

Abstract
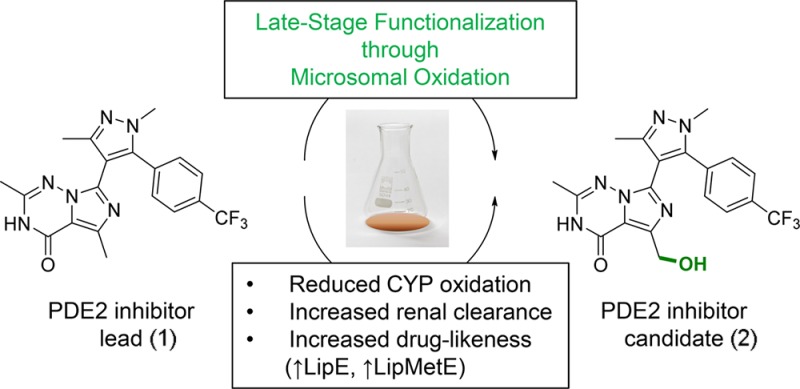
Late-stage oxidation using liver microsomes was applied to phosphodiesterase 2 inhibitor 1 to reduce its clearance by cytochrome P450 enzymes, introduce renal clearance, and minimize the risk for victim drug–drug interactions. This approach yielded PF-06815189 (2) with improved physicochemical properties and a mixed metabolic profile. This example highlights the importance of C–H diversification methods to drug discovery.
Keywords: late-stage oxidation, liver microsomes, renal clearance, drug−drug interactions, PDE2
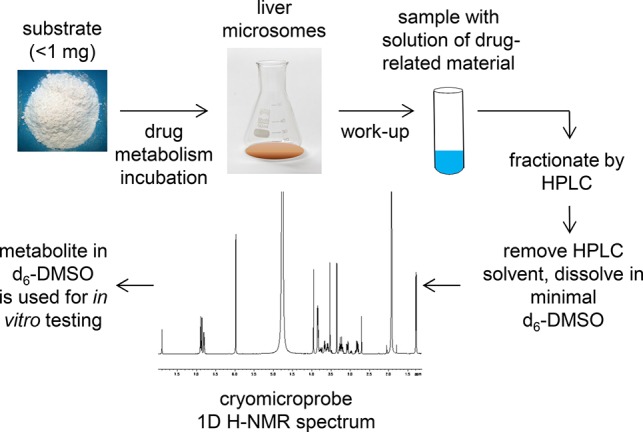
Late-stage C–H diversification has gained considerable attention in drug discovery.1,2 By adding small substituents to pre-existing drug scaffolds, these methods have the ability to accelerate analogue generation, therefore potentially reducing the high cost of pharmaceutical research.3 Examples of C–H functionalizations applied to drug molecules include the directed palladium-catalyzed functionalization of celecoxib.4 Rhodium-stabilized carbenoids facilitated the site-specific C–H insertion at the N-methyl functionality of various pharmaceuticals such as sercloremine.5 Radical-mediated functionalization of heterocylic drugs (e.g., caffeine) has been achieved using zinc sulfinate salts.6 Iridium-catalyzed C–H borylation has been applied to a c-Met kinase inhibitor.7 Hydroxylation using iron catalysis has been used for the diversification of amino acids and peptides.8 Alternative oxidation methods include biocatalysis with recombinant cytochrome P450 (CYP) enzymes and microbial preparations, electrochemistry, as well as biomimetic catalysis.9,10 We have previously described methods for metabolite biosynthesis that leverage various in vitro drug metabolism systems (e.g., liver microsomes) and quantitative nuclear magnetic resonance (NMR) spectroscopy using cryomicroprobe technology coupled with a mathematically inserted internal standard signal.11 In addition to utilizing this approach for the preparation of drug metabolites, we now use it routinely as a means for late-state lead diversification (see workflow in Scheme 1) and have recently disclosed the diversification of several known drugs to generate analogues with improved metabolic stability.12 This approach is particularly attractive if hydroxylation of the lead molecule does not negatively impact the analogue’s target organ distribution and may not be applicable to drugs that require central activity. Creation of small quantities of such products, quantified by NMR in DMSO-d6 solutions, permits these materials to be rapidly tested for target potency and examined in other in vitro assays. We herein report the application of this late-stage functionalization platform to the Pfizer phosphodiesterase 2A (PDE2A) inhibitor program: Oxidation of triazinone lead 1 identified potential clinical candidate 7-(1,3-dimethyl-5-(4-(trifluoromethyl)phenyl)-1H-pyrazol-4-yl)-5-(hydroxymethyl)-2-methylimidazo[5,1-f][1,2,4]triazin-4(3H)-one (PF-06815189, 2), which maintained parent potency but significantly reduced CYP metabolism. Its low oxidative turnover and high polarity introduced the desired amount of renal elimination in three preclinical species, minimizing the clinical victim drug–drug interaction (DDI) risk. This example therefore highlights the power of C–H diversification methods as a strategic tool to rapidly discover molecules with enhanced druglike properties since it allows generation of in vitro data without the need for de novo synthesis.
