

Abstract
AIM
To investigate the effect of metformin on activated hepatic stellate cells (HSCs) and the possible signaling pathways involved.
METHODS
A fibrotic mouse model was generated by intraperitoneal injection of carbon tetrachloride (CCl4) and subsequent treatment with or without metformin. The level of fibrosis was detected by hematoxylin-eosin staining, Sirius Red staining, and immunohistochemistry. The HSC cell line LX-2 was used for in vitro studies. The effect of metformin on cell proliferation (CCK8 assay), motility (scratch test and Transwell assay), contraction (collagen gel contraction assay), extracellular matrix (ECM) secretion (Western blot), and angiogenesis (ELISA and tube formation assay) was investigated. We also analyzed the possible signaling pathways involved by Western blot analysis.
RESULTS
Mice developed marked liver fibrosis after intraperitoneal injection with CCl4 for 6 wk. Metformin decreased the activation of HSCs, reduced the deposition of ECM, and inhibited angiogenesis in CCl4-treated mice. Platelet-derived growth factor (PDGF) promoted the fibrogenic response of HSCs in vitro, while metformin inhibited the activation, proliferation, migration, and contraction of HSCs, and reduced the secretion of ECM. Metformin decreased the expression of vascular endothelial growth factor (VEGF) in HSCs through inhibition of hypoxia inducible factor (HIF)-1α in both PDGF-BB treatment and hypoxic conditions, and it down-regulated VEGF secretion by HSCs and inhibited HSC-based angiogenesis in hypoxic conditions in vitro. The inhibitory effects of metformin on activated HSCs were mediated by inhibiting the Akt/mammalian target of rapamycin (mTOR) and extracellular signal-regulated kinase (ERK) pathways via the activation of adenosine monophosphate-activated protein kinase (AMPK).
CONCLUSION
Metformin attenuates the fibrogenic response of HSCs in vivo and in vitro, and may therefore be useful for the treatment of chronic liver diseases.
Keywords: Hepatic stellate cell, Intrahepatic vascular resistance, Angiogenesis, Contraction, Liver fibrosis, Adenosine monophosphate-activated protein kinase
Core tip: Activation of hepatic stellate cells (HSCs) contributes to liver fibrosis and portal hypertension. In this study, we examined the effect of metformin on activated HSCs in vivo and in vitro. Metformin decreased the activation of HSCs, reduced the deposition of extracellular matrix (ECM), and inhibited angiogenesis in CCl4-treated mice. Moreover, metformin inhibited the activation, proliferation, motility, and contraction of activated HSCs, reduced the secretion of ECM, and decreased HSC-based angiogenesis, thus providing a new therapeutic approach to the treatment of liver fibrosis and portal hypertension.
INTRODUCTION
Liver fibrosis is a common pathological condition resulting from chronic liver injury stemming from a variety of etiological factors. Hepatic stellate cells (HSCs) play a key role in the progression of liver fibrosis, and are thought to be its primary effector cells[1]. In chronic liver diseases (CLDs), quiescent HSCs are activated and change to myofibroblast-like cells, which are proliferative, contractile, and secrete increased levels of more extracellular matrix (ECM)[2]. Angiogenesis is widely noted in CLDs, and influences liver fibrosis and portal hypertension (PHT)[3]. HSCs are liver-specific pericytes that participate in angiogenesis and sinusoidal remodeling. The primary pathological feature of sinusoidal remodeling is sinusoidal capillarization and coverage of the vessels with contractile HSCs[4]. HSCs reduce the diameter of sinusoids after contraction, causing a functional change in modulating the hepatic tone and increasing intrahepatic vascular resistance (IHVR), ultimately contributing to PHT[5]. HSCs occupy a crossroad at the intersection between inflammation, angiogenesis, and fibrosis[6], and activation of HSCs is a key event mediating increased IHVR[7]. Thus, activated HSCs are a potent therapeutic target for the treatment of CLDs.
Metformin is the first-line drug recommended for the treatment of diabetes. Previous studies have demonstrated that metformin has a wide range of pharmacological activities beyond its antidiabetic effects. The beneficial effects of metformin in hepatic disorders have been previously confirmed for reducing fibrosis[8,9], IHVR, and therefore PHT in cirrhosis[10], and decreasing hepatocellular carcinoma risk[11]. Metformin is a potent therapeutic approach for CLDs, but the mechanisms underlying its effects are still unclear, especially in the treatment of PHT. Further studies are needed to investigate the effect of metformin in CLDs.
Platelet-derived growth factor (PDGF) signaling is among the most well characterized pathways of HSC activation. It induces activation of the extracellular signal-regulated kinase (ERK) and the Akt/mammalian target of rapamycin (mTOR) pathways, which are associated with cellular proliferation and migration[12]. Studies have also linked ERK and mTOR signaling to vascular endothelial gowth factor (VEGF) expression during angiogenesis[13,14]. Activation of adenosine monophosphate-activated protein kinase (AMPK) inhibits the proliferation and migration of HSCs induced by PDGF, and this effect is related to the inhibition of the Akt and ERK pathways[15]. Metformin is known to activate AMPK, therefore, we speculated that metformin may regulate the fibrogenic response of HSCs and have an anti-angiogenic effect.
In the present study, we investigated the effect of metformin on activated HSCs. The inhibitory effects of metformin on the activation, proliferation, motility, contraction, and ECM secretion of HSCs and HSC-based angiogenesis were evaluated. We also investigated the underlying mechanisms, with a focus on AMPK and the downstream AKT/mTOR and ERK signaling pathways.
MATERIALS AND METHODS
Animals
Thirty male C57BL/6 mice weighing 20-22 g were purchased from the Central Animal Care Facility of Shandong University and randomly divided into three groups (a control group, a CCl4 group, and a metformin group, n = 10 in each group). The animals were housed in an airconditioned room at 2325 °C with a light/dark (12 h:12 h) cycle for one week prior to the initiation of the experiment. All animals received appropriate care during the study, with free access to chow and water. The liver fibrosis model was induced by intraperitoneal injection of carbon tetrachloride (CCl4, 1 μL/g, Sinopharm, Beijing, China) dissolved 1:1 (v/v) in olive oil twice per week, while the control mice were injected with olive oil alone. Mice in the metformin group were treated with metformin (Sigma-Aldrich, Saint Louis, MO, United States) in drinking water (1 g/L) at the same time. All mice were sacrificed at the end of 6 wk. A portion of liver tissue was fixed in 4% paraformaldehyde and then embedded in paraffin. The other liver tissues were stored at -80 °C.
Cell culture
The HSC cell line LX-2 (a kind gift from Professor Wei-fen Xie, Changzheng Hospital, the Second Military Medical University) and human umbilical vascular endothelial cells (HUVECs, ATCC, Manassas, VA, United States) were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco, Grand Island, NY, United States) supplemented with 10% fetal bovine serum (FBS; Gibco) in an incubator at 37 °C with 5% CO2 and 90% humidity.
CCK-8 assay
First, 5 × 103 LX-2 cells were seeded in 96-well plates and incubated overnight, and then the medium was changed to fresh medium containing different concentrations of metformin. After incubation for 24 h, 10 μL of CCK-8 (Dojindo, Japan) was added to each well. The optical density (OD) values were measured every 30 min with a spectrophotometer (Thermo Fisher, Finland) at 450 nm. The OD values at 2 h were chosen for analysis.
Migration and invasion assay
A scratch test was used for HSC migration assay. Cells (5 × 105) were seeded in 6-well plates, incubated overnight to cover the full plate, and then serum-starved for 8 h. After making scratch wounds, plates were washed three times with PBS. Cells were treated with or without 10 ng/mL PDGF-BB (PeproTech, Rocky Hill, NJ, United States) for 24 h. Different concentrations of metformin were added to the medium 2 h before the PDGF-BB addition. Images were acquired at 0 and 24 h. The Transwell (8 μm pore size, Costar) assay was used to test the invasive ability of HSCs. HSCs were serum-free for 6 h and then harvested. Cells (1 × 105) in 100 μL serum-free medium were seeded in the upper chambers with the Matrigel (BD Bioscience, Bedford, MA, United States) membrane and different concentrations of metformin, and the lower chambers were loaded with DMEM with or without 10% FBS. After incubation for 24 h, cells that migrated through the membrane were fixed and stained with hematoxylin. Cell numbers were counted under a microscope (Olympus, Japan).
