AND(≤5% decline from baseline or increased from baseline) | 15% – <25% at week 48 AND(≤ 5% decline from baseline or increased from baseline) | CD4% < 15% at week 48 AND≥ 5% increase from baseline | > 5% decrease from baseline (irrespective of CD4% at week 48);OR < 15% at week 48 AND(≤ 5% decline from baseline or < 5% increase from baseline) |
The Division of AIDS Table for Grading the Severity of Adult and Pediatric Adverse Events, Version 1.0, dated December 2004 clarification August 2009 is used (http://rsc.tech-res.com/safetyandpharmacovigilance)
Based on WHO criteria
Children were classified in the “Non-Responder” outcome category if they died before 48 weeks or were classified as “Non-Responders” for one or more of the five component outcomes. Among those not classified as “Non-Responders”, children were classified as “Poor Responders” if they were “Poor Responders” for one or more of the five component outcomes. Among those not classified as “Non-Responders” or “Poor Responders”, children were classified as “Partial Responders” if one or more of the five component outcomes were so classified.
The remaining children were classified as “Responders” and thus were “Responders” for all five component outcomes.
For example, a child would be classified as an overall “responder” at week 48 if they were alive and were a responder in all five components (HIV-1 RNA ≤ 400 copies/mL at week 48 and no low-level viral rebounds after achieving HIV-1 RNA ≤ 400 copies/mL; no grade 3 or 4 adverse events (AEs) and no change to ART regimen due to AEs; no hospitalizations; weight z-score ≥ −1 at week 48 and less than an 0.5 decline from baseline; and CD4% ≥ 25% at week 48 and less than 5% decline from baseline). A child would be classified as an overall “non-responder” if they had died or were classified as a non-responder in any of the five components. There were very few children who were lost to follow-up therefore for simplicity in our example the assigned categories for these children were based on their status as of their last study visit.
The final overall risk/benefit category captures trade-offs among all efficacy and tolerability components. For example, a participant who achieved and maintained HIV-1 RNA < 400 copies/mL but whose growth profile was sub-optimal would be assigned to a worse category than a participant with a low but detectable viral load (400 copies/mL < HIV-1 RNA < 4000 copies/mL) and a significantly improved growth trajectory.
With this overall, composite, patient-level outcome, proportions of study participants classified into each of the four categories can be compared between treatments using standard statistical methods. To formally compare the proportions across the ordered categories we used the Cochran-Armitage test for trend. Alternatively, a logistic regression model with proportional odds could be used or odds ratios could be calculated among groups of categories (i.e. the odds of being classified as a “responder” or “partial responder” on one treatment vs. the other).
IMPAACT P1060
To illustrate this approach, we used data from the IMPAACT P1060 clinical trial. P1060 was a randomized trial designed to compare LPV/r- with NVP-based ART. The primary study endpoint was treatment failure defined as a confirmed plasma HIV-1 RNA level less than one log10 copies/mL below the baseline value at 12 to 24 weeks after treatment was initiated, a confirmed HIV-1 RNA level ≥400 copies/mL at 24 weeks, or permanent discontinuation of the randomized NVP or LPV/r component of the study treatment by 24 weeks (for any reason including death). Secondary endpoints included changes in immunologic (assessed by CD4%) and growth (assessed by World Health Organization (WHO) weight and height-for-age- z-scores) outcomes, both crucial in HIV-1 infected infants in resource-limited settings. The risk/benefit outcome was used to retrospectively classify participant response to initiating ART in P1060 and results were compared to those from the study’s primary and secondary outcomes.
Results
P1060 primary and secondary study outcomes
Among the 451 participants who started treatment in P1060, the primary week 24 study endpoint for virologic failure/going off study treatment was met by 28.1% of participants in the NVP arm and 12.3% in the NVP arm (a difference of 15.9%, 95% confidence interval (CI): 8.3%, 23.4%, p=0.004 in favor of LPV/r). In contrast, average increases by week 48 in CD4% and weight growth were higher in the NVP arm: CD4%: 15.2% for the NVP arm and 13.6% for the LPV/r arm (a difference of 1.6%, 95% CI: −0.3, 3.4, p=0.09); weight-for-age z-scores: 1.19 for the NVP arm and 0.83 for the LPV/r arm (a difference of 0.36, 95% CI: 0.06, 0.65, p=0.017). Mean CD4% and weight z-scores by study visit from entry to week 48 are shown in Figure 1. Mean values for both outcomes were higher in the NVP arm at all time points. There were 20 deaths (15 in the NVP arm and 5 in the LPV/r arm) and 23 (5%, 12 in the NVP arm and 11 in the LPV/r arm) participants were lost-to-follow-up before week 48.
