

Abstract
Farnesoid X receptor (FXR) plays a major role in the control of cholesterol metabolism. Antagonizing transcriptional activity of FXR is an effective means to treat the relevant metabolic syndrome. Some of antagonists so far have the charged functions; however, they may negatively affect the pharmacokinetics. We describe herein a structure–activity relationship (SAR) exploration of nonacidic FXR antagonist 6 focusing on two regions in the structure and biological evaluation of nonacidic 10 with the characteristic N-acylated piperidine group obtained from SAR studies. As the robust affinity to FXR is feasible with our nonacidic analogue, 10 is among the most promising candidates for in vivo testing.
Keywords: FXR antagonists, benzimidazole scaffold, N-acylated piperidine, triglyceride accumulation
Farnesoid X receptor (FXR) has been characterized as the bile acid nuclear receptor.1−3 It plays a pivotal role in regulating bile acids and lipid homeostasis by regulating the expression of several key genes involved in bile acid (BA) synthesis, metabolism, and transport in the liver.4 Specific BAs bind and activate FXR, the most potent being chenodeoxycholic acid (CDCA), which is an endogenous ligand of FXR and the primary BA found in human bile.1 CDCA transcriptionally represses the expression of cholesterol 7α-hydroxylase (CYP7A1), the rate-limiting enzyme during bile acid synthesis, by inducing that of small heterodimer partner (SHP) in liver.5 FXR induces bile acid transporter, bile salt export pump (BSEP), which transports bile acid from hepatocytes to bile canaliculi and induces organic solute transporter α(OSTα).6
GW4064,7 which is used as a reference FXR agonist, had been published in 2000 (Figure 1). GW4064 and its derivatives with an acidic function tend to be biased against poor selectivity.8,9 Recently, nonacidic FXR agonists have been reported.10
Figure 1.

Structures of GW4064 and representative nonsteroidal FXT antagonists.
Nonsteroidal FXR antagonists (1, 2)11,12 are used for the study in the treatment of hyperlipidemia (Figure 1). These studies are based on the following reports: FXR promotes adipocyte differentiation and controls adipose cell function.13−15 Additionally, naturally occurring FXR antagonist, guggulsterone, inhibits adipocyte differentiation and induces apoptosis in 3T3-L1 cell.16 Other nonsteroidal FXR antagonists (3, 4) have also been published (Figure 1).17,18 Some antagonists contain an acidic moiety as part of the pharmacophore. The acidic components have a tendency for the promiscuous interaction with off-targets and are susceptible to the conversion to acyl-glucuronides or acyl-CoA thioesters.19,20 Nonacidic antagonists would avoid unexpected toxicity and metabolites.
Early in the research process, a novel chemotype 5 with the acidic moiety as a hit compound was found to show moderate antagonistic activity against FXR21 (Figure 2). As a hit-to-lead approach,21 a chemical modification was initiated based on the structure of 5 to increase its antagonistic potency against FXR. Eventually, the potency of nonacidic analogue 6 was not substantially different from that of 5 in FXR time-resolved fluorescence resonance energy transfer (TR-FRET) binding assay. It was, however, found that the moderate potency of 6 in the luciferase reporter gene assay could be obtained in the absence of the acidic moiety of our new chemotype. The primary structure–activity relationship (SAR) data enable further insights: (1) the IC50 values increased with the enhancement of the hydrophobicity in region A; and (2) replacing the carboxylic acid of 5 with nonacidic groups in region B had an impact on FXR antagonism and will be a clue to development of nonacidic FXR antagonists. Thus, we developed a potent nonacidic FXR antagonist by focusing on regions A and B in the structure of 6 as the lead pharmacophore and also evaluated triglyceride accumulation in 3T3-L1 adipocytes.
Figure 2.

Hit and lead compounds (5 and 6), and regions for optimization.
The designed compounds listed in Tables 1 and 2 were synthesized via the procedures shown in Schemes S1–S4 (Supporting Information (SI)). The structure of the prepared compounds was confirmed by spectroscopic and analytical techniques. The potent FXR antagonist, T3 (Takeda Pharmaceutical Co.),22 used as a reference compound (1), was also prepared from commercially available starting materials. Characterization of 1 by 1H NMR, 13C NMR, and HR-MS is available in the SI.
Table 1. FXR Antagonistic Activity and Cytotoxicity of 6–11.