Collagen gel contraction assay
Rat tail tendon collagen type I was obtained from Sybio (Hangzhou, China). The collagen gel was prepared in 24-well plates. We used 0.1 mol/L NaOH to adjust the pH and 10 × PBS to adjust the solution to physiological strength. The mixed solution (500 μL) was added to each plate and incubated at 37 °C for 1 h to allow gelatinization. LX-2 cells (1 × 105) in 1000 μL of medium were seeded on the gel and incubated overnight. Cells were starved for 8 h in DMEM, and then the DMEM was replaced with fresh medium with 1% FBS and different concentrations of metformin. PDGF-BB (10 ng/mL) was added to the medium, except in the control group, 2 h after metformin addition. The tip of a 200 μL pipette was used to gently detach the gel from the plates. After incubation for 24 h, the areas of the gels were measured.
Enzyme-linked immunosorbent assay
First, 4 × 105 LX-2 cells were seeded in 6-well plates and incubated overnight. Cells were starved in DMEM for 8 h. The DMEM was changed to 1 mL of fresh medium with 1% FBS and different concentrations of metformin. Cobalt (II) chloride hexahydrate (CoCl2· 6H2O, 150 μmol/L, Sigma-Aldrich, Saint Louis, MO, United States) was added to the medium, except for the control group, 2 h after metformin addition. After 12 h of incubation, the supernatant was collected and centrifuged at 1000 rpm for 4 min. VEGF was measured with an ELISA kit (Boster, Wuhan, China). The ELISA protocol was performed according to the manufacturer’s instructions.
Tube formation assay
A 96-well plate was coated with 50 μL of Matrigel, and then placed in an incubator at 37 °C for 1 h. Cells were treated in the same way as in the ELISA assay, and the supernatant was collected. Conditioned medium was generated from supernatant diluted 4:1 (v/v) in DMEM with 10% FBS. HUVECs were harvested and suspended in the conditioned medium. HUVECs (2 × 104) in 100 μL of conditioned medium were seeded in 96-well plates and incubated at 37 °C. The cells were monitored every 2 h for 12 h under a microscope. Images of the tube formation were acquired at 8 h.
Western blot analysis
Total protein was extracted with RIPA buffer, and the protein concentration was measured by the bicinchoninic acid method. Equal amounts of proteins were loaded and separated by SDS-PAGE, and then transferred onto a PVDF membrane. The membrane was blocked in TBST buffer with 5% non-fat milk for 1 h and incubated with different antibodies overnight at 4 °C. Primary antibodies against α-SMA (14395-1-AP), fibronectin (66042-1-IG), and collagen type I (14695-1-AP) were obtained from Proteintech (Wuhan, China). Primary antibodies against p-ERK1/2 (#4376), p-Akt (#4060), p-AMPK (#2535), p-mTOR (#5536), ERK1/2 (#4695), Akt (#4691), AMPK (#5832), and mTOR (#2983) were obtained from CST (Boston, MA, United States). Primary antibody against VEGF (ab46154) was obtained from Abcam (Cambridge, CA, United States). Primary antibody against HIF-1α (NB100-105) was obtained from Novus (Littleton, CO, United States). Primary antibody against glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and horseradish peroxidase (HRP)-conjugated secondary antibody were obtained from Zhongshan Golden Bridge (Beijing, China). The HRP-conjugated secondary antibodies were goat anti-rabbit or anti-mouse antibody depending on the primary antibodies. AICAR (an AMPK activator) and rapamycin (an mTOR inhibitor) were obtained from Selleck (Houston, TX, United States). PD98059 (an ERK inhibitor) and LY294002 (an AKT inhibitor) were obtained from MCE (Monmouth Junction, NJ, United States). Antibody bands were detected by enhanced chemiluminescence with Amersham Imager 600 (GE Healthcare, United States). GAPDH in the same membrane was used as an internal control, and all bands were normalized to its expression.
Reverse transcription-polymerase chain reaction
Total RNA was extracted with TRIzol reagent (Takara, Japan) from frozen liver tissues and was reverse-transcribed to cDNA using an RT reagent kit (Takara, Japan). Amplifications were detected using a the SYBR Premix Ex Taq kit (Takara, Japan) on a LightCycler 480 Real-Time PCR system (Roche Diagnostics, United States). The primers used in this study are presented in Table 1. Expression of target genes was normalized to expression of GAPDH by the 2-ΔCT method.
Table 1.
The primers used for RT-PCR analysis
| Primer (Mouse) | Sequence (5’-3’) |
| GAPDH F | AAATGGTGAAGGTCGGTGTGAAC |
| GAPDH R | CAACAATCTCCACTTTGCCACTG |
| α-SMA F | GACAATGGCTCTGGGCTCTGTA |
| α-SMA R | TTTGGCCCATTCCAACCATTA |
| COL1A1 F | GACATGTTCAGCTTTGTGGACCTC |
| COL1A1 R | GGGACCCTTAGGCCATTGTGTA |
α-SMA: Alpha-smooth muscle actin; COL1A1: Collagen type 1 alpha 1.
Histopathological and immunohistochemical analyses
Liver specimens embedded in paraffin were cut into 4 μm-thick sections. The specimens were stained with hematoxylin and eosin and Sirius Red. Immunohistochemistry (IHC) was performed using a 2-step plus Poly-HRP Anti-Mouse/Rabbit IgG Detection system (Zhongshan Golden Bridge, Beijing, China), according to the manufacturer’s instructions. Sections were incubated with antibodies against α-SMA, fibronectin, and VEGF, and the blots were developed with a DAB kit (Zhongshan Golden Bridge, Beijing, China).
Statistical analysis
All data are presented as the mean ± SEM from at least three independent experiments. Statistics were analyzed using GraphPad Prism 5.0 and SPSS19.0 software. Statistical significance was determined by one-way analysis of variance (ANOVA) followed by Dunnett’s test. A P-value < 0.05 was considered statistically significant.
RESULTS
Metformin decreases the activation of HSCs, reduces the deposition of ECM, and inhibits angiogenesis in CCl4-treated mice
Liver specimens from mice exposed to CCl4 showed hepatocellular degeneration with excessive accumulation of connective tissue, the formation of fibrotic septa, and infiltration of inflammatory cells. Metformin treatment attenuated the fibrotic level of the fibrotic tissue, the appearance of degenerated hepatocytes, and inflammatory cell infiltration (Figure 1A). Increased collagen deposition was observed in CCl4-induced fibrotic mice, which could be suppressed by metformin (Figure 1B). A similar effect of metformin on fibronectin was seen in IHC (Figure 1D). As shown in Figure 1E, fibrotic mice expressed more VEGF, indicating more intrahepatic angiogenesis than the control group. Treatment with metformin significantly suppressed expression of VEGF.
Figure 1.