Scheme 1. Workflow of Late-Stage Lead Diversification Microsomal Screen.
PDE2A is one of 11 gene families of cyclic nucleotide-specific phosphodiesterases.13 It is a cyclic guanosine monophosphate (cGMP) activated enzyme that hydrolyses the secondary messengers cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) to the corresponding acyclic nucleotides (AMP and GMP). Under most conditions, it is cAMP hydrolysis that mediates effects of PDE2A inhibition to its corresponding acyclic nucleotide (AMP). Endothelial cells express phosphodiesterases, including PDE3, PDE5, and PDE2. These phosphodiesterases exert a coordinated effect on cAMP and cGMP levels and, by inhibiting PDE2, it is possible to raise cAMP levels in endothelial cells, ultimately leading to a reduction in vascular permeability. PDE2 inhibitors may, therefore, be a therapy for indications resulting from a dysregulation of vascular permeability such as stroke, sepsis, and acute lung injury.14
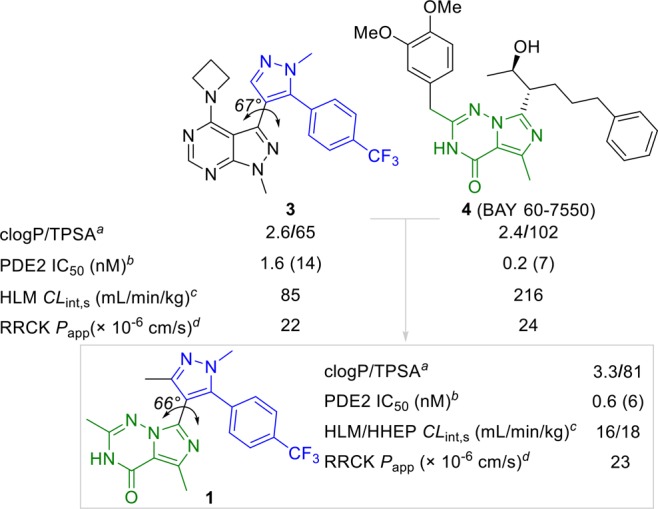
We recently disclosed pyrazolopyrimidine 3 as a brain penetrant PDE2 inhibitor for the treatment of neurological disorders (Scheme 2).15 Compound 3 binds to PDE2 analogously to cAMP, as described by Helal et al.15 In the binding of 4 (BAY 60-7550) to PDE2, however, the orientation of Gln859 is switched and resembles the cGMP binding mode.16−18 To design a proprietary inhibitor that binds the PDE2 enzyme similarly to 4, we substituted the pyrazolopyrimidine moiety in 3 with the triazinone core of 4, yielding analogue 1 (Scheme 2; see Scheme 3 for the synthesis of 1).
Scheme 2. Design Strategy to PDE2 Inhibitor 1.
Topological polar surface area.
Number of replicates in parentheses.
Intrinsic scaled clearance.
Apparent passive permeability in the Ralph Russ Canine Kidney cell line.19
Scheme 3. Synthesis of 1 and Biocatalytic Oxidation to 2.

Reagents and conditions: (a) Br2, DMF, 0–25 °C, 65%; (b) POCl3, NEt3, toluene, reflux; (c) pyrrolidine, NEt3, CH2Cl2, 0–25 °C, 77%, two steps; (d) Pd(dppf)Cl2·CH2Cl2, Na2CO3, dioxane/H2O, 110 °C, 62%; (e) 1 M HCl, THF, reflux, 86%; (f) S. aerocolonigenes ATCC 39243 in Iowa medium, DMSO, 30 °C, 7 days, 20%.
