

Abstract
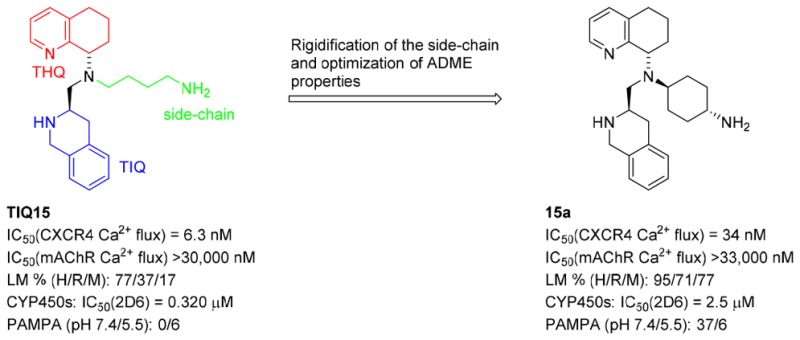
A structure–activity relationship study of potent TIQ15-derived CXCR4 antagonists is reported. In this investigation, the TIQ15 side-chain was constrained to improve its drug properties. The cyclohexylamino congener 15a was found to be a potent CXCR4 inhibitor (IC50 = 33 nM in CXCL12-mediated Ca2+ flux) with enhanced stability in liver microsomes and reduced inhibition of CYP450 (2D6). The improved CXCR4 antagonist 15a has potential therapeutic application as a single agent or combinatory anticancer therapy.
Keywords: CXCR4, TIQ15, anticancer drugs, antagonist
One of the most commonly investigated chemokine receptors is CXCR4 (CXC chemokine receptor 4), a seven transmembrane G protein-coupled receptor whose only endogenous ligand is CXCL12 (CXC chemokine ligand 12), also known as stromal cell-derived factor 1 (SDF-1).1 CXCL12 regulates the trafficking of CXCR4+ leukocytes and hematopoetic stem cells under normal physiological conditions (homeostasis). The binding of CXCL12 induces CXCR4+ leukocytes to migrate along chemokine gradients toward sites with high concentrations of CXCL12 (liver, lungs, kidneys, heart, skin, and bone marrow).2 CXCR4 also plays a crucial role in pathological conditions, such as cancer, HIV/AIDS, and autoimmune diseases,3 as well as in important normal and aberrant physiological processes, such as chemotaxis, metastasis, angiogenesis, and tumor growth. In addition, CXCR4 is highly overexpressed in more than 23 hematopoietic and solid tumor types.4 Overexpression of CXCR4 in pancreatic adenocarcinoma,5 soft-tissue sarcoma,6 and ovarian cancers7 serves as biomarkers that indicate a poor prognosis for clinical outcome. The effects of CXCL12 binding to CXCR4 can be divided in two groups: (a) direct autocrine and paracrine effects, which activate specific pro-survival signaling pathways in the cancer cells, and (b) indirect effects, which include CXCR4+ cell migration to CXCL12 expressing sites, e.g., cancer metastasis.2,8 The CXCR4/CXCL12 axis accounts for approximately 75% of the metastatic phenotype of all cancers.9 Inhibition of the CXCL12 and CXCR4 interactions could decrease organ-targeted metastasis. For example, the small molecule CXCR4 inhibitor, AMD3100 (Figure 1), has been used in animal models to suppress metastasis.10 AMD3100 is also used for hematopoietic stem cell mobilization for transplantations in the case of non-Hodgkin lymphoma and multiple myeloma (FDA approved) and for chemotherapeutic sensitization in the treatment of hematologic cancers.11−13
Figure 1.

Small molecule CXCR4 antagonists.