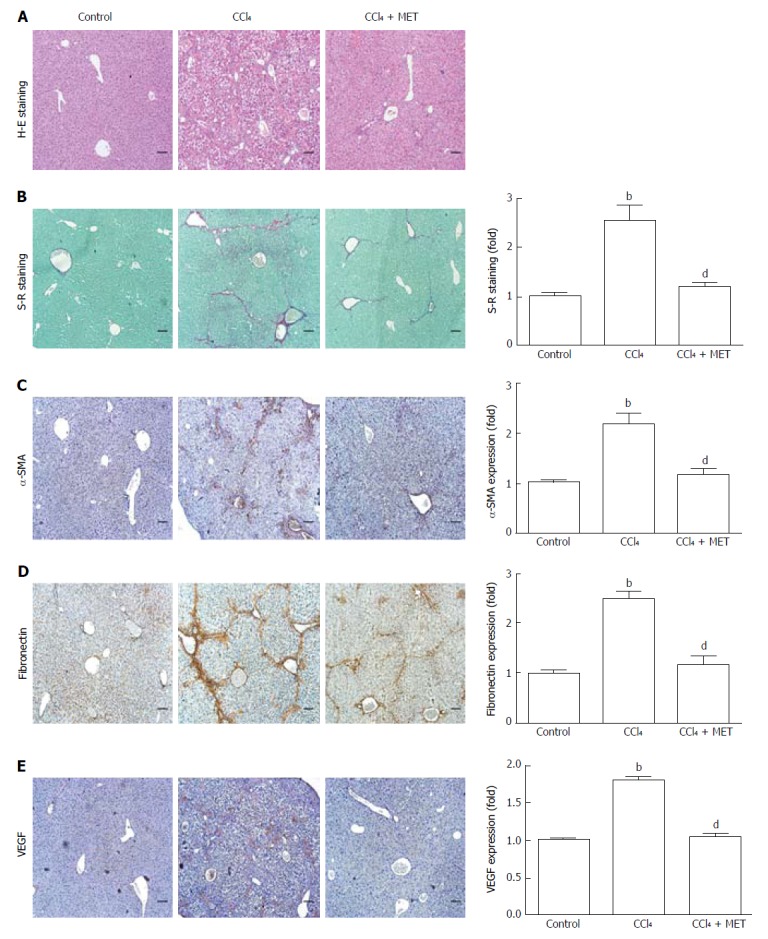
Effect of metformin in CCl4-induced fibrotic mice. A fibrotic mouse model was induced by intraperitoneal injection of CCl4 (1 μL/g) dissolved in olive oil (CCl4:olive oil = 1:1, v/v) twice per week for 6 weeks. A and B: Histological changes were assessed by hematoxylin-eosin (H-E) staining and Sirius Red (S-R) staining (100 × magnification); C-E: Expression levels of α-SMA, fibronectin, and VEGF in the liver tissues were measured by immunohistochemistry (100 × magnification). Sirius Red staining was analyzed with ImageJ and immunohistochemical staining was analyzed with Image-Pro Plus 6.0. (Scale bar = 200 μm, n = 5, bP < 0.01 vs the control group, dP < 0.01 vs the CCl4 group).
Mice exposed to CCl4 increased α-SMA at both the protein and mRNA levels, while co-treatment with metformin reduced this effect (Figure 2A and B), which was also confirmed by IHC (Figure 1C). The CCl4-induced increase in collagen I mRNA expression was reduced by co-treatment with metformin. Taken together, metformin decreased the activation of HSCs, reduced the deposition of ECM, and inhibited angiogenesis in CCl4-treated mice. Therefore, metformin attenuated CCl4-induced liver fibrosis in mice.
Figure 2.
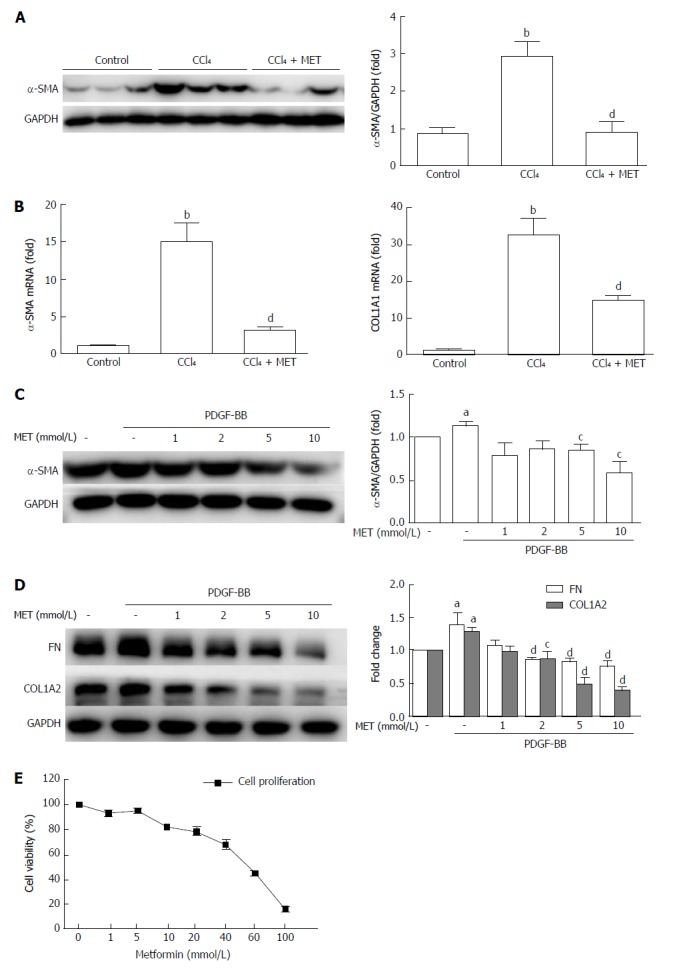
Effect of metformin on the activation, proliferation, and extracellular matrix secretion of hepatic stellate cells. A: Measurement of α-SMA levels in murine liver tissues by Western blot; B: Measurement of hepatic α-SMA and collagen type I mRNA expression levels by quantitative real-time PCR (n = 5, bP < 0.01 vs the control group, dP < 0.01 vs the CCl4 group); C and D: HSCs were treated with or without 10 ng/mL PDGF-BB for 24 h, and the effect of metformin (1, 2, 5, and 10 mmol/L) on the expression levels of α-SMA, collagen type I, and fibronectin (FN) were measured by Western blot (aP < 0.05 vs the control group, cP < 0.05 and dP < 0.01 vs the PDGF-BB only group); E: HSCs were treated with a series of concentrations ranging from 1 mmol/L to 100 mmol/L of metformin for 24 h, and the proliferation was measured by CCK-8 assays. HSCs: Hepatic stellate cells.
Metformin inhibits the proliferation of activated HSCs
HSCs were treated with different concentrations (1 mmol/L to 100 mmol/L) of metformin for 24 h (Figure 2E). The proliferation of HSCs was inhibited by metformin in a dose-dependent manner, and the IC50 was 50.01 mmol/L.
Metformin suppresses the activation of HSCs and decreases the expression of ECM in vitro
The protein levels of α-SMA, collagen type I, and fibronectin were measured by Western blot (Figure 2C and D). PDGF-BB up-regulated the expression of α-SMA, while treatment with metformin at 5 mmol/L and 10 mmol/L suppressed this increase, from 113.5% ± 4.66% to 84.87% ± 6.63% and 58.79% ± 12.64%, respectively (P < 0.05). Collagen type I and fibronectin are the major components of the ECM, and HSCs expressed increased levels of these protein after co-incubation with PDGF-BB. Metformin decreased the protein levels at doses of 2, 5, and 10 mmol/L. These results indicated that metformin suppressed the activation of HSCs and the secretion of ECM in vitro.
Metformin decreases the migration and invasion of HSCs
The migration rate of HSCs was significantly increased by PDGF-BB treatment compared with that of the control group (26.38% ± 2.98% to 48.05% ± 3.67%, P < 0.01). Treatment with metformin at 5 mmol/L and 10 mmol/L reduced PDGF-BB-induced migration, from 48.05% ± 3.67% to 21.67% ± 2.73% and 14.99% ± 0.25% (P < 0.01), respectively (Figure 3A and C). As shown in Figure 3B, cells that migrated through the matrigel membrane decreased from 1352% ± 62.87% to 748.0% ± 76.18%, 453.0% ± 4.58%, and 190.0% ± 14.73% (P < 0.01) compared with the control group when treated with metformin at 1, 5, and 10 mmol/L, respectively (Figure 3D). These findings indicated that metformin decreased the motility of HSCs.
Figure 3.
