

Abstract
The diagnosis and study of the fragile X–related disorders is complicated by the difficulty of amplifying the long CGG/CCG-repeat tracts that are responsible for disease pathology, the potential presence of AGG interruptions within the repeat tract that can ameliorate expansion risk, the occurrence of variable DNA methylation that modulates disease severity, and the high frequency of mosaicism for both repeat number and methylation status. These factors complicate patient risk assessment. In addition, the variability in these parameters that is seen when patient cells are grown in culture requires their frequent monitoring to ensure reproducible results in a research setting. Many existing assays have the limited ability to amplify long alleles, particularly in a mixture of different allele sizes. Others are better at this, but are too expensive for routine use in most laboratories or for newborn screening programs and use reagents that are proprietary. We describe herein a set of assays to routinely evaluate all of these important parameters in a time- and cost-effective way.
CME Accreditation Statement: This activity (“JMD 2016 CME Program in Molecular Diagnostics”) has been planned and implemented in accordance with the Essential Areas and policies of the Accreditation Council for Continuing Medical Education (ACCME) through the joint providership of the American Society for Clinical Pathology (ASCP) and the American Society for Investigative Pathology (ASIP). ASCP is accredited by the ACCME to provide continuing medical education for physicians.
The ASCP designates this journal-based CME activity (“JMD 2016 CME Program in Molecular Diagnostics”) for a maximum of 36 AMA PRA Category 1 Credit(s)™. Physicians should claim only credit commensurate with the extent of their participation in the activity.
CME Disclosures: The authors of this article and the planning committee members and staff have no relevant financial relationships with commercial interests to disclose.
The fragile X–related disorders are diseases resulting from the expansion of a CGG/CCG-repeat tract in the 5′ untranslated region of the FMR1 gene (Mendelian Inheritance in Man no. 309550).1 These disorders include fragile X–associated primary ovarian insufficiency and fragile X–associated tremor/ataxia syndrome (Mendelian Inheritance in Man no. 300623) that are seen in carriers of alleles with 55 to 200 repeats, so-called premutation (PM) alleles. Fragile X syndrome (Mendelian Inheritance in Man no. 300624), the most common heritable cause of intellectual disability, is seen in carriers of full mutation (FM) alleles, alleles that have >200 repeats. The differences in pathology seen in carriers of FM and PM alleles stem from the fact that fragile X–associated tremor/ataxia syndrome and fragile X–associated primary ovarian insufficiency arise from some as yet unresolved deleterious consequence of the expression of FMR1 transcripts with large repeat numbers, whereas fragile X syndrome results from repeat-mediated gene silencing. However, the situation is complicated because the repeat tract is unstable, undergoing expansions and contractions in both the germline and somatic cells. Thus, patients can be mosaic for a combination of FM and PM alleles. Furthermore, the extent of gene silencing may vary, with even some PM carriers showing significant methylation of their FMR1 allele.2 In addition, many alleles contain one or more AGG interruptions at the 5′ end of the repeat tract that significantly affect expansion risk.3, 4, 5, 6, 7
Knowing the total number of repeats, the number of AGG interruptions, and the methylation status of the FMR1 gene are thus important for a proper understanding of an individual's risk of transmission of larger alleles to their offspring and to their personal risk of disease pathology. It is also important for laboratory studies because these parameters are expected to affect many cellular properties, such as cell viability and gene expression. We have previously demonstrated that the CGG/CCG-repeats can be unstable in tissue culture, in one case changing over time from having a single PM allele to having multiple larger alleles that extended into the full mutation range.8 Given the increased use of tissue culture models for studying repeat instability and disease pathology and the development of cell-based assays for high-throughput screening for therapeutics,9, 10, 11 the careful monitoring of cells in culture is essential. However, Southern blotting requires large amounts of DNA, is time consuming, is relatively insensitive, and only allows limited resolution of CGG/CCG-repeat sizes. PCR-based diagnostic strategies are complicated by the fact the individual strands of the CGG/CCG-repeats form secondary structures, some of which are stable, particularly when methylated.12, 13, 14, 15, 16 This makes PCR through the repeats relatively inefficient, resulting in diminishing yields of larger alleles. This can cause the extent of mosaicism in the culture to be underestimated and larger alleles to go undetected.
Several commercial kits are currently available for the PCR-based detection of FMR1 alleles in patient cells.17 However, their expense limits their routine use in a research setting, and they each have some methodological limitations. The compositions of their reagents are also proprietary. Several other assays have been proposed, but many are limited either in terms of the information they provide or by virtue of their use of specialized equipment or technologies not commonly available in basic research laboratories.18, 19, 20, 21, 22, 23 We describe herein a set of assays for the accurate determination of the repeat size, the number of AGG-interruptions, and the extent of methylation of most PM and FM alleles that is both robust and inexpensive enough for routine research use and has the potential to be optimized for use in a clinical setting as well.
Materials and Methods
Cell Lines/DNA
The cell lines and DNAs used in this study are listed in Table 1. The lymphoblastoid cell lines GM06897 and GM04025 were obtained from the Coriell Institute for Medical Research (Camden, NY). C10700 and C10259 FM fibroblasts were provided by Carl Dobkin (Institute for Basic Research in Developmental Disabilities, Staten Island, NY), the SC120 PM and SC128 FM induced pluripotent stem cell lines were provided by Phil Schwartz (CHOC Research Institute, Orange, CA), the F006 PM fibroblasts were from Deborah Hall and Elizabeth Berry-Kravis (Rush University Medical Center, Chicago, IL), the SB5 normal female fibroblast line was from Kenneth Fischbeck (National Institutes of Neurological Diseases and Stroke, Bethesda, MD), and the H1 normal embryonic stem cell line was from Barbara Mallon (NIH Stem Cell Unit, NIH, Bethesda, MD). The HT-51E cell line is an induced pluripotent stem cell line derived from F006 PM fibroblasts in our laboratory. The FX-1, FX-2, FX-3, and FX-4 blood and saliva samples were obtained from Carolyn Smith (National Institute of Mental Health, Bethesda, MD). The DNA samples F10149, F15608, F23656, F23297, F23690, F23856, and F20729, obtained from blood, were provided by Sally Nolin (Institute for Basic Research in Developmental Disabilities), and the A8018, A8020, and A7306 DNA samples, obtained from blood, were provided by Ariane Soldatos (Undiagnosed Diseases Program, NIH, Bethesda, MD). The NA09237 DNA from the lymphoblastoid cells from a FM carrier, GM09237, was obtained from Coriell Institute for Medical Research. The fragile X syndrome embryonic stem cells, WCMC37, were obtained from Nikica Zaninovic (Weill Cornell Medical College of Cornell University, New York, NY). The MC37B subclone was derived from this cell line by isolating and expanding individual colonies. The repeat size for previously undescribed cell lines was verified by Southern blotting and PCR using the Asuragen kit by an independent laboratory (Carl Dobkin). The primers used in this study were obtained from Life Technologies (Grand Island, NY) and are listed in Table 2. Genomic DNA from cell lines was prepared using standard procedures. Genomic DNA from human saliva was collected in an OGR-500 (Oragene, DNA Genotek, Kanata, ON, Canada) and purified using the prepIT-L2P reagent (DNA Genotek), as instructed by the manufacturer. DNA quantification was performed on a Denovix DS-11 spectrophotometer (Denovix, Wilmington, DE).
Table 1.