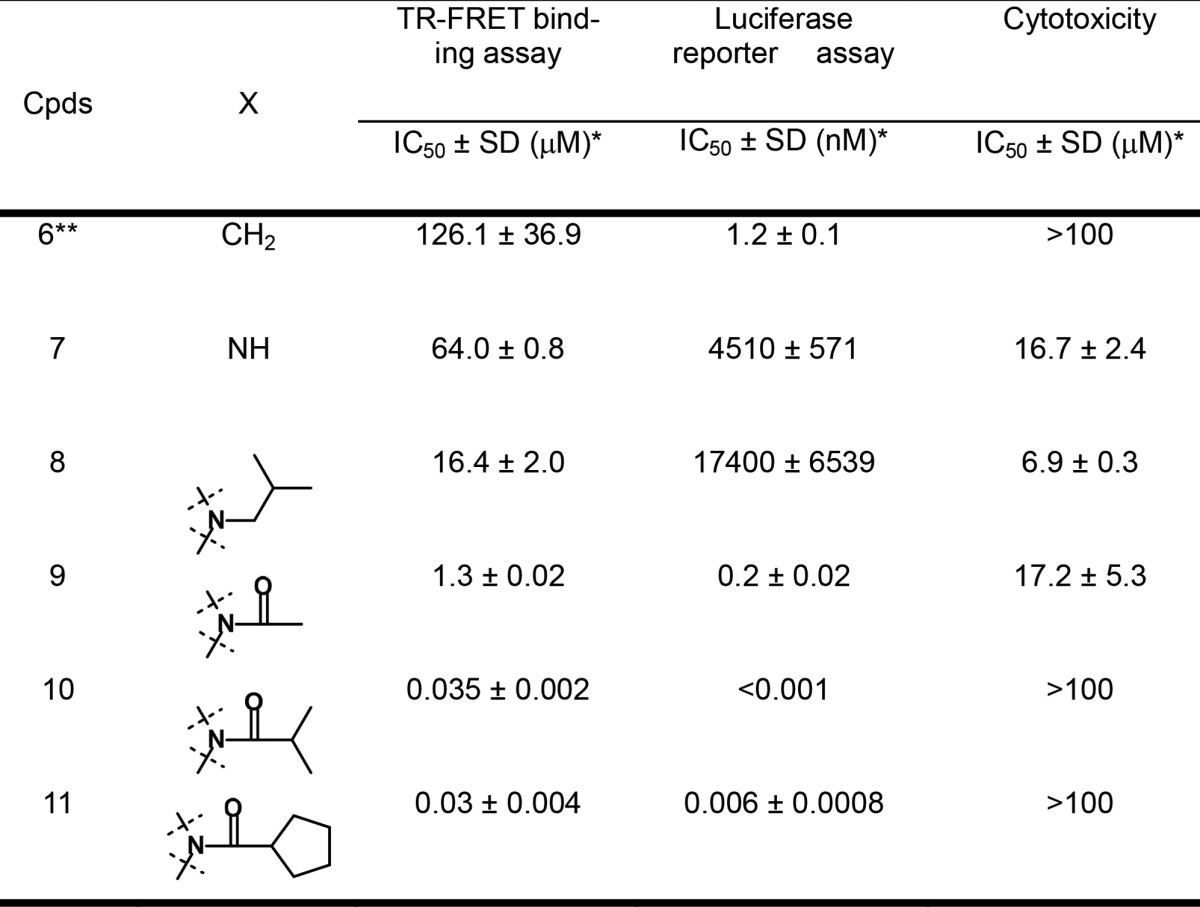
The IC50 values are presented as the means ± SD of three independent experiments.
Reference (21).
Table 2. FXR Antagonistic Activity and Cytotoxicity of 12–17.
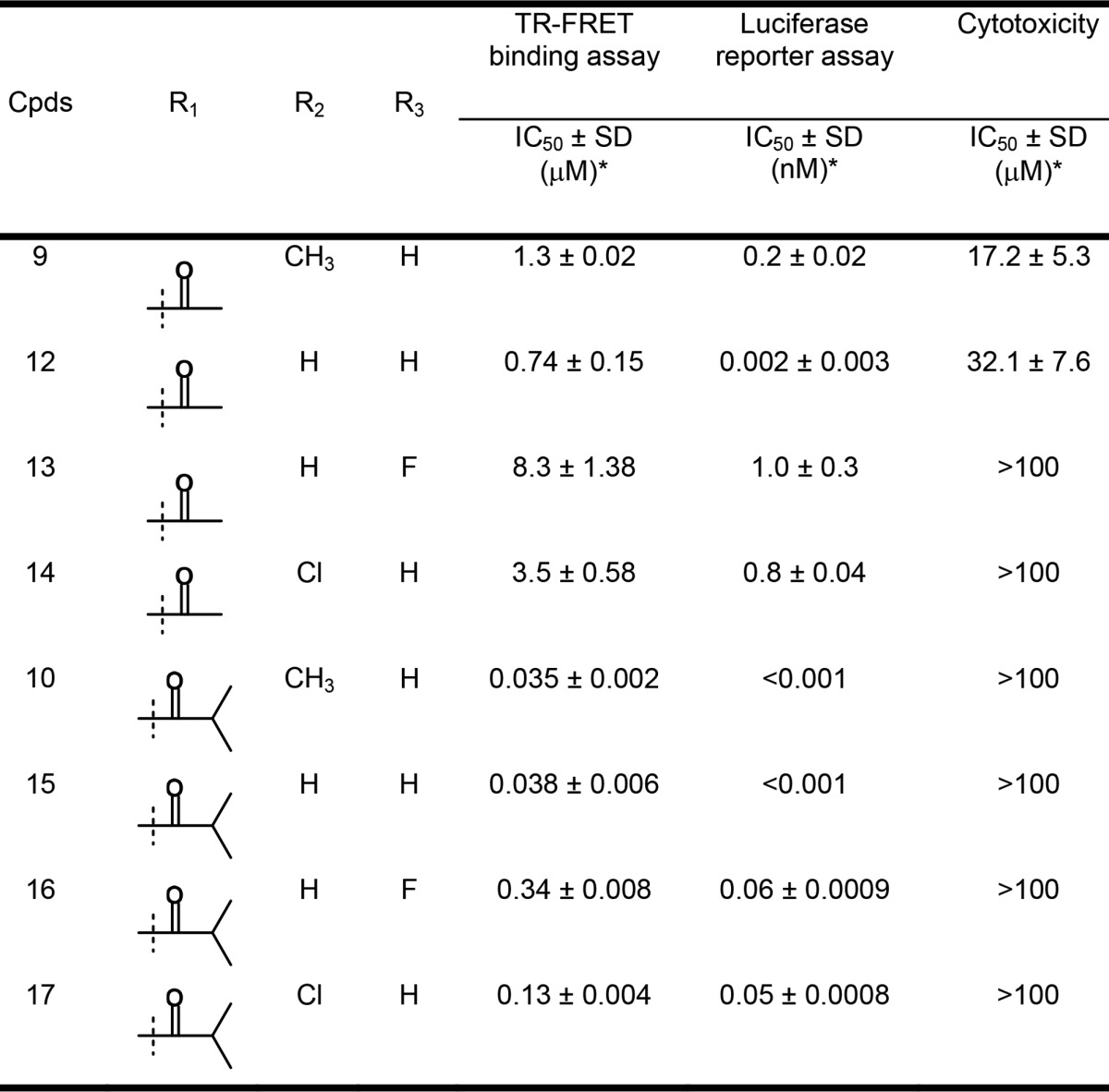
The IC50 values are presented as the means ± SD of three independent experiments.
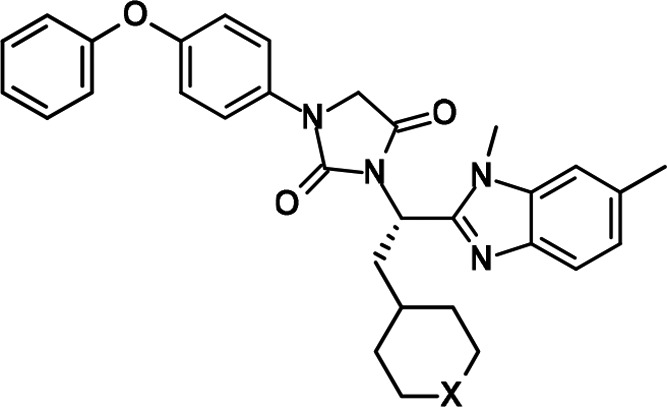
The first step in tackling derivatization of region A in 6 was carried out by replacing the cyclohexyl component with N-substituted piperidine while keeping the methyl group in region B (R1 = CH3 and R2 = H) (Figure 2). The piperidine derivative, which can be relatively and easily synthesized from commercial pyridine derivative, is appropriate for the investigation of structural diversity of region A. The newly synthesized analogues were evaluated by a FXR TR-FRET binding assay and a luciferase reporter assay (Table 1). Additionally, the antagonistic rate of the analogues is shown in Table S1 (SI). The modification on the nitrogen atom in piperidine made a profound difference in the antagonism for FXR.