Cancer growth is promoted by several CXCR4 downstream signaling pathways, e.g., activation of MAPK (mitogen activated protein kinase) and PI3K (phosphinositide 3 kinase)/AKT (protein kinase B) pathways that induce cell proliferation. Activation of MAPK also upregulates CXCR4 expression, creating a positive feedback loop for cancer progression.2 Deactivation of canonical Wnt signaling pathway and activation of NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) also result in cell proliferation, but the mechanism of action is through suppression of cell apoptosis. In addition to stimulating cancer growth, CXCL12 binding induces intracellular calcium flux, and inhibition of this activity is a common measurement for CXCR4 antagonist potency. The small molecule CXCR4 antagonist, AMD3100, interrupts CXCR4 signaling, inhibiting the progression of a variety of cancers, including breast cancer,14 UV-induced skin cancer,15 prostate cancer,16 and ovarian cancer.17 In addition, promising results have been achieved using AMD3100 in combination with 1,3-bis(2-chloroethyl)-1-nitrosourea chemotherapy for glioblastomas.18 AMD3100 exhibited an anti-immunosuppression effect as a single agent in the case of CXCR4+/CXCL12+ ovarian cancer,19 as well as synergy with immunotherapeutics for CXCR4low pancreatic ductal adenocarcinoma20 and CXCR4+/CXCL12+ hepatocellular carcinoma.21 Taken together, these preclinical studies represent a proof of concept that CXCR4 antagonists can suppress CXCR4+ tumors, either as a single agent or in combination with immunotherapeutics.
Several potent small molecule CXCR4 antagonists (e.g., AMD3100, AMD11070, GSK812397, and IT1t; Figure 1) are known.22 Recently, TIQ15, a potent CXCR4 antagonist (IC50 = 3 nM for CXCL12-induced Ca2+ flux and IC50 = 5 nM for MAGI HIV-1IIIB), was reported by our group. Similar activities were observed for inhibition of cAMP production (IC50 = 19 nM) and β-arrestin recruitment (IC50 = 15 nM), substantiating the applicability of the Ca2+ flux assay as an indicator of CXCR4 downstream signaling. TIQ15 showed a good safety profile with low cytotoxicity (TC50 = 47 μM; TC50 = 50% cytotoxicity). In addition, this compound did not potently target other chemokine receptors, nor the muscarinic acetylcholine receptor (mAChR), which we used as a preliminary indication of off-target activity.
Although TIQ15 exhibited good metabolic stability in human liver microsomes, stability in rodent liver microsomes was unsatisfactory, effectively precluding the use of preclinical rodent models for studying in vivo efficacy. In addition, reinvestigation of CYP450 inhibitory activity revealed a significant inhibition of CYP450 (2D6) (IC50 = 320 nM), which would severely limit its use in combination therapies.
Since most oral drugs are absorbed by passive transcellular absorption, the use of a parallel artificial membrane permeability assay (PAMPA) seemed well-suited for assessing the permeability of our compounds.23 While the studies by Truax et al. reported good water solubility for TIQ15 (>100 μg/mL in pH 7.4 buffer) and good oral availability in rats (F = 63%),22 TIQ15 exhibited low intestinal permeability (PAMPA values close to zero), probably as a consequence of the primary and secondary amino groups, which are protonated at physiological pH. Thus, we sought to reduce the pKA of the nitrogens to increase compound lipophilicity and permeability. Simultaneously, the use of bulky groups to hinder solvation of the protonated amine might improve intestinal permeability.
Since prior studies suggested that rigidification of the butylamine side-chain of the structurally related CXCR4 antagonist, AMD11070, enhanced its activity, we decided to assess the effects of rigidifying it in our series.24 Compounds in which an olefin was introduced to increase rigidity were synthesized from the common intermediate, secondary amine 1, and the respective side-chains 2a–i, 8a–d, and 12. The diastereomerically pure amine 1 was synthesized following the procedure developed in our group,22 and the side-chain precursors were either purchased or synthesized as described in the Supporting Information (Scheme 1). The alkenyl side-chains were attached by alkylation of 1 with bromides 2a–g, followed by removal of the phthalimide and Boc protecting groups to afford the final compounds 4a–g in good yields. The compounds 4h–i were synthesized by alkylation of 1 with either (E)- or (Z)-((4-bromo-3-fluoro-2-methylbut-2-en-1-yl)oxy)(tert-butyl)dimethylsilane (2h–i), followed sequentially by TBS deprotection, Mitsunobu reaction with phthalimide, and global deprotection. Compounds 6 and 7 were prepared from amine 5 by reductive amination with 4,4-difluorocyclohexanone or by a reaction with (CH3)3SiNCO, respectively, followed by CF3COOH-induced Boc deprotection.