FMR1 Allele Characterization
| Sample ID | Sex | Repeat no.∗ | % Methylation | AGGs |
|---|---|---|---|---|
| GM06897 | M | 240-570 (Smear) | 5 | Y |
| GM09145 | M | 660 | 97 | N |
| GM04025 | M | 645 | 110/77† | N |
| GM03200 | M | 570 | 112 | N |
| GM09237 | M | 940 | 111 | N |
| GM07294 | M | 745 | 94 | N |
| C10700 | M | 350, 780, 1075 | 89 | Y |
| C10259 | M | 930 | 102 | Y |
| SC128 | M | 210 | 98 | N |
| FX-1 | M | 218, 340, 410, 510 | 57 | N |
| FX-2 | M | 240, 250, 410 | 75 | Y |
| FX-3 | M | 202, 310, 360, 490, 560, 750 | 102 | N |
| FX-4 | M | 164, 177, FM smear | 43 | N |
| WCMC37 | M | 410, 510 | 85 | N |
| MC37B‡ | M | 100, 185, 330, 410, 510 | 110 | N |
| F20729 | M | 285, >400 | 106 | Y |
| F23856 | F | 30, >200 | 53 | Y, Y |
| F23690 | F | 31, 240, 250, 310 | 63 | Y, Y |
| F10149 | M | 94 | 3 | Y |
| F15608 | M | 101 | 9 | Y |
| F006 | F | 29, 92 | 55 | Y, Y |
| HT-51E§ | F | 29, 92 | 47 | Y, Y |
| SC120 | M | 97 | 4 | N |
| A8018¶ | F | 30, 30 | 55 | Y, Y |
| A8020¶ | M | 52 | 1 | Y |
| A7306 | F | 30, 52 | 64 | Y, Y |
| F23297 | F | 23, 31 | 50 | Y, Y |
| F23656 | F | 24, 30 | 63 | Y, Y |
| SB5 | F | 30, 30 | 48 | Y, Y |
| H1 | M | 31 | 6 | Y |
| GM06865 | M | 30 | 5 | Y |
F, female; M, male; FM, full mutation; N, no AGG interruptions; Y, at least one interruption in each allele.
All full mutation allele sizes are approximate because of the limits of resolution of agarose gels and capillary gel electrophoresis.
The different methylation percentages reflect the extent of methylation in untreated cells versus cells treated with 5-azadeoxycytidine.
Subclone of WCMC37 embryonic stem cell line.
HT-51E is an induced pluripotent stem cell line derived from F006 fibroblasts.
A8018 and A8020 are the mother and father of the A7306 daughter.
Table 2.
DNA Sequences
| Primer/fragment | Sequence |
|---|---|
| Not_FraxC | 5′-AGTTCAGCGGCCGCGCTCAGCTCCGTTTCGGTTTCACTTCCGGT-3′ |
| Not_FraxR4 | 5′-CAAGTCGCGGCCGCCTTGTAGAAAGCGCCATTGGAGCCCCGCA-3′ |
| Fmr1_CCG5 | 5′-AGCGTCTACTGTCTCGGCACTTGCCCGCCGCCGCCGCCG-3′ |
| Fmr1_CGG5 | 5′-AGCGTCTACTGTCTCGGCACTTGCCGGCGGCGGCGGCGG-3′ |
| FMR1 exon 1 (forward) | 5′-GAACAGCGTTGATCACGTGAC-3′ |
| FMR1 exon 1 (reverse) | 5′-GTGAAACCGAAACGGAGCTGA-3′ |
| GAPDH exon1 (forward) | 5′-TCGACAGTCAGCCGCATCT-3′ |
| GAPDH intron1 (reverse) | 5′-CTAGCCTCCCGGGTTTCTCT-3′ |
| Internal methylation control fragment | 1 GCTCAGCTCC GTTTCGGTTT CACTTCCGGT AGCACGTCGC GGGGCGCCGT 51 AACGGCGAAT CGAGGGCGCG CACCGCCGTG ACATCGGCGT CATTCGGTCG 101 CATCACGGTT GTCGCCGGTG CGCGGTCGGA AAAGCGGGCG CGAGGCAGCG 151 GCGACGGCGC GAGTGAATCT AACGGGCGAC AACGCGTTCG GCCTCGGAGT 201 GGCGGCCACG GGTATGCGGG GCTCCAATGG CGCTTTCTAC AAG |
Sequences in bold are not homologous to the FMR1 gene. The primer sequences are underlined, whereas the 2 HpaII sites within the amplicon are italicized.
FMR1 Allele Assays
These assays are all PCR based. The primers used for these assays are listed in Table 2.
Repeat Number Assay (RPT-PCR)
Genomic DNA was predigested with HindIII, which is not methylation sensitive and does not cut within the PCR amplicon. The resultant smaller genomic DNA fragments are more efficiently denatured in the PCR. DNA (600 ng) was made to a final volume of 45 μL of a solution containing 50 mmol/L Tris-HCl (pH 8.9), 1.5 mmol/L MgCl2, 22 mmol/L (NH4)2SO4, 0.2% Triton X-100, and 1 μL of FastDigest-HindIII (Life Technologies, Carlsbad, CA). A PCR master mix was made up containing 50 mmol/L Tris-HCl (pH 8.9), 1.5 mmol/L MgCl2, 22 mmol/L (NH4)2SO4, 0.2% Triton X-100, 0.67 μmol/L of the primers Not_FraxC and Not_FraxR4, 3.3 mol/L betaine (Sigma, St. Louis, MO), 2.67% dimethyl sulfoxide (Sigma), and 0.27 mmol/L each dNTP (New England Biolabs, Ipswich, MA). Phusion DNA polymerase (New England Biolabs) was added to 2.7 U/100 μL. Five microliters of the digestion mixes was mixed on ice with 15 μL of the PCR master mix, giving a final composition of 50 mmol/L Tris-HCl (pH 8.9), 1.5 mmol/L MgCl2, 22 mmol/L (NH4)2SO4, 0.2% Triton X-100, 0.5 μmol/L each primer, 2.5 mol/L betaine, 2% dimethyl sulfoxide, 0.2 mmol/L each dNTP, and 0.4 U Phusion polymerase. The samples were then loaded onto a preheated (>70°C) PCR block (C1000-Touch; Bio-Rad, Hercules, CA) and subjected to the following cycles of heating and cooling: 98°C for 3 minutes, 30 × (98°C for 30 seconds, 64°C for 30 seconds, 72°C for 210 seconds), and 72°C for 10 minutes. The PCR products were resolved on agarose gels and visualized with ethidium bromide (Life Technologies) or SYBR Gold (Thermo Fisher Scientific, Waltham, MA), according to standard procedures. Because the region flanking the repeat in the PCR product consists of a total of 269 bases, the repeat number associated with each PCR product can be derived using the following formula: N ∼ (fragment size − 269)/3.
When a fluorescently labeled primer was used, the samples could also be resolved by high-resolution capillary electrophoresis on an ABI Genetic Analyzer (Thermo Fisher Scientific) and the PCR products analyzed using GeneMapper software version 5 (Thermo Fisher Scientific). Because CGG repeats migrate slightly faster than similarly sized fragments in typical molecular weight ladders used in capillary electrophoresis, like the Genescan 500 ROX size standard, it is necessary, as is the case with commercially available kits, to use a standard curve generated using alleles of known repeat number to obtain accurate results. For our standards, we used five alleles whose repeat size had been previously determined using a commercially available protocol by another laboratory (Carl Dobkin, unpublished data). The alleles had repeat numbers ranging from 23 to 177. Some of the electrophoretograms shown herein were generated using an R script incorporating the plotabif and peakabif modules from the seqinR suite.24
Instead of the standard cycle parameters described above, the following heat pulse-extension regimen was used for those rare samples that were not amplified in the standard RPT-PCR assay: 98°C for 3 minutes, 30 × [98°C for 30 seconds, 64°C for 30 seconds, ramp at 0.6°C/seconds to 72°C, hold 2 seconds, 7 × (86°C for 2 seconds, 72°C for 2 seconds), 7 × (89°C for 2 seconds, 72°C for 2 seconds), 7 × (90°C for 2 seconds, 72°C for 2 seconds)], 72°C for 10 minutes.25
Triplet-Primed PCR Assay for AGG Interruptions (TP_RPT-PCR)
For determination of the AGG-interspersion pattern, the repeat-containing primers, Fmr1_CCG5 or Fmr1_CGG5, were added to the RPT-PCR mix to a final concentration of 0.033 μmol/L (1:20 ratio to external primers) and the samples analyzed as described above.