Docking of 1 into PDE2 suggests that its triazinone core has interactions similar to those found in the X-ray cocrystal structure of 4 (Figure 1).16 These include hydrogen bonds with Gln812, Gln859, Tyr655, and the backbone carbonyl of Asp808 and aromatic stacking and van der Waals interactions with the hydrophobic clamp residues Phe862 and Ile826. The p-trifluorophenyl group of 1 occupies a pocket distinct to PDE2 and necessary for selectivity over other PDEs.15 The additional methyl group on the pyrazole ring of 1, relative to 3, reduced the strain energy to adopt the bound conformation by enforcing a local minimum structure with a dihedral angle (pyrazole–imidazotriazinone) of ∼45°. This conformation requires less than 1 kcal/mol to adopt the bound conformation dihedral angle of ∼66°, mimicking the low-energy conformation of 3 (dihedral angle of 67°), optimal for the interaction of the p-trifluorophenyl group with the selectivity pocket. In contrast, without the additional methyl group, the local minimum structure has a dihedral angle of 0.0° and requires ∼3 kcal/mol to adopt the bound conformation.
Figure 1.
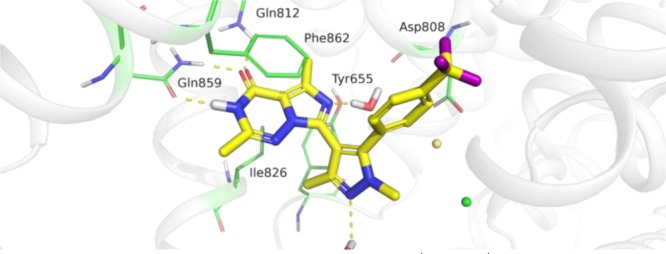
Modeled pose of compound 1 (yellow) in PDE2 showing key residue interactions (green sticks). Hydrogen-bond interactions are indicated by yellow dashed lines.
As shown in Scheme 2, compound 1 was characterized by a favorable potency (PDE2 IC50 = 0.6 nM) and moderate metabolic stability in human liver microsomes and hepatocytes (HLM/HHEP CLint,s = 16/18 mL/min/kg). Its lipophilic efficiency20 (LipE = 5.9) and lipophilic metabolism efficiency21 (LipMetE = 1.9) parameters also indicated an appropriate level of potency and metabolic stability for a lead compound. Triazinone 1 was evaluated in a panel of human recombinant CYPs, suggesting that CYP3A4 was the main cytochrome involved in the metabolism of 1. The in vivo rat clearance profile of 1 did also not show any extra-hepatic clearance pathways. Taken together, the in vitro and in vivo clearance data indicated that 1 has a distinct probability for exclusive clearance by CYP3A4, resulting in a risk for clinical victim DDI.22
To improve metabolic stability and lower DDI risk, a design strategy was implemented that reduced the lipophilicity of 1 in order to minimize oxidative turnover by CYP enzymes and increase renal elimination, giving a PDE2 inhibitor with a mixed clearance profile. Renal clearance typically increases with reduced lipophilicity and passive permeability, since both properties minimize passive reabsorption through the nephron.23,24 Since a hydroxy group reduces lipophilicity by more than half a log unit, a late-stage oxidation strategy was used to rapidly prepare hydroxylated analogues of 1 with appropriate lipophilicity.