Scheme 1. Synthesis of Compounds 4a–i, 6, and 7.

Reagents and conditions: (i) 0.1 equiv of KI, 1.5 equiv of Et(iPr)2N, acetonitrile, 60–78%; (ii) 1.5 equiv of 1 M n-Bu4N+F– (THF), 53%; (iii) 1.2 equiv of phthalimide, 1.15 equiv of PPh3, 1.2 equiv of DIAD, THF, 78–100%; (iv) 8 equiv of 24 wt % N2H4 in water, MeOH, quant.; (v) 30 equiv of CF3COOH, CH2Cl2, 43–100%; (vi) 1.3 equiv of 4,4-difluorocyclohexanone, 1.2 equiv of CH3COOH, 1.5 equiv of NaBH(OAc)3, DCE, 81%; (vii) 1.3 equiv of 85 wt % (CH3)3SiNCO, 2 equiv of Et(iPr)2N, THF, 86%.
Next, saturated carbocyclic side-chains with increased rigidity were installed. Alkyl halides were found to react significantly slower in SN2 reactions as compared to allyl halides; thus, we employed reductive amination reactions for the synthesis of these analogs. To that end, compounds 10a–d were prepared by reductive amination of amine 1 with aldehydes 8a–d as shown in Scheme 2.
Scheme 2. Synthesis of Compounds 10a–d and 15a–b.

Reagents and conditions: (i) 1.3 equiv of CH3COOH, 1.25 equiv of STAB, DCE, 76–94%; (ii) 8 equiv of 24 wt % N2H4 in water, MeOH, quant.; (iii) 30 equiv of CF3COOH, CH2Cl2, 49–100%; (iv) 1.5 equiv of CH3COOH, 1.5 equiv of NaBH(OAc)3, DCE, 72%; (v) 1.5 equiv of Ti(OiPr)4, 2 equiv of NaBH(OAc)3, CH2Cl2, 92–94%.
Applying the same synthetic approach, no desired product was detected in the synthesis of 15a and 15b as a result of a bulky cyclohexyl side chain. The order of reductive aminations was therefore changed. Instead of reductive amination between secondary amine 1 and cyclohexanone 12, the cyclohexanone was allowed to react with amine 11 to afford cis- and trans-amines 13a–b. The trans-geometry was assigned from X-ray crystal structure of 13a (see Supporting Information). Then, amine 13a–b was employed in a reductive amination with the more reactive and less bulky aldehyde 14. This reductive amination required the use of Ti(OiPr)4 to facilitate formation of the imine intermediate, which was reduced in situ by NaBH(OAc)3. The desired compounds 15a–b were obtained after Boc deprotection.
Table 1 shows the results of our investigation into the effects of various alkenyl side-chains on TIQ15. The degree of CXCR4 antagonism was measured by inhibition of CXCL12-mediated intracellular calcium flux. Muscarinic receptor (mAChR) off-target activity was similarly measured using the same cell line (CCRF-CEM). The introduction of Z- or E-alkene isomers rigidified the butyl amine side-chain of TIQ15. The decrease of CXCR4 Ca2+ flux activity for the E-isomer 4b suggested that the active binding conformation of TIQ15 is closer to the cis- and gauche-conformations. Further study of the drug properties for both isomers revealed negligible alteration of the metabolic stability in liver microsomes. Significant improvement of the PAMPA values, however, was observed due to lower basicity of allyl amines. Unfortunately, the introduction of the alkene motif slightly increased inhibition of the CYP450 (3A4 BFC) isoform as well as mAChR inhibition. In contrast, other CYP450 isoforms (1A2, 2C19, 2C9, 3A4 BZR, 2C8, 2B6, 2A6) were not inhibited. As a result of its potency, the Z-isomer of 4a was chosen for further investigation. Gratifyingly, cyclohexenyl analog (4c) showed dramatic improvement in off-target mAChR activity but, unfortunately, at the expense of CXCR4 inhibition. The loss of CXCR4 activity suggested either a conformational change to a less active binding pose or increased steric interaction with the receptor in the binding pocket due to the cylcohexyl ring.
