

Abstract
Background:
The BCL-2-specific BH3-mimetic ABT-199 (venetoclax) has been reported to be principally active against favourable-risk multiple myeloma (MM) cells, prompting efforts to extend its activity to include more resistant, higher-risk MM subsets.
Methods:
Effects of the CDK9 inhibitor flavopiridol (FP; alvocidib) on responses to ABT-199 were examined in MM cells. Cell death and protein expression were evaluated by western blot and immunofluorescence. Xenograft models were used to study combination effects in vivo.
Results:
FP synergistically increased ABT-199 lethality in both ABT-199-sensitive and insensitive MM cells. FP blocked CDK9 activation/positive transcription elongation factor B phosphorylation, downregulated MCL-1, increased BCL-2/MCL-1 ratios, and upregulated BIM. MCL-1 ectopic expression or knockdown in MM cells significantly diminished or increased ABT-199 sensitivity, respectively. CDK9 knockdown triggered MCL-1 downregulation and increased ABT-199 activity, whereas BIM knockdown significantly reduced FP/ABT-199 lethality. FP also enhanced ABT-199 lethality in unfavourable prognosis primary MM cells. HS-5 cell co-culture failed to protect MM cells from the FP/ABT-199 regimen, suggesting circumvention of microenvironmental signals. Finally, FP/ABT-199 significantly increased survival in systemic xenograft and immune-competent MM models while exhibiting minimal toxicity.
Conclusions:
These findings argue that CDK9 inhibitors, for example, FP may increase the antimyeloma activity of ABT-199, including in unfavourable-risk MM minimally responsive to ABT-199 alone.
Keywords: CDK9 inhibitor, ABT-199, flavopiridol, MCL-1, BCL-2, myeloma
Multiple myeloma (MM) is a lymphoproliferative neoplasm involving mature plasma cells that afflicts >30 000 patients per year in the United States (Siegel et al, 2016) and considerably more worldwide. Despite advances, for example, novel proteasome inhibitors (PIs; Muchtar et al, 2016), immunomodulatory agents (Dimopoulos et al, 2014), and antibodies (for example, CD38; van de Donk et al, 2016), resistance invariably supervenes (Lauring et al, 2008), and most patients die of their disease. Currently, major challenges include intrinsic, pre-existing high-risk disease (Usmani et al, 2015) and acquired resistance (Zhu et al, 2014; Dolloff, 2015). Recently, the International Myeloma Working Group (IMWG) developed MM risk stratification schemes that predict therapeutic responses and survival (Chng et al, 2014). For example, patients with t(11;14) translocations have a favourable prognosis (for example, survival ⩾6 yrs), whereas patients with t(4;14) or del(17p), plasma cell leukaemia (PCL), or who are PI-refractory do worse (for example, survival ⩽2 yrs), although patients with t(4;14) are currently considered intermediate risk because of bortezomib responsiveness (Palumbo et al, 2015). Consequently, more effective treatments are clearly needed for higher-risk groups.
MM, like other neoplastic diseases, exhibits deregulation of the BCL-2 family of apoptotic regulatory proteins, prompting development of BH3-mimetics against B-cell malignancies such as CLL and MM. ABT-199 (venetoclax), a selective BCL-2 inhibitor, has recently been approved for relapsed del17p CLL. In contrast to ABT-737, which inhibits both BCL-2 and BCL-XL, ABT-199 is platelet-sparing, affording a wider therapeutic index (Schoenwaelder and Jackson, 2012). However, preclinically, ABT-199 is primarily active against karyotypically lower-risk MM subtypes, for example, t(11;14) with high BCL-2/MCL-1 ratios, and less effective against subtypes with relatively low BCL-2/MCL-1 ratios, for example, t(4;14) (Gong et al, 2016), consistent with the role of MCL-1 in intrinsic resistance. Very recently, this finding has been extended to the clinic (Kumar et al, 2017; Punnoose et al, 2016). This observation raises the possibility that interventions that increase the BCL-2/MCL-1 ratio might potentiate ABT-199 activity in the setting of intrinsically resistant, high-risk MM cells.
Cell cycle deregulation in neoplastic cells spurred the development of cyclin-dependent kinase inhibitors (CDKIs), for example, the pan-CDKI flavopiridol (FP; alvocidib), the first to enter the clinic (Ambrosini et al, 2008). However, CDKIs exhibit “off-target” effects, including DNA repair disruption (Chao et al, 2000). Moreover, inhibitors of CDK7 and 9, components of the positive transcription elongation factor B, which phosphorylates the carboxy-terminal domain of RNA Pol II, repress transcription (Gojo et al, 2002). Such CDKIs downregulate multiple short-lived proteins implicated in MM cell survival and proliferation, including MCL-1 and cyclin D1 (Hofmeister et al, 2014). CDKIs have limited MM single-agent activity (Hofmeister et al, 2014), although in a phase I FP trial a confirmed minimal response was observed in a patient with high (IMWG)/intermediate (mSMART) risk t(4;14) MM (Hofmeister et al, 2014).
Previously, we reported that FP increased lethality of the BCL-2/BCL-XL inhibitor navitoclax in AML cells through a mechanism involving MCL-1 downregulation (Chen et al, 2012). However, as ABT-199 does not target BCL-XL, another determinant of ABT-199 sensitivity (Punnoose et al, 2016; Touzeau et al, 2014), it is uncertain whether it would interact similarly with FP. In addition, the activity of a transcriptional inhibitory CDKI/ABT-199 strategy has not yet been evaluated in MM models, particularly in less favourable MM subtypes intrinsically resistant to ABT-199 alone. Here we report that FP markedly enhances ABT-199 activity in multiple MM models in vitro and in vivo, as well as in primary MM cells, including those insensitive to ABT-199 alone. Our results also demonstrate that FP circumvents resistance to ABT-199 through MCL-1 downregulation and BIM upregulation, and overcomes cytoprotective microenvironmental factors.