Assay for the Number of Uninterrupted CGG/CCG-Repeats at the 3′ End of the Repeat Tract (AGG_RPT-PCR)
A 10 μL aliquot of the standard RPT-PCR was digested with 2 U EciI or mock digested in a final volume of 20 μL containing 2 μL 10× CutSmart buffer (New England Biolabs). Digestion was performed for 1 hour and the enzyme inactivated by incubation at 80°C for 20 minutes. The extent of EciI digestion can be assessed by inclusion of M13 DNA in the digestion mix or in a digestion performed in parallel. The location of the first A in the repeat tract is given by the formula: N1 ∼ (B1 − 144)/3, where N1 is the number of the repeat corresponding to the first AGG interruption, B1 is the length of the approximately 170 bp digestion product, and 144 corresponds to the sum of the 135 bp from the 5′ flanking region in the PCR product and the nine additional bases resulting from EciI cleavage downstream of the AGG. The number of uninterrupted repeats at the 3′ end is given by the formula: N2 ∼ (B2 − 125)/3, where N2 is the number of uninterrupted repeats at the 3′ end of the repeat tract, B2 is the length of the second digestion product, and 125 corresponds to the number of bases in the 3′ flanking region of the PCR fragment minus the bases resulting from EciI cleavage downstream of the A. For more precise analysis of the AGG interruption status, the RPT-PCR assay was performed using a fluoroscein (FAM)-labeled Not_FraxC primer and a hexachlorofluorescein (HEX)-labeled Not_FraxR4 primer. The products of the subsequent EciI digest were resolved by capillary electrophoresis. As with the standard RPT-PCR assay, an accurate determination of the number of uninterrupted repeats requires the use of an appropriate set of standards. In this case, it would be samples for which the interspersion pattern was known.
Methylation-Sensitive Repeat PCR (MS_RPT-PCR)
For analysis of the methylation status of the region containing the repeat, a methylation-sensitive enzyme, HpaII, was included in the predigestion step of the RPT-PCR described above. Specifically, a 20 μL aliquot of the digestion mix containing HindIII was transferred to a new tube to which 10 U HpaII (New England Biolabs) was added. Equivalent aliquots of the mix with and without HpaII were digested overnight at 37°C. Both samples were then processed for the RPT-PCR as described above. An internal control for HpaII digestion was generated as follows. A 243-bp fragment with the same GC content as the FMR1 CGG/CCG-flanking sequence was synthesized. This fragment contained two HpaII sites and can be amplified using the Not_FraxC and Not_FraxR4 primers to give a 271-bp PCR fragment. The sequence of this region is shown in Table 2. The fragment was cloned into the pJAZZ-OK (Lucigen Corp., Middleton, WI) vector to generate pJZ-FMRSyn11. This plasmid can be added to genomic samples at approximately one fifth genome equivalent before predigestion and PCR. This internal control plasmid is available on request.
Quantitative FMR1 Promoter Methylation Assay (qMS-PCR)
DNA samples at a concentration of 10 ng/μL in a total volume of 100 μL in TE buffer were first sonicated using a Bioruptor (Diagenode, Inc., Denville, NJ) on the medium setting with cycles of 30 seconds on/30 seconds off for a total of 5 minutes. This resulted in DNA fragments that were 0.5 to 1 kb in length. Three hundred nanograms of each DNA sample was then either mock digested or digested overnight with HpaII in CutSmart buffer (New England Biolabs) in a total volume of 50 μL. Quantitative PCR was then performed in a final volume of 20 μL using 2 μL of the mock digested and digested DNA (13 ng), 0.4 μL each of 10 μmol/L stocks of the FMR1 ex 1 (F) and FMR1 ex 1 (R) primers (Table 2),26 and 10 μL of Power SYBR Green master mix (Thermo Fisher Scientific) on a StepOne Plus (Thermo Fisher Scientific) PCR machine. Cycling parameters were 10 minutes at 95°C, followed by 40 cycles of 15 seconds at 95°C and 60 seconds at 60°C. The extent of DNA methylation was determined by averaging the results of five technical replicates and calculating the difference in the Ct values of the digested and mock digested samples using the ΔΔCt method. A control for HpaII digestion was generated by amplifying a region of the GAPDH gene that contains an unmethylated HpaII site using the same thermal cycling condition as described above. The sequence of primers, GAPDH exon1 (forward), and GAPDH intron1 (reverse) that were used to amplify the GAPDH control region is listed in Table 2.
Results
Robust PCR Assay for the Determination of the CGG/CCG-Repeat Number in FMR1 Alleles
We have previously described a PCR-based assay useful for the analysis of CGG/CCG-repeat numbers in the PM range.8 However, this assay was unable to amplify alleles in the FM range, particularly when the alleles were methylated. In an effort to improve the yield of PCR product from large methylated FMR1 alleles, we tested several DNA polymerases reported to be effective at the amplification of difficult templates, including Phusion DNA polymerase (New England Biolabs) and the Expand High Fidelity enzyme mix (Roche, Indianapolis, IN). These polymerases were tested with a variety of buffers, including those supplied by the manufacturers. These buffers were tested in the absence or presence of betaine, dimethyl sulfoxide, glycerol, 7-deaza-dGTP, and single-stranded DNA binding protein, compounds previously reported to facilitate the amplification of regions of high G + C-content.27, 28 The best results were obtained with Phusion DNA polymerase and K+-free buffers containing betaine and dimethyl sulfoxide. The requirement for a K+-free buffer likely reflects the ability of the repeats to form a quadruplex, a K+-stabilized secondary structure, as we demonstrated previously.12 Addition of glycerol, single-stranded DNA binding protein, and 7-deaza-dGTP gave no appreciable improvement in PCR yield. A further increase in the yield of PCR product was achieved by predigesting the template DNA with HindIII, an enzyme that is methylation insensitive and does not cut within the amplicon of interest. We then optimized the buffer conditions so that the digested material could be used directly in the PCR without purification. Using the resultant protocol, as outlined in Materials and Methods, we were able to amplify an allele with approximately 940 repeats (Figure 1A). For comparison, the results obtained using the Expand High Fidelity enzyme mix in the same buffer are also shown (Roche) (Figure 1A). PM and FM carriers are often mosaic for alleles with a variety of repeat sizes, and the difficulty in amplifying large alleles leads to the preferential amplification of smaller alleles in some assays. However, our RPT-PCR assay is able to amplify a FM allele when it represented just 5% of alleles in a mixture with either normal or PM alleles (Supplemental Figure S1). A more accurate determination of repeat size can be obtained using high-resolution capillary electrophoresis (Figure 1B). In this case, standards generated from FMR1 alleles with known repeat numbers would need to be used (see Materials and Methods). Gray zone alleles (repeat numbers between 45 and 54) can also be distinguished from normal alleles using a 2% agarose gel as in this example of a mother (A8018) with two normal alleles, a father (A8020) with a gray zone allele, and their daughter (A7306) with a normal allele and a gray zone allele (Figure 1C).
Figure 1.

The use of HiFi DNA polymerase and Phusion DNA polymerase in the amplification of normal (N), gray zone (GZ), premutation (PM), and full mutation (FM) alleles. Repeat amplification was performed on various samples using the protocol described in Materials and Methods and either HiFi DNA polymerase or Phusion DNA polymerase. A: The products of the PCRs resolved by electrophoresis on a 1% agarose gel. Water (lane 1), DNA from a normal embryonic stem cell line (H1) (lane 2), from SC120, a PM induced pluripotent stem cell (iPSC) line (lane 3), from SC128, a FM iPSC line (lane 4), and from two FM cell lines, GM04025 (lane 5) and GM09237 (lane 6). The molecular weight marker (MW) was a 1-kb ladder. The asterisks indicate the repeat numbers corresponding to the amplified alleles. B: The electropheretograms produced on capillary electrophoresis for SC128 and GM09237 together with the LIZ1200 molecular weight marker. C: The results obtained for the amplification of DNA from a female (F) carrier of two normal alleles (30 repeats each), a male (M) carrier of a gray zone allele (GZ; 51 repeats), and their daughter who carries a normal allele and the paternal gray zone allele. Electrophoresis was performed on a 2% agarose gel.
The RPT-PCR assay is also robust enough to reliably amplify mosaic FM alleles from saliva samples with minimal purification (Supplemental Figure S2A). In one case, FX-4, two PM alleles and a faint, broadly distributed, smear of PCR products in the FM range could be seen on agarose gels (Supplemental Figure S2A) but that are clearly visible in capillary electrophoretograms (Supplemental Figure S2C).