Positive ionizable functions in the piperidine ring of 7 and 8 revealed an increase in the binding activity to FXR compared to 6. The piperidine of 7 was acylated to evaluate electrically neutral analogues. This campaign led to the discovery of key analogues 9–11, which exhibited significant improvement in the binding activity. The N-acylated analogue 9, for instance, appeared to bind nearly 12-fold greater than 8. In addition, replacing the acetyl group of 8 with bulky acyl groups (10 and 11) showed the binding activity approximately 40-times higher than that of 9 in the binding assay (0.035 ± 0.002 and 0.03 ± 0.004 μM, respectively), differing from those of 2 and 3 (IC50: 8.96 ± 3.62 μM12 and 7.5 nM,17 respectively). Analogue 10 dose-dependently competed against the binding of GW4064 to FXR (Figure S1 in SI). However, the potency of 10 in the binding assay is marginally weaker than that of 1, which is the potent FXR antagonist.11 In the luciferase assay, the acylated compounds (9–11) exhibited inhibition against FXR; in particular, 10 and 11 displayed potent FXR antagonism (<0.001 and 0.006 ± 0.0008 nM, respectively). Analogue 10 had substantial changes (more than a 1000-fold increase) in its antagonist activity compared to nonacylated piperidines 7 and 8. It turns out that the bulky acyl moiety (10 and 11) in region A is a key structural element and contributes substantially to potency.