Materials and methods
Cell lines and reagents
Human NCI-H929 (t(4;14)), U266(t(11;14)), OPM2 (t(4;14)), KMS11 (t(4;14), t(8;14) and t(14;16)), and RPMI8226 (t(14;16) and t(8;22)) cells were from ATCC and were maintained as described previously (Chen et al, 2009). Bortezomib-resistant cells (PS-R) were generated by continuously culturing U266 cells in increasing concentrations of bortezomib, beginning at 0.5– 20 nM, and maintained in a medium containing 15 nM bortezomib. Another bortezomib-resistant RPMI8226 (8226/V10R) subline was similarly established and maintained in 10 nM bortezomib. A revlimid-resistant RPMI8226 (8226/R10R) cell line was maintained in 10 μM revlimid (Dai et al, 2002). Melphalan-resistant RPMI8226 (8226/LR5) subline was maintained as before (Dai et al, 2002). KMS-28PE (t(4;14)) was purchased from the JCRB cell bank (JCRB1191). U266/MCL-1 cells were established by stably transfecting full-length human MCL-1 cDNA (Chen et al, 2009).
All experiments utilised logarithmically growing cells (3–5 × 105 cells per ml). MycoAlert (Lonza, Allendale, NJ, USA) assays were performed to demonstrate the absence of mycoplasma contamination.
ABT-199 was a gift from AbbVie (Chicago, IL, USA). The pan-CDK inhibitor FP was purchased from Selleck (Houston, TX, USA). The caspase inhibitor Z-VAD-FMK was obtained from Enzo Life Sciences Inc., Farmingdale, NY, USA. All drugs were dissolved in DMSO, aliquoted, and stored at −80 °C. Final DMSO concentrations did not exceed 0.1%.
Stromal cell co-culture of MM
The human stromal cell line HS-5 was obtained from ATCC. HS-5 cells were maintained in the RPMI1640 medium containing 10% FBS and were subcultured twice weekly by trypsinisation at a subculture ratio of 1 : 5. For co-culture experiments, HS-5 cells were cultured for 6 h before seeding MM cells.
RNA interference, CRISPR/cas9 plasmids, immunoblotting analysis, immunofluorescence, and analysis of cell death
Isolation of primary MM cells
Bone marrow samples from MM patients undergoing diagnostic aspirations at the VCU Health Massey Cancer Center were obtained with informed consent and approval of the VCU IRB (protocol #HM3340). CD138+ cells were separated using a MACS magnetic separation technique (Miltenyi Biotech, San Diego, CA, USA) as per the manufacturer’s instructions. The purity (>90%) of CD138+ and viability (>95%) were determined with flow cytometry and trypan blue exclusion, respectively. Normal CD34+ haematopoietic progenitor cells were isolated from cord blood (CB) samples similarly. Isolated cells were cultured in RPMI1640 medium containing 10% FBS.
Animal studies
All animal studies were performed under protocol AD10000035, approved by our local IACUC, and regulated by VCU’s Animal Care and Use Program, which is AAALAC-accredited (#000036), USDA-registered (#52-R-0124), and has an approved PHS Assurance on file (#A3281-01). The two models employed included an orthotopic murine model (NOD/SCID-γ mice (Jackson Laboratories) injected intravenously via tail vein with 5 × 106 PS-R (bortezomib-resistant U266) stably transfected with a construct encoding luciferase) and an immunocompetent model (C57BL/KaLwR1 mice injected intravenously via tail vein with 5 × 106 murine MM 5TGM1 cells stably expressing luciferase, provided by Dr. Babatunde O Oyajobi, UT Health San Antonio; Oyajobi et al, 2003). C57BL/KaLwRijHsd breeding pairs were originally obtained from Harlan Europe (now Envigo, Horst, Netherlands), and a specific pathogen-free colony is maintained at VCU by Dr Jolene Windle.
Treatment was administered by gavage after luciferase activity was detected. ABT-199 was formulated in 10% EtOH/30% polyethylene glycol 400/60% phosal 50 propylene glycol PG; v/v/v). FP in DMSO was diluted in 0.9% saline and administered via intraperitoneal (i.p.) injection. Control animals were injected with equal volumes of vehicle.
Mice were monitored for tumour growth with an IVIS 200 imaging system (Xenogen Corporation, Alameda, CA, USA). Measurement of animal body weight was performed Q.O.D. throughout the study to monitor toxicity.
Statistical analysis
Values represent the means±s.d. for at least three independent experiments performed in triplicate. The significance of differences between experimental variables was determined using the Student’s t-test or One-way ANOVA with Tukey–Kramer multiple comparisons test. The significance of P-values are *P<0.05, **P<0.01, or ***P<0.001 wherever indicated. Analysis of synergism was performed by Median Dose Effect analysis using the software Calcusyn (Biosoft, Ferguson, MO, USA). Kaplan–Meier analysis of mouse survival performed with GraphPad Prism 6 software (La Jolla, CA, USA).
Results
FP/ABT-199 co-administration induces apoptosis in unfavourable-risk MM cells
As reported (Touzeau et al, 2014), primary MM cells with either no FISH anomalies or favourable FISH profiles (for example, t(11;14)) underwent marked apoptosis following exposure to ABT-199 (200 nM), whereas little cell death was observed in cells with unfavourable karyotypes (del 13, del 17p; Supplementary Figure 1). Co-exposure (24 h) of intermediate risk H929 cells (t(4;14)) to minimally toxic FP concentrations (75–100 nM) significantly increased subtoxic ABT-199 concentration lethality (Figure 1A). These interactions were synergistic by median dose effect analysis with CI values <1.0 in both H929 and OPM-2 (t(4;14)) cells (Figure 1B). Combined treatment increased caspase activation and PARP cleavage in H929 cells (Figure 1C). Synergism was also observed in U266 cells, which despite the t(11;14) karyotype, are relatively insensitive to ABT-199 (Gong et al, 2016). Synergistic interactions or markedly enhanced lethality were also observed in multiple other intermediate or high-risk models (for example, KMS11(t(4;14)), OPM2(t(4;14)), RPMI8226 (t(14;16) and t(8;22)), and KMS28-PE (t(4;14); Bergsagel and Kuehl, 2001; Supplementary Figure 2).