It has previously been reported that the incorporation of a series of short heat pulses to the PCR cycling parameters improves the ability to amplify long repeats.25 By combining a modified version of these cycling parameters with our reaction mix we were able to visualize alleles in cell lines that were resistant to our standard RPT-PCR cycling. Specifically, we were able to visualize all three alleles present in a mosaic cell line containing approximately 350, approximately 780, and approximately 1075 repeats (Supplemental Figure S2B), whereas with the standard RPT-PCR only the smallest allele was amplified. The results we obtained with PCR are consistent with results obtained by Southern blotting.9 We were also able to visualize an allele with approximately 900 repeats that we were unable to visualize with the standard PCR cycling. The Southern blotting data for this cell line were also congruent with the size of the PCR product obtained.9 Because we are able to amplify a slightly larger allele that has no AGG interruptions using conventional cycling (GM09237), it is unclear why we were unable to do so with this sample. However, because the use of the heat pulse protocol significantly increases the amplification time, it may not be necessary for routine amplification of smaller alleles.
FMR1 Methylation Assays
The RPT-PCR assay can be readily modified to allow assessment of the methylation status of the FMR1 gene by incorporating the methylation-sensitive restriction enzyme HpaII into the predigestion step. HpaII has two recognition sites in the amplicon generated by the PCR, one of which is located within the region to which the forward primer hybridizes. Predigestion with HpaII eliminates unmethylated alleles. This allows the methylated alleles to be identified and their proportion to be ascertained by comparison to the sample without HpaII digestion. This methylation-specific repeat PCR (MS_RPT-PCR) assay was suitable for the semiquantitative analysis of methylation in both males and females (Figure 2). Digestion by HpaII can be monitored, if necessary, using a synthetic 243-bp internal control fragment that we generated. This fragment, whose sequence is shown in Table 2, can be amplified using the same PCR primers used for the RPT-PCR assay. The resultant PCR product is smaller than a normal FMR1 allele but has the same G + C content and also has two HpaII sites. This plasmid can be added at a low concentration (0.2 pg or approximately one fifth genome equivalent to 100 ng genomic DNA) to the predigestion mix to verify that the HpaII digestion was complete (Supplemental Figure S3).
Figure 2.

Analysis of the methylation status of the repeat region in DNA from carriers of normal (N), premutation (PM), and full mutation (FM) FMR1 alleles. The methylation status of the repeat region was assessed using DNAs predigested with or without HpaII before the PCR, as described in Materials and Methods. The resultant products were resolved on a 1% agarose gel. The + sign indicates those reactions that were predigested with HpaII in addition to HindIII. Molecular weight marker (MW)1 and MW2 are 100-bp and 1-Kb molecular weight ladders, respectively. A: Water (lane 1), DNA from a normal embryonic stem cell (ESC) line (H1) (lane 2), from SC120, a PM induced pluripotent stem cell (iPSC) line (lane 3), from SC128, a FM iPSC line (lane 4), and from two FM cell lines, GM04025 (lane 5) and GM09237 (lane 6). B: Analysis of DNA from a fragile X syndrome ESC WCMC37, MC37B, a subclone isolated from this cell line, and from two female (F) FM carriers F23856 and F23690. M, male.
The MS_RPT-PCR assay has the advantage of being able to correlate specific alleles with their methylation status. So, for example, we can see that although the parental embryonic stem cell line WCMC37 contained two methylated FM alleles, a subclone isolated from that cell line contained a mixture of two unmethylated PM alleles, two methylated FM alleles, and one unmethylated FM allele (Figure 2B).
Although this assay is useful for correlating methylation with a particular allele, there are situations where having a more quantitative measurement of the extent of DNA methylation that is not based on analysis of the repeat itself would be useful. Although several assays have been described for this purpose, many of them have limited sensitivity, some make use of specialized equipment and sometimes involve modification steps that are time consuming and can be prone to failure or are cost-prohibitive for routine use. To generate a rapid and cost-effective assay for methylation of the FMR1 promoter, we opted to develop a simple methylation-sensitive PCR strategy that we call quantitative methylation-sensitive PCR (qMS-PCR) that obviates the need for bisulfite modification or TaqMan primers. We used a primer pair that amplifies a region upstream of the repeat that contains a HpaII site.26 This allows us to interrogate the methylation status of the template by digestion of the genomic DNA before PCR. PCR using these primers generated a single product with no primer dimers, which allowed us to use SYBR Green dye to monitor the levels of the PCR product. Because the repeat has a strong effect on the PCR efficiency of most primer pairs adjacent to it, we included a sonication step before the HpaII digestion that minimized this effect. The digested samples were then subjected to real-time PCR for both the FMR1 promoter region and a region of the unmethylated GAPDH gene as a control for HpaII digestion, and the data processed as described in Materials and Methods. The amount of PCR product produced after HpaII digestion of samples from normal males (GM06865, H1), a male gray zone allele carrier (A8020), and three male PM carriers (SC120, F15608, F10149) was 1% to 9% of that seen without HpaII digestion, consistent with little, if any, methylation (Table 1). As expected, DNA from four unaffected females, a female heterozygous for a gray zone allele, and females heterozygous for PM alleles all showed methylation levels of approximately 50% (range, 48% to 64%) (Table 1). Two female carriers of FM alleles, F23690 and F23856, also showed methylation levels of 63% and 53%, respectively (ie, levels that fall within the range seen in normal females) (Table 1). Because a level of approximately 75% methylation would be expected if the FM allele was fully methylated in all cells and the normal allele was fully methylated in 50% of cells, this would be consistent with some skewed X inactivation. This was consistent with what was seen by the standard MS_RPT-PCR assay where HpaII digestion results in the loss of >50% of the normal allele (Figure 2B).
In contrast, four FM male cell lines that appear fully methylated by Southern blotting and our standard MS_RPT-PCR assay (SC128, GM04025, GM07294 and GM09145) (Figure 2A and data not shown) showed methylation levels of >90% in the qMS-PCR assay (Table 1). Occasionally, the extent of methylation determined by this assay exceeded 100% (Table 1). However, the associated %CV of this assay is <1 (ie, well within acceptable limits for most qPCR applications),29 and a greater precision is difficult to attain when small changes in Ct values are being measured. Nonetheless, this level of accuracy is comparable to or better than that reported by some other methods for determining methylation status.30, 31, 32 To test this assay further, we mixed different proportions of DNA from a male cell line with a completely unmethylated normal FMR1 allele (GM06865) and DNA from a male cell line with a completely methylated FM allele (GM09145). This assay showed a linear relationship between the percentage methylation as measured in this assay and the proportion of DNA from the methylated cell line that was present in the samples (R2 = 0.9933) (Figure 3).
Figure 3.

Analysis of different ratios of methylated and unmethylated alleles using the qMS-PCR assay. DNA from a fully methylated full mutation carrier (GM09145) and a full unmethylated normal allele (GM06865) was mixed at different ratios before sonication. The DNA mixtures were then either mock digested or digested with HpaII and the amount of DNA corresponding to the 5′ end of the FMR1 gene analyzed by real-time PCR, as described in Materials and Methods. The percentage of uncut FMR1 DNA (ie, the percentage methylated) was then plotted as a function of the relative amount of the original methylated and unmethylated DNA (closed circles). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH; open circles) was analyzed in parallel as a control for HpaII digestion.
One mosaic male FM patient, FX-1, showed 57% methylation in the qMS-PCR assay (Table 1). This is consistent with the fact that in the standard MS_RPT-PCR assay, there was an apparent decrease in the yield of PCR product after HpaII digestion (Supplemental Figure S2). A second male, FX-4, showed a methylation level of 43% in the qMS-PCR assay (Table 1). This patient showed clear evidence of two methylated PM alleles and a HpaII-sensitive smear in FM range in the MS_RPT-PCR assay (Supplemental Figure S2A). A similar distribution of alleles was also seen in blood using a commecially available assay (Carolyn Smith, personal communication).
DNA from a male FM cell line (GM06897) was found to be completely unmethylated in our qMS-PCR assay (Table 1). Although this cell line was fully methylated when it was deposited in the Coriell repository (Camden, NY), it was also found to be unmethylated in our MS_RPT PCR assay (Supplemental Figure S4A) and by sequencing of bisulphite-modified DNA from immediately upstream of the repeats (Supplemental Figure S4B). This cell line still has repeats in the FM range, although the repeat number ranges from approximately 240 to approximately 570 (Supplemental Figure S4A). Again, the broad range of repeat sizes is consistent with the observation that unmethylated alleles are more unstable than methylated ones.33, 34, 35, 36
Although the FM allele from untreated GM04025 cells was fully methylated in the qMS-PCR assay, DNA isolated from this cell line after 3 days of treatment with 10 μmol/L of 5-azadeoxycytidine, a DNA methyltransferase inhibitor, showed a methylation level of approximately 77% (Table 1). Pyrosequencing of an adjacent DNA region containing 22 CpGs showed methylation of individual CpG residues ranging from 60% to 80%.37 Thus, the qMS-PCR assay can also be used for monitoring the extent of demethylation in response to compounds that target epigenetic modifications.