Table 1. Strained TIQ-15 analogs.

| CXCR4 Ca2+ flux | mAChR M3 Ca2+ flux | PAMPA, nm/s |
CYP450 (2D6) | CYP 450 (3A4 BFC) | metabolic
stability, % remaining |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| entry | compd | IC50, nMa | IC50, μM | pH = 7.4 | pH = 5.5 | IC50, μM | IC50, μM | mouse | rat | human |
| 1 | TIQ15 | 6.3 | >30 | 0 | 6 | 0.32 | >20 | 17 | 37 | 77 |
| 2 | 4a | 10 | 0.61 | 570 | 39 | 0.33 | 13 | 4 | 4 | 100 |
| 3 | 4b | 130 | >16 | 830 | 260 | 0.51 | 9.3 | 11 | 34 | 100 |
| 4 | 4c | 230 | 23 | e | 170 | 0.18 | 11 | 2 | 1 | 65 |
| 5 | 4d | 8.6 | 0.032 | 11 | 4 | <0.027 | >20 | 2 | 1 | 100 |
| 6 | 4e | 28 | 1.1 | 250 | 83 | 0.088 | >20 | 2 | 2 | 96 |
| 7 | 4f | 80 | 9.6 | 550 | 280 | 0.19 | 13 | 1 | 6 | 51 |
| 8b | 4g | 1,800 | >17 | 700 | 0 | 0.11 | 1.8 | 15 | 61 | 35 |
| 9 | 4h | 28 | 0.77 | 490 | 50 | 0.059 | 15 | 2 | 2 | 100 |
| 10c | 4i | 44 | 8.8 | 220 | 0 | 0.14 | 2.4 | 61 | 72 | 97 |
| 11d | 6 | 98 | 11 | 13 | 0 | 0.54 | 12 | 3 | 1 | 44 |
| 12 | 7 | 210 | >16 | 110 | 13 | 0.47 | 19 | 2 | 2 | 73 |
See Supporting Information for standard deviations.
IC50 = 18 μM CYP450 (2C8).
IC50 = 19 μM CYP450 (2C19).
Some agonist activity.
Value was not obtained.
To avoid steric congestion, the Z-alkene was substituted with a smaller methyl group (4d and 4e). A slight improvement in CXCR4 activity was achieved in the case of 3-methyl substituted analog 4d. Unfortunately, the methyl group increased the inhibitory activity of CYP450 (2D6) and the binding affinity toward mAChR to 30 nM range, while no other CYP450 isoforms were inhibited. To avoid CYP450 (2D6) inhibition, it has been suggested to lower aliphatic amine pKA, overall lipophilicity, and topographical polar surface area.25 To achieve these properties, fluorine substitution was proposed (entries 7–10). The substitution would lower the pKA of the terminal amine, which would also likely improve the PAMPA values, which suffered after introducing the methyl substitution. Despite improvements in mAChR activity and permeability values (Table 1, 4f–i), the CYP450 (2D6) values stayed in nanomolar range and CYP450 (3A4) values went to single digit micromolar range. In addition, a 3-fluoro substitution decreased the CXCR4 Ca2+ flux activity. Interestingly, the fluoro substitution on 4i increased the metabolic stability in liver microsomes in mice and rats. Alternatively, a general trend of multiple CYP inhibition was recognized for the trans-fluoro alkenes 4g and 4i.