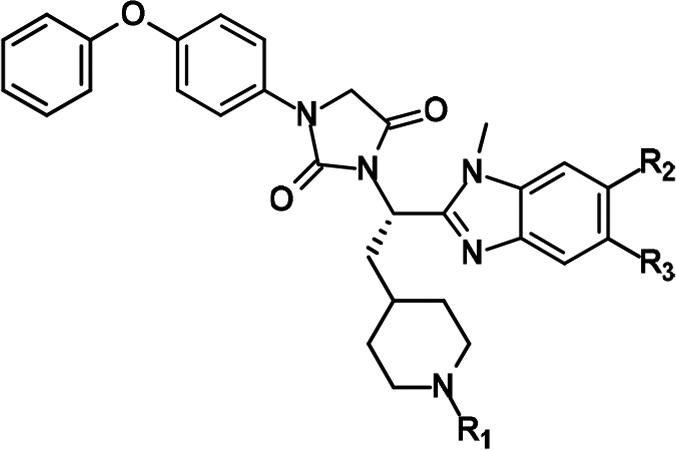
To pursue nonacidic substituents other than CH3 (R1 = CH3, R2 = H) on benzimidazole, we successively modified the substituents on benzimidazole (region B) by keeping an acetyl or isobutyryl moiety and by anticipating a synergistic effect through a combination of regions A and B (Table 2). In the binding assays, removal of methyl group (12) enhanced inhibitory activity against FXR relative to 9, in contrast to analogues bearing fluoride (13) or chloride (14) substituents on benzimidazole, which were less active than 9. The same observations were made with 12–14 in the luciferase assay, showing that both substitutions (13 and 14) are less favorable in region B. The two series (9, 12, 13, 14 and 10, 15, 16, 17) shows almost the same SAR in both assays. For instance, the IC50 values of 13 and 14 in the binding assay are reduced about <10 and 2–3 times as compared with that of parent compound 9, respectively, and these trends are shared with 16 and 17 versus 10. However, only the IC50 value of 12 in the reporter gene assay (0.002 ± 0.003 nM) largely deviates from the SAR. In the combination of regions A and B reported here, the substituent pattern (R1–R3) of 9, 10, and 15 facilitated the favorable interaction with FXR. Notably, robust potency was observed for the substituent pattern of 15, being nearly equipotent with 10.
To clarify the effects exerted by N-acyl piperidine of 10 toward FXR, we ran a molecular docking simulation (AutoDock Vina 1.1.2)23 with 10. Our computer-assisted modeling studies suggested that the binding mode of 10 (yellow) complexed with LBD of FXR was shown to be similar to that of 6 (pink) other than around N-acyl piperidine as depicted in Figure 3A. The obvious difference is that some leeway exists for the isobutyryl group of 10 to interact. Additionally, modeling studies predicted that the carbonyl group of isobutyryl in 10 is able to establish a hydrogen bond with His298 (Figure 3B). The interaction of an N-acyl group is viewed to affect the antagonism for FXR. Indeed, comparison of the IC50 values obtained for N-acylated piperidines 10 or 15 with those for 8 (absence of N-acyl group) clearly demonstrated that the N-acylated piperidine ring is a key structural element and contributes to potency. In terms of the SAR of region B, the results obtained with 13, 14, 16, and 17 reveal that the hydrogen atom and methyl group in region B cannot be successfully replaced by fluoride or chloride. This insight into region B was not obtained by modeling studies. The structures of 10 and 15 are thus combined with the synergism exerted by the appropriate moieties in two distinct regions. Eventually, both of the analogues inhibited FXR at low nanomolar IC50 values in the binding assay and revealed robust antagonism against FXR.
Figure 3.
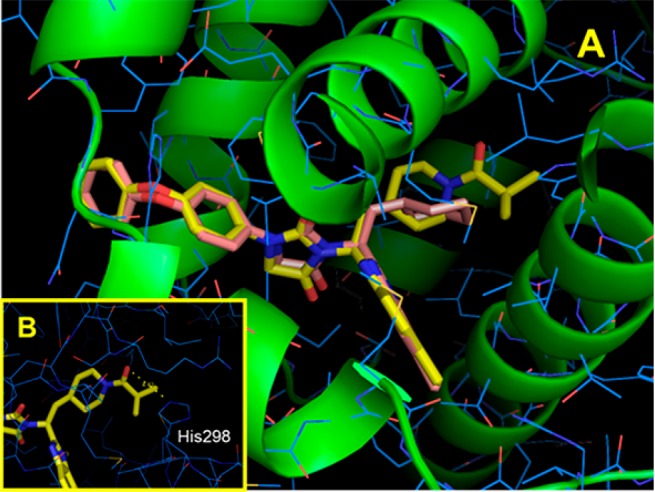
FXR model complexed with 6 and 10. (A) Complex model of hFXRα-LBD homodimer (PDB ID: 4OIV, blue wires and green ribbons) with 10 (yellow) and 6 (pink) was built using AutoDock Vina1.1.2. (B) Hydrogen bond between the carbonyl group on piperidine of 10 and His298 (yellow dashed line).
Large difference in the IC50 values of 10 and 15 reported for the binding and reporter assays was observed. This would be due to the proteins used. FXR is used for the luciferase assay; in contrast, a partial sequence of FXR (ligand binding domain, LBD) is employed for the binding assay. It is obvious that the three-dimensional structures of FXR and FXR-LBD would not be identical; however, a clear SAR on the analogues with N-acyl group was distinctly observed in each assay and the changes in the IC50 values in both assays are nearly identical. In contrast, since 6, 7, and 8 have no N-acyl group, there will be a large difference in binding to both proteins with the three-dimensional structures.