Modification of the RPT-PCR Assay to Examine the AGG Interruption Pattern in the FMR1 Gene (TP_RPT-PCR)
The RPT-PCR assay could also be readily modified to generate a variant of the previously described triplet-primed (TP) PCR38 to examine the AGG-interruption pattern present in some FMR1 alleles. The modification involved the addition of a third primer, Fmr1_CCG5, containing five repeats of the trinucleotide CCG at the 3′ end, into the reaction mix rather than a primer containing CGG-repeats, as previously described.32 Like the CGG primer, the CCG primer hybridizes throughout the repeat tract except in regions containing an AGG interruption. This results in a continuous distribution of PCR products with dips visible in the GeneMapper profile corresponding to the position of any AGG interruptions, where no primer binding, and thus no primer extension, occurred. However, the use of the CCG primer should give better resolution of the AGG interruption pattern on longer alleles because the AGG interruptions are generally located at the 5′ end of the repeat and thus the CCG repeat–primed products flanking the AGG interruption are smaller than the equivalent repeat-primed products for the CGG primer. This allows them to be more easily amplified and resolved by capillary electrophoresis. The use of both flanking primers rather than a single flanking primer39 allows the full length of the repeat tract to be determined simultaneously if necessary.
The results obtained for three PM carriers using the CCG and CCG repeat primers are shown in Figure 4. A 20:1 ratio of the external primer pair to the CCG repeat–containing primer gave the clearest results in our assay, showing both the interruption pattern and a small amount of the full-length allele that could be seen in the same scan (Figure 4A). The first sample had no interruptions, the second had single interruption at position 10, and the third had interruptions at position 10 and position 20, consistent with previous testing (S. Nolin, personal communication). In contrast, results obtained with a (CGG)5 primer were less distinct at all ratios of the third primer (Figure 4B), although they worked well for smaller alleles (data not shown).
Figure 4.

Determination of the AGG interruption pattern in three premutation (PM) carriers by TP_RPT-PCR. The AGG interruption pattern was assessed by PCR using three primers, Not_FraxC, Not_FraxF, and either Fmr1_CCG5 (A) or Fmr1_CGG5 (B), as described in Materials and Methods. The arrowheads in A and B indicate the full-length PCR product. The arrows in A indicate the location of the AGG interruptions. The inset in A shows a magnification of the region close to the 5′ end of the repeat. The brackets indicate the location of the interruptions. The insets in B show the same samples amplified with a 40:1 ratio of the external primers to the repeat containing primer. M, male; RFU, relative fluoresence units.
Modification of the RPT-PCR Assay to Determine the Number of Uninterrupted Repeats at 3′ End of the Repeat Tract (AGG_RPT-PCR)
Although the determination of the AGG interruption pattern in males using the CCG repeat–primed PCR assay is straightforward, the presence of the normal allele can complicate the interpretation of the results in female carriers of PM or gray/intermediate zone alleles. However, evidence suggests that the threshold for repeat instability is approximately 34 uninterrupted repeats40 and that the number of pure CGG repeats at the 3′ end of the repeat tract of a PM allele is the single factor that predicts the risk of large expansions, particularly in alleles with 45 to 69 repeats.5 We therefore modified the RPT-PCR assay to more easily determine this parameter in complex samples. In this assay, the product of the RPT-PCR is digested with the restriction enzyme, EciI, that recognizes the sequence GGCGGA(N)11. It thus cuts a CGG repeat tract with an AGG interruption 11 bp downstream of the A (ie, three repeats downstream of the AGG interruption). This generates cleavage products that can readily be identified on agarose gels. Because the distance between the 5′ end of the PCR fragment and the beginning of the repeat is 135 bp and the first 5′ AGG interruption is generally at the 10th or 11th triplet of the repeat,3, 6, 40 digestion of a fragment with one or more AGG interruptions would generate a band of approximately 170 bp representing the sum of the 135 bases in the 5′ flanking region, the number of bases in the repeat tract up to the first A along with the 11 bp resulting from EciI cleavage downstream of the A. A second band corresponding to the 3′ end of the PCR product should also be clearly visible (Figure 5A). Fragments corresponding to the region between consecutive AGGs would be approximately 30 bp long and as such would be difficult to detect on an agarose gel. However, their presence could be inferred from the difference between the full-length fragment and the sum of the lengths of the 5′ and 3′ fragments. Because the number of uninterrupted repeats that confers a risk of expansion is thought to be >34,5, 40 it should in principle be possible to ascertain all at-risk alleles using this strategy, because the vast majority of normal alleles contain 30 repeats or less.
Figure 5.

Analysis of the number of uninterrupted repeats at the 3′ end of the repeat tract using the AGG_RPT-PCR assay. A: Schematic representation of the basis of the AGG_RPT-PCR assay for AGG interruptions showing the FAM and HEX labeled strands generated by PCR that is the substrate for EciI digestion. B: Analysis of interruption status in A7306, a female carrier of a normal allele (30 repeats) and a gray zone allele (52 repeats) and HT-51E, a female carrier of a normal (29 repeats) and a PM allele (92 repeats). The products of the reaction were resolved by electrophoresis on a 2% agarose gel. C: Capillary electrophoretograms of the reaction products for HT-51E in which a FAM-labeled forward primer and a HEX-labeled reverse primer were used for the PCR. The CCG-rich strand of each fragment is shown in red and the CGG-rich strand is shown in blue. In the absence of EciI, the red and blue peaks correspond to the individual strands of the full-length PCR fragment. These fragments do not comigrate because the CGG strand has a higher mobility than the CCG strand in this gel system. After EciI digestion, the red peaks correspond to the CGG strand of the 3′ end of the repeat tract and the blue peaks correspond to the CCG strand of the 5′ end of the repeat tract. F, female; GZ, gray zone allele; MW, 100-bp molecular weight ladder; PM, premutation.
The use of this assay to analyze female carriers of gray zone and PM alleles is illustrated in Figure 5, B and C. One female, A7306, has a normal allele of 30 repeats and a gray zone allele with 52 repeats with two interruptions in each allele as ascertained by sequencing of the parental alleles (Supplemental Figure S5). Both alleles were sensitive to EciI, resulting in an approximately 175-bp band and a smaller amount of an approximately 225-bp band (Figure 5B). The larger band corresponds to approximately 30 uninterrupted repeats at the 3′ end of the repeat tract of the gray zone allele, consistent with the sequencing data. The smaller band represents the fragment generated from the 5′ end of both alleles, along with the fragment from the 3′ end of the normal allele that has nine repeats. HT-51E has a normal allele of 29 repeats and a PM allele of 92 repeats. EciI digestion generated a band of approximately 175 bp and a band of approximately 350 bp that migrates slightly faster than the undigested normal allele. EciI cleavage of the PM allele would be consistent with the presence of at least one AGG interruption and the size of the digestion product is consistent with an uninterrupted 3′ tract of approximately 75 repeats (Figure 5B).
The use of primer pairs with different fluorescent labels allows both the number of pure repeats at the 5′ end of the repeat tract and the number of pure repeats at the 3′ end of the repeat tract to be accurately determined in the same electrophoresis run. The FAM-labeled forward primer labels the CGG strand of the repeat, whereas the HEX-labeled reverse primer labels the CCG strand (Figure 5A). Both strands migrate faster than a random sequence of the same length in capillary electrophoresis, with the CGG strand migrating faster than the CCG strand. Thus, separate red and blue peaks are seen for each allele in the undigested sample. However, the repeat numbers determined by reference to the appropriate FAM and HEX-labeled markers should correlate. After EciI digestion, each allele produces a single peak in the FAM channel corresponding to the 5′ end of the repeat and a single peak in the HEX channel corresponding to the 3′ end of the repeat.