Figure 1.
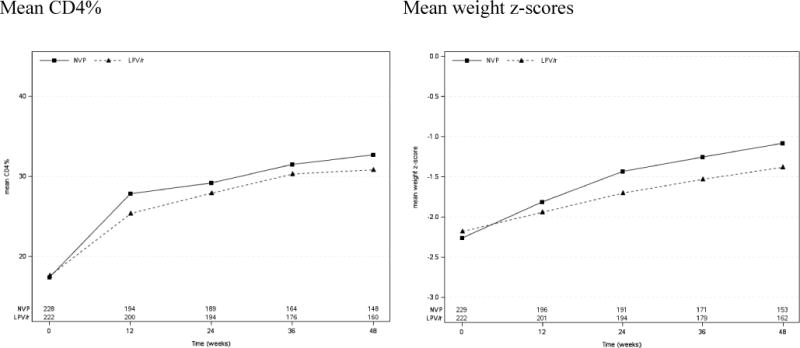
Mean CD4% and weight z-scores by treatment group
Risk/benefit outcome
Shown in Table 2 are the distributions by treatment group of the levels of response in each of the six components and the overall risk/benefit outcome. Corresponding with the primary analyses, the LPV/r arm had higher percentages of participants classified as “responders” for the HIV-1 RNA component (66% vs. 52% in the NVP arm, Cochran-Armitage test for trend across 4 responder levels p<0.001) and the toxicity/tolerability component (60% vs. 53% for the NVP arm, p=0.012). In contrast with the primary analyses, for the CD4% and weight growth components, the LPV/r arm did as well as or better than the NVP arm. For weight growth, 79% of those in the LPV/r arm were classified as “responders” or “partial responders” versus 77% in NVP arm (p=0.80) and for CD4%, these percentages were 94% for LPV/r and 87% for NVP (p=0.029 in favor of LPV/r). For the overall risk/benefit outcome, which also captured information on hospitalizations, the LPV/r arm had a higher proportion of “responders” (18% vs. 12%) and “partial responders” (30% vs. 21%) than the NVP arm (p=0.002).
Table 2.
Distribution of the overall risk/benefit outcome and its defining components by treatment group (n (%))
| Randomized Treatment | Cochran-Armitage trend test | |||
|---|---|---|---|---|
| Outcome | Category | NVP (N=229) | LPV/r (N=222) | p-value |
| Overall Outcome Category | Responder | 28 (12) | 39 (18) | 0.002 |
| Partial Responder | 49 (21) | 66 (30) | ||
| Poor Responder | 28 (12) | 28 (13) | ||
| Non-responder | 124 (54) | 89 (40) | ||
| Outcome category for HIV-1 RNA | Responder | 118 (52) | 147 (66) | <0.001 |
| Partial Responder | 17 (7) | 20 (9) | ||
| Poor Responder | 3 (1) | 3 (1) | ||
| Non-responder | 91 (40) | 52 (23) | ||
| Outcome category for toxicities | Responder | 121 (53) | 133 (60) | 0.012 |
| Partial Responder | 59 (26) | 67 (30) | ||
| Poor Responder | 30 (13) | 10 (5) | ||
| Non-responder | 19 (8) | 12 (5) | ||
| Outcome category for hospitalizations | Responder | 153 (67) | 176 (79) | 0.001 |
| Partial Responder | 7 (3) | 13 (6) | ||
| Poor Responder | 52 (23) | 23 (10) | ||
| Non-responder | 17 (7) | 10 (5) | ||
| Outcome category for weight-for-age z-score | Responder | 120 (52) | 116 (52) | 0.80 |
| Partial Responder | 57 (25) | 59 (27) | ||
| Poor Responder | 16 (7) | 15 (7) | ||
| Non-responder | 36 (16) | 32 (14) | ||
| Outcome category for CD4% | Responder | 164 (72) | 169 (76) | 0.029 |
| Partial Responder | 35 (15) | 40 (18) | ||
| Poor Responder | 5 (2) | 3 (1) | ||
| Non-responder | 25 (11) | 10 (5) | ||
Discussion
We have proposed using outcomes to evaluate treatments in clinical trials that capture information on efficacy and toxicity in order to assess an overall risk/benefit profile. We illustrated the approach using existing data from a completed trial in HIV-1 infected children. In this study, although LPV/r-based ART was superior for the primary efficacy outcome, there was some evidence of better outcomes for NVP-based treatment for two secondary endpoints. Our example study was not designed or analyzed using a risk/benefit outcome but using this approach retrospectively provided additional insight to published results. By carefully defining what an optimal response to treatment would be at an individual patient-level, and comparing proportions of participants with this response, the LPV/r-based ART turned out to be as good as or better than the NVP-based ART for growth and immunologic outcomes – a finding inconsistent with the more standard population-averaged comparisons. The LPV/r-based ART also emerged as superior on the overall risk/benefit outcome.