Tetrazolium (MTT) colorimetric assays are the most common employed for the detection of cytotoxicity or cell viability following exposure to toxic substances,24 and the cytotoxic activities of 6 and 7–17 were thus evaluated (Tables 1 and 2). Analogues 10 and 15, which differ from 9 and 12 only by substituents on the nitrogen atom in piperidine, respectively, exhibited no cytotoxicity. Relatively strong antagonism in the luciferase reporter gene assay given by 12 was anticipated to be overestimated by cytotoxicity. The results obtained in the MTT assay suggested that cytotoxicity tends to be partially affected by varying in the substituents on piperidine.
To gain insights into the antagonism of 10, we then investigated the downstream genes of FXR by real-time reverse transcription PCR (real-time RT-PCR). The cells cultured in six-well plates were treated with different concentrations of 10 in the presence or absence of endogenous agonist, CDCA (Figure 4A,C,E) and synthetic agonist, GW4064 (Figure 4B,D,F). After 24 h, cells were collected. CDCA and GW4064 induced the expression of BSEP, SHP, and OSTα.5,6 Interestingly, 10 equipotently suppressed the effect of CDCA; namely, by decreasing the expression levels of three target genes at a low dose (0.02 μM). Similar to 10, the 1,3,4-trisubstituted-pyrazolone derivative (2) inhibited the activation of promoters related to FXR activated by CDCA.12 The effect of GW4064 tended to be inhibited dose-dependently by 10. Altogether, these data indicate the effect of 10 on downstream targeted gene expressions through FXR inactivation.
Figure 4.

mRNA expression of CDCA- or GW4064-modulated FXR target genes was antagonized by 10. Huh-7 cells were treated with different concentrations of 10 in the presence of 10 μM CDCA (A,C,E) or 1 μM GW4064 (B,D,F). Data are presented as means ± SD from n = 3 experiments (*p < 0.01 vs CDCA or GW4064 only). N.C.: Negative control.
CDCA-induced FXR activity was prevented by 10 in the cell-based luciferase assay (Table 1). Likewise, 10 inhibited the synthetic agonist GW4064-stimulated FXR activity (Figure S2A). We next attempted to see whether 10 interacts with eight other nuclear receptors including retinoid X receptor α (RXRα) (Figure S2B), peroxisome proliferator-activated receptor α, γ, and δ (PPAR α, γ, and δ) (Figure S2C–E), liver X receptor α and β (LXR α and β) (Figure S2F,G), vitamin D receptor (VDR) (Figure S2H), and retinoic acid receptor α (RARα) (Figure S2I) according to published methods.21,25,26 Analogue 10 (1 μM) had no agonistic (alone) or antagonistic (plus the corresponding agonists) effect toward any of these receptors. Compound 2 reported previously had no effect on six nuclear receptors other than FXR.12 Furthermore, we showed no effect of 10 on PXR target gene, CYP3A4 (Figure S3).
TGR5 receptor is the first known G-protein-coupled receptors specific for deoxycholic acid and lithocholic acid (LCA) but not for CDCA.27 It has been reported that dual activation of FXR and TGR5 has a high profile in nonalcoholic fatty liver disease (NAFLD) and atherosclerosis.28 The combined FXR antagonist/TGR5 agonist compounds derived from FXR modulators have been also developed.29 In this study, no cross-reactivity between 10 and TGR5 was observed (Figure S2J). The results in a panel of the receptors revealed that the nonacidic analogue 10 had neither agonist nor antagonist properties for RXRα, PPAR α, δ, γ, LXR α, β, VDR, RARα, PXR, and TGR5.
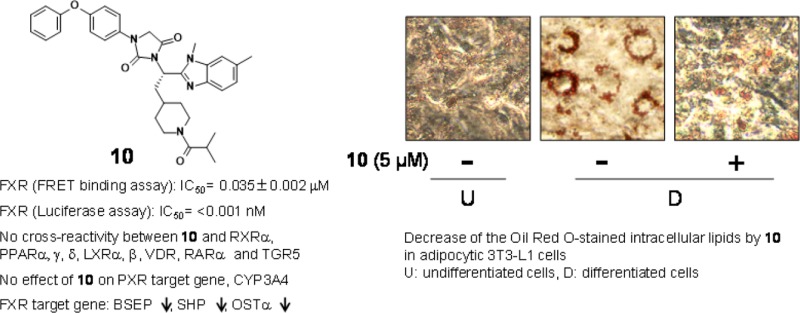
Mouse 3T3-L1 adipocytes have been extensively employed to examine the cellular and molecular mechanism of adipocyte differentiation.30 FXR expression was induced during adipocyte differentiation in them in order to investigate the antiadipogenic effect of 10 on 3T3-L1 adipocytes. Prior to adipocyte differentiation, we investigated cell toxicity of 10 in 3T3-L1 cells. Cells were cultured for 6 days in Dulbecco’s modified Eagle’s medium (DMEM) containing various concentrations of 10 (0–10 μM); no significant toxic effects were observed up to 10 μM in 3T3-L1 cells (Figure 5A).
Figure 5.