In the case of the female PM carrier, HT-51E, two red peaks and two blue peaks are seen in the sample that was not treated with EciI (Figure 5C). These peaks correspond to the full-length CCG (red) and CGG strands (blue) of the normal and PM allele. After digestion, two peaks are seen in the HEX channel of the EciI-digested sample (Figure 5C). These correspond to the 3′ ends of the normal and PM allele, respectively. A single peak is seen in the FAM channel corresponding to the 5′ end of both the normal and the PM allele (Figure 5C). This is consistent with the results of the CCG-primed TP_RPT-PCR that demonstrates that although two different alleles are present, they both possess the same AGG interruption pattern (Supplemental Figure S6).
Discussion
We have developed a set of simple, sensitive, and relatively inexpensive assays that can be used to monitor repeat length, the AGG interruption pattern, and the extent of methylation of most FMR1 alleles on a routine basis in a research laboratory setting. These assays permit the determination of all of the critical factors that impact expansion risk and determine clinical involvement even in individuals whose allele size or mosaicism for these factors make this challenging (Table 1). The workflow for these assays is illustrated in Figure 6 and can be completed within a time frame similar to that of the more recent commercial assays and is comparable in terms of hands-on time required. The basic assays for repeat number (RPT-PCR), methylation status (MS_RPT-PCR), and the number of uninterrupted repeats (AGG_RPT-PCR) using agarose gel electrophoresis cost well under the $5.00 mark typically considered the threshold for population-based screening.
Figure 6.

Diagram illustrating the work-flow for the analysis of FMR1 alleles. RPT-PCR/MS_RPT-PCR enables the determination of repeat size and gives an indication of the methylation status for full mutation (FM) carriers. It can also reveal skewed X-inactivation in female premutation (PM) and FM carriers. Agarose gel electrophoresis can be used for rapid evaluation of alleles followed by capillary electrophoresis for samples for which an accurate repeat number is necessary. PM alleles can be further processed by either TP_RPT-PCR or AGG_RPT-PCR to determine AGG interruption status or number of uninterrupted CGG repeats at 3′ end of the repeat tract. The remaining RPT-PCR product can be used without further purification for the AGG_RPT-PCR assay if necessary, thus saving time and sample. FM alleles or alleles that were not amplified in the RPT-PCR (with or without a heat pulse) should then be processed by qMS-PCR to quantitate the amount of methylation.
The RPT-PCR assay can readily detect alleles with >900 repeats even in samples with multiple large alleles (Figure 1 and Supplemental Figure S2B). It is sensitive enough to detect 5% to 10% of FM alleles present in a mixture of normal or PM alleles (Supplemental Figure S1); this assay is robust enough to be used on samples like saliva with minimal purification (Supplemental Figure S2A). The ability to use saliva samples simplifies sample collection in the field and is particularly advantageous for collecting DNA from FM carriers who often find blood draws difficult. The TP_RPT PCR allows the unambiguous determination of the AGG interspersion pattern that is particularly useful for males (Figure 4). The AGG_RPT-PCR may be more useful for females because it can be used to determine the number of uninterrupted CGG repeats at the 3′ end of the repeat tract of each allele (Figure 5).
The MS_RPT-PCR allows specific alleles to be correlated with their methylation status. This can be important for females with skewed XCI (Figure 2) or in the case of patient FX-4, who is unusual in having two methylated PM alleles and multiple unmethylated FMs (Supplemental Figure S2). Furthermore, subculturing of fragile X syndrome embryonic stem cells frequently results in both contractions and the loss of methylation from smaller alleles (Figure 2B). This variability in repeat size and methylation status is also seen in differentiated cells in culture, as illustrated by the case of GM06897, a lymphoblastoid cell line that was methylated when deposited in the Coriell Collection but that is now completely unmethylated (Table 1 and Supplemental Figure S4). Thus, having an inexpensive assay suitable for routine use in laboratories working with cells such as these is critical.
The qMS-PCR assay offers an advantage over the MS_RPT-PCR in giving a more quantitative readout of methylation status than the MS_RPT-PCR. It is able to clearly distinguish between normal males and females, between normal and methylated FM alleles, and between FM alleles with and without treatment with a demethylating agent. It can also reveal some cases of skewed X inactivation in female FM carriers (Table 1). However, because in females with FM or large PM alleles and preferential inactivation of the normal allele, the percentage methylation could vary anywhere from 50% if the PM/FM allele were completely unmethylated, to 100% if this allele was completely methylated, both the MS_RPT-PCR and the qMS-PCR assay should be used. The qMS-PCR assay is also useful in cases such as FX-4 (Supplemental Figure S2), where the heterogeneity of large unmethylated alleles may make the proportion of such alleles difficult to assess. In addition, qMS-PCR can also reliably determine even small changes in methylation, resulting from treatment with DNA demethylating agents (Table 1). Thus, this assay may be useful not only for assessing patient methylation levels, but also for addressing basic research questions, including the mechanism of gene silencing and the identification of epigenetic modifiers that may reactivate silenced FM alleles in tissue culture.
In conclusion, the assays we have developed can be used to monitor in a cost-effective manner many of the key factors that impact expansion risk and disease severity. They are also cheaper and rapid enough to be used in the laboratory setting for the routine monitoring of cell lines and to test compounds that affect repeat stability and methylation status.
Footnotes
Supported by a National Institute of Diabetes and Digestive Kidney Diseases, NIH intramural program (DK057808; K.U.).
B.E.H. and Y.Z. contributed equally to this work.
Disclosures: The bisulfite modification data for the GM06897 cell line were obtained by Rea Biacsi (National Institute of Diabetes and Digestive Kidney Diseases, Bethesda, MD) as part of another study.
Supplemental material for this article can be found at http://dx.doi.org/10.1016/j.jmoldx.2016.06.001.
Supplemental Data
Supplemental Figure S1.

Amplification of full mutation (FM) alleles in the presence of various amounts of normal or premutation (PM) alleles. DNA from a carrier of a completely methylated FM allele with a repeat number of approximately 660 repeats (GM04025) was mixed with different amounts of DNA from a carrier of either a normal (N) allele (H1) with 30 repeats (A) or a PM allele (SC120) with 97 repeats (B). The DNA sample was then analyzed using the RPT-PCR assay with and without HpaII digestion, as described in Materials and Methods.
Supplemental Figure S2.

Amplification of DNA from challenging templates. A: Amplification of full mutation (FM) alleles from saliva. Genomic DNA was prepared from saliva DNA obtained from male FM carriers FX-1, FX-2, and FX-4, and the repeat size analyzed with and without HpaII digestion, as described in Materials and Methods. The PCR products produced from purified DNA from a sample with 97 repeats (SC120) and 940 repeats (NA09237) are shown for comparison. Molecular weight marker (MW)1 and MW2 are 100-bp and 1-kb molecular weight ladders, respectively. The bracket indicates a smear of products in the FM range that was reproducibly obtained for this sample. This smear is less apparent after HpaII digestion, suggesting that although the premutation (PM) alleles are methylated, the FM alleles are not. Analysis of this DNA by qMS-PCR confirmed that >50% of the alleles in this sample are unmethylated (Table 1). B: Amplification of large methylated FM alleles using the pulse extension modification. Our standard RPT-PCRs were performed using a pulse extension protocol for the amplification, as described in Materials and Methods. The resultant products were resolved by agarose gel electrophoresis, and the DNA visualized using SYBR Gold staining. MW1 is a 1-kb molecular weight ladder. The arrowheads indicate alleles that are only seen using the pulse PCR protocol. Lane 1, Water; lane 2, NA09237; lane 3, C10700; and lane 4, C10259. C: Capillary electrophoretogram of the RPT-PCR products for patient FX-4 demonstrating the presence of FM alleles. M, male.
Supplemental Figure S3.
