

Abstract
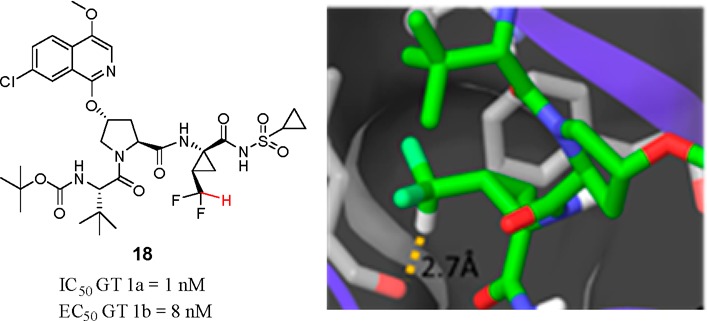
The design and synthesis of potent, tripeptidic acylsulfonamide inhibitors of HCV NS3 protease that contain a difluoromethyl cyclopropyl amino acid at P1 are described. A cocrystal structure of 18 with a NS3/4A protease complex suggests the presence of a H-bond between the polarized C–H of the CHF2 moiety and the backbone carbonyl of Leu135 of the enzyme. Structure–activity relationship studies indicate that this H-bond enhances enzyme inhibitory potency by 13- and 17-fold compared to the CH3 and CF3 analogues, respectively, providing insight into the deployment of this unique amino acid.
Keywords: Hepatitis C virus, NS3 protease, enzyme and replicon inhibitor, difluoromethylcyclopropyl amino acid, difluoromethyl, tripeptide acylsulfonamide, hydrogen-bond donor
Treatment for hepatitis C virus (HCV) has evolved significantly in recent years with the approval of direct-acting antiviral agents (DAAs) that provide improved cure rates and reduced duration of treatment when compared to earlier interferon-based regimens.1 Proof-of-concept for curative therapy with DAA treatment was established in clinical trials using the combination of the NS3 protease inhibitor asunaprevir (1) and the NS5A inhibitor daclatasvir (2) (Figure 1), the first interferon-free combination to be approved worldwide.2,3
Figure 1.
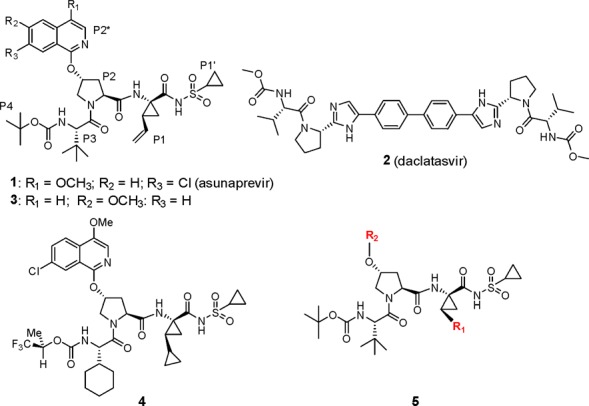
Structures of asunaprevir (1), daclatasvir (2), BMS-605339 (3), BMS-890068 (4), and P1 cyclopropyl prototype 5, which form the basis of this study.
The discovery of 1 has been described in detail, and this compound was a replacement for BMS-605339 (3), which exhibited a cardiovascular (CV) signal in the clinic at unexpectedly low plasma concentrations based on profiling in preclinical studies.4,5 The structural changes that differentiate 1 from 3 are subtle and limited to the P2* region of these molecules, with repositioning of the CH3O at C4 and the introduction of a Cl substituent at C7 of the isoquinoline ring, providing a compound free of a CV signal in both preclinical and clinical studies. As 1 was progressing through clinical trials, BMS-890068 (4) was identified as a back-up compound that, based on preclinical PK data, offered the potential to provide once-daily dosing while maintaining the potency and favorable preclinical CV profile found in 1.6 Among the structural changes that differentiated 4 from 1, modifications to the P1 position proved pivotal, with the bis-cyclopropyl group at the P1 position of 4 leading to enhanced absorption that translated into a significant improvement in the pharmacokinetic (PK) profile in rats. This observation underscored the potential value in exploring additional structural modifications at P1, as represented generically by 5 (Figure 1).