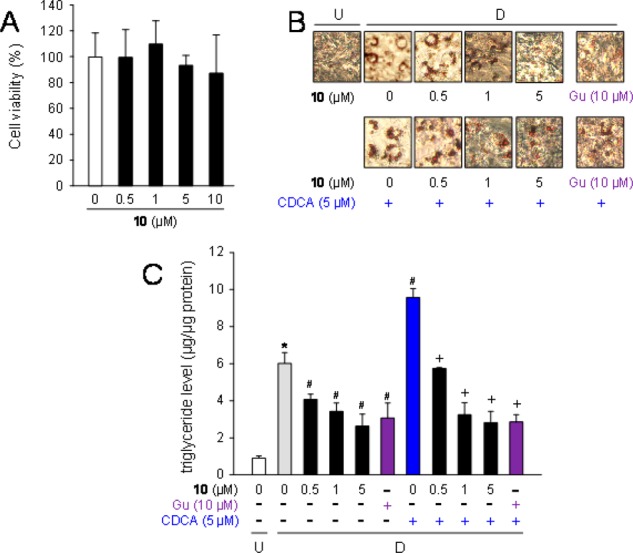
Suppression of lipid accumulation by 10 in adipocytes. (A) Cell toxicity of 10 on 3T3-L1 adipose cells. The cells were incubated for 6 days in DMEM with various concentrations of 10 (0–10 μM), and cell toxicity was measured. Data represent means ± SD (n = 3 independent experiments). (B) Staining of intracellular lipids by Oil Red O in 3T3-L1 cells. Cells (undifferentiated cells: U) were differentiated into adipocytes [D] for 6 days in DMEM with 10 (0–5 μM) or guggulsterone (Gu: 10 μM) together without (upper) or with CDCA (5 μM; lower). Intracellular lipid droplets were stained with Oil Red O. Data are representative of n = 3 repetitions. (C) Decrease of the intracellular triglyceride level by 10 in 3T3-L1 cells. The cells (undifferentiated cells: U; white column) were differentiated [D] into adipocytes for 6 days in DMEM without (gray column) or with 10 (0.5, 1, or 5 μM; black columns), or guggulsterone (Gu: 10 μM; purple columns) together without or with CDCA (5 μM; blue column). Data are presented as means ± SD from n = 3 experiments. *p < 0.01 (vs undifferentiated cells), #p < 0.01 (vs vehicle-treated differentiated cells), and +p < 0.01 (vs CDCA-treated differentiated cells).
Subsequently, the effect of 10 on adipogenesis in 3T3-L1 cells was examined. Cells were differentiated into adipocytes for 6 days in DMEM with various concentrations of 10 (0–5 μM) or guggulsterone (Gu: 10 μM) (top pictures, Figure 5B). The number of lipid droplets in the cells was increased during adipogenesis. Results from Oil Red O staining indicated that exposure to 10 (0–5 μM) dose-dependently reduced the number of lipid droplets (lower pictures, Figure 5B). Guggulsterone also decreased the number of cells. In addition, the cells were differentiated for 6 days in DMEM containing CDCA with or without 10 (0–5 μM). The number of lipid droplets in the CDCA-treated differentiated cells increased greater than in the vehicle-treated differentiated cells. Intracellular lipids were decreased by the cotreatment with 10 in a dose-dependent manner or with guggulsterone.
To further confirm the decrease of the lipids in adipocytes by the treatment with 10, we measured the intracellular triglyceride levels (Figure 5C). The level in the differentiated cells increased about 5.9-fold in comparison to the undifferentiated cells. Analogue 10 dose-dependently antagonized the effect of CDCA on triglyceride accumulation, indicating that it decreased lipid accumulation in 3T3-L1 cells.
Lead optimization efforts for our FXR antagonist series began with SAR exploration starting from the previously reported benzimidazole analogue 6. Replacing the cyclohexyl group of 6 with N-acylated piperidine in region A resulted in the discovery of a key inhibitor, 10, which possessed robust potency as a FXR antagonist. Concurrent with these investigations, the SAR exploration of substituents on benzimidazole of 6 was conducted, which led to the discovery of 10 and 15; both were potent in the binding and cell assays. The rationale might involve the carbonyl of the isobutyryl group of piperidine 10 participating in hydrogen bond interaction with His298 of FXR-LBD (Figure 3), and the isopropyl moiety of isobutyryl group might affect the putative hydrophobic interaction with LBD (9 vs 10 and 11, Table 1). Furthermore, 10 reversed the regulation of CDCA and GW4064 on FXR target gene expressions (BSEP, SHP, and OSTα). The nonacidic analogue also had greater affinity toward FXR over TGR5 and other nuclear receptors. Moreover, analogue 10 inhibits differentiation of preadipocytes and reduces intracellular triglyceride content in 3T3-L1 adipocytes. We envision that the N-acylated piperidine moiety holds great potential for structural modifications and on the design of nonacidic FXR antagonists. Since the robust affinity to FXR is feasible with our nonacidic analogue, additional studies on hyperlipidemia relevant in vivo biological activities are in progress, and the results will be published in due course.
Acknowledgments
We are grateful to Dr. Lawrence H. Lazarus for critically reading the manuscript. We thank Professor Hidetoshi Tahara for data collection.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00363.