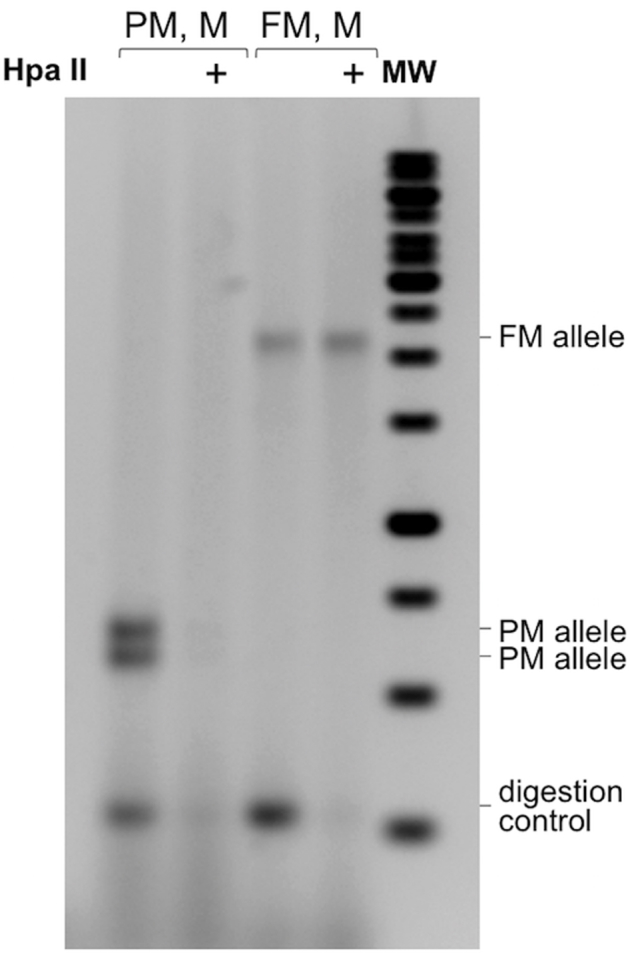
Use of an internal control for HpaII digestion in the repeat methylation assay (MS_RPT-PCR). Example of the MS_RPT-PCR assay performed using the 243-bp synthetic internal control fragment shown in Table 2 and DNA from a premutation (PM) carrier with two PM alleles and DNA from a carrier of a full mutation (FM) allele. M, male; MW, molecular weight marker.
Supplemental Figure S4.

The methylation status of GM03200 and GM06897, two full mutation (FM) cell lines used in this study. A: PCR analysis of the repeat without and with (+) predigestion with HpaII. The asterisks indicate the migration of alleles with repeats corresponding to 570 and 240 repeats, respectively. B: Sequence of bisulfite-modified DNA from a region upstream of the repeat (corresponding to chromosome X:146993340-146993381 of the hg19 build) in GM06897 confirming that the potentially methylatable CpG residues, indicated by the red arrowheads, were converted to Ts by the bisulfite treatment and were thus unmethylated in the original DNA sample. M, male; UFM, unmethylated full mutation.
Supplemental Figure S5.

The DNA sequence of repeat tract in A8018 and A8020, the parents of A7306. PCR products were generated using the protocol described in Materials and Methods, purified using QIAquick PCR purification column (Qiagen), and sequenced using the FraxC primer.
Supplemental Figure S6.
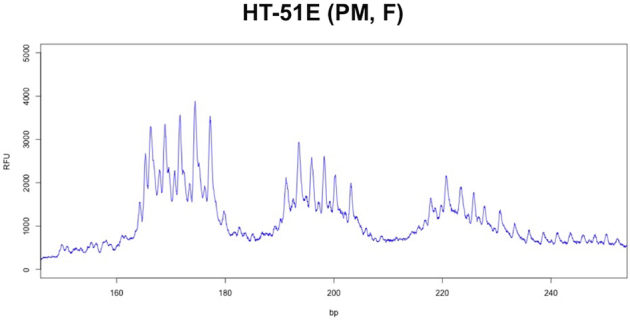
The normal and premutation (PM) alleles of HT51-E both contain two AGG interruptions with the same interspersion pattern. TP_RPT-PCR for HT51-E (compare to the pattern seen for the mono-allelic F15608 in Figure 4A). bp, base pairs; F, female; RFU, relative fluorescence units; TP, triplet-primed.
References
- 1.Chonchaiya W., Schneider A., Hagerman R.J. Fragile X: a family of disorders. Adv Pediatr. 2009;56:165–186. doi: 10.1016/j.yapd.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pretto D.I., Mendoza-Morales G., Lo J., Cao R., Hadd A., Latham G.J., Durbin-Johnson B., Hagerman R., Tassone F. CGG allele size somatic mosaicism and methylation in FMR1 premutation alleles. J Med Genet. 2014;51:309–318. doi: 10.1136/jmedgenet-2013-102021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nolin S.L., Glicksman A., Ersalesi N., Dobkin C., Brown W.T., Cao R., Blatt E., Sah S., Latham G.J., Hadd A.G. Fragile X full mutation expansions are inhibited by one or more AGG interruptions in premutation carriers. Genet Med. 2015;17:358–364. doi: 10.1038/gim.2014.106. [DOI] [PubMed] [Google Scholar]
- 4.Latham G.J., Coppinger J., Hadd A.G., Nolin S.L. The role of AGG interruptions in fragile X repeat expansions: a twenty-year perspective. Front Genet. 2014;5:244. doi: 10.3389/fgene.2014.00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nolin S.L., Sah S., Glicksman A., Sherman S.L., Allen E., Berry-Kravis E., Tassone F., Yrigollen C., Cronister A., Jodah M., Ersalesi N., Dobkin C., Brown W.T., Shroff R., Latham G.J., Hadd A.G. Fragile X AGG analysis provides new risk predictions for 45-69 repeat alleles. Am J Med Genet A. 2013;161A:771–778. doi: 10.1002/ajmg.a.35833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yrigollen C.M., Martorell L., Durbin-Johnson B., Naudo M., Genoves J., Murgia A., Polli R., Zhou L., Barbouth D., Rupchock A., Finucane B., Latham G.J., Hadd A., Berry-Kravis E., Tassone F. AGG interruptions and maternal age affect FMR1 CGG repeat allele stability during transmission. J Neurodev Disord. 2014;6:24. doi: 10.1186/1866-1955-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yrigollen C.M., Durbin-Johnson B., Gane L., Nelson D.L., Hagerman R., Hagerman P.J., Tassone F. AGG interruptions within the maternal FMR1 gene reduce the risk of offspring with fragile X syndrome. Genet Med. 2012;14:729–736. doi: 10.1038/gim.2012.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lokanga R.A., Entezam A., Kumari D., Yudkin D., Qin M., Smith C.B., Usdin K. Somatic expansion in mouse and human carriers of fragile X premutation alleles. Hum Mutat. 2013;34:157–166. doi: 10.1002/humu.22177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumari D., Bhattacharya A., Nadel J., Moulton K., Zeak N.M., Glicksman A., Dobkin C., Brick D.J., Schwartz P.H., Smith C.B., Klann E., Usdin K. Identification of fragile X syndrome specific molecular markers in human fibroblasts: a useful model to test the efficacy of therapeutic drugs. Hum Mutat. 2014;35:1485–1494. doi: 10.1002/humu.22699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumari D., Swaroop M., Southall N., Huang W., Zheng W., Usdin K. High-throughput screening to identify compounds that increase fragile X mental retardation protein expression in neural stem cells differentiated from fragile X syndrome patient-derived induced pluripotent stem cells. Stem Cells Transl Med. 2015;4:800–808. doi: 10.5966/sctm.2014-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaufmann M., Schuffenhauer A., Fruh I., Klein J., Thiemeyer A., Rigo P., Gomez-Mancilla B., Heidinger-Millot V., Bouwmeester T., Schopfer U., Mueller M., Fodor B.D., Cobos-Correa A. High-throughput screening using iPSC-derived neuronal progenitors to identify compounds counteracting epigenetic gene silencing in fragile X syndrome. J Biomol Screen. 2015;20:1101–1111. doi: 10.1177/1087057115588287. [DOI] [PubMed] [Google Scholar]
- 12.Usdin K., Woodford K.J. CGG repeats associated with DNA instability and chromosome fragility form structures that block DNA synthesis in vitro. Nucleic Acids Res. 1995;23:4202–4209. doi: 10.1093/nar/23.20.4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woodford K., Weitzmann M.N., Usdin K. The use of K(+)-free buffers eliminates a common cause of premature chain termination in PCR and PCR sequencing. Nucleic Acids Res. 1995;23:539. doi: 10.1093/nar/23.3.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitas M., Yu A., Dill J., Haworth I.S. The trinucleotide repeat sequence d(CGG)15 forms a heat-stable hairpin containing Gsyn. Ganti base pairs. Biochemistry. 1995;34:12803–12811. doi: 10.1021/bi00039a041. [DOI] [PubMed] [Google Scholar]