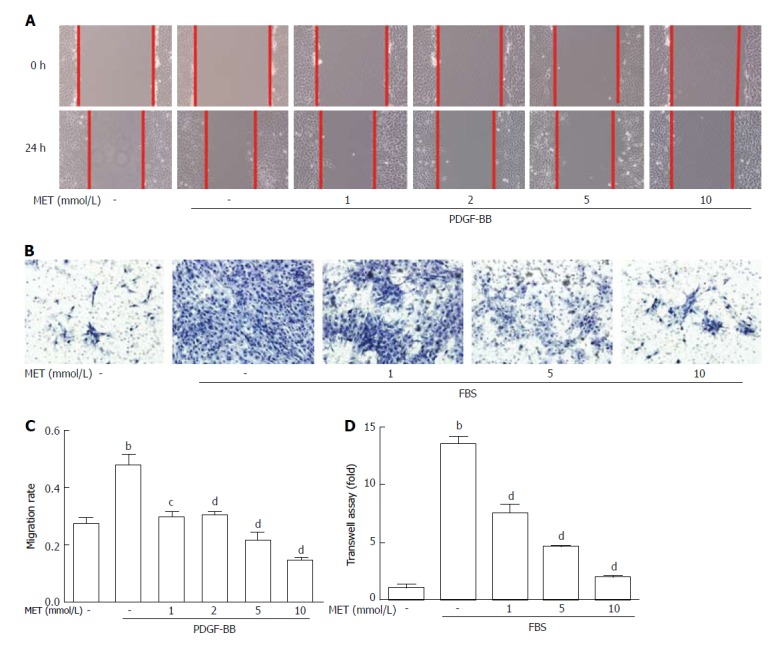
Effect of metformin on hepatic stellate cell migration and invasion. Scratch tests were used to determine cell migration, and Transwell assays were used to evaluate cell invasion. A: HSCs were scraped and then incubated with or without PDGF-BB (10 ng/mL) and metformin (1, 2, 5, and 10 mmol/L). Images were acquired at 0 and 24 h (100 × magnification); B: HSCs were seeded in the upper chamber with a Matrigel membrane, and various concentrations of metformin (0, 1, 5, and 10 mmol/L) were added to the medium. The lower chambers were loaded with DMEM with or without 10% FBS. Cells that migrated through the membrane were fixed and stained with hematoxylin at 24 h; C: The migration ability was quantified by measuring the distance of the scratch edge; D: Cells that migrated through the membrane were counted and quantified. (bP < 0.01 vs the control group, cP < 0.05 and dP < 0.01 vs the PDGF-BB or FBS only groups). HSCs: Hepatic stellate cells.
Metformin inhibits the contraction of HSCs
We assessed the inhibitory effect of metformin on the contractility of HSCs by collagen gel contraction assay. PDGF-BB caused a significant increase in cell contractility, while co-culture with metformin neutralized these effects (Figure 4A and C). PDGF-BB treatment enhanced the contraction rate of HSCs from 47.43% ± 2.13% to 70.25% ± 1.35% (P < 0.01), while treatment with metformin at 1, 5, and 10 mmol/L attenuated the contraction rate to 49.70% ± 6.59% (P < 0.05), 44.73% ± 4.65%, and 42.26% ± 3.28% (P < 0.01), respectively.
Figure 4.
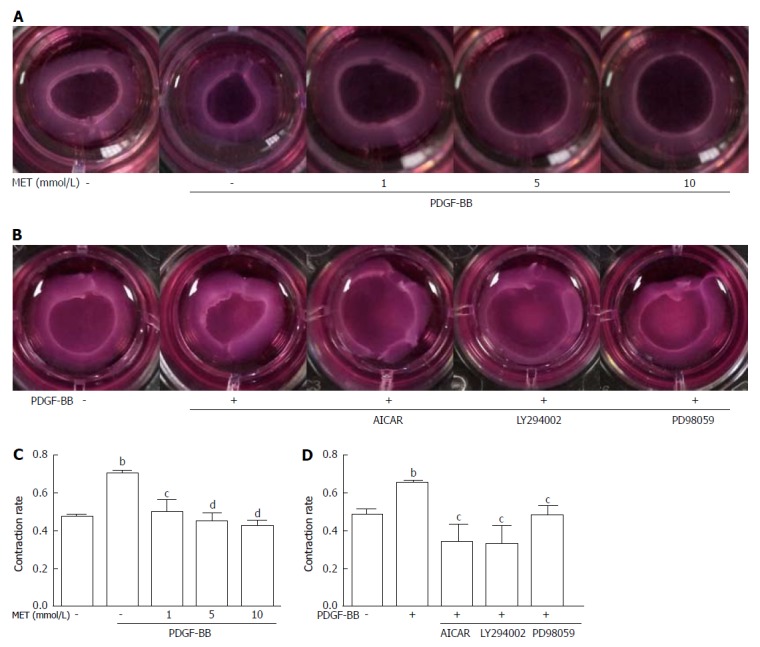
Effect of metformin on hepatic stellate cell contraction. Collagen gels were prepared in 24-well plates. A: HSCs were seeded on the collagen gel with or without PDGF-BB (10 ng/mL) and metformin (1, 5, and 10 mmol/L); B: Metformin was changed to AICAR (500 μmol/L), LY294002 (20 μmol/L), and PD98059 (10 μmol/L); C and D: After incubation for 24 h, the areas of the collagen gel were measured for analysis. (bP < 0.01 vs the control group, cP < 0.05 and dP < 0.01 vs the PDGF-BB only group). HSCs: Hepatic stellate cells.
Metformin decreases the expression of VEGF in HSCs through inhibition of HIF-1α in both PDGF-BB and hypoxic conditions
CoCl2· 6H2O (150 μmol/L) was added to the medium to mimic hypoxic conditions[16,17]. HSCs expressed more VEGF when incubated with PDGF-BB or CoCl2 compared with the control group, and this effect was associated with an increased level of HIF-1α (Figure 5A and B). Metformin decreased the level of HIF-1α and further reduced the expression of VEGF in HSCs. Treatment with metformin at 5 and 10 mmol/L had an inhibitory effect on VEGF expression in both PDGF-BB and hypoxic conditions.
Figure 5.
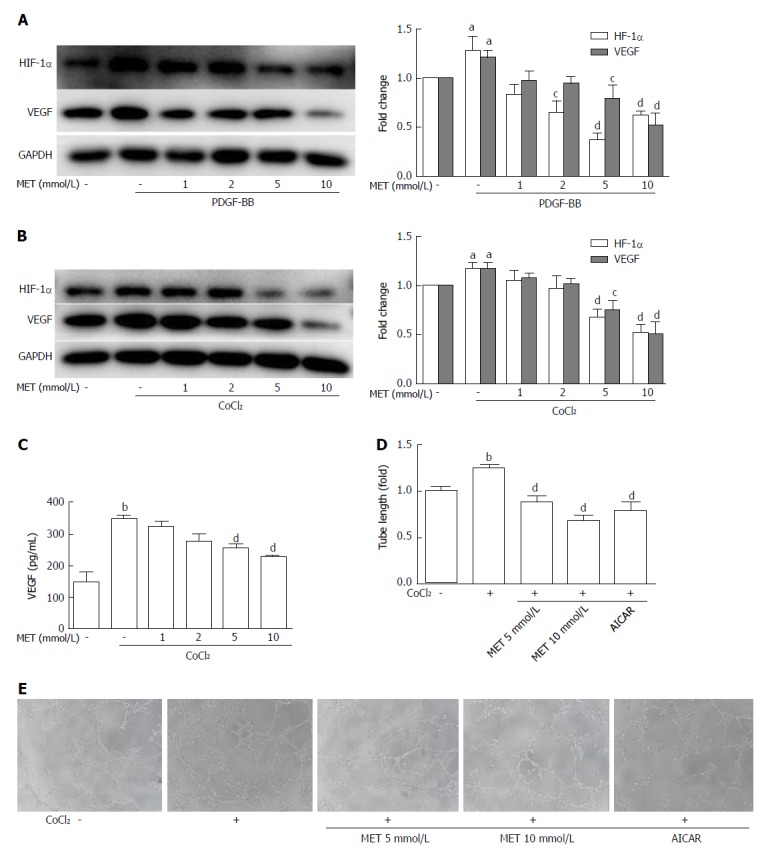
Effect of metformin on VEGF expression and secretion of hepatic stellate cells and angiogenesis in vitro. A and B: HSCs were incubated with or without PDGF-BB (10 ng/mL) for 24 h or CoCl2 (150 μmol/L) for 12 h and metformin (1, 2, 5, and 10 mmol/L). The expression levels of HIF-1α and VEGF were measured by Western blot analysis, and the results were quantified; C: Cells were treated as in panel B, and the supernatant was collected. The protein level of VEGF was measured by ELISA assay; D and E: HSCs were pretreated with metformin (5 and 10 mmol/L) or AICAR (500 μmol/L) for 2 h, and then incubated with or without CoCl2 (150 μmol/L) for 12 h. The supernatant was collected and diluted 4:1 (v/v) in DMEM with 10% FBS to form conditioned medium. HUVECs were harvested and suspended in the conditioned medium, and then seeded on Matrigel. Images were acquired at 8 h (100 × magnification), and tube lengths were calculated with ImageJ and quantified. aP < 0.05 and bP < 0.01 vs the control group, cP < 0.05 and dP < 0.01 vs the PDGF-BB or CoCl2 only groups. HSCs: Hepatic stellate cells.