Dose-dependent manner of 10 in TR-FRET and the affinity between 10 and other nuclear receptors, TGR5; synthesis and characterization data for 7–17; antagonistic rate of 6–17; analytical data of 1 and 6, purity determination (1, 6–17), and biological protocols (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Makishima M.; Okamoto A. Y.; Repa J. J.; Tu H.; Learned R. M.; Luk A.; Hull M. V.; Lustig K. D.; Mangelsdorf D. J.; Shan B. Identification of a nuclear receptor for bile acids. Science 1999, 284, 1362–1365. 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- Parks D. J.; Blanchard S. G.; Bledsoe R. K.; Chandra G.; Consler T. G.; Kliewer S. A.; Stimmel J. B.; Willson T. M.; Zavacki A. M.; Moore D. D.; Lehmann J. M. Bile acids: Natural ligands for an orphan nuclear receptor. Science 1999, 284, 1365–1368. 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- Wang H.; Chen J.; Hollister K.; Sowers K. C.; Forman B. M. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell 1999, 3, 543–553. 10.1016/S1097-2765(00)80348-2. [DOI] [PubMed] [Google Scholar]
- Sinal C. J.; Tohkin M.; Miyata M.; Ward J. M.; Lambert G.; Gonzalez F. J. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 2000, 102, 731–744. 10.1016/S0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- Goodwin B.; Jones S. A.; Price R. R.; Watson M. A.; McKee D. D.; Moore L. B.; Galardi D.; Wilson J. G.; Lewis M. C.; Roth M. E.; Maloney P. R.; Willson T. M.; Kliewer S. A. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol. Cell 2000, 6, 517–526. 10.1016/S1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- Dawson P. A.; Lan T.; Rao A. Bile acid transporters. J. Lipid Res. 2009, 50, 2340–2357. 10.1194/jlr.R900012-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloney P. R.; Parks D. J.; Haffner C. D.; Fivush A. M.; Chandra G.; Plunket K. D.; Creech K. L.; Moore L. B.; Wilson J. G.; Lewis M. C.; Jones S. A.; Willson T. M. Identification of a chemical tool for the orphan nuclear receptor FXR. J. Med. Chem. 2000, 43, 2971–2974. 10.1021/jm0002127. [DOI] [PubMed] [Google Scholar]
- Akwabi-Ameyaw A.; Bass J. Y.; Caldwell R. D.; Caravella J. A.; Chen L.; Creech K. L.; Deaton D. N.; Jones S. A.; Kaldor I.; Liu Y.; Madauss K. P.; Marr H. B.; McFadyen R. B.; Miller A. B.; Navas F. III; Parks D. J.; Spearing P. K.; Todd D.; Williams S. P.; Wisely G. B. Conformationally constrained farnesoid X receptor (FXR) agonists: Naphthoic acid-based analogs of GW 4064. Bioorg. Med. Chem. Lett. 2008, 18, 4339–4343. 10.1016/j.bmcl.2008.06.073. [DOI] [PubMed] [Google Scholar]
- Singh N.; Yadav M.; Singh A. K.; Kumar H.; Dwivedi S. K. D.; Mishra J. S.; Gurjar A.; Manhas A.; Chandra S.; Yadav P. N.; Jagavelu K.; Siddiqi M. I.; Trivedi A. K.; Chattopadhyay N.; Sanyal S. Synthetic FXR Agonist GW4064 Is a Modulator of Multiple G Protein–Coupled Receptors. Mol. Endocrinol. 2014, 28, 659–673. 10.1210/me.2013-1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flesch D.; Cheung S.-Y.; Schmidt J.; Gabler M.; Heitel P.; Kramer J.; Kaiser A.; Hartmann M.; Lindner M.; Lüddens-Dämgen K.; Heering J.; Lamers C.; Lüddens H.; Wurglics M.; Proschak E.; Schubert-Zsilavecz M.; Merk D. Nonacidic Farnesoid X Receptor Modulators. J. Med. Chem. 2017, 60, 7199–7205. 10.1021/acs.jmedchem.7b00903. [DOI] [PubMed] [Google Scholar]
- Amano Y.; Shimada M.; Miura S.; Adachi R.; Tozawa R. Effects of a farnesoid X receptor antagonist on hepatic lipid metabolism in primates. Eur. J. Pharmacol. 2014, 723, 108–115. 10.1016/j.ejphar.2013.10.048. [DOI] [PubMed] [Google Scholar]
- Huang H.; Yu Y.; Gao Z.; Zhang Y.; Li C.; Xu X.; Jin H.; Yan W.; Ma R.; Zhu J.; Shen X.; Jiang H.; Chen L.; Li J. Discovery and optimization of 1,3,4-trisubstituted-pyrazolone derivatives as novel, potent, and nonsteroidal farnesoid X receptor (FXR) selective antagonists. J. Med. Chem. 2012, 55, 7037–7053. 10.1021/jm3002718. [DOI] [PubMed] [Google Scholar]
- Rizzo G.; Disante M.; Mencarelli A.; Renga B.; Gioiello A.; Pellicciari R.; Fiorucci S. The farnesoid X receptor promotes adipocyte differentiation and regulates adipose cell function in vivo. Mol. Pharmacol. 2006, 70, 1164–1173. 10.1124/mol.106.023820. [DOI] [PubMed] [Google Scholar]