The S1 subsite of HCV NS3 protease is a shallow, lipophilic pocket defined by Leu135, Phe154, and Ala157, and the P1 moieties of potent inhibitors are lipophilic in nature.7 The carbonyl moiety of Leu135, which lies at the base of the pocket, has been recognized as a potential H-bond acceptor in interactions with bound ligands. The hexameric peptide 6, which incorporates a CHF2-substituted amino acid at P1, was reported as a moderately potent inhibitor of NS3 protease (Figure 2).8 The CHF2-based P1 functionality in 6 was elegantly designed as a bioisosteric replacement of the P1 cysteine that is present in endogenous substrates and inhibitor 7.9 A cocrystal structure of an analogue of 6 with a NS3/4A protease complex indicated the presence of a H-bond between the terminal CHF2 moiety at P1 and the carbonyl oxygen atom of Leu135.9 The role of a CHF2 as a H-bond donor had previously been recognized, but its application in the context of 6 demonstrated its potential in drug design.10−12 In this Letter, efforts to employ a P1 CHF2-bearing amino acid moiety in the context of tripeptide-based acyl sulfonamides as inhibitors of the NS3 protease are described.
Figure 2.
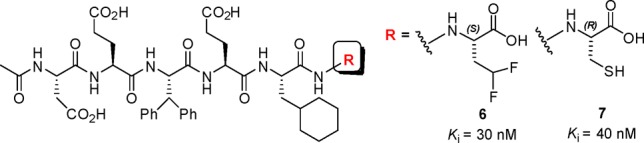
Hexameric peptide-based NS3 inhibitors.
Incorporation of a P1 CHF2CH2 moiety in a tripeptidic acylsulfonamide series provided diastereoisomers 12 and 13, both of which were poorly active NS3 inhibitors. The ethyl homologues 14 and 15 offered similar enzyme inhibitory activity, suggesting minimal potency contribution from the terminal CHF2 group in 12 and 13. A P1 CHF2CH2 group was also examined in the context of 16, which, while of similar activity to 12 and 13, was cocrystallized with a NS3/4A protease complex (Figure 3b), allowing comparison with 1 (Figure 3a).13 Note that while 16 was synthesized and tested as a mixture of diastereoisomers epimeric at P1, only the (S)-stereoisomer was observed in the cocrystal structure.
Figure 3.
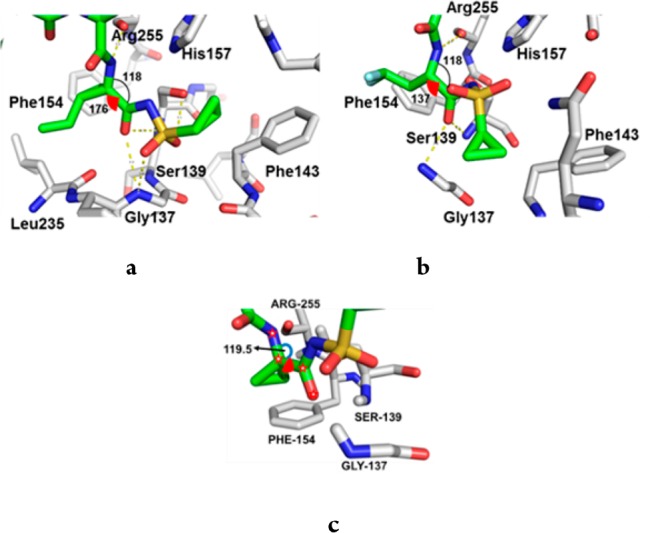
(a) Cocrystal structure of 1 bound to the NS3/4A protease complex illustrating interactions between the P1 and P1′ elements of the inhibitor and the enzyme subpockets. (b) Similar perspective from the cocrystal structure of 16 bound to the NS3/4A protease complex. (c) CADD model of 11 bound to the NS3/4A protease complex. The arc represents the bite angle, while the red wedge depicts the dihedral angle.