Analogue 1 was subjected to metabolism by a panel of liver microsomal samples from several species, including human, and heterologously expressed common human P450 enzymes. The metabolite profiles were analyzed and compared by HPLC–MS, and it was determined that cynomolgus monkey liver microsomes offered the best promise of delivering new products with a good level of substrate turnover. A monkey liver microsomal incubation was therefore carried out at a substrate scale of 600 nmol in an incubation volume of 30 mL. From this single incubation, three new products were isolated: two monohydroxyl products (2 and 5) and one dihydroxy product (6, Table 1). Notably, these analogues maintain good inhibitory potency at PDE2 (PDE2 IC50 = 0.4–7.8 nM) and have higher metabolic stability (HLM CLint,s < 9.2 mL/min/kg) than parent analogue 1. Analogue 2 has the most favorable balance of potency and metabolic stability with significant improvements in both LipE (LipE = 7.4) and LipMetE (LipMetE = 2.4) values relative to lead 1, reflecting that hydroxylation of 1 enhanced drug-likeness. All three hydroxylated analogues are also in the appropriate low lipophilicity space (clogP = 0.7–2.0, TPSA = 101–121) for renal clearance, and analogue 2 has a 3-fold decreased passive permeability value relative to 1 (2: RRCK Papp = 8 × 10–6 cm/s vs 1: RRCK Papp = 23 × 10–6 cm/s), reducing the extent of reabsorption through the nephron and thus favoring renal clearance. Compound 2 also has good selectivity over other phosphodiesterases with, at least, a 1000-fold margin (PDE5/2 selectivity: 2439 nM/0.4 nM). Broader selectivity screening with 2 demonstrated no significant activities (i.e., IC50 or EC50 > 10 μM) in a broad spectrum BioPrint panel, in which the Na+ channel (site 2) had the greatest activity (17% inhibition of binding at 10 μM).
Table 1. In Vitro PDE2 Potency and Absorption, Distribution, Metabolism, and Excretion (ADME) Characteristics of Analogues Obtained through C–H Oxidation.
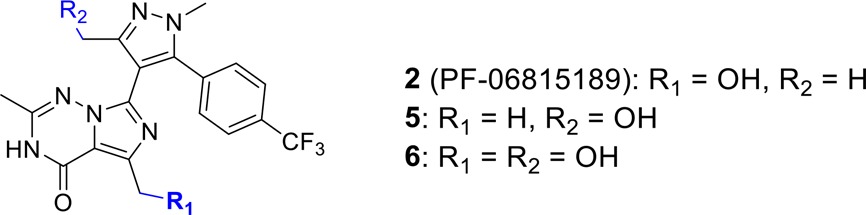
| 2 | 5 | 6 | |
|---|---|---|---|
| clogP/TPSAa | 2.0/101 | 2.0/101 | 0.7/121 |
| PDE2 IC50 (nM)b | 0.4 (5) | 6.9 (4) | 7.8 (4) |
| HLM/HHEP CLint,sc (mL/min/kg) | <8.0/<0.36 | <8.0/– | <9.2/– |
| RRCK Pappd (×10–6 cm/s) | 8 | 17 | 5 |
Topological polar surface area.
Number of replicates in parentheses.
Intrinsic scaled clearance.
Apparent passive permeability in the Ralph Russ Canine Kidney cell line.19
The small-scale microsomal biosynthesis enabled rapid identification of inhibitor 2 with desired attributes, resulting in accelerated progress on the PDE2 program. To access this new lead on scale for in vivo studies, a two-pronged approach was implemented reliant either on a biocatalytic or purely synthetic strategy. In the biocatalysis route, switching the microsomal to a more established, and cost-effective, microbial oxidation enabled the synthesis of hundreds of milligrams of 2 from 1, the synthesis of which is shown in Scheme 3. Bromination of known imidazotriazinone 7(25) gave compound 8, and subsequent chlorination with phosphoryl chloride and a SnAr reaction with pyrrolidine then furnished triazene 9. Pyrrolidine served as a protection group to enhance the ensuing cross-coupling reaction. Thus, reaction of 9 with boronate 10 under standard Suzuki conditions, followed by pyrrolidine removal under acidic conditions, furnished 1. More than 350 microorganisms were screened for the microbial oxidation of 1 to 2, and the optimized conditions utilized a 7-day incubation of 1 with Saccharothrix aerocolonigenes to yield 2 in 20% yield.
A purely synthetic approach was used to access multigram quantities of 2 for in vivo studies and relied on symmetrical imidazole 11 (Scheme 4). The electron-withdrawing nature of its diester groups facilitated anion formation and subsequent amination under mild conditions. Thus, amination of 11 with hydroxylamine O-sulfonic acid gave aminoimidazole 12 in 78% yield. Hydroxylamine O-sulfonic acid was selected as the aminating reagent due its superior safety profile based on differential scanning calorimetry and availability from commercial sources.26 Coupling of aminoimidazole 12 with ethyl acetimidate under basic conditions produced imidazotriazinone smoothly. Next, halogenation gave the desired bromide 13 in high yield. In analogy to the synthesis of 4, triazinone 13 was then masked as the corresponding pyrrolidine–triazene, this time via the activated triazole intermediate27 due to the high reactivity and instability of the corresponding triazene chloride. Suzuki cross-coupling with boronate 10 then gave intermediate 14 in excellent yield. Reduction of the ester group with lithium borohydride to 15 and removal of the pyrrolidine protecting group with hydrochloric acid, furnished candidate 2. Hence, this method afforded the target in eight steps from the diester–imidazole with a 26% overall yield.