Our next approach involved modifying the terminal amino group by introducing heteroatom-containing motifs, which usually reduce the activity against CYP450 enzymes.26 4,4-Difluorocyclohexyl and urea groups were installed, moving mAChR activity into the micromolar range. Unfortunately, these modifications decreased CXCR4 activity. Although the PAMPA values were better for alkene side-chain analogs, the continued off-target activity required further investigation into other constrained side chains. Replacing unsaturation with saturation and decreasing lipophilicity usually improves the metabolic stability of compounds metabolized by CYP450 (2D6) and CYP450 (3A4), the enzymes responsible for most of drug metabolism.26
Next, CXCR4 inhibitory activity of carbocyclic side-chain analogs were investigated (Table 2). In order to maintain the favored cis-configuration, a cyclopropane ring was introduced (compounds 10a and 10b). Both of the cis-isomers were synthesized and resolved, and both were found to be potent CXCR4 antagonists. The absolute stereochemistry of the cyclopropane rings was not assigned due to almost identical activity profiles for both diastereomers. mAChR activity was greatly improved, although the PAMPA values slightly suffered. The absence of a double bond avoided the inhibition of CYP450 with the exception of CYP450 (2D6). Next, the cyclopropanes were replaced by cyclohexanes in hopes of improving the permeability and to decrease the affinity toward CYP450 (2D6). To avoid introducing new stereocenters, 1,4-cyclohexanes were selected. The trans-cyclohexane 15a was more potent than the cis-analog 15b in CXCR4 Ca2+ flux assay, although 15a was 4-fold less active compared to cyclopropyl congeners. No mAChR activity was observed for 15a, and the CYP450 (2D6) inhibitory activity was greatly improved. Interestingly, the inhibitory activity of 15b at CYP450 (2D6) was much higher, most likely due to the relative positions of the axial amino group and the lipophilic cyclohexyl ring. Compound 15a stands out due to its metabolic stability in rat and mouse liver microsomes, allowing evaluation of preclinical antitumor efficacy of this compound in animal models. The introduction of one or two methylene spacers in the side-chain (compounds 10c and 10d) did not improve the overall drug profile.
Table 2. Drug Properties of Cyclic Side-Chain Congeners of TIQ15.

| CXCR4 Ca2+ flux | mAChR M3 Ca2+ flux | PAMPA, nm/s |
CYP450 (2D6) | CYP 450 (3A4 BFC) | metabolic
stability, % remaining |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| entry | compd | IC50, nMa | IC50, μM | pH = 7.4 | pH = 5.5 | IC50, μM | IC50, μM | mouse | rat | human |
| 1b | 10a | 6.4 | >33 | 130 | 8 | 0.54 | >20 | 1 | 1 | 100 |
| 2 | 10b | 13 | 13 | d | 88 | 0.27 | >20 | 4 | 1 | 100 |
| 3 | 10c | 280 | >33 | 12 | 0 | 0.76 | 10 | 44 | 51 | 39 |
| 4c | 10d | 340 | 10 | d | 0 | 12 | 12 | 92 | 86 | 63 |
| 5 | 15a | 34 | >33 | 37 | 6 | 2.5 | >20 | 77 | 71 | 95 |
| 6 | 15b | 1,800 | >33 | 0 | 10 | 0.081 | >20 | 56 | 24 | 86 |
See Supporting Information for standard deviations.
Ymax for mAChR is 51%.
IC50 = 9.1 μM CYP450 (2C19), 4.1 μM CYP450 (2C8).
Value was not obtained.