In the cocrystal structures, a significant point of differentiation between 1 and 16 was the distinct binding mode of the acyl sulfonamide moieties. One of the diastereotopic sulfone oxygens of 1 established a H-bond with the backbone N–H of Gly137 in the oxyanion hole, while the other engaged both the backbone N–H and the side chain hydroxyl of the catalytic Ser139.5 In addition, the terminal cyclopropyl ring of 1 effectively interfaced with the small, well-defined S1′ pocket. However, these interactions were not recapitulated in the cocrystal structure of 16, which presumably accounts, in part, for its poor inhibitory activity. The reduction in activity observed for 16 was not unexpected since structure–activity relationship (SAR) studies had established the importance of a cyclopropyl group at the P1 position, as exemplified by 9 and 10, which were poor NS3/4A inhibitors with IC50 values of 33 and 2.2 μM, respectively.14 An analysis of the binding mode of 1 (Figure 3a) indicated that the bite angle, defined by the N and C substituents attached to the α-carbon of the P1 amino acid, is optimal in the cyclopropyl series. This angle controls the relative positions of the H-bond donor (N–H) and H-bond acceptor (C=O) that flank the cyclopropyl ring (Figure 3a). The spatial complementarity of these elements to the H-bond acceptor (Arg255) and donors (Ser139 and Gly137) within the enzyme is essential to the efficiency of H-bonding and hence activity. The measured bite angle for the bound structure of 1 is ∼118° (Figure 3a), which is, interestingly, the same as that found (118°) in the single crystal X-ray structure of 1, suggestive of an effective preorganization for optimal binding with the protein.4 The bite angle for 16 when bound to the enzyme is also 118° (Figure 3b);13 however, the average bite angle for a prototypical acyclic amino acid is ∼108°, which suggests that upon binding 16 requires significant reorganization to enable optimal H-bonding with the enzyme.15
The cyclopropyl ring system at P1 also provides an optimal dihedral angle between the backbone N and carbonyl O moieties that flank the P1 cyclopropyl. In the bound state, the P1 dihedral angle in 1 is ∼176°, rendering it near planar (Figure 3a), which compares with the ∼160° dihedral angle measured in the single crystal X-ray structure of 1.4 Coupled with the bite angle analysis, this observation reinforces the critical role of the cyclopropyl group in preorganizing the P1 moiety for optimal binding with the enzyme. Not surprisingly, small structural modifications can result in a significant loss in activity, exemplified by the P1 spirocyclobutyl derivative 17, which is 6-fold less potent than its matched pair 11 (Figure 3c).
Given the critical nature of the cyclopropyl group at P1, the potential of a CHF2 substituent was given consideration. A model of the proposed compound 5 (R1 = CHF2) bound to the NS3/4A protease complex suggested the potential for a H-bonding interaction between the C–H of the CHF2 group and the carbonyl oxygen atom of Leu135. This model relied upon the (1R, 2R) stereochemistry of the CHF2-substituted cyclopropyl amino acid moiety analogous to that found in 1 and 3.
The synthesis of the desired P1 difluoromethylcyclopropyl amino acid is presented in Scheme 1.16,17 The commercially available CHF2CHO derivative 25 was subjected to a Horner–Wadsworth–Emmons homologation with 24 to provide the unsaturated amino acid derivative 26,18 with the Z-olefin geometry assigned by 1H NMR.19 The carbamate N atom of 26 was protected with a Boc moiety to provide intermediate 27, which underwent cyclopropanation upon exposure to (CH3)3SO+I– and NaH, providing 28 as a mixture of stereoisomers (syn/anti = 3.5:1). De-esterification of 28 using NaOH/MeOH afforded 29, with the relative stereochemistry of the cyclopropane ring substituents confirmed by a single crystal X-ray structure determination (Figure 4). Amino acid 29 was incorporated into the tripeptide acylsulfonamide series using the previously described chemistry outlined in Scheme 2, with the final compound isolated as a mixture of stereoisomers 18 and 19. A single crystal X-ray structure of 18 confirmed the absolute stereochemistry of the P1 moiety as (1R, 2R), consistent with that found at the P1 position of 1.