Figure 1.

ABT-199 interacts synergistically with FP to induce apoptosis in unfavourable risk multiple myeloma cells. (A) NCI-H929 cells were incubated with ABT-199±FP for 24 h, after which cell death was analysed by flow cytometry after staining with 7-AAD. **P<0.01; (B) H929 and OPM-2 cells were exposed (24 h) to varying concentrations of FP±ABT-199 at a fixed ratio (1 : 5), after which the percentage of Annexin V+ cells was determined. Combination Index (CI) values less than 1.0 denote a synergistic interaction. The results are representative of three separate experiments; (C) H929 cells were treated with 500 nM ABT-199±100 nM FP for 24 h, after which slides were prepared and stained with antibodies against activated caspase-3, followed by Alexa Fluor 594-conjugated secondary antibody with DAPI counterstaining (left panels, × 200 magnification). Upper panel: activated caspase-3 (red); lower panel: activated caspase-3 and DAPI (blue) merged. H929 cells were incubated with ABT-199±FP for 24 h, after which cleavage of caspase-3 and PARP was monitored by immunoblotting analysis (right panel). CF=cleaved fragment. α-tubulin was assayed to ensure equivalent loading and transfer. A full colour version of this figure is available at the British Journal of Cancer journal online.
FP downregulates MCL-1 and upregulates BIM, events that contribute functionally to potentiation of ABT-199 lethality
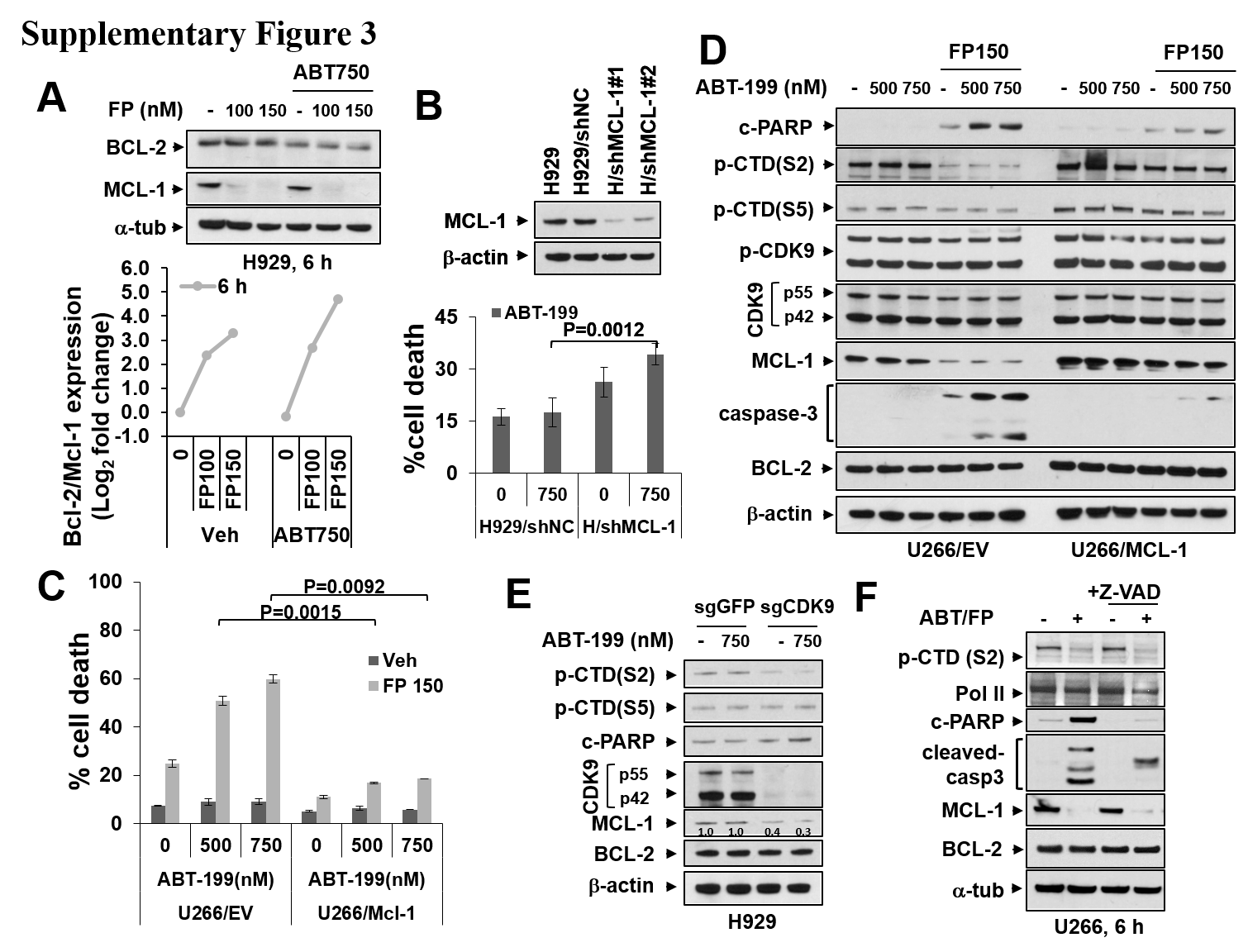
FP downregulated MCL-1 expression in ABT-199-insensitive U266 cells 6 h after exposure but had little effect on BCL-2 expression (Figure 2A, upper panel). Consequently, ratios of BCL-2 to MCL-1 protein levels were sharply increased following FP exposure (Figure 2A, lower panel). Parallel results were observed in H929 cells (Supplementary Figure 3A). To assess the functional contribution of MCL-1 expression, U266 cells transiently expressing MCL-1 shRNA were employed (Figure 2B, upper panel). U266/shMCL-1 cells were significantly more sensitive to ABT-199 than their empty-vector counterparts (Figure 2B, lower panel). Parallel results were observed in H929 cells (Supplementary Figure 3B). Conversely, U266 cells ectopically expressing MCL-1 displayed less MCL-1 downregulation after FP/ABT-199 exposure, and significantly reduced apoptosis (Supplementary Figure 3C), as well as caspase-3 cleavage (Supplementary Figure 3D). Finally, a CRISPR-Cas9 gene-editing technique was employed to target CDK9 in both U266 and H929 cells. Notably, CDK9 knockdown diminished p-CTD(S2) phosphorylation, downregulated MCL-1, and increased caspase activation following ABT-199 exposure in both U266 and H929 cells (Figure 2C and Supplementary Figure 3E). In addition, CTD phosphorylation was inhibited by FP after 12 h treatment of U266 cells (Figure 2D) and H929 cells (6 and 9 h; Supplementary Figure 4A), arguing that MCL-1 is a client of the CDK9/RNA Pol II pathway. Finally, the pan-caspase inhibitor Z-VAD-FMK blocked PARP and caspase-3 cleavage but not CTD phosphorylation or MCL-1 downregulation, arguing against the caspase dependence of MCL-1 downregulation (Supplementary Figure 3F). Collectively, these findings indicate that CDK9 inhibition and MCL-1 downregulation by FP contribute functionally to potentiation of ABT-199 lethality.