Scheme 4. Synthesis of Candidate 2, Leveraging the Symmetry of Diester–Imidazole 11.

Reagents and conditions: (a) K2CO3, H2O/EtOH, H2NOSO3H, 0–25 °C, 78%; (b) DIPEA, 2-Me-THF, reflux; (c) NBS, HOAc/MeCN, 70 °C, 48%, 2 steps; (d) 1,2,4-triazole, POCl3, NEt3, CH2Cl2; (e) pyrrolidine, NEt3, CH2Cl2, 25 °C, 86%, 2 steps; (f) 10, Pd(dppf)Cl2·CH2Cl2, Na2CO3, dioxane/H2O, 110 °C, 91%; (g) LiBH4, THF, 50 °C, 98%; (h) 1 M HCl, THF, reflux, 92%.
The in vivo efficacy of compound 2 was assessed in the guinea pig Miles Assay, in which histamine, injected intradermally, increases vascular permeability as measured by the extravasation of Evan’s blue dye-labeled albumin from blood to skin.28 As shown in Figure 2, 2 was effective at reducing histamine-mediated increases in vascular permeability at all doses tested.
Figure 2.

Administration of 2 significantly reduced histamine-induced extravasation of Evan’s Blue dye in guinea pig skin (*p < 0.05, **p < 0.01, ***p < 0.005).
Due to its favorable pharmacological and in vitro ADME profile, the pharmacokinetic attributes of 2 were assessed in rat, dog and nonhuman primate (NHP). As shown in Table 2, 2 is characterized by low plasma clearance (CLp), low steady-state volume of distribution (Vss), short half-life (t1/2), and good bioavailability (%F = 50–79%). Compound 2 also had the desired renal clearance component in all three preclinical species, with 22, 48, and 26% renal clearance in rat, dog, and NHP, respectively. In accordance with the design strategy of introducing renal clearance by increasing compound polarity and minimizing compound reabsorption through the nephron, the renal excretion was passive in nature as indicated by the renal clearance being at or below the glomerular filtration rate. Integration of the in vitro and in vivo properties leads to predicted human pharmacokinetics for 2 that are characterized by a low plasma clearance (2.9 mL/min/kg), moderate volume of distribution (0.9 L/kg), and a moderate t1/2 of 3.7 h. Notably, a daily dose of only 1.3 mg is required to achieve an average PDE2 IC90 coverage over 24 h, reflecting the exquisite drug-like attributes of 2. Compound 2 was evaluated in 2-week rat and dog exploratory toxicology studies with adequate exposure multiples over the PDE2 IC50 and has been considered for indications caused by impaired vascular permeability.
Table 2. Preclinical Pharmacokinetics of Compound 2a.
| CLpb (mL/min/kg) | Vssc (L/kg) | t1/2d (h) | %Fe | % renal clearancef | |
|---|---|---|---|---|---|
| rat | 28 | 1.5 | 1.2 | 50 | 22 |
| dog | 1.3 | 0.4 | 4.8 | 79 | 48 |
| NHP | 4.0 | 0.8 | 3.6 | 61 | 26 |
N = 2 animals/dose in all studies.
Observed plasma clearance.
Steady-state volume of distribution.
Half-life.
Bioavailability.
Percent dose recovered in urine (0–24 h).
In summary, we have successfully applied a late-stage functionalization strategy using microsomal oxidation to the PDE2 inhibitor program at Pfizer. As reflected by the significant increases in the LipE and LipMetE values, this approach improved the drug-likeness of the early lead 1 and allowed rapid identification of the potential clinical candidate 2. Importantly, this hydroxylation also introduced the desired amount of renal clearance, minimizing the risk for clinical victim drug–drug interactions. This work therefore highlights the impact late-stage diversification technologies can have on drug discovery by accelerating the speed to novel analogues and fine-tuning properties to achieve an optimal balance of in vitro and in vivo ADME and potency attributes.