Scheme 1. Synthesis of the N-Boc Difluoromethylcyclopropyl Amino Acid 29.

Reagents and conditions: (a) KOtBu, THF, −78 °C to rt, 44%; (b) separation by a SFC column; (c) (Boc)2O, DMAP, rt, 2 h,THF, 85%; (d) (CH3)3SO+I–, NaH, DMSO, 80 °C, 2 h, 48%; (e) NaOH, MeOH, rt, 18 h, 87%.
Figure 4.
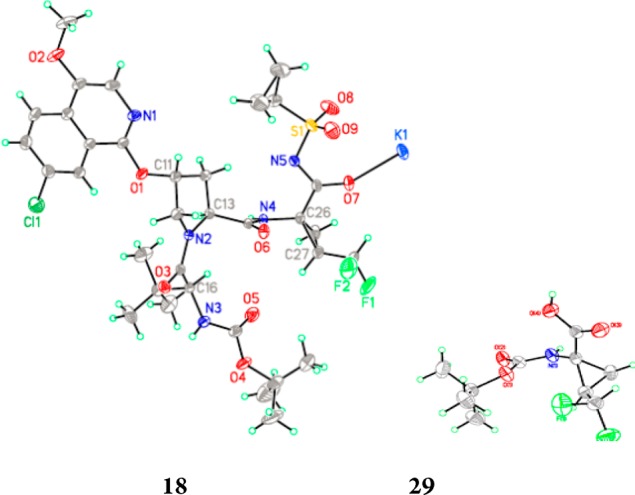
Single crystal X-ray structures of 18 and 29 (racemate).
Scheme 2. Synthesis of the Tripeptide Acylsulfonamide Derivatives 18 and 19.

Reagents and conditions: (a) CDI, DBU, THF, reflux, 84%; (b) HCl/dioxane, rt, 100%; (c) HATU, 4-methylmorhholine, DCM, rt, 78%.
Compound 18 was found to be a potent inhibitor of HCV NS3/4A protease, IC50 = 1 nM, with low nanomolar antiviral activity observed in the cell-based replicon assay (EC50 = 8 nM). A cocrystal structure of 18 with the NS3/4A protease complex indicated that 18 bound to the enzyme analogously to 1 (Figure 5).13 The P1 bite angle for 18 was measured as 115.6°, while the dihedral angle between the backbone N and carbonyl O that flanks the P1 cyclopropyl was measured as −174.7°. Moreover, the CHF2 group projected toward Leu135, with the C–H of the CHF2 group positioned 2.7 Å from the carbonyl O atom, a distance consistent with a H-bond. In the single crystal X-ray structure of 18, the P1 bite angle (115.06°) and the dihedral angle (178.7°) were similar to that found in the cocrystal structure, once again illustrating the role of the cyclopropyl moiety in preorganizing the P1 site.
Figure 5.
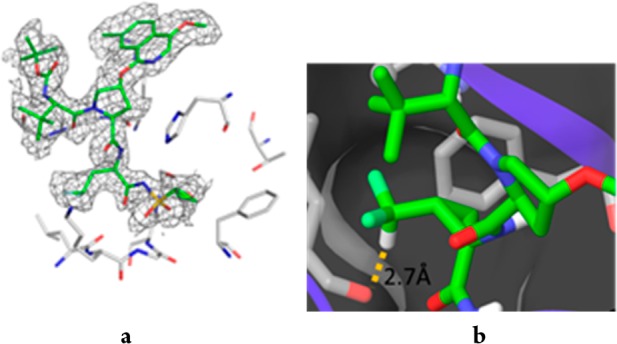
(a) Cocrystal structures of 18 bound to HCV NS3/4A. (b) Details of the P1 subsite of 18 bound to the NS3/4A protease complex.