- 15.Chen X., Mariappan S.V., Catasti P., Ratliff R., Moyzis R.K., Laayoun A., Smith S.S., Bradbury E.M., Gupta G. Hairpins are formed by the single DNA strands of the fragile X triplet repeats: structure and biological implications. Proc Natl Acad Sci U S A. 1995;92:5199–5203. doi: 10.1073/pnas.92.11.5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Renciuk D., Zemanek M., Kejnovska I., Vorlickova M. Quadruplex-forming properties of FRAXA (CGG) repeats interrupted by (AGG) triplets. Biochimie. 2009;91:416–422. doi: 10.1016/j.biochi.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 17.Juusola J.S., Anderson P., Sabato F., Wilkinson D.S., Pandya A., Ferreira-Gonzalez A. Performance evaluation of two methods using commercially available reagents for PCR-based detection of FMR1 mutation. J Mol Diagn. 2012;14:476–486. doi: 10.1016/j.jmoldx.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 18.Adler K., Moore J.K., Filippov G., Wu S., Carmichael J., Schermer M. A novel assay for evaluating fragile X locus repeats. J Mol Diagn. 2011;13:614–620. doi: 10.1016/j.jmoldx.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Godler D.E., Slater H.R., Amor D., Loesch D.Z. Methylation analysis of fragile X-related epigenetic elements may provide a suitable newborn screening test for fragile X syndrome. Genet Med. 2010;12:595. doi: 10.1097/GIM.0b013e3181f07088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Godler D.E., Slater H.R., Bui Q.M., Ono M., Gehling F., Francis D., Amor D.J., Hopper J.L., Hagerman R., Loesch D.Z. FMR1 intron 1 methylation predicts FMRP expression in blood of female carriers of expanded FMR1 alleles. J Mol Diagn. 2011;13:528–536. doi: 10.1016/j.jmoldx.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Teo C.R., Rajan-Babu I.S., Law H.Y., Lee C.G., Chong S.S. Methylation-specific triplet-primed PCR and melting curve analysis as a rapid screening tool for identifying actionable FMR1 genotypes. Clin Chem. 2013;59:1668–1670. doi: 10.1373/clinchem.2013.206771. [DOI] [PubMed] [Google Scholar]
- 22.Inaba Y., Schwartz C.E., Bui Q.M., Li X., Skinner C., Field M., Wotton T., Hagerman R.J., Francis D., Amor D.J., Hopper J.L., Loesch D.Z., Bretherton L., Slater H.R., Godler D.E. Early detection of fragile X syndrome: applications of a novel approach for improved quantitative methylation analysis in venous blood and newborn blood spots. Clin Chem. 2014;60:963–973. doi: 10.1373/clinchem.2013.217331. [DOI] [PubMed] [Google Scholar]
- 23.Rajan-Babu I.S., Law H.Y., Yoon C.S., Lee C.G., Chong S.S. Simplified strategy for rapid first-line screening of fragile X syndrome: closed-tube triplet-primed PCR and amplicon melt peak analysis. Expert Rev Mol Med. 2015;17:e7. doi: 10.1017/erm.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Charif D., Lobry J.R. Seqin{R} 1.0-2: a contributed package to the {R} project for statistical computing devoted to biological sequences retrieval and analysis. In: Bastolla U., Porto M., Roman H.E., Vendruscolo M., editors. Structural Approaches to Sequence Evolution: Molecules, Networks, Populations. Springer Verlag; New York: 2007. pp. 207–232. [Google Scholar]
- 25.Orpana A.K., Ho T.H., Stenman J. Multiple heat pulses during PCR extension enabling amplification of GC-rich sequences and reducing amplification bias. Anal Chem. 2012;84:2081–2087. doi: 10.1021/ac300040j. [DOI] [PubMed] [Google Scholar]
- 26.Groh M., Lufino M.M., Wade-Martins R., Gromak N. R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet. 2014;10:e1004318. doi: 10.1371/journal.pgen.1004318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frey U.H., Bachmann H.S., Peters J., Siffert W. PCR-amplification of GC-rich regions: “slowdown PCR”. Nat Protoc. 2008;3:1312–1317. doi: 10.1038/nprot.2008.112. [DOI] [PubMed] [Google Scholar]
- 28.Rapley R. Enhancing PCR amplification and sequencing using DNA-binding proteins. Mol Biotechnol. 1994;2:295–298. doi: 10.1007/BF02745882. [DOI] [PubMed] [Google Scholar]
- 29.Bookout A.L., Cummins C.L., Kramer M.F., Pesola J.M., Mangelsdorf D.J. High-throughput real-time quantitative reverse transcription PCR. Curr Protoc Mol Biol. 2006;Chapter 15:Unit 15.8. doi: 10.1002/0471142727.mb1508s73. [DOI] [PubMed] [Google Scholar]
- 30.Quillien V., Lavenu A., Karayan-Tapon L., Carpentier C., Labussiere M., Lesimple T., Chinot O., Wager M., Honnorat J., Saikali S., Fina F., Sanson M., Figarella-Branger D. Comparative assessment of 5 methods (methylation-specific polymerase chain reaction, MethyLight, pyrosequencing, methylation-sensitive high-resolution melting, and immunohistochemistry) to analyze O6-methylguanine-DNA-methyltranferase in a series of 100 glioblastoma patients. Cancer. 2012;118:4201–4211. doi: 10.1002/cncr.27392. [DOI] [PubMed] [Google Scholar]
- 31.Dahl C., Guldberg P. A ligation assay for multiplex analysis of CpG methylation using bisulfite-treated DNA. Nucleic Acids Res. 2007;35:e144. doi: 10.1093/nar/gkm984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen L., Hadd A., Sah S., Filipovic-Sadic S., Krosting J., Sekinger E., Pan R., Hagerman P.J., Stenzel T.T., Tassone F., Latham G.J. An information-rich CGG repeat primed PCR that detects the full range of fragile X expanded alleles and minimizes the need for southern blot analysis. J Mol Diagn. 2010;12:589–600. doi: 10.2353/jmoldx.2010.090227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wohrle D., Salat U., Hameister H., Vogel W., Steinbach P. Demethylation, reactivation, and destabilization of human fragile X full-mutation alleles in mouse embryocarcinoma cells. Am J Hum Genet. 2001;69:504–515. doi: 10.1086/322739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wohrle D., Salat U., Glaser D., Mucke J., Meisel-Stosiek M., Schindler D., Vogel W., Steinbach P. Unusual mutations in high functioning fragile X males: apparent instability of expanded unmethylated CGG repeats. J Med Genet. 1998;35:103–111. doi: 10.1136/jmg.35.2.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pieretti M., Zhang F.P., Fu Y.H., Warren S.T., Oostra B.A., Caskey C.T., Nelson D.L. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell. 1991;66:817–822. doi: 10.1016/0092-8674(91)90125-i. [DOI] [PubMed] [Google Scholar]
- 36.Vincent A., Heitz D., Petit C., Kretz C., Oberle I., Mandel J.L. Abnormal pattern detected in fragile-X patients by pulsed-field gel electrophoresis. Nature. 1991;349:624–626. doi: 10.1038/349624a0. [DOI] [PubMed] [Google Scholar]
- 37.Kumari D., Usdin K. Polycomb group complexes are recruited to reactivated FMR1 alleles in Fragile X syndrome in response to FMR1 transcription. Hum Mol Genet. 2014;23:6575–6583. doi: 10.1093/hmg/ddu378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tassone F., Pan R., Amiri K., Taylor A.K., Hagerman P.J. A rapid polymerase chain reaction-based screening method for identification of all expanded alleles of the fragile X (FMR1) gene in newborn and high-risk populations. J Mol Diagn. 2008;10:43–49. doi: 10.2353/jmoldx.2008.070073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lyon E., Laver T., Yu P., Jama M., Young K., Zoccoli M., Marlowe N. A simple, high-throughput assay for Fragile X expanded alleles using triple repeat primed PCR and capillary electrophoresis. J Mol Diagn. 2010;12:505–511. doi: 10.2353/jmoldx.2010.090229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eichler E.E., Holden J.J., Popovich B.W., Reiss A.L., Snow K., Thibodeau S.N., Richards C.S., Ward P.A., Nelson D.L. Length of uninterrupted CGG repeats determines instability in the FMR1 gene. Nat Genet. 1994;8:88–94. doi: 10.1038/ng0994-88. [DOI] [PubMed] [Google Scholar]