Acknowledgments
We thank the Pfizer ADME technology group for generating the in vitro data in Table 1, Brian Samas and Ivan Samardjiev for generating the X-ray single-crystal structure of compound 2, Matthew Teague for obtaining HRMS data, and Usa Reilly and Manjinder Lall for helpful discussions regarding this work.
Biography
Antonia F. Stepan graduated from the ETH Zurich and received her Ph.D. with Professor Steven V. Ley. Following postdoctoral research with Professor K. C. Nicolaou, Antonia joined Pfizer in 2008. She served as a chemistry and research project leader on discovery programs and advanced drug candidates to clinical candidate nomination and beyond. In 2017, Antonia joined Boehringer Ingelheim as a project leader. Antonia is passionate about discovering treatments for neurological and immunological disorders, as well as utilizing novel technologies (e.g., C−H functionalization reactions, sp3-rich fragments) in medicinal chemistry. She was recognized as an ACS “Young Investigator in Medicinal Chemistry” in 2015.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00343.
Dihedral angle calculation for 1, experimental procedures for the late-stage oxidation of 1, experimental procedures for the synthesis of 2, in vitro and in vivo compound characterization, single-crystal X-ray structure of 2, rat and dog toxicology study data of 2, recombinant CYP assay data for 1 (PDF)
Author Present Address
Medicinal Chemistry, Boehringer Ingelheim Pharma GmbH & Co. KG, 88307 Biberach an der Riss, Germany.
The authors declare no competing financial interest.
Supplementary Material
References
- Beck E. M.; Stepan A. F.; Webb D.. C–H Activation Approaches to Molecules; Royal Society of Chemistry, 2016; pp 274–383. [Google Scholar]
- Cernak T.; Dykstra K. D.; Tyagarajan S.; Vachal P.; Krska S. W. The medicinal chemist’s toolbox for late stage functionalization of drug-like molecules. Chem. Soc. Rev. 2016, 45, 546–576. 10.1039/C5CS00628G. [DOI] [PubMed] [Google Scholar]
- Kola I.; Landis J. Opinion: Can the pharmaceutical industry reduce attrition rates?. Nat. Rev. Drug Discovery 2004, 3, 711–716. 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- Dai H.-X.; Stepan A. F.; Plummer M. S.; Zhang Y.-H.; Yu J.-Q. Divergent C-H functionalizations directed by sulfonamide pharmacophores: Late-stage diversification as a tool for drug discovery. J. Am. Chem. Soc. 2011, 133, 7222–7228. 10.1021/ja201708f. [DOI] [PubMed] [Google Scholar]
- He J.; Hamann L. G.; Davies H. M. L.; Beckwith R. E. J. Late-stage C-H functionalization of complex alkaloids and drug molecules via intermolecular rhodium-carbenoid insertion. Nat. Commun. 2015, 6, 5943. 10.1038/ncomms6943. [DOI] [PubMed] [Google Scholar]
- Fujiwara Y.; Dixon J. A.; O’Hara F.; Funder E. D.; Dixon D. D.; Rodriguez R. A.; Baxter R. D.; Herle B.; Sach N.; Collins M. R.; Ishihara Y.; Baran P. S. Practical and innate carbon-hydrogen functionalization of heterocycles. Nature (London, U. K.) 2012, 492, 95–99. 10.1038/nature11680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen M. A.; Hartwig J. F. Iridium-catalyzed C-H borylation of heteroarenes: Scope, regioselectivity, application to late-stage functionalization, and mechanism. J. Am. Chem. Soc. 2014, 136, 4287–4299. 10.1021/ja412563e. [DOI] [PubMed] [Google Scholar]