Figure 2.

FP downregulates MCL-1 expression and upregulates BIM that contributes functionally to potentiation of ABT-199 lethality. (A) U266 cells were treated with ABT-199±FP for 6 h, after which immunoblotting analysis was performed to monitor the levels of MCL-1 and BCL-2 (upper panel). The ratio of BCL-2/MCL-1 was quantified by densitometry (lower panel). The results are representative of three separate experiments; (B) U266 cells were infected with shMCL-1 lentivirus particles to target MCL-1 (shMCL-1#1 using one viral dose, shMCL-1#2 using two viral doses) or control particles (shNC) according to the manufacturer’s instructions. Following 48 h infection, MCL-1 protein levels were assessed by immunoblotting (upper panel), and cells were further treated with ABT-199 (500 and 750 nM) for further 24 h. Cell death was analysed by flow cytometry after staining with 7-AAD, with knockdown cells showing MCL-1 downregulation and significantly greater death than control cells (lower panel). The results are representative of three separate experiments; (C) U266 cells were infected with lentivirus encoding Cas9 and sgRNA targeting GFP or CDK9. Following 48 h infection, cells were treated with ABT-199 (500 and 750 nM) for 24 h. Immunoblotting analysis was carried out to monitor p-CTD(S2), p-CTD(S5), CDK9, MCL-1, BCL-2, and cleaved PARP; (D) U266 cells were incubated with varying concentrations of ABT-199±FP (150 nM) for 12 h. Immunoblot analysis was performed to monitor p-CTD(S2), p-CTD(S5), RNA Pol II, MCL-1, BCL-2, Bik, and cleaved PARP (left panel). Meanwhile, NOXA, PUMA, BMF, HRK, BCL-XL, and three isoforms (EL, L, and S) of BIM were monitored (right panel); (E) U266 cells were stably transfected with constructs encoding shRNA targeting BIM (shBIM) or scrambled sequence as a negative control (shNC). Cells were treated with ABT-199 (750 nM)±FP (150 nM) for 12 h. Immunoblot analysis was carried out to monitor the three isoforms (EL, L, and S) of BIM, p-CTD(S2), p-CTD(S5), MCL-1, BCL-2, and cleaved caspase-3 and PARP. α-tubulin or β-actin were assayed to ensure equivalent loading and transfer.
Our studies with pan-BH3-mimetics (for example, obatoclax) suggested that upregulation of BIM contributed to enhanced lethality with FP (Chen et al, 2012). Consistently, FP sharply downregulated MCL-1 in H929 cells at early intervals, for example, 6 and 9 h (Supplementary Figure 4A). Moreover, administration of FP alone or with ABT-199 triggered BIM upregulation (particularly BIM-L) in both H929 and U266 cells (Figure 2D and Supplementary Figure 4B). Notably, shRNA knockdown of BIM in U266 cells largely abrogated apoptosis (Supplementary Figure 4C) as well as PARP and caspase-3 cleavage (Figure 2E). Multiple other BCL-2 family members were monitored in response to the FP/ABT-199 regimen, including NOXA, PUMA, MBF, HRK, and BCL-XL, and BIK (Figure 2D). However, no major changes were observed in the expression of these proteins following treatment in U266 and H929 cells (Figure 2D and Supplementary Figure 4A). These findings argue that FP-mediated BIM upregulation contributes functionally to potentiation of ABT-199 lethality.
The FP-ABT-199 combination circumvents various forms of MM-related drug resistance and microenvironment-driven resistance
The activity of the FP/ABT-199 regimen was then tested against various drug-resistant MM cells. Highly bortezomib-resistant PS-R cells (Kuhn et al, 2009) were as sensitive to FP/ABT-199 as their bortezomib-sensitive U266 counterparts (Figure 3A). The FP/ABT-199 regimen was also equally effective in bortezomib-resistant V10R (Dai et al, 2002), revlimid-resistant R10R (Dai et al, 2002), and melphalan-resistant LR5 8226 (Dai et al, 2002) cells compared to their sensitive 8226 counterparts (Figure 3B; P>0.05 in each case). Of note, MCL-1 downregulation and BIM upregulation were also observed in bortezomib-resistant V10R 8226 cells (data not shown).
Figure 3.