The team performed several sensitivity analyses. Instead of using a Cochran-Armitage trend test to compare the profiles of response for the overall risk/benefit outcome between treatment arms, a model-based analysis using a proportional odds model was used ((5, 6)). Additionally, the team applied three different scoring systems based on the final consensus grid, and analyzed them as continuous variables. Finally, we used a single summary measure of the difference between treatments based on the “general risk difference” ((7–9)). The general risk difference measures the difference in proportion of participants expected to have a better response on LPV/r versus NVP from the proportion expected to have a better response on NVP versus LPV/r. All analyses supported the superiority of LPV/r versus NVP with respect to the patient level risk/benefit outcome. These analyses should help alleviate concern in the pediatric HIV-1 community as to the best initial choice of ART regimen for HIV-1 infected infants and children in resource-limited settings.
In our illustrative example, the overall risk/benefit outcome measure was constructed retrospectively via consensus by four clinicians involved in the treatment of pediatric HIV-1 infection in resource-limited settings in which the trial was undertaken. Developing a clear definition of the risk/benefit outcome is not straightforward. Even among these four clinicians there was extensive debate on which outcomes should be included and how each level of response should be defined, and they anticipate that not all their colleagues will agree with the proposed definitions. Ideally, outcome measures would be defined by a broader panel of clinical experts not associated with any specific clinical trial before a study is developed and initially use the definitions in clinical trials as secondary outcomes, perhaps adapting the definitions as more is learned. This would allow the same outcome to be used across clinical trials and that results of studies using the accepted risk/benefit outcome measure definition would be generally accepted in the field. As the construction of the risk/benefit outcome may involve subjective components (e.g.: clinical response), use of double-blind design or blinded adjudication committees should also be considered (10, 11). Evans et al. provide a useful summary of suggestions for the risk/benefit outcome construction. Such an outcome should at minimum consider the relative timing of events, the severity of events, the censoring of events from competing risks and the challenge of relative interpretation (11).
We used an example in pediatric HIV-1 to illustrate the approach, but creation of a risk/benefit outcome is relevant in any disease area or study population. To ensure applicability of outcomes for use by clinicians in a general clinical setting, the components should be based on information collected in the clinic. This was largely the case with the components used in the example study we used, which included HIV-1 RNA (used to monitor efficacy in clinical trials and the clinic), adverse events and hospitalizations (information that would be available in the clinic), weight (always collected in pediatric visits) and CD4% (perhaps not as regularly collected in resource-limited settings). The rules for defining levels of response should also be straightforward for application in the clinic. The outcomes chosen in this example were either based on measurements at week 48, events that occurred between entry and week 48, or changes from baseline to week 48, which would all be simple for a clinician to assess in the clinic. Finally, the outcomes (efficacy and toxicity) themselves would need to be carefully chosen and be relevant to the disease and the anticipated types of toxicities. In the example study, adverse events were generic (any labs, signs, symptoms or diagnoses above a certain level of severity). With some treatments, there could be specific side effects that could be parsed out into separate components of the overall risk/benefit outcome.