Metformin down-regulates VEGF secretion by HSCs and inhibits angiogenesis in hypoxic conditions in vitro
The VEGF protein level in the supernatant was increased from 148.96 ± 50.62 pg/mL to 343.52 ± 25.91 pg/mL (P < 0.01) when CoCl2 (150 μmol/L) was added to the medium, but co-culture with metformin at 5 mmol/L and 10 mmol/L decreased VEGF levels to 254.40 ± 16.91 pg/mL and 229.04 ± 1.62 pg/mL, respectively (P < 0.01) (Figure 5C). Tube formation of HUVECs on Matrigel can be used to analyze angiogenesis in vitro. HUVECs were cultured in conditioned medium on Matrigel-coated plates. The conditioned medium from CoCl2-treated HSCs significantly increased tube formation, while conditioned medium from HSCs co-treated with CoCl2 and metformin decreased tube formation. AICAR mimicked the effect of metformin on tube formation (Figure 5D and E).
Metformin inhibits the fibrogenic response of HSCs through inhibition of the Akt/mTOR and ERK pathways via the activation of AMPK
Metformin increased the phosphorylation of AMPK in a dose-dependent manner (Figure 6C). After stimulation with PDGF-BB, the levels of p-Akt, p-mTOR, and p-ERK were significantly increased compared with those of the control group, while co-treatment with metformin decreased these effects (Figure 6A and E). The Akt/mTOR and ERK pathways are associated with cell proliferation, migration, and phenotypic change in HSCs. To further confirm these effects, we used various indicated inhibitors to treat HSCs (Figure 7A and C). LY294002 (an Akt inhibitor, 20 μmol/L) and rapamycin (an mTOR inhibitor, 100 nmol/L) inhibited the activation of HSCs, decreased ECM secretion, and reduced the expression of HIF-1α and VEGF. Moreover, LY294002 inhibited the contraction of HSCs (Figure 4B and D). PD98059 (an ERK inhibitor, 10 μmol/L) had a similar effect as LY294002, except that it could not decrease the secretion of collagen type I. Additionally, AICAR (500 μmol/L), another AMPK activator, mimicked the effect of metformin. In conclusion, PDGF-BB increased the fibrogenic response of HSCs through activating the downstream Akt/mTOR and ERK pathways, while metformin inhibited these effects via activation of AMPK.
Figure 6.
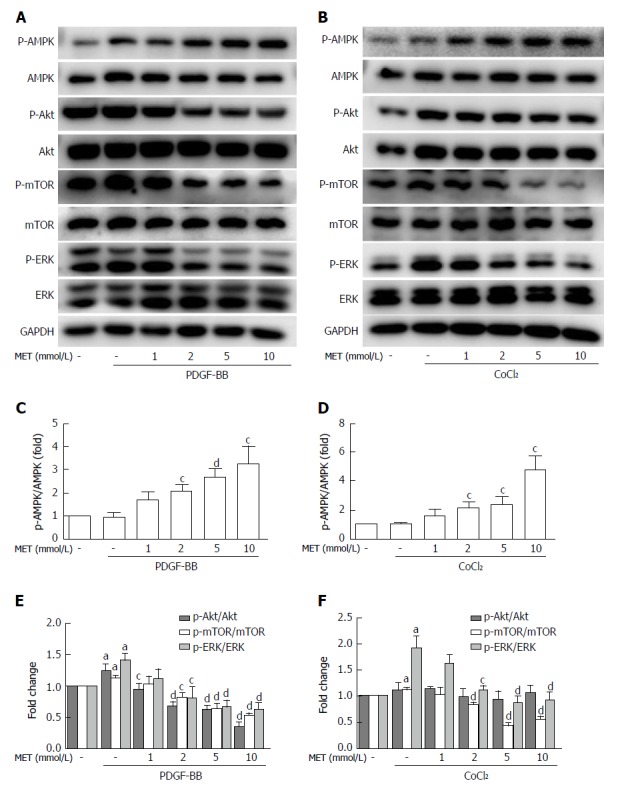
Effect of metformin on AMPK, Akt/mTOR, and ERK signaling in hepatic stellate cells. A and B: HSCs were pretreated with metformin (1, 2, 5 and 10 mmol/L) for 2 h and then incubated with PDGF-BB (10 ng/mL) for 24 h or CoCl2 (150 μmol/L) for 12 h. AMPK, Akt/mTOR, and ERK signaling pathways were assessed by Western blot analysis; C and D: The results were quantified. aP < 0.05 vs the control group, cP < 0.05 and dP < 0.01 vs the PDGF-BB or CoCl2 only groups. HSCs: Hepatic stellate cells.
Figure 7.
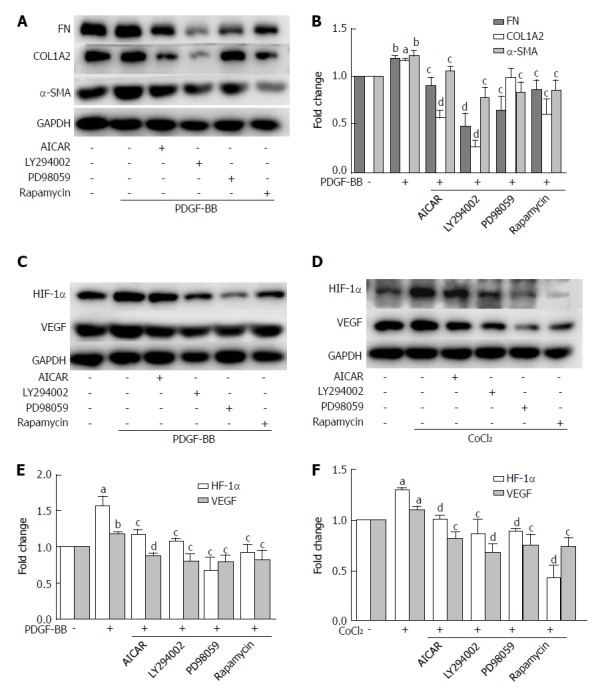
The inhibitory effects of metformin on activated hepatic stellate cells were associated with activation of AMPK and subsequent down-regulation of the Akt/mTOR and ERK signaling pathways. A and B: HSCs were pretreated with AICAR (500 μmol/L), LY294002 (20 μmol/L), PD98059 (10 μmol/L), or rapamycin (100 nmol/L) for 2 h and then incubated with PDGF-BB (10 ng/mL) for 24 h. Expression levels of fibronectin (FN), collagen type I (COL1A2), and α-SMA were measured by Western blot analysis and the results were quantified; C and D: HSCs were pretreated with AICAR (500 μmol/L), LY294002 (20 μmol/L), PD98059 (10 μmol/L), or rapamycin (100 nmol/L) for 2 h and then incubated with or without CoCl2 (150 μmol/L) for 12 h. Expression levels of HIF-1α and VEGF were measured by Western blot analysis; E and F: The results were quantified. (aP < 0.05 and bP < 0.01 vs the control group, cP < 0.05 and dP < 0.01 vs the PDGF-BB or CoCl2 only groups). HSCs: Hepatic stellate cells.