The FP/ABT-199 regimen circumvents various forms of MM-related drug- and microenvironment-related resistance. (A) U266 or PS-R cells were incubated with 500 nM ABT-199±100 nM FP or 10 nM bortezomib (btz) for 24 h, followed by flow cytometry after staining with 7-AAD; (B) 8226 or 8266 cells resistant to bortezomib (V10R), revlimid (R10R), or melphalin (LR5) were treated with 500 nM ABT-199±100 nM FP for 24 h. Cell death was then determined by flow cytometry after staining with 7-AAD; (C) Luciferase-labelled U266 (upper panel) or PS-R (lower panel) cells co-cultured±BM stromal HS-5 cells were incubated with 750 nM ABT-199+100 nM FP or 10 nM bortezomib for 24 h. Cell viability was determined by bioluminescence assay; (D) GFP-labelled U266 (upper panel) or PS-R (lower panel) cells co-cultured±BM stromal HS-5 cells were treated with 750 nM ABT-199±100 nM FP for 24 h. Cells were stained with 7-AAD to monitor MM (GFP+) cell death (× 200 magnification). Veh, vehicle; BF, bright field. P-values indicate no significant difference between groups. The results are representative of three separate experiments. A full colour version of this figure is available at the British Journal of Cancer journal online.
HS-5 co-culture studies were performed to determine whether stromal factors ameliorated FP/ABT-199 lethality. Co-culture of luciferase-labelled U266 cells with HS-5 cells failed to prevent diminished viability following FP/ABT-199 24 h exposure (Figure 3C, upper panel). Fluorescence microscopy revealed a marked increase in red staining (7-AAD uptake) after drug treatment in GFP-labelled U266 cells (Figure 3D, upper panel). Parallel results were obtained with luciferase- or GFP-labelled bortezomib-resistant PS-R cells co-cultured with HS-5 cells (lower panels, Figure 3C and D), suggesting that the FP/ABT-199 regimen can circumvent stromal cell-related forms of resistance.
The FP/ABT-199 regimen is active against unfavourable prognosis primary cells
The ability of the FP/ABT-199 regimen to induce cell death in primary CD138+ cells was then examined. Exposure (24 h) to 75 nM FP+200 nM ABT-199 robustly induced apoptosis (green staining; activated caspase-3) in primary cells without FISH abnormalities or favourable aberrations (t(11;14); Figure 4A). It also effectively induced apoptosis in primary specimens with unfavourable risk features (for example, del17p, PCL, Figure 4B). Notably, four of five specimens were obtained from patients with relapsed/refractory disease.
Figure 4.

The FP/ABT-199 regimen is highly active against unfavourable prognosis primary MM cells. Isolated primary CD138+ cells from a patient with either no (A, left panel) or favourable (for example, t(11;14)) (A, right panel) FISH abnormalities were exposed to 200 nM ABT-199±75 nM FP for 24 h, after which slides were prepared and stained with antibody against activated caspase-3, followed by Alexa Fluor 488-conjugated secondary antibody with DAPI counterstaining (× 200 magnification). Parallel studies were performed on three samples obtained from patients with unfavourable karyotypes (del 17p; B, left and central panels) or PCL (B, right panel). FISH, fluorescence in situ hybridisation; del, deletion; PCL, plasma cell leukaemia; Dx, newly diagnosed; RR, relapsed or refractory. A full colour version of this figure is available at the British Journal of Cancer journal online.
The FP/ABT-199 regimen is active against primary CD138+ MM cells and primitive progenitor cell-enriched CD138−/CD19+/CD20+/CD27+ populations but not normal CD34+ cells
The effects of ABT-199±FP were investigated in a larger number of primary specimens (N=23). Those with unfavourable karyotypic features (Supplementary Table 1) are colour-coded in Figure 5A. Combined treatment very significantly reduced the survival of cells compared to single-agent treatment (24 h; P<0.0001 and 0.0016; Figure 5A). In the subset of specimens in which primitive progenitor cells could be identified (CD138−/CD19+/CD20+/CD127+; N=18), combined treatment significantly reduced survival (P<0.0074 and 0.0001; Figure 5B). In contrast, FP did not increase ABT-199 lethality in normal CD34+ CB cells (Figure 5C).
Figure 5.

The FP/ABT-199 regimen is active against primary CD138+ MM cells and diminishes the primitive progenitor cell enriched CD138-/CD19+/CD20+/CD27+ population while displaying minimal toxicity towards normal CD34+ cells. (A) Isolated CD138+ MM cells from a total of 23 MM patients were exposed to 200 nM ABT-199±75 nM FP for 24 h, after which the percentage of Annexin V+ cells was determined by flow cytometry. Eight patients were carrying cancer hotspot mutations (colours) as indicated in Supplementary Table 1; (B) Additional experiments were carried out with 18 of the primary MM samples. Viability of the CD138-/CD19+/CD20+/CD27+ cells population, which is enriched for MM progenitor cells, was determined by 7-AAD staining and analysed by multicolour flow cytometry; (C, left panel) A representative normal CB CD34+ cells were exposed (24 h) to 200 nM ABT-199±75 nM FP, and viability determined by flow cytometry (7-AAD/Annexin V+; C, right panel). Parallel experiments carried out with five CB CD34+ samples indicate no differences between groups. A full colour version of this figure is available at the British Journal of Cancer journal online.
The FP/ABT-199 regimen is active in an intravenous BM-homing murine model
To assess the in vivo relevance of these findings, NOD/SCID-γ mice were inoculated with fluorescently labelled PS-R cells. After luciferase signals became visible, FP (3 mg kg−1, i.p.)±ABT-199 (100 mg kg−1, p.o.) was administered daily for 7 days, followed by FP (5 mg kg−1, i.p)±ABT-199 (100 mg kg−1, p.o.) 5 days a week for an additional 4 weeks. Combined treatment sharply reduced tumour burden (Figure 6A) and significantly prolonged survival compared to single-agent treatment (P<0.0001). The median survival was 79 days for animals in the combined treatment group, vs 62, 63, and 71 days for those in vehicle, ABT-199, or FP groups, respectively (Figure 6B). Combined treatment did not induce weight loss (Figure 6C) or other toxicities, for example, behavioural changes, fur loss and so on.
Figure 6.