Compound 18 was 13-fold more active than the CH3 analogue 20 and 17-fold more active than the corresponding CF3 homologue 21. Illustrating the importance of the correct positioning of the CF2H moiety, the one carbon homologue 22 was 14-fold less active than 18, but similar in potency to the ethyl analogue 23, while the CHF2 moiety in 19, a diastereoisomer of 18, projects in a manner that does not enable H-bonding within the S1 pocket. These SAR points indicate the importance of the C–H H-bond between the P1 CHF2 group in 18 and the C=O of Leu135. The H-bonding potential of the P1 amino acid deployed in 18 bears similarity to serine and cysteine but is distinct by virtue of its conformational rigidity as well as a more lipophilic disposition. For example, the cLogP value of 29 is 1.4, while that for the N-Boc serine is −0.12 and that for the N-Boc cysteine is 1.0, suggesting potential as a replacement for serine or cysteine in cases where enhanced rigidity and lipophilicity may provide an advantage.
The t1/2 of 18 in human liver microsomes (HLMs) was 120 min, while permeability across a confluent Caco-2 layer was low, <15 nm/s (Pc A-B); 47 nm/s (Pc B-A) (Table 1). Following IV administration, 18 demonstrated a short t1/2, which was driven by moderate clearance, while the oral bioavailability was poor (F < 1%), consistent with the low Caco-2 cell permeability. These PK parameters were inferior compared to those recorded for the P1 bis-cyclopropyl analogue 8, which had an oral bioavailability of 34% and a prolonged plasma t1/2 of 6 h, further underscoring the significance of small structural changes on the PK profile of analogues within this series. The reduced exposure observed with 18 compared to 8 suggests that the P1 moiety in 8 is important for optimal absorption and that the lipophilicity intrinsic to the cyclopropyl group of 8 may more effectively balance the polarity associated with the adjacent acylsulfonamide moiety than does the CHF2 in 18. Consistent with this hypothesis, the cLogP value for 18 was 3.2, while that for 8 was 0.3 Log unit greater at 3.5. Likewise, the ethyl derivative 23, cLogP = 3.7, demonstrated improved PK compared to 18. Ultimately, the poor oral bioavailability of 18 limited the progression of the CHF2-based chemotype in this acyclic series, and efforts were diverted to the bis-cyclopropyl chemotype, which, as previously noted, yielded a back-up candidate to 1. However, the potential of the CHF2-substitued cyclopropyl amino acid moiety has been further confirmed by its presence in the recently approved HCV NS3 inhibitors voxilaprevir and glecaprevir.22,23 While details of these research efforts have yet to be provided, the presence of the CHF2-substituted cyclopropyl-based P1 moiety in these marketed drugs coupled with the design insights provided herein suggest the potential utility of this interesting and useful amino acid.
Table 1. Virology and PK Parameters for a Series of P1-Modified Tripeptidic HCV NS3 Protease Inhibitors.

GT-1a = genotype-1a; HCV NS3 enzyme inhibitory activity was assessed according to the conditions previously described.20
GT-1b = genotype-1b; HCV replicon inhibitory activity was assessed in the presence of 10% fetal bovine serum (FBS) according to the conditions previously described.21
IV/PO dose: 5/15 mg/kg, n = 3. Vehicle: PEG-400/ethanol (90/10, v/v).
Oral bioavailability.
Pharmacokinetic area under curve.
Pharmacokinetic clearance.
Plasma half-life.