Metformin decreases VEGF expression by activated HSCs by down-regulating the mTOR/HIF-1α and ERK/HIF-1α pathways under hypoxic conditions
The levels of p-mTOR and p-ERK were significantly increased when compared with the control group under hypoxic conditions, while no change was found in the phosphorylation of Akt (Figure 6B and D). Metformin increased the phosphorylation of AMPK and inhibited the activation of p-mTOR and p-ERK. PD98059 and rapamycin decreased the expression of HIF-1α and VEGF. LY294002 inhibited the activation of p-Akt and the downstream p-mTOR, which therefore decreased the expression of HIF-1α and VEGF. AICAR had a similar effect as metformin under these conditions (Figure 7D and F). These results indicated that metformin decreased VEGF expression by activated HSCs by down-regulating the mTOR/HIF-1α and ERK/HIF-1α signaling pathways under hypoxic conditions.
DISCUSSION
The prime determinant of PHT is increased IHVR, which is thought to be generated by the following two factors: structural (distortion of the liver vascular architecture caused by fibrosis, scarring, and nodule formation) and functional (hepatic sinusoidal cellular alterations that promote constriction of the hepatic sinusoids) components[18]. Research has shown that activation of HSCs is a key event mediating augmented IHVR[7]. We designed this study to investigate the role of metformin in activated HSCs. We found that metformin could attenuate the fibrogenic response of HSCs and decrease IHVR. Our research indicated that metformin treatment may be a potent therapeutic approach to treating liver fibrosis and PHT.
First, we used a fibrotic mouse model to determine whether metformin had effects on liver fibrosis. Mice injected with CCl4 for 6 wk developed marked fibrosis compared with the control group, while co-treatment with metformin attenuated histopathologic features of fibrosis. α-SMA, a marker of HSC activation, was inhibited by metformin at both the protein and mRNA levels. Sirius Red staining showed that collagen deposition was also decreased, as well as the mRNA level of collagen type I. Moreover, VEGF expression was up-regulated in fibrotic mice, which was decreased by metformin treatment. Therefore, metformin could alleviate the progression of liver fibrosis in fibrotic mice. A recent study also showed that metformin mitigated CCl4-induced liver fibrosis in mice. The anti-fibrogenic response was reported to primarily involve suppression of ECM deposition, and this effect might have resulted from suppressed TGF-β1/Smad3 signaling[8]; this was supported by a previous in vitro study[9]. In our study, we found that metformin could also inhibit the angiogenesis in liver fibrosis, indicating that metformin may attenuate liver fibrosis in other ways. The PDGF signaling pathway is among the most well characterized pathways involved in HSC activation; PDGF-BB is the most potent stimulator of HSC growth and intracellular signaling[1], and blocking of PDGF signaling ameliorates experimental liver fibrogenesis[19]. As described above, we speculated that metformin could regulate the fibrogenic response of HSCs and have an anti-angiogenic effect via PDGF and its downstream pathways. Therefore, we performed an in vitro study to further explain the effect of metformin on activated HSCs and investigate the possible signaling pathways involved.
We used PDGF-BB to stimulate HSCs in vitro. PDGF-BB up-regulated the expression of α-SMA, as well as type I collagen and fibronectin, in HSCs, while these protein levels were decreased when treated with metformin. These results are in agreement with our animal experiments. Caligiuri showed that activation of AMPK modulated the activated phenotype change of HSCs caused by PDGF-BB[15]. In this study, PDGF induced activation of the downstream molecules ERK and Akt/mTOR in activated HSCs, which are associated with cellular proliferation, migration, and phenotype changes. Metformin inhibited the activation of ERK and Akt/mTOR by activating AMPK. To further analyze the role of the signaling pathways, we used various indicated inhibitors to determine whether the signaling pathways could affect activated HSCs. LY294002 and rapamycin inhibited the expression of α-SMA, collagen I, and fibronectin. PD98059 had a similar effect, except for the expression of collagen type I. Furthermore, AICAR, another AMPK activator, could imitate the effect of metformin on activated HSCs. Metformin inhibited activation and ECM secretion of HSCs. This effect was mediated by the activation of AMPK, thereby inhibiting the activation of ERK and Akt/mTOR by PDGF-BB.
In liver cirrhosis, an imbalance between vasoconstrictors and vasodilators makes HSCs more contractile, which increases IHVR and aggravates PHT. Metformin has been reported to attenuate contractile responses in rat aortas[20], and to reduce pulmonary artery contraction in pulmonary arterial hypertension[21]. Therefore, we used a collagen gel contraction assay to evaluate the effect of metformin on the contraction of HSCs. Our results showed that metformin inhibited the contraction of HSCs caused by PDGF-BB. The RhoA/Rock pathway is the contractile pathway in vascular smooth muscle that is also expressed in HSCs[22]. Sorafenib can down-regulate Rho kinase by inhibiting the ERK pathway in secondary biliary cirrhotic rats and further reduce portal pressure[23]. Metformin can decrease the activation of ERK caused by PDGF-BB; therefore, we speculated that the inhibitory effect of metformin on the contraction of HSCs were due in part to the inhibition of the ERK pathway. To confirm this effect, we used PD98059 to treat HSCs stimulated with PDGF-BB. PD98059 inhibited the contraction of HSCs, as expected. There have also been studies that linked the inhibition of the Akt pathway to attenuation of contraction[24,25], and this effect may be associated with the Akt/L-type calcium channel and the Akt/RhoA/Rho kinase pathways[26,27]. In our research, LY294002 could also inhibit the contraction of HSCs, indicating that the Akt pathway was also involved in modulating the contraction of HSCs. In addition, AICAR had a similar effect to metformin on the contraction of HSCs. Thus, metformin inhibits the contraction of activated HSCs, which can decrease IHVR and lower portal pressure.
PDGF can promote HSCs to develop an angiogenic phenotype via modulating HSC-based vascular tube formation and increasing coverage of sinusoids, with resulting effects on vascular permeability, vessel mutation, and portal pressure regulation[6,28]. Activated HSC recruitment to liver sinusoidal epithelial cells is an important step in sinusoidal remodeling, and PDGF may be the most important growth factor in this process[29]. In our study, metformin decreased the motility of HSCs. Moreover, metformin decreased the level of VEGF secreted by HSCs, and inhibited angiogenesis in vitro. Taken together, we showed that metformin could inhibit the angiogenic properties of HSCs.
VEGF plays a predominant role in the initial stages of angiogenesis[3]. Reports have shown that PDGF can increase the HIF-1α and VEGF protein levels in activated HSCs[30]. PDGF can also stimulate VEGF expression and HSC-driven vascularization through signals mediated by ERK and mTOR[31]. In our study, the AKT/mTOR and ERK pathways were up-regulated by PDGF and caused increased levels of HIF-1α and VEGF in HSCs. The activation of AMPK by metformin decreased the up-regulation of VEGF by PDGF. This result was partly in agreement with the research by Zhang et al[32], who found that curcumin interrupted the PDGF-βR/ERK and mTOR pathways, leading to reductions in VEGF expression in HSCs. As hypoxia is the most potent stimulus for VEGF expression, we further used CoCl2 to mimic a hypoxic environment. Hypoxia stabilized HIF-1α and up-regulated the expression of VEGF in our study. The protein level of VEGF in the HSCs and the medium was significantly higher than that in the control group, and the phosphorylation of ERK and mTOR was also increased. Co-treatment with the AMPK activator metformin inhibited the increase of HIF-1α, VEGF, and the activation of ERK and mTOR. In addition, AICAR, LY294002, PD98059, and rapamycin could also inhibit the expression of HIF-1α and VEGF. These results indicated that metformin could decrease the VEGF levels secreted by HSCs, and this effect was partly mediated by the ERK/HIF-1α and mTOR/HIF-1α pathways. Finally, we used tube formation assays to analyze angiogenesis in vitro. When HUVECs were cultured with conditioned medium from HSCs treated with metformin under hypoxic conditions, tube formation was less than that in medium without metformin. AICAR had a similar effect to metformin. Therefore, metformin could inhibit PDGF and hypoxia-induced VEGF expression in HSCs, thus decreasing HSC-based angiogenesis. These effects were mediated through the inhibition of the ERK/HIF-1α and mTOR/HIF-1α pathways by activation of AMPK.