The FP/ABT-199 regimen is active in an intravenous BM-homing murine model. (A) NOD/SCID-γ mice were injected intravenously via tail vein with 5 × 106 PS-R cells stably expressing luciferase. After signals were visible (for example, 10 days after injection of tumour cells), FP (3 mg kg−1, i.p.)±ABT-199 (100 mg kg−1, p.o.) was administered daily for the first 7 days, followed by FP (5 mg kg−1, i.p)±ABT-199 (100 mg kg−1, p.o.) 5 days a week for an additional 4 weeks; n=5 per group. Control animals were administered equal volumes of vehicle. Tumour cells were monitored every other day after i.p. injection with 150 mg kg−1 luciferin using the IVIS 200 imaging system. Veh, vehicle; (B) Survival of the animals was determined by Kaplan–Meier analysis. Inset, median survival days. P-value indicates significant difference between groups; (C) Mice did not display significant body weight loss (⩾20% of initial weight) or other signs of toxicity due to the treatment. A full colour version of this figure is available at the British Journal of Cancer journal online.
Finally, parallel studies were performed in an immunocompetent model. C57BL/KaLwR1 mice were injected intravenously with murine MM 5TGM1 cells stably expressing luciferase. FP (3 mg kg−1, i.p.)±ABT-199 (100 mg kg−1, p.o.) were administered daily 5 days a week for 4 weeks. As shown in Supplementary Figure 5A, combined administration suppressed tumour growth, manifested by clearly diminished intensity and significantly prolonged survival compared to single agents (P<0.001, Supplementary Figure 5B), but did not lead to increased toxicity, for example, weight loss (Supplementary Figure 5C). Specifically, the median survival was 69 days for animals in the combined treatment group, vs 39, 49, and 53 days for those in vehicle, ABT-199, or FP groups, respectively (Supplementary Figure 5B; P<0.001). These findings, along with those indicating sparing of normal cells (Figure 5C), raise the possibility of selective lethality of BCL-2 and MCL-1 disruption towards MM cells.
Discussion
Apoptosis is strictly regulated by BCL-2 family members, consisting of pro- and anti-apoptotic proteins. The former contain activator (for example, BIM, PUMA, and BID) or sensitiser (for example, BAD) BH3-only proteins as well as multidomain effectors (for example, BAK and BAX; Chipuk et al, 2008), which trigger mitochondrial membrane permeabilisation, cytochrome c release, and initiator caspases (for example, caspase-3, -7, and -9) activation (Green and Kroemer, 2004). Multidomain anti-apoptotic proteins (for example, BCL-2, BCL-XL, MCL-1, A1 and so on) neutralise sensitiser, activator, and/or effector pro-apoptotic proteins. Both relative levels and direct interactions between these pro- and anti-apoptotic proteins determine cell fate (Morales et al, 2011). Apoptotic signalling pathways include the intrinsic (mitochondrial) and extrinsic (death receptor) pathways. MM has been associated with deregulation of both the intrinsic and extrinsic pathways (Oancea et al, 2004). Notably, the short-lived (2–3 h) anti-apoptotic protein MCL-1 plays a particularly important role in MM cell survival (Zhang et al, 2002), and cooperates with BCL-XL to tether/inactivate pro-apoptotic BAK. Moreover, BCL-2 family deregulation (for example, BCL-2 and MCL-1) may be particularly relevant to less favourable-risk MM.
Structure-based fragment design spurred development of BH3 mimetics, which neutralise multidomain anti-apoptotic proteins. These include the pan-BH3-mimetic obatoclax (Nguyen et al, 2007), no longer in clinical use, and the BCL-2/BCL-XL antagonist navitoclax (ABT-263; Tse et al, 2008). However, thrombocytopaenia and other toxicities have focused attention on ABT-199, a specific BCL-2 antagonist, which is relatively platelet-sparing (Souers et al, 2013). Significantly, ABT-199 has recently received FDA and EMA approval in a B-cell malignancy (del(17p) CLL; Cang et al, 2015) and breakthrough therapy designations with rituximab in relapsed CLL and in combination with 5-azacytidine in AML. Notably, ABT-737 (a navitoclax precursor) variably inhibited MM cell survival in preclinical studies (Bodet et al, 2011), although susceptibility correlated strongly with favourable prognostic subtypes (for example, t(11;14)) and high BCL-2/MCL-1 ratios (Bodet et al, 2011), reflecting BCL-2 addiction and ABT-737 inactivity against MCL-1. Despite its lack of BCL-XL-inhibitory activity, ABT-199 was as or more potent against MM cells (cultured or primary) than ABT-737 (Touzeau et al, 2014). However, like ABT-737 (Bodet et al, 2011), it exhibited activity against favourable prognosis (for example, t(11;14)) cells with high BCL-2/MCL-1 ratios, but limited activity against less favourable or intermediate prognostic category cells (for example, t(4;14)) with relatively low BCL-2/MCL-1 or BCL-XL ratios (Punnoose et al, 2016; Touzeau et al, 2014). This suggests that favourable prognosis MM (for example, t(11;14)) may represent a biomarker for ABT-199 susceptibility in MM (Touzeau et al, 2014). In fact, early clinical results reveal some single-agent activity in this setting, although initial encouraging results have been reported for ABT-199 with bortezomib/dexamethasone in non-t(11;14) MM (Kumar et al, 2016), suggesting a role for combination strategies (Hebraud et al, 2015). More recently, results from a single-agent venetoclax study revealed that patients with the t(11;14) translocation are most more likely to respond to this regimen (Kumar et al, 2017). However, as favourable prognosis subtypes are likely to respond to multiple other regimens (Albershardt et al, 2011), efforts to enhance ABT-199 activity in less favourable subtypes appear justified.