In summary, the incorporation of a difluoromethyl cyclopropyl amino acid in the tripeptide 18 conferred 13-fold higher potency than the corresponding des-fluoro analogue 20. A cocrystal structure of 18 with an NS3/4A protease complex construct indicated the presence of a H-bond between the C–H of the CHF2 group and the carbonyl oxygen atom of Leu135. While the PK profiles of this series were poor, the results demonstrate the potential of this amino acid moiety as a tool to drive activity through the use of the CHF2 group as a H-bond donor.
Acknowledgments
We thank the following individuals for their roles they played in enabling the work described in this paper: DDS-BBRC, John Sack, Xiaohu Huang, Jingfang Cutrone, Dieter Drexler, Edward S. Kozlowski, Christopher J. Poronsky, Julia M. Nielson, and Kap-Sun Yeung.
Glossary
ABBREVIATIONS
- AUC
area under the curve
- Cl
clearance
- CV
cardiovascular
- DAA
direct-acting antiviral agent
- HCV
hepatitis C virus
- HLM
human liver microsomes
- PK
pharmacokinetic
- PO
per os
- SAR
structure–activity relationship
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00503.
Synthetic methods and characterization data for compounds 18 and 26–31, and methods for in vitro, in vivo, and pharmacokinetic assays (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Li G.; De Clercq E. Current therapy for chronic hepatitis C: the role of direct-acting antivirals. Antiviral Res. 2017, 142, 83–122. 10.1016/j.antiviral.2017.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lok A. S.; Gardiner D. F.; Lawitz E.; Martorell C.; Everson G. T.; Ghalib R.; Reindollar R.; Rustgi V.; McPhee F.; Wind-Rotolo M.; Persson A.; Zhu K.; Dimitrova D. I.; Eley T.; Guo T.; Grasela D. M.; Pasquinelli C. Preliminary study of two antiviral agents for hepatitis C genotype 1. N. Engl. J. Med. 2012, 366, 216–224. 10.1056/NEJMoa1104430. [DOI] [PubMed] [Google Scholar]
- Poole R. M. Daclatasvir + asunaprevir: first global approval. Drugs 2014, 74, 1559–1571. 10.1007/s40265-014-0279-4. [DOI] [PubMed] [Google Scholar]
- Scola P. M.; Sun L.-Q.; Wang A. X.; Chen J.; Sin N.; Venables B. L.; Sit S.-Y.; Chen Y.; Cocuzza A.; Bilder D. M.; D’Andrea S. V.; Zheng B.; Hewawasam P.; Tu Y.; Friborg J.; Falk P.; Hernandez D.; Levine S.; Chen C.; Yu F.; Sheaffer A. K.; Zhai G.; Barry D.; Knipe J. O.; Han Y.-H.; Schartman R.; Donoso M.; Mosure K.; Sinz M. W.; Zvyaga T.; Good A. C.; Rajamani R.; Kish K.; Tredup J.; Klei H. E.; Gao Q.; Mueller L.; Colonno R. J.; Grasela D. M.; Adams S. P.; Loy J.; Levesque P. C.; Sun H.; Shi H.; Sun L.; Warner W.; Li D.; Zhu J.; Meanwell N. A.; McPhee F. The discovery of asunaprevir (BMS-650032), an orally efficacious NS3 protease inhibitor for the treatment of hepatitis C virus infection. J. Med. Chem. 2014, 57, 1730–1752. 10.1021/jm500297k. [DOI] [PubMed] [Google Scholar]