In conclusion, metformin can inhibit the activation, proliferation, motility, and contraction of HSCs, reduce the secretion of ECM, attenuate HSC angiogenic properties, and decrease HSC-based angiogenesis. Metformin has effects on both structural and functional components of IHVR, suggesting a novel therapeutic approach for the treatment of liver fibrosis and PHT.
ARTICLE HIGHLIGHTS
Research background
Activation of hepatic stellate cells (HSCs) contributes to liver fibrosis and portal hypertension, and it is a therapeutic target for the treatment of chronic liver diseases (CLDs). Previous studies have demonstrated that metformin has a wide range of pharmacological activities beyond its antidiabetic effects. It may therefore represent a potent therapeutic approach to CLDs, but the mechanisms underlying its effects are still unclear.
Research motivation
This study was performed to investigate the effect of metformin on activated HSCs and clarify its molecular mechanisms.
Research objectives
The inhibitory effects of metformin on the activation, proliferation, motility, contraction, extracellular matrix (ECM) secretion of HSCs and HSC-based angiogenesis were evaluated. We also characterized its underlying mechanisms with a focus on AMPK and downstream AKT/mTOR and ERK signaling pathways.
Research methods
The effect of metformin on activated HSCs were investigated in vivo and in vitro. A fibrotic mouse model was treated with or without metformin, and the effect of metformin on liver fibrosis was evaluated. The HSC cell line LX-2 was used for in vitro studies. The effect of metformin on cell proliferation was detected by CCK8 assay. Cell motility was measured by scratch tests and Transwell assays. Collagen gel contraction assays were performed to assess the effect of metformin on cell contraction. Expression of α-SMA, collagen type I, and fibronectin was determined by Western blot analysis. We also analyzed the effect of metformin on HSC-based angiogenesis in both PDGF and hypoxic conditions. The phosphorylation levels of AMPK, AKT, mTOR, and ERK were measured by Western blot analysis. We also used the indicated pharmacologic inhibitors and agonists to confirm our findings.
Research results
Metformin decreased the activation of HSCs, reduced the deposition of ECM, and inhibited angiogenesis in fibrotic mice. PDGF-BB promoted the fibrogenic response of HSCs, while metformin inhibited the activation, proliferation, migration, and contraction of HSCs, reduced their secretion of ECM, and decreased HSC-based angiogenesis. These inhibitory effects were mediated by inhibition of the Akt/mTOR and ERK pathways via the activation of AMPK.
Research conclusions
Metformin attenuates the fibrogenic response of HSCs in vivo and in vitro, and may therefore be useful for the treatment of chronic liver diseases.
Research perspective
This study investigated the inhibitory effect of metformin on activated HSCs and the possible signaling pathways involved. The results strongly confirmed the potential use of metformin for the treatment of CLDs. In future studies, we will provide more evidence for the use of metformin, especially in the treatment of portal hypertension. The effect of metformin on liver sinusoidal endothelial cells will also be analyzed.
ACKNOWLEDGMENTS
We gratefully thank Dr. Edward C Mignot, Shandong University, for linguistic advice.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: China
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Supported by National Natural Science Foundation of China, No. 81370590.
Institutional review board statement: The study was reviewed and approved by the Institutional Review Board of Shandong Provincial Hospital Affiliated to Shandong University.
Institutional animal care and use committee statement: The consent procedure and study protocol were approved by the Animal Medical Ethics Committee of Shandong Provincial Hospital Affiliated to Shandong University (No. 2017-228).
Conflict-of-interest statement: The authors declare no conflict of interest related to this manuscript.
Data sharing statement: No additional unpublished data are available.
Peer-review started: November 21, 2017
First decision: December 6, 2017
Article in press: December 26, 2017
P- Reviewer: Shin T, Siddiqui I S- Editor: Gong ZM L- Editor: Wang TQ E- Editor: Li D
Contributor Information
Zhen Li, Department of Gastroenterology, Shandong Provincial Hospital Affiliated to Shandong University, Jinan 250021, Shandong Province, China; Shandong Provincial Engineering and Technological Research Center for Liver Disease Prevention and Control, Jinan 250021, Shandong Province, China.
Qian Ding, Department of Gastroenterology, Shandong Provincial Hospital Affiliated to Shandong University, Jinan 250021, Shandong Province, China.
Li-Ping Ling, Department of Gastroenterology, Shandong Provincial Hospital Affiliated to Shandong University, Jinan 250021, Shandong Province, China; Shandong Provincial Engineering and Technological Research Center for Liver Disease Prevention and Control, Jinan 250021, Shandong Province, China.
Ying Wu, Department of Gastroenterology, Shandong Provincial Hospital Affiliated to Shandong University, Jinan 250021, Shandong Province, China; Shandong Provincial Engineering and Technological Research Center for Liver Disease Prevention and Control, Jinan 250021, Shandong Province, China.
Dong-Xiao Meng, Department of Gastroenterology, Shandong Provincial Hospital Affiliated to Shandong University, Jinan 250021, Shandong Province, China; Shandong Provincial Engineering and Technological Research Center for Liver Disease Prevention and Control, Jinan 250021, Shandong Province, China.
Xiao Li, Department of Gastroenterology, Shandong Provincial Hospital Affiliated to Shandong University, Jinan 250021, Shandong Province, China; Shandong Provincial Engineering and Technological Research Center for Liver Disease Prevention and Control, Jinan 250021, Shandong Province, China.
Chun-Qing Zhang, Department of Gastroenterology, Shandong Provincial Hospital Affiliated to Shandong University, Jinan 250021, Shandong Province, China. zhangchunqing_sdu@163.com.