The results of the present study indicate that FP markedly increases the antimyeloma activity of ABT-199 in MM cells, including higher-risk subtypes that display limited single-agent ABT-199 sensitivity, due, at least in part, to higher MCL-1 expression. The mechanisms underlying this phenomenon seem likely to be multifactorial, including MCL-1 downregulation and BIM upregulation in cooperation with disabling of BCL-2. Furthermore, analogous mechanisms appear to be operative in bortezomib-resistant cells. In this context, a strong rationale exists for employing transcriptional repressor CDKIs, to improve the anti-MM activity of BH3 mimetics, including in less responsive subtypes. For example, we (Chen et al, 2007) and other groups (Touzeau et al, 2011) have shown that CDKIs, for example, FP, roscovitine, among others, interact synergistically with the BCL-2/XL antagonist ABT-737 in various tumour cells, including AML and NHL (Yecies et al, 2010). This process involves downregulation of MCL-1, mimicking NOXA (Gomez-Bougie et al, 2007), combined with BCL-XL inhibition, mimicking BAD, thus releasing pro-apoptotic BAK from both anti-apoptotic proteins. In addition, FP sharply increased the anti-MM activity of the pan-BH3-mimetic obatoclax both in vitro and in vivo (Chen et al, 2012). However, whether such findings could be extended to a selective, clinically relevant BCL-2 antagonist such as ABT-199, which does not target BCL-XL, particularly in MM cells intrinsically resistant to ABT-199 or to agents such as bortezomib has not been determined. In this context, potentiation of ABT-199 activity by another CDKI (dinaciclib) in a NHL model has been reported (Phillips et al, 2015). The observations that FP downregulated MCL-1 and that MCL-1 knockdown or ectopic expression reciprocally modified ABT-199 sensitivity argues that MCL-1 neutralisation plays a significant mechanistic role in the combination. This interpretation is corroborated by the finding that CDK9 knockdown phenocopied the effects of FP by diminishing MCL-1 expression and increasing ABT-199 lethality.
Aside from downregulating MCL-1, CDKIs like FP may act through other mechanisms to potentiate MM cell killing. These include upregulation of pro-apoptotic proteins (for example, BIM and Bik; Chen et al, 2014), and disruption of the cytoprotective IRE-1α arm of the unfolded protein response (UPR; Nguyen and Grant, 2014). The mechanism by which CDKIs upregulate pro-apoptotic proteins such as BIM is currently unknown. However, the ability of BIM shRNA knockdown to protect cells from FP/ABT-199 lethality supports an important functional contribution of BIM upregulation to synergistic interactions. In addition to the absolute levels of pro- and anti-apoptotic proteins like BIM and MCL-1, their subcellular distribution and interactions may also determine cell fate (Morales et al, 2011). Finally, we recently reported that CDK9 knockdown or transcriptional repressor CDKIs converted ABT-737-treated MM cells to an autophagy-inefficient state by blocking induction of the cargo-loading protein p62/SQSTM1, leading to cell death (Chen et al, 2014). As disruption of BCL-2/beclin-1 binding triggers autophagy (Marquez and Xu, 2012), the possibility exists that similar mechanisms contribute to FP/ABT-199 interactions. Studies designed to test this possibility are currently underway.
Microenvironmental factors represent an important contributor to drug resistance in numerous tumour types, including MM (Podar et al, 2009). In particular, the transcription factor Stat3 has been strongly linked to stromal cell resistance in MM (Loffler et al, 2007). Interestingly, downstream cytoprotective targets of Stat3 include BCL-2, BCL-XL, and MCL-1, among other proteins. Significantly, stromal cells (for example, HS-5 cells were unable to protect MM cells, including highly bortezomib-resistant cells, from the FP/ABT-199 regimen. Moreover, this regimen was highly active against multiple drug-resistant sublines and cells intrinsically resistant to ABT-199, for example, unfavourable-risk subtypes. Such findings suggest that mechanisms involved in FP/ABT-199 synergism are potentially relevant to multiple forms of intrinsic and adaptive drug resistance.
Importantly, the FP/ABT-199 regimen was active against primary CD138+ cells, including those expressing higher-risk features and which displayed minimal sensitivity to ABT-199 alone. In marked contrast, identical regimens were relatively non-toxic to normal haematopoietic cells. The basis for this selectivity is uncertain, but may reflect the addiction of MM cells to MCL-1 for survival (De Veirman et al, 2015). In any event, FP/ABT-199 regimen activity in both immune-deficient and immunocompetent MM models with minimal toxicity argues that this strategy may preferentially target MM cells.
In summary, the present results indicate that the CDKI FP sharply increases ABT-199 killing of MM cells, including those with less favourable prognostic features exhibiting minimal sensitivity to ABT-199 alone. This regimen involves downregulation of MCL-1 accompanied by BIM upregulation, is active in the presence of multiple resistance mechanisms (including stroma-mediated), and targets primary MM cells but not normal haematopoietic cells. Importantly, the regimen is well tolerated and significantly improves survival in several preclinical animal MM models. Given the promise of ABT-199 in another B-cell malignancy (CLL; Bojarczuk et al, 2016), efforts to improve its efficacy in MM, particularly higher-risk MM, through CDK9 inhibition appear warranted.
Acknowledgments
This work was supported by awards P50 CA142509-01 to SG, RZO, and YD, and CA100866, UH2TR001373 and CA167708 to SG from the National Institutes of Health; Plasmid preparation was performed at the VCU Macromolecule Core Facility, supported, in part, with funding from NIH-NCI Cancer Center Core Grant 5P30CA016059-29. Services in support of the research project were generated by the VCU Massey Cancer Center Transgenic/Knockout Mouse Shared Resource, supported, in part, with funding from NIH-NCI Cancer Center Support Grant P30 CA016059. We also want to thank Lora Kramer for help in preparing the manuscript.
Author contributions
LZ and YZ designed and performed research, and analysed data. MK performed research. DS, JL, RZO, and YD helped design research. SG, LZ, and YZ designed research, analysed data, and wrote the manuscript.