- Scola P. M.; Wang A. X.; Good A. C.; Sun L.-Q.; Combrink K. D.; Campbell J. A.; Chen J.; Tu Y.; Sin N.; Venables B. L.; Sit S.-Y.; Chen Y.; Cocuzza A.; Bilder D. M.; D’Andrea S.; Zheng B.; Hewawasam P.; Ding M.; Thuring J.; Li J.; Hernandez D.; Yu F.; Falk P.; Zhai G.; Sheaffer A. K.; Chen C.; Lee M. S.; Barry D.; Knipe J. O.; Li W.; Han Y.-H.; Jenkins S.; Gesenberg C.; Gao Q.; Sinz M. W.; Santone K. S.; Zvyaga T.; Rajamani R.; Klei H. E.; Colonno R. J.; Grasela D. M.; Hughes E.; Chien C.; Adams S.; Levesque P. C.; Li D.; Zhu J.; Meanwell N. A.; McPhee F. Discovery and early clinical evaluation of BMS-605339, a potent and orally efficacious tripeptidic acylsulfonamide NS3 protease inhibitor for the treatment of hepatitis C virus infection. J. Med. Chem. 2014, 57, 1708–1729. 10.1021/jm401840s. [DOI] [PubMed] [Google Scholar]
- Sun L.-Q.; Mull E.; Zheng B.; D’Andrea S.; Zhao Q.; Wang A. X.; Sin N.; Venables B. L.; Sit S.-Y.; Chen Y.; Chen J.; Cocuzza A.; Bilder D. M.; Mathur A.; Rampulla R.; Chen B.-C.; Palani T.; Ganesan S.; Arunachalam P. N.; Falk P.; Levine S.; Chen C.; Friborg J.; Yu F.; Hernandez D.; Sheaffer A. K.; Knipe J. O.; Han Y.-H.; Schartman R.; Donoso M.; Mosure K.; Sinz M. W.; Zvyaga T.; Rajamani R.; Kish K.; Tredup J.; Klei H. E.; Gao Q.; Ng A.; Mueller L.; Grasela D. M.; Adams S.; Loy J.; Levesque P. C.; Sun H.; Shi H.; Sun L.; Warner W.; Li D.; Zhu J.; Wang Y.-K.; Fang H.; Cockett M. I.; Meanwell N. A.; McPhee F.; Scola P. M. Discovery of a potent acyclic, tripeptidic, acyl sulfonamide inhibitor of hepatitis C virus NS3 protease as a back-up to asunaprevir with the potential for once-daily dosing. J. Med. Chem. 2016, 59, 8042–8060. 10.1021/acs.jmedchem.6b00821. [DOI] [PubMed] [Google Scholar]
- Kwong A. D.; Kim J. L.; Rao G.; Lipovsek D.; Raybuck S. A. Hepatitis C virus NS3/4A protease. Antiviral Res. 1998, 40, 1–18. 10.1016/S0166-3542(98)00043-6. [DOI] [PubMed] [Google Scholar]
- Narjes F.; Brunetti M.; Colarusso S.; Gerlach B.; Koch U.; Biasiol G.; Fattori D.; De Francesco R.; Matassa V. G.; Steinkuehler C. α-Ketoacids are potent slow binding inhibitors of the hepatitis C virus NS3 protease. Biochemistry 2000, 39, 1849–1861. 10.1021/bi9924260. [DOI] [PubMed] [Google Scholar]
- Narjes F.; Koehler K. F.; Koch U.; Gerlach B.; Colarusso S.; Steinkühler C.; Brunetti M.; Altamura S.; De Francesco R.; Matassa V. G. A designed P1 cysteine mimetic for covalent and non-covalent inhibitors of HCV NS3 protease. Bioorg. Med. Chem. Lett. 2002, 12, 701–704. 10.1016/S0960-894X(01)00842-3. [DOI] [PubMed] [Google Scholar]
- Erickson J. A.; McLoughlin J. I. Hydrogen bond donor properties of the difluoromethyl group. J. Org. Chem. 1995, 60, 1626–1631. 10.1021/jo00111a021. [DOI] [Google Scholar]
- Zafrani Y.; Yeffet D.; Sod-Moriah G.; Berliner A.; Amir D.; Marciano D.; Gershonov E.; Saphier S. Difluoromethyl bioisostere: examining the “lipophilic hydrogen bond donor” concept. J. Med. Chem. 2017, 60, 797–804. 10.1021/acs.jmedchem.6b01691. [DOI] [PubMed] [Google Scholar]
- Sessler C. D.; Rahm M.; Becker S.; Goldberg J. M.; Wang F.; Lippard S. J. CF2H, a hydrogen bond donor. J. Am. Chem. Soc. 2017, 139, 9325–9332. 10.1021/jacs.7b04457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The coordinates of the complex of HCV NS3 protease with compounds 16 (PDBID 6BQJ) and 18 (PDBID 6BQK) have been deposited in the Protein Data Bank (www.rcsb.org).