References
- 1.Lee UE, Friedman SL. Mechanisms of hepatic fibrogenesis. Best Pract Res Clin Gastroenterol. 2011;25:195–206. doi: 10.1016/j.bpg.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Elpek GÖ. Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: An update. World J Gastroenterol. 2014;20:7260–7276. doi: 10.3748/wjg.v20.i23.7260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fernández M, Semela D, Bruix J, Colle I, Pinzani M, Bosch J. Angiogenesis in liver disease. J Hepatol. 2009;50:604–620. doi: 10.1016/j.jhep.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 4.Thabut D, Shah V. Intrahepatic angiogenesis and sinusoidal remodeling in chronic liver disease: new targets for the treatment of portal hypertension? J Hepatol. 2010;53:976–980. doi: 10.1016/j.jhep.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 5.Gracia-Sancho J, Maeso-Díaz R, Fernández-Iglesias A, Navarro-Zornoza M, Bosch J. New cellular and molecular targets for the treatment of portal hypertension. Hepatol Int. 2015;9:183–191. doi: 10.1007/s12072-015-9613-5. [DOI] [PubMed] [Google Scholar]
- 6.Bocca C, Novo E, Miglietta A, Parola M. Angiogenesis and Fibrogenesis in Chronic Liver Diseases. Cell Mol Gastroenterol Hepatol. 2015;1:477–488. doi: 10.1016/j.jcmgh.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fernandez M. Molecular pathophysiology of portal hypertension. Hepatology. 2015;61:1406–1415. doi: 10.1002/hep.27343. [DOI] [PubMed] [Google Scholar]
- 8.Fan K, Wu K, Lin L, Ge P, Dai J, He X, Hu K, Zhang L. Metformin mitigates carbon tetrachloride-induced TGF-β1/Smad3 signaling and liver fibrosis in mice. Biomed Pharmacother. 2017;90:421–426. doi: 10.1016/j.biopha.2017.03.079. [DOI] [PubMed] [Google Scholar]
- 9.Lim JY, Oh MA, Kim WH, Sohn HY, Park SI. AMP-activated protein kinase inhibits TGF-β-induced fibrogenic responses of hepatic stellate cells by targeting transcriptional coactivator p300. J Cell Physiol. 2012;227:1081–1089. doi: 10.1002/jcp.22824. [DOI] [PubMed] [Google Scholar]
- 10.Tripathi DM, Erice E, Lafoz E, García-Calderó H, Sarin SK, Bosch J, Gracia-Sancho J, García-Pagán JC. Metformin reduces hepatic resistance and portal pressure in cirrhotic rats. Am J Physiol Gastrointest Liver Physiol. 2015;309:G301–G309. doi: 10.1152/ajpgi.00010.2015. [DOI] [PubMed] [Google Scholar]
- 11.Chen HP, Shieh JJ, Chang CC, Chen TT, Lin JT, Wu MS, Lin JH, Wu CY. Metformin decreases hepatocellular carcinoma risk in a dose-dependent manner: population-based and in vitro studies. Gut. 2013;62:606–615. doi: 10.1136/gutjnl-2011-301708. [DOI] [PubMed] [Google Scholar]
- 12.Pinzani M. PDGF and signal transduction in hepatic stellate cells. Front Biosci. 2002;7:d1720–d1726. doi: 10.2741/A875. [DOI] [PubMed] [Google Scholar]
- 13.Berra E, Pagès G, Pouysségur J. MAP kinases and hypoxia in the control of VEGF expression. Cancer Metastasis Rev. 2000;19:139–145. doi: 10.1023/a:1026506011458. [DOI] [PubMed] [Google Scholar]
- 14.Karar J, Maity A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front Mol Neurosci. 2011;4:51. doi: 10.3389/fnmol.2011.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caligiuri A, Bertolani C, Guerra CT, Aleffi S, Galastri S, Trappoliere M, Vizzutti F, Gelmini S, Laffi G, Pinzani M, et al. Adenosine monophosphate-activated protein kinase modulates the activated phenotype of hepatic stellate cells. Hepatology. 2008;47:668–676. doi: 10.1002/hep.21995. [DOI] [PubMed] [Google Scholar]
- 16.Tadakawa M, Takeda T, Li B, Tsuiji K, Yaegashi N. The anti-diabetic drug metformin inhibits vascular endothelial growth factor expression via the mammalian target of rapamycin complex 1/hypoxia-inducible factor-1α signaling pathway in ELT-3 cells. Mol Cell Endocrinol. 2015;399:1–8. doi: 10.1016/j.mce.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 17.Al Qahtani A, Holly J, Perks C. Hypoxia negates hyperglycaemia-induced chemo-resistance in breast cancer cells: the role of insulin-like growth factor binding protein 2. Oncotarget. 2017;8:74635–74648. doi: 10.18632/oncotarget.20287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garcia-Tsao G, Abraldes JG, Berzigotti A, Bosch J. Portal hypertensive bleeding in cirrhosis: Risk stratification, diagnosis, and management: 2016 practice guidance by the American Association for the study of liver diseases. Hepatology. 2017;65:310–335. doi: 10.1002/hep.28906. [DOI] [PubMed] [Google Scholar]
- 19.Borkham-Kamphorst E, Weiskirchen R. The PDGF system and its antagonists in liver fibrosis. Cytokine Growth Factor Rev. 2016;28:53–61. doi: 10.1016/j.cytogfr.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 20.Pyla R, Osman I, Pichavaram P, Hansen P, Segar L. Metformin exaggerates phenylephrine-induced AMPK phosphorylation independent of CaMKKβ and attenuates contractile response in endothelium-denuded rat aorta. Biochem Pharmacol. 2014;92:266–279. doi: 10.1016/j.bcp.2014.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Agard C, Rolli-Derkinderen M, Dumas-de-La-Roque E, Rio M, Sagan C, Savineau JP, Loirand G, Pacaud P. Protective role of the antidiabetic drug metformin against chronic experimental pulmonary hypertension. Br J Pharmacol. 2009;158:1285–1294. doi: 10.1111/j.1476-5381.2009.00445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trebicka J, Hennenberg M, Laleman W, Shelest N, Biecker E, Schepke M, Nevens F, Sauerbruch T, Heller J. Atorvastatin lowers portal pressure in cirrhotic rats by inhibition of RhoA/Rho-kinase and activation of endothelial nitric oxide synthase. Hepatology. 2007;46:242–253. doi: 10.1002/hep.21673. [DOI] [PubMed] [Google Scholar]
- 23.Hennenberg M, Trebicka J, Stark C, Kohistani AZ, Heller J, Sauerbruch T. Sorafenib targets dysregulated Rho kinase expression and portal hypertension in rats with secondary biliary cirrhosis. Br J Pharmacol. 2009;157:258–270. doi: 10.1111/j.1476-5381.2009.00158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu H, Chen Z, Liu J, Liu L, Gao Y, Dou D. Endothelium-independent hypoxic contraction of porcine coronary arteries may be mediated by activation of phosphoinositide 3-kinase/Akt pathway. Vascul Pharmacol. 2014;61:56–62. doi: 10.1016/j.vph.2014.03.005. [DOI] [PubMed] [Google Scholar]
- 25.Liegl R, Wertheimer C, Kernt M, Docheva D, Kampik A, Eibl-Lindner KH. Attenuation of human lens epithelial cell spreading, migration and contraction via downregulation of the PI3K/Akt pathway. Graefes Arch Clin Exp Ophthalmol. 2014;252:285–292. doi: 10.1007/s00417-013-2524-z. [DOI] [PubMed] [Google Scholar]
- 26.Carnevale D, Vecchione C, Mascio G, Esposito G, Cifelli G, Martinello K, Landolfi A, Selvetella G, Grieco P, Damato A, et al. PI3Kγ inhibition reduces blood pressure by a vasorelaxant Akt/L-type calcium channel mechanism. Cardiovasc Res. 2012;93:200–209. doi: 10.1093/cvr/cvr288. [DOI] [PubMed] [Google Scholar]
- 27.Miao L, Dai Y, Zhang J. Mechanism of RhoA/Rho kinase activation in endothelin-1- induced contraction in rabbit basilar artery. Am J Physiol Heart Circ Physiol. 2002;283:H983–H989. doi: 10.1152/ajpheart.00141.2002. [DOI] [PubMed] [Google Scholar]
- 28.Coulon S, Heindryckx F, Geerts A, Van Steenkiste C, Colle I, Van Vlierberghe H. Angiogenesis in chronic liver disease and its complications. Liver Int. 2011;31:146–162. doi: 10.1111/j.1478-3231.2010.02369.x. [DOI] [PubMed] [Google Scholar]
- 29.Lee JS, Semela D, Iredale J, Shah VH. Sinusoidal remodeling and angiogenesis: a new function for the liver-specific pericyte? Hepatology. 2007;45:817–825. doi: 10.1002/hep.21564. [DOI] [PubMed] [Google Scholar]
- 30.Aleffi S, Navari N, Delogu W, Galastri S, Novo E, Rombouts K, Pinzani M, Parola M, Marra F. Mammalian target of rapamycin mediates the angiogenic effects of leptin in human hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2011;301:G210–G219. doi: 10.1152/ajpgi.00047.2010. [DOI] [PubMed] [Google Scholar]
- 31.Zhang F, Kong D, Chen L, Zhang X, Lian N, Zhu X, Lu Y, Zheng S. Peroxisome proliferator-activated receptor-γ interrupts angiogenic signal transduction by transrepression of platelet-derived growth factor-β receptor in hepatic stellate cells. J Cell Sci. 2014;127:305–314. doi: 10.1242/jcs.128306. [DOI] [PubMed] [Google Scholar]
- 32.Zhang F, Zhang Z, Chen L, Kong D, Zhang X, Lu C, Lu Y, Zheng S. Curcumin attenuates angiogenesis in liver fibrosis and inhibits angiogenic properties of hepatic stellate cells. J Cell Mol Med. 2014;18:1392–1406. doi: 10.1111/jcmm.12286. [DOI] [PMC free article] [PubMed] [Google Scholar]