Footnotes
Supplementary Information accompanies this paper on British Journal of Cancer website (http://www.nature.com/bjc)
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- Albershardt TC, Salerni BL, Soderquist RS, Bates DJ, Pletnev AA, Kisselev AF, Eastman A (2011) Multiple BH3 mimetics antagonize antiapoptotic MCL1 protein by inducing the endoplasmic reticulum stress response and up-regulating BH3-only protein NOXA. J Biol Chem 286(28): 24882–24895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosini G, Seelman SL, Qin LX, Schwartz GK (2008) The cyclin-dependent kinase inhibitor flavopiridol potentiates the effects of topoisomerase I poisons by suppressing Rad51 expression in a p53-dependent manner. Cancer Res 68(7): 2312–2320. [DOI] [PubMed] [Google Scholar]
- Bergsagel PL, Kuehl WM (2001) Chromosome translocations in multiple myeloma. Oncogene 20(40): 5611–5622. [DOI] [PubMed] [Google Scholar]
- Bodet L, Gomez-Bougie P, Touzeau C, Dousset C, Descamps G, Maiga S, Avet-Loiseau H, Bataille R, Moreau P, Le Gouill S, Pellat-Deceunynck C, Amiot M (2011) ABT-737 is highly effective against molecular subgroups of multiple myeloma. Blood 118(14): 3901–3910. [DOI] [PubMed] [Google Scholar]
- Bojarczuk K, Sasi BK, Gobessi S, Innocenti I, Pozzato G, Laurenti L, Efremov DG (2016) BCR signaling inhibitors differ in their ability to overcome Mcl-1-mediated resistance of CLL B cells to ABT-199. Blood 127(25): 3192–3201. [DOI] [PubMed] [Google Scholar]
- Cang S, Iragavarapu C, Savooji J, Song Y, Liu D (2015) ABT-199 (venetoclax) and BCL-2 inhibitors in clinical development. J Hematol Oncol 8: 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao SH, Fujinaga K, Marion JE, Taube R, Sausville EA, Senderowicz AM, Peterlin BM, Price DH (2000) Flavopiridol inhibits P-TEFb and blocks HIV-1 replication. J Biol Chem 275(37): 28345–28348. [DOI] [PubMed] [Google Scholar]
- Chen S, Dai Y, Harada H, Dent P, Grant S (2007) Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res 67(2): 782–791. [DOI] [PubMed] [Google Scholar]
- Chen S, Dai Y, Pei XY, Grant S (2009) Bim upregulation by histone deacetylase inhibitors mediates interactions with the Bcl-2 antagonist ABT-737: evidence for distinct roles for Bcl-2, Bcl-xL, and Mcl-1. Mol Cell Biol 29(23): 6149–6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Dai Y, Pei XY, Myers J, Wang L, Kramer LB, Garnett M, Schwartz DM, Su F, Simmons GL, Richey JD, Larsen DG, Dent P, Orlowski RZ, Grant S (2012) CDK inhibitors upregulate BH3-only proteins to sensitize human myeloma cells to BH3 mimetic therapies. Cancer Res 72(16): 4225–4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Zhou L, Zhang Y, Leng Y, Pei XY, Lin H, Jones R, Orlowski RZ, Dai Y, Grant S (2014) Targeting SQSTM1/p62 induces cargo loading failure and converts autophagy to apoptosis via NBK/Bik. Mol Cell Biol 34(18): 3435–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipuk JE, Fisher JC, Dillon CP, Kriwacki RW, Kuwana T, Green DR (2008) Mechanism of apoptosis induction by inhibition of the anti-apoptotic BCL-2 proteins. Proc Natl Acad Sci USA 105(51): 20327–20332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chng WJ, Dispenzieri A, Chim CS, Fonseca R, Goldschmidt H, Lentzsch S, Munshi N, Palumbo A, Miguel JS, Sonneveld P, Cavo M, Usmani S, Durie BG, Avet-Loiseau H International Myeloma Working G (2014) IMWG consensus on risk stratification in multiple myeloma. Leukemia 28(2): 269–277. [DOI] [PubMed] [Google Scholar]
- Dai Y, Landowski TH, Rosen ST, Dent P, Grant S (2002) Combined treatment with the checkpoint abrogator UCN-01 and MEK1/2 inhibitors potently induces apoptosis in drug-sensitive and -resistant myeloma cells through an IL-6-independent mechanism. Blood 100(9): 3333–3343. [DOI] [PubMed] [Google Scholar]
- De Veirman K, Van Ginderachter JA, Lub S, De Beule N, Thielemans K, Bautmans I, Oyajobi BO, De Bruyne E, Menu E, Lemaire M, Van Riet I, Vanderkerken K, Van Valckenborgh E (2015) Multiple myeloma induces Mcl-1 expression and survival of myeloid-derived suppressor cells. Oncotarget 6(12): 10532–10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimopoulos MA, Leleu X, Palumbo A, Moreau P, Delforge M, Cavo M, Ludwig H, Morgan GJ, Davies FE, Sonneveld P, Schey SA, Zweegman S, Hansson M, Weisel K, Mateos MV, Facon T, Miguel JF (2014) Expert panel consensus statement on the optimal use of pomalidomide in relapsed and refractory multiple myeloma. Leukemia 28(8): 1573–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolloff NG (2015) Emerging therapeutic strategies for overcoming proteasome inhibitor resistance. Adv Cancer Res 127: 191–226. [DOI] [PubMed] [Google Scholar]
- Gojo I, Zhang B, Fenton RG (2002) The cyclin-dependent kinase inhibitor flavopiridol induces apoptosis in multiple myeloma cells through transcriptional repression and down-regulation of Mcl-1. Clin Cancer Res 8(11): 3527–3538. [PubMed] [Google Scholar]
- Gomez-Bougie P, Wuilleme-Toumi S, Menoret E, Trichet V, Robillard N, Philippe M, Bataille R, Amiot M (2007) Noxa up-regulation and Mcl-1 cleavage are associated to apoptosis induction by bortezomib in multiple myeloma. Cancer Res 67(11): 5418–5424. [DOI] [PubMed] [Google Scholar]
- Gong JN, Khong T, Segal D