Extensive structure–activity relationship studies of TIQ15 side chains were performed in the course of this study. Rigidification of the side-chain by an alkene moiety and the measurements of the CXCR4 activity suggest that the cis geometry is closer to the bioactive binding pose. Although the alkene moiety improved cell permeability, persistent off-target activity countered the positive effects. Allylic amine analogs also exhibited poor liver microsomal activity in all three animal models, which was resolved when alkenes were replaced with cyclohexanes, leading to potent compound 15a. The TIQ15 congener 15a has high liver microsome stability and improved off-target profile such as inhibition of CYP450 (2D6). The low intestinal permeability of 15a could potentially be improved by applying a prodrug strategy on the terminal amino group.
Acknowledgments
We thank Dr. John Bacsa, Emory X-ray Crystallography Facility, for the X-ray structural analysis. We also acknowledge the use of the Rigaku SYNERGY diffractometer, supported by the National Science Foundation under grant CHE-1626172.
Glossary
ABBREVIATIONS
- MAGI
multinuclear activation of galactosidase inhibitor assay
- HIV1IIIB
CXCR4 tropic HIV isolate
- cAMP
cyclic adenosine monophosphate
- DIAD
diisopropyl azodicarboxylate
- Boc
tert-butyloxycarbonyl
- Phth
phthalimide
- TBS
tert-butyldimethylsilyl
- DCE
1,2-dichloroethane
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00406.
Experimental details for all compound synthesis, bioassays, and compound characterization (PDF)
Author Contributions
E.J., E.J.M., R.J.W., H.H.N., Y.A.T., B.M.K., V.M.T., M.B.K., K.M.K., L.J.W., and D.C.L. synthesized and characterized the compounds discussed in this manuscript. T.W., C.S.S., M.E.C., and G.M.S. designed and performed all biological assays. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This study was financed by Bristol-Myers Squibb Research & Development center.
The authors declare the following competing financial interest(s): D.C.L. is the principle investigator on a research grant from Bristol-Myers Squibb Research and Development to Emory University. D.C.L., L.J.W., E.J.M., E.J., H.H.N., Y.A.T., R.J.W., V.T.T., and M.B.K. are co-inventors on Emory-owned Intellectual Property that includes CXCR4 antagonists.
Supplementary Material
References
- Ganju R. K.; Brubaker S. A.; Meyer J.; Dutt P.; Yang Y.; Qin S.; Newman W.; Groopman J. E. The α-chemokine, stromal cell-derived factor-1α, binds to the transmembrane G-protein-coupled CXCR-4 receptor and activates multiple signal transduction pathways. J. Biol. Chem. 1998, 273 (36), 23169–23175. 10.1074/jbc.273.36.23169. [DOI] [PubMed] [Google Scholar]
- Guo F.; Wang Y.; Liu J.; Mok S. C.; Xue F.; Zhang W. CXCL12/CXCR4: a symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. Oncogene 2016, 35 (7), 816–826. 10.1038/onc.2015.139. [DOI] [PubMed] [Google Scholar]
- Weitzenfeld P.; Ben-Baruch A. The chemokine system, and its CCR5 and CXCR4 receptors, as potential targets for personalized therapy in cancer. Cancer Lett. 2014, 352 (1), 36–53. 10.1016/j.canlet.2013.10.006. [DOI] [PubMed] [Google Scholar]