- Rancourt J.; Cameron D. R.; Gorys V.; Lamarre D.; Poirier M.; Thibeault D.; Llinas-Brunet M. Peptide-based inhibitors of the hepatitis C virus NS3 protease: structure-activity relationship at the C-terminal position. J. Med. Chem. 2004, 47, 2511–2522. 10.1021/jm030573x. [DOI] [PubMed] [Google Scholar]
- Messai A.; Benali-Cherif R.; Jeanneau E.; Benali-Cherif N. Structure and thermal analysis of amino acids. Acta Crystallogr., Sect. E: Struct. Rep. Online 2012, E68, o1307–o1308. 10.1107/S1600536812013682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A. X.; Zheng B. Z.; D’Andrea S.; Zhao Q.; Scola P. M.. Preparation of proline-containing tripeptides as hepatitis C virus inhibitors. World Patent Application, 2008064066, 2008.
- Wan W.; Gao Y.; Jiang H.; Hao J. Fluoroalkyl substituted (Z)-dehydro α-amino ester as a building block for the fluorine-containing cyclopropyl α-amino esters and dihydrooxazole. J. Fluorine Chem. 2008, 129, 510–514. 10.1016/j.jfluchem.2008.03.008. [DOI] [Google Scholar]
- Lamar J.; Hu J.; Bueno A. B.; Yang H.-C.; Guo D.; Copp J. D.; McGee J.; Gitter B.; Timm D.; May P.; McCarthy J.; Chen S.-H. Phe*-Ala-based pentapeptide mimetics are BACE inhibitors: P2 and P3 SAR. Bioorg. Med. Chem. Lett. 2004, 14, 239–243. 10.1016/j.bmcl.2003.09.084. [DOI] [PubMed] [Google Scholar]
- Hu Z.; Han W. An efficient chiral synthesis of fluoro-containing amino acids: N-benzyloxycarbonyl-2-amino-4,4-difluorobutyric acid methyl ester and its analogs. Tetrahedron Lett. 2008, 49, 901–902. 10.1016/j.tetlet.2007.11.163. [DOI] [Google Scholar]
- Wang X. A.; Sun L.-Q.; Sit S.-Y.; Sin N.; Scola P. M.; Hewawasam P.; Good A. C.; Chen Y.; Campbell J. A.. Hepatitis C virus inhibitors. U.S. Patent 6,995,174, 2006.
- Sheaffer A. K.; Lee M. S.; Hernandez D.; Chaniewski S.; Yu F.; Falk P.; Friborg J.; Zhai G.; McPhee F. Development of a chimeric replicon system for phenotypic analysis of NS3 protease sequences from HCV clinical isolates. Antiviral Ther. 2011, 16, 705–718. 10.3851/IMP1825. [DOI] [PubMed] [Google Scholar]
- Voaklander R.; Jacobson I. M. Sofosbuvir, velpatasvir and voxilaprevir combination for the treatment of hepatitis C. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 789–795. 10.1080/17474124.2017.1351295. [DOI] [PubMed] [Google Scholar]
- Poordad F.; Felizarta F.; Asatryan A.; Sulkowski M. S.; Reindollar R. W.; Landis C. S.; Gordon S. C.; Flamm S. L.; Fried M. W.; Bernstein D. E.; Lin C.-W.; Liu R.; Lovell S. S.; Ng T. I.; Kort J.; Mensa F. J. Glecaprevir and pibrentasvir for 12 weeks for hepatitis C virus genotype 1 infection and prior direct-acting antiviral treatment. Hepatology 2017, 66, 389–397. 10.1002/hep.29081. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.