- Osberger T. J.; Rogness D. C.; Kohrt J. T.; Stepan A. F.; White M. C. Oxidative diversification of amino acids and peptides by small-molecule iron catalysis. Nature (London, U. K.) 2016, 537, 214–219. 10.1038/nature18941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T.; Liu Y.; Yang X.; Martin G. E.; Yao H.; Shang J.; Bugianesi R. M.; Ellsworth K. P.; Sonatore L. M.; Nizner P.; Sherer E. C.; Hill S. E.; Knemeyer I. W.; Geissler W. M.; Dandliker P. J.; Helmy R.; Wood H. B. Definitive metabolite identification coupled with Automated Ligand Identification System (ALIS) technology: A novel approach to uncover structure-activity relationships and guide drug design in a Factor IXa inhibitor program. J. Med. Chem. 2016, 59, 1818–1829. 10.1021/acs.jmedchem.5b01293. [DOI] [PubMed] [Google Scholar]
- Cusack K. P.; Koolman H. F.; Lange U. E. W.; Peltier H. M.; Piel I.; Vasudevan A. Emerging technologies for metabolite generation and structural diversification. Bioorg. Med. Chem. Lett. 2013, 23, 5471–5483. 10.1016/j.bmcl.2013.08.003. [DOI] [PubMed] [Google Scholar]
- Walker G. S.; Bauman J. N.; Ryder T. F.; Smith E. B.; Spracklin D. K.; Obach R. S. Biosynthesis of drug metabolites and quantitation using NMR spectroscopy for use in pharmacologic and drug metabolism studies. Drug Metab. Dispos. 2014, 42, 1627–1639. 10.1124/dmd.114.059204. [DOI] [PubMed] [Google Scholar]
- Obach R. S.; Walker G. S.; Brodney M. A. Biosynthesis of fluorinated analogs of drugs using human cytochrome P450 enzymes followed by deoxyfluorination and quantitative nuclear magnetic resonance spectroscopy to improve metabolic stability. Drug Metab. Dispos. 2016, 44, 634–646. 10.1124/dmd.116.069310. [DOI] [PubMed] [Google Scholar]
- Azevedo M. F.; Faucz F. R.; Bimpaki E.; Horvath A.; Levy I.; de Alexandre R. B.; Ahmad F.; Manganiello V.; Stratakis C. A. Clinical and molecular genetics of the phosphodiesterases (PDEs). Endocr. Rev. 2014, 35, 195–233. 10.1210/er.2013-1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran P.; Rengarajan T.; Thangavel J.; Nishigaki Y.; Sakthisekaran D.; Sethi G.; Nishigaki I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. 10.7150/ijbs.7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helal C. J.; Arnold E. P.; Boyden T. L.; Chang C.; Chappie T. A.; Fennell K. F.; Forman M. D.; Hajos M.; Harms J. F.; Hoffman W. E.; Humphrey J. M.; Kang Z.; Kleiman R. J.; Kormos B. L.; Lee C.-W.; Lu J.; Maklad N.; McDowell L.; Mente S.; O’Connor R. E.; Pandit J.; Piotrowski M.; Schmidt A. W.; Schmidt C. J.; Ueno H.; Verhoest P. R.; Yang E. X. Application of structure-based design and parallel chemistry to identify a potent, selective, and brain penetrant Phosphodiesterase 2A inhibitor. J. Med. Chem. 2017, 60, 5673–5698. 10.1021/acs.jmedchem.7b00397. [DOI] [PubMed] [Google Scholar]
- Zhu J.; Yang Q.; Dai D.; Huang Q. X-ray crystal structure of Phosphodiesterase 2 in complex with a highly selective, nanomolar inhibitor reveals a binding-induced pocket important for selectivity. J. Am. Chem. Soc. 2013, 135, 11708–11711. 10.1021/ja404449g. [DOI] [PubMed] [Google Scholar]
- Mikami S.; Sasaki S.; Asano Y.; Ujikawa O.; Fukumoto S.; Nakashima K.; Oki H.; Kamiguchi N.; Imada H.; Iwashita H.; Taniguchi T. Discovery of an orally bioavailable, brain-penetrating, in vivo active Phosphodiesterase 2A inhibitor lead series for the treatment of cognitive disorders. J. Med. Chem. 2017, 60, 7658–7676. 10.1021/acs.jmedchem.7b00709. [DOI] [PubMed] [Google Scholar]