The use of composite outcomes in clinical trials is not a new idea. The construction of four ordered “response” categories for the patient-level risk/benefit outcome in this example HIV-1 study was based on each individual patient’s data for several clinically important outcomes observed during 48 weeks of follow-up. This extends a common practice in studies of some diseases of using a well-defined “response” categorization as the study outcome, for example categories of tumor response in oncology studies. It also expands the use of composite outcomes in some studies, for example an outcome which includes different types of cardiovascular events to measure treatment efficacy. Of note however, the choice of component outcomes in our application included ones which might measure the beneficial effects of treatments on the disease being treated, as well as ones which might measure the adverse effects or toxicities of treatment. Finkelstein and Schoenfeld proposed a methodology of how to combine mortality with other longitudinal measures (12). Rather than defining an ordered categorical response, one could define a numerical scoring system to order the participant’s responses to treatment (11, 13–15). A more continuous study outcome may result in greater statistical power to detect differences in treatment effects, but the challenge would be defining clinically meaningful differences in these potentially artificial scores. This approach would also be more difficult to extend to general clinical practice. There are also methodological extensions to the simple approach taken in this paper addressing how to better handle participants lost-to-follow-up (9). Finally, the primary endpoint in the HIV-1 example study we used (virologic failure or going off study treatment) was a composite endpoint made up of efficacy and drug tolerability, which is a miniature version of what we are proposing.
Some argue that studies should be designed with efficacy as the primary endpoint and toxicity/tolerability as secondary outcomes. However, drugs that are highly efficacious are not always the most practical (effective) to use in general clinical practice if a patient cannot tolerate the drug. Choice of a pure efficacy endpoint or a composite risk/benefit outcome will depend on the goals of the individual clinical trial, with composite outcomes more relevant in large Phase III/IV studies and pure efficacy endpoints for Phase II designs.
The goal of this article was to point out the pitfalls of relying on marginal, population-averaged estimates of treatment effects and compartmentalized efficacy and safety outcomes as is the current convention in the design and analysis of most clinical trials. We instead propose the use of composite risk/benefit outcomes when evaluating treatments. These outcomes might better capture risk/benefit profiles at the individual patient-level, and provide more informative data on which to base decisions in general clinical practice.
Acknowledgments
We thank Sean Brummel, PhD, Scott Evans, PhD and David Shapiro, PhD from Harvard T.H Chan School of Public Health for helpful review and comments on the manuscript. We thank the children, their families, and the care providers who agreed to participate in the IMPAACT P1060 trial and place their trust in the site study teams.
Funding/support
This research was supported by the R01 AI024643 grant from the National Institutes of Health. Overall support for the International Maternal Pediatric Adolescent AIDS Clinical Trials (IMPAACT) Network was provided by the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health (NIH) under Award Numbers UM1AI068632 (IMPAACT LOC), UM1AI068616 (IMPAACT SDMC) and UM1AI106716 (IMPAACT LC), with co-funding from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) and the National Institute of Mental Health (NIMH). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Trial registration: ClinicalTrials.gov Identifier: NCT00307151
Conflict of interest disclosures: The authors declare that there are no conflicts of interest.
References