- Fiorucci S.; Rizzo G.; Antonelli E.; Renga B.; Mencarelli A.; Riccardi L.; Morelli A.; Pruzanski M.; Pellicciari R. Cross-Talk between farnesoid-X-receptor (FXR) and peroxisome proliferator-activated receptor contributes to the antifibrotic activity of FXR ligands in rodent models of liver cirrhosis. J. Pharmacol. Exp. Ther. 2005, 315, 58–68. 10.1124/jpet.105.085597. [DOI] [PubMed] [Google Scholar]
- Feng S.; Reuss L.; Wang Y. Potential of natural products in the inhibition of adipogenesis through regulation of PPARγ expression and/or its transcriptional activity. Molecules 2016, 21, 1278. 10.3390/molecules21101278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.-Y.; Della-Fera M. A.; Baile C. A. Guggulsterone inhibits adipocyte differentiation and induces apoptosis in 3T3-L1 cells. Obesity 2008, 16, 16–22. 10.1038/oby.2007.24. [DOI] [PubMed] [Google Scholar]
- Xu Y. Recent progress on bile acid receptor modulators for treatment of metabolic diseases. J. Med. Chem. 2016, 59, 6553–6579. 10.1021/acs.jmedchem.5b00342. [DOI] [PubMed] [Google Scholar]
- Yu D. D.; Lin W.; Forman B. M.; Chen T. Identification of trisubstituted-pyrazol carboxamide analogs as novel and potent antagonists of farnesoid X receptor. Bioorg. Med. Chem. 2014, 22, 2919–2938. 10.1016/j.bmc.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metabolism, Pharmacokinetics and Toxicity of Functional Groups: Impact of Chemical Building Blocks on ADMET; Smith D. A., Ed.; RSC Drug Discovery; Royal Society of Chemistry: Cambridge, U.K., 2010. [Google Scholar]
- Skonberg C.; Olsen J.; Grimstrup Madsen K.; Hansen S. H.; Grillo M. P. Metabolic activation of carboxylic acids. Expert Opin. Drug Metab. Toxicol. 2008, 4, 425–438. 10.1517/17425255.4.4.425. [DOI] [PubMed] [Google Scholar]
- Teno N.; Iguchi Y.; Yamashita Y.; Mori N.; Une M.; Nishimaki-Mogami T.; Gohda K. Discovery and optimization of benzimidazole derivatives as a novel chemotype of farnesoid X receptor (FXR) antagonists. Bioorg. Med. Chem. 2017, 25, 1787–1794. 10.1016/j.bmc.2017.01.040. [DOI] [PubMed] [Google Scholar]
- Kitamura S.; Hosono H.; Miura S.; Aoki K.. Preparation of azole compounds as FXR inhibitors. Jpn. Kokai Tokkyo Koho (2008), JP 2008273847, 13 Nov 2008.
- Trott O.; Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David T. V.; Philip S.; Dominic S.; Anne M.; Angela P.; Michael R. B. Tetrazolium-based assays for cellular viability: a critical examination of selected parameters affecting formazan production. Cancer Res. 1991, 51, 2515–2520. [PubMed] [Google Scholar]
- Tamehiro N.; Sato Y.; Suzuki T.; Hashimoto T.; Asakawa Y.; Yokoyama S.; Kawanishi S.; Ohno Y.; Inoue K.; Nagao T.; Nishimaki-Mogami T. Riccardin C: A natural product that functions as a liver X receptor (LXR)α agonist and an LXRβ antagonist. FEBS Lett. 2005, 579, 5299–5304. 10.1016/j.febslet.2005.08.054. [DOI] [PubMed] [Google Scholar]
- Iguchi Y.; Yamaguchi M.; Sato H.; Kihira K.; Nishimaki-Mogami T.; Une M. Bile alcohols function as the ligands of membrane-type bile acid-activated G protein-coupled receptor. J. Lipid Res. 2010, 51, 1432–1441. 10.1194/jlr.M004051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C.; Gioiello A.; Noriega L.; Strehle A.; Oury J.; Rizzo G.; Macchiarulo A.; Yamamoto H.; Mataki C.; Pruzanski M.; Pellicciari R.; Auwerx J.; Schoonjans K. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009, 10, 167–177. 10.1016/j.cmet.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadhav K.; Xu Y.; Zhang Y. Dual activation of bile acid receptors FXR and TGR5 plays a protective role in non-alcoholic fatty liver disease and atherosclerosis. FASEB J. 2016, 30, 870–874. [Google Scholar]
- Lamers C.; Merk D.; Gabler M.; Flesch D.; Kaiser A.; Schubert-Zsilavecz M. SAR studies on FXR modulators led to the discovery of the first combined FXR antagonistic/TGR5 agonistic compound. Future Med. Chem. 2016, 8, 133–148. 10.4155/fmc.15.178. [DOI] [PubMed] [Google Scholar]
- Cariou B.; van Harmelen K.; Duran-Sandoval D.; van Dijk T. H.; Grefhorst A.; Abdelkarim M.; Caron S.; Torpier G.; Fruchart J. C.; Gonzalez F. J.; Kuipers F.; Staels B. The farnesoid X receptor modulates adiposity and peripheral insulin sensitivity in mice. J. Biol. Chem. 2006, 281, 11039–11049. 10.1074/jbc.M510258200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.