- Balkwill F. Cancer and the chemokine network. Nat. Rev. Cancer 2004, 4 (7), 540–550. 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- Maréchal R.; Demetter P.; Nagy N.; Berton A.; Decaestecker C.; Polus M.; Closset J.; Devière J.; Salmon I.; van Laethem J. L. High expression of CXCR4 may predict poor survival in resected pancreatic adenocarcinoma. Br. J. Cancer 2009, 100 (9), 1444–1451. 10.1038/sj.bjc.6605020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda Y.; Tateishi N.; Matono H.; Matsuura S.; Yamamaoto H.; Tamiya S.; Yokoyama R.; Matsuda S.; Iwamoto Y.; Tsuneyoshi M. Chemokine receptor CXCR4 expression is correlated with VEGF expression and poor survival in soft-tissue sarcoma. Int. J. Cancer 2009, 124 (8), 1852–1859. 10.1002/ijc.24128. [DOI] [PubMed] [Google Scholar]
- Popple A.; Durrant L. G.; Spendlove I.; Rolland P.; Scott I. V.; Deen S.; Ramage J. M. The chemokine, CXCL12, is an independent predictor of poor survival in ovarian cancer. Br. J. Cancer 2012, 106 (7), 1306–1313. 10.1038/bjc.2012.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teicher B. A.; Fricker S. P. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin. Cancer Res. 2010, 16 (11), 2927–2931. 10.1158/1078-0432.CCR-09-2329. [DOI] [PubMed] [Google Scholar]
- Cojoc M.; Peitzsch C.; Trautmann F.; Polishchuk L.; Telegeev G. D.; Dubrovska A. Emerging targets in cancer management: Role of the CXCL12/CXCR4 axis. OncoTargets Ther. 2013, 6, 1347–1361. 10.2147/OTT.S36109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida D.; Onoue T.; Kuribayashi N.; Tomizuka Y.; Tamatani T.; Nagai H.; Miyamoto Y. Blockade of CXCR4 in oral squamous cell carcinoma inhibits lymph node metastases. Eur. J. Cancer 2011, 47 (3), 452–459. 10.1016/j.ejca.2010.09.028. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Recent advances on the use of the CXCR4 antagonist plerixafor (AMD3100, Mozobil) and potential of other CXCR4 antagonists as stem cell mobilizers. Pharmacol. Ther. 2010, 128 (3), 509–518. 10.1016/j.pharmthera.2010.08.009. [DOI] [PubMed] [Google Scholar]
- Weisberg E.; Azab A. K.; Manley P. W.; Kung A. L.; Christie A. L.; Bronson R.; Ghobrial I. M.; Griffin J. D. Inhibition of CXCR4 in CML cells disrupts their interaction with the bone marrow microenvironment and sensitizes them to nilotinib. Leukemia 2012, 26 (5), 985–990. 10.1038/leu.2011.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azab A. K.; Runnels J. M.; Pitsillides C.; Moreau A.-S.; Azab F.; Leleu X.; Jia X.; Wright R.; Ospina B.; Carlson A. L.; Alt C.; Burwick N.; Roccaro A. M.; Ngo H. T.; Farag M.; Melhem M. R.; Sacco A.; Munshi N. C.; Hideshima T.; Rollins B. J.; Anderson K. C.; Kung A. L.; Lin C. P.; Ghobrial I. M. CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood 2009, 113 (18), 4341–4351. 10.1182/blood-2008-10-186668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefort S.; Thuleau A.; Kieffer Y.; Sirven P.; Bieche I.; Marangoni E.; Vincent-Salomon A.; Mechta-Grigoriou F. CXCR4 inhibitors could benefit to HER2 but not to triple-negative breast cancer patients. Oncogene 2017, 36 (9), 1211–1222. 10.1038/onc.2016.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarchio S. N. E.; Scolyer R. A.; Beaugie C.; McDonald D.; Marsh-Wakefield F.; Halliday G. M.; Byrne S. N. Pharmacologically antagonizing the CXCR4-CXCL12 chemokine pathway with AMD3100 inhibits sunlight-induced skin cancer. J. Invest. Dermatol. 2014, 134 (4), 1091–1100. 10.1038/jid.2013.424. [DOI] [PubMed] [Google Scholar]