- 1.Palumbo MD Paul, Lindsey, ScD Jane C, Hughes, PhD Michael D, Cotton, MMed, PhD Mark F, Bobat, MD Raziya, Meyers, MD Tammy, Bwakura-Dangarembizi, MD Mutsawashe, Chi, MD Benjamin H, Musoke, MB, ChB Philippa, Kamthunzi, MD Portia, Schimana, MD Werner, Purdue PD Lynette, Eshleman, MD, PhD Susan H, Abrams, MD Elaine J, Millar, BA Linda, Petzold, PhD Elizabeth, Mofenson, MD Lynne M, Jean-Philippe, MD Patrick, Violari FCPaed Avy. Antiretroviral Treatment for Children with Peripartum Nevirapine Exposure. N Engl J Med. 2010;363(16):1510–20. doi: 10.1056/NEJMoa1000931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Violari FCP Avy, Lindsey, ScD Jane C, Hughes, PhD Michael D, Mujuru, MD Hilda A, Barlow-Mosha, MD Linda, Kamthunzi, MD Portia, Chi MD Benjamin H, Cotton, MMed Mark F, Moultrie, MD Harry, Khadse MD Sandhya, Schimana, MD Werner, Bobat MD Raziya, Purdue, PharmD Lynette, Eshleman, MD, PhD Susan H, Abrams, MD Elaine J, Millar, BA Linda, Petzold, PhD Elizabeth, Mofenson MD Lynne M, Jean-Philippe MD Patrick, Palumbo, MD Paul. Nevirapine versus Ritonavir-Boosted Lopinavir for HIV-Infected Children. N Engl J Med. 2012;366(25):2380–9. doi: 10.1056/NEJMoa1113249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lindsey S Jane C, Hughes, PhD Michael D, Violari, MD Avy, Eshleman, MD, PhD Susan H, Abrams, MD Elaine J, Bwakura-Dangarembizi, MD Mutsa, Barlow-Mosha, MD Linda, Kamthunzi, MD Portia, Sambo, M Med Pauline M, Cotton, M Med Mark F, Moultrie, MD Harry, Khadse, MD Sandhya, Schimana, MD Werner, Bobat, MD Raziya, Zimmer, BS Bonnie, Petzold, PhD Elizabeth, Mofenson, MD Lynne M, Jean-Philippe, MD Patrick, Palumbo, MD Paul, for the P1060 Study Team Predictors of virologic and clinical response to nevirapine versus lopinavir/ritonavir-based antiretroviral therapy in young children with and without prior nevirapine exposure for the prevention of mother-to-child HIV transmission. Pediatr Infect Dis. 2014;33(8):846–54. doi: 10.1097/INF.0000000000000337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuhn L, Coovadia A, Strehlau R, Martens L, Hu C-C, Meyers T, et al. Switching children previously exposed to nevirapine to nevirapine-based treatment after initial suppression with a protease-inhibitor-based regimen: long-term follow-up of a randomised, open-label trial. The Lancet Infectious Diseases. 2012;12(7):521–30. doi: 10.1016/S1473-3099(12)70051-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Agresti A. An Introduction to Categorical Data Analysis. (2nd) 2007 [Google Scholar]
- 6.Hosmer DW, Lemeshow S. Applied Logistic Regression. (2nd) 2000 [Google Scholar]
- 7.DeLong DMD Elizbeth R, Clarke-Pearson Daniel L. Comparing the areas under two or more correlated receiver operating characteristics curves: a nonparametric approach. Biometrics. 1988;44:837–45. [PubMed] [Google Scholar]
- 8.Halperin Max, H MI, Thall Peter F. Distribution-Free Confidence Intervals for a Parameter of Wilcoxon-Mann- Whitney Type for Ordered Categories and Progressive Censoring. Biometrics. 1989;45(2):509–21. [PubMed] [Google Scholar]
- 9.Claggett B, Tian L, Castagno D, Wei LJ. Treatment selections using risk-benefit profiles based on data from comparative randomized clinical trials with multiple endpoints. Biostatistics. 2015;16(1):60–72. doi: 10.1093/biostatistics/kxu037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Evans S, Ting N. Fundamental Concepts for New Clinical Trialists. 2016 [Google Scholar]
- 11.Evans SR, Rubin D, Follmann D, Pennello G, Huskins WC, Powers JH, et al. Desirability of Outcome Ranking (DOOR) and Response Adjusted for Duration of Antibiotic Risk (RADAR) Clinical infectious diseases: an official publication of the Infectious Diseases Society of America. 2015;61(5):800–6. doi: 10.1093/cid/civ495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finkelstein DM, Schoenfeld DA. Combining mortality and longitudinal measures in clinical trials. Statistics in medicine. 1999;18:1341–54. doi: 10.1002/(sici)1097-0258(19990615)18:11<1341::aid-sim129>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 13.Chuang-Stein C. A new proposal for benefit-less-risk analysis in clinical trials. Control Clin Trials. 1994;15(1):30–43. doi: 10.1016/0197-2456(94)90026-4. [DOI] [PubMed] [Google Scholar]
- 14.Follmann D, Wittes J. The use of subjective rankings in clinical trials with application to cardiovascular disease. Statistics in medicine. 1992 doi: 10.1002/sim.4780110402. [DOI] [PubMed] [Google Scholar]
- 15.Stone A, Chuang-Stein C. Strong control over multiple endpoints: are we adding value to the assessment of medicines? Pharmaceutical statistics. 2013;12(4):189–91. doi: 10.1002/pst.1574. [DOI] [PubMed] [Google Scholar]