- Mikami S.; Nakamura S.; Ashizawa T.; Nomura I.; Kawasaki M.; Sasaki S.; Oki H.; Kokubo H.; Hoffman I. D.; Zou H.; Uchiyama N.; Nakashima K.; Kamiguchi N.; Imada H.; Suzuki N.; Iwashita H.; Taniguchi T. Discovery of clinical candidate n-((1s)-1-(3-fluoro-4-(trifluoromethoxy)phenyl)-2-methoxyethyl)-7-methoxy-2-oxo-2, 3-dihydropyrido[2,3-b]pyrazine-4(1h)-carboxamide (tak-915): A highly potent, selective, and brain-penetrating Phosphodiesterase 2A inhibitor for the treatment of cognitive disorders. J. Med. Chem. 2017, 60, 7677–7702. 10.1021/acs.jmedchem.7b00807. [DOI] [PubMed] [Google Scholar]
- Di L.; Whitney-Pickett C.; Umland J. P.; Zhang H.; Zhang X.; Gebhard D. F.; Lai Y.; Federico J. J.; Davidson R. E.; Smith R.; Reyner E. L.; Lee C.; Feng B.; Rotter C.; Varma M. V.; Kempshall S.; Fenner K.; El-kattan A. F.; Liston T. E.; Troutman M. D. Development of a new permeability assay using low-efflux MDCKII cells. J. Pharm. Sci. 2011, 100, 4974–4985. 10.1002/jps.22674. [DOI] [PubMed] [Google Scholar]
- Ryckmans T.; Edwards M. P.; Horne V. A.; Correia A. M.; Owen D. R.; Thompson L. R.; Tran I.; Tutt M. F.; Young T. Rapid assessment of a novel series of selective CB2 agonists using parallel synthesis protocols: A Lipophilic Efficiency (LipE) analysis. Bioorg. Med. Chem. Lett. 2009, 19, 4406–4409. 10.1016/j.bmcl.2009.05.062. [DOI] [PubMed] [Google Scholar]
- Stepan A. F.; Kauffman G. W.; Keefer C. E.; Verhoest P. R.; Edwards M. Evaluating the differences in cycloalkyl ether metabolism using the design parameter ″Lipophilic Metabolism Efficiency″ (LipMetE) and a matched molecular pairs analysis. J. Med. Chem. 2013, 56, 6985–6990. 10.1021/jm4008642. [DOI] [PubMed] [Google Scholar]
- Ogilvie B. W.; Parkinson A. Drugs as victims and perpetrators and the pharmacokinetic concept of maximum exposure. Handb. Metab. Pathways Xenobiot. 2014, 1, 103–123. 10.1002/9781118541203.xen0004. [DOI] [Google Scholar]
- Lombardo F.; Obach R. S.; Varma M. V.; Stringer R.; Berellini G. Clearance mechanism assignment and total clearance prediction in human based upon in silico models. J. Med. Chem. 2014, 57, 4397–4405. 10.1021/jm500436v. [DOI] [PubMed] [Google Scholar]
- Varma M. V.; Steyn S. J.; Allerton C.; El-Kattan A. F. Predicting clearance mechanism in drug discovery: Extended Clearance Classification System (ECCS). Pharm. Res. 2015, 32, 3785–3802. 10.1007/s11095-015-1749-4. [DOI] [PubMed] [Google Scholar]
- Heim-Riether A.; Healy J. A novel method for the synthesis of imidazo[5,1-f][1,2,4]triazin-4(3h)-ones. J. Org. Chem. 2005, 70, 7331–7337. 10.1021/jo051014w. [DOI] [PubMed] [Google Scholar]
- Tran T. P.; Fisher E. L; Wright A. S.; Yang J.. Concise Synthesis of Versatile Imidazo[5,1-f][1,2,4]triazin-4(3H)-ones. Org. Process Res. Dev. 2018, DOI: 10.1021/acs.oprd.7b00333. [DOI]
- Knutsen L. J. S.; Judkins B. D.; Newton R. F.; Scopes D. I. C.; Klinkert G. Synthesis of imidazo-fused bridgehead-nitrogen 2′-deoxyribo-c-nucleosides: Coupling-elimination reactions of 2,5-anhydro-3,4,6-tri-o-benzoyl-d-allonic acid. J. Chem. Soc., Perkin Trans. 1 1985, 621–630. 10.1039/P19850000621. [DOI] [Google Scholar]
- Miles A. A.; Miles E. M. Vascular reactions to histamine, histamine-liberator and leukotaxine in the skin of guinea-pigs. J. Physiol. 1952, 118, 228–257. 10.1113/jphysiol.1952.sp004789. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.