- Kang Su C. H. O.; So Jung Y.; Joo Yong L. E. E.; Nam Hoon C. H. O.; Young Deuk C.; Yun Seob S.; Sung Joon H. Inhibition of tumor growth and histopathological changes following treatment with a chemokine receptor CXCR4 antagonist in a prostate cancer xenograft model. Oncol. Lett. 2013, 6 (4), 933–938. 10.3892/ol.2013.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray P.; Lewin S. A.; Mihalko L. A.; Schmidt B. T.; Luker K. E.; Luker G. D. Noninvasive imaging reveals inhibition of ovarian cancer by targeting CXCL12-CXCR4. Neoplasia 2011, 13 (12), 1152–1161. 10.1593/neo.111076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redjal N.; Chan J. A.; Segal R. A.; Kung A. L. CXCR4 inhibition synergizes with cytotoxic chemotherapy in gliomas. Clin. Cancer Res. 2006, 12 (22), 6765–6771. 10.1158/1078-0432.CCR-06-1372. [DOI] [PubMed] [Google Scholar]
- Righi E.; Kashiwagi S.; Yuan J.; Santosuosso M.; Leblanc P.; Ingraham R.; Forbes B.; Edelblute B.; Collette B.; Xing D.; Kowalski M.; Mingari M. C.; Vianello F.; Birrer M.; Orsulic S.; Dranoff G.; Poznansky M. C. CXCL12/CXCR4 blockade induces multimodal antitumor effects that prolong survival in an immunocompetent mouse model of ovariancancer. Cancer Res. 2011, 71 (16), 5522–5534. 10.1158/0008-5472.CAN-10-3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig C.; Jones J. O.; Kraman M.; Wells R. J. B.; Deonarine A.; Chan D. S.; Connell C. M.; Roberts E. W.; Zhao Q.; Caballero O. L.; Teichmann S. A.; Janowitz T.; Jodrell D. I.; Tuveson D. A.; Fearon D. T. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti–PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (50), 20212–20217. 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Ramjiawan R. R.; Reiberger T.; Ng M. R.; Hato T.; Huang Y.; Ochiai H.; Kitahara S.; Unan E. C.; Reddy T. P.; Fan C.; Huang P.; Bardeesy N.; Zhu A. X.; Jain R. K.; Duda D. G. CXCR4 inhibition in tumor microenvironment facilitates anti-programmed death receptor-1 immunotherapy in sorafenib-treated hepatocellular carcinoma in mice. Hepatology 2015, 61 (5), 1591–1602. 10.1002/hep.27665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truax V. M.; Zhao H.; Katzman B. M.; Prosser A. R.; Alcaraz A. A.; Saindane M. T.; Howard R. B.; Culver D.; Arrendale R. F.; Gruddanti P. R.; Evers T. J.; Natchus M. G.; Snyder J. P.; Liotta D. C.; Wilson L. J. Discovery of tetrahydroisoquinoline-based CXCR4 antagonists. ACS Med. Chem. Lett. 2013, 4 (11), 1025–1030. 10.1021/ml400183q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerns E. H.; Di L. Pharmaceutical profiling in drug discovery. Drug Discov. Today 2003, 8 (7), 316–323. 10.1016/S1359-6446(03)02649-7. [DOI] [PubMed] [Google Scholar]
- Skerlj R.; Bridger G.; McEachern E.; Harwig C.; Smith C.; Wilson T.; Veale D.; Yee H.; Crawford J.; Skupinska K.; Wauthy R.; Yang W.; Zhu Y.; Bogucki D.; Di Fluri M.; Langille J.; Huskens D.; De Clercq E.; Schols D. Synthesis and SAR of novel CXCR4 antagonists that are potent inhibitors of T tropic (X4) HIV-1 replication. Bioorg. Med. Chem. Lett. 2011, 21 (1), 262–266. 10.1016/j.bmcl.2010.11.023. [DOI] [PubMed] [Google Scholar]
- de Groot M. J.; Wakenhut F.; Whitlock G.; Hyland R. Understanding CYP2D6 interactions. Drug Discovery Today 2009, 14 (19), 964–972. 10.1016/j.drudis.2009.07.005. [DOI] [PubMed] [Google Scholar]
- Sun H.; Veith H.; Xia M.; Austin C. P.; Huang R. Predictive models for cytochrome P450 isozymes based on quantitative high throughput screening data. J. Chem. Inf. Model. 2011, 51 (10), 2474–2481. 10.1021/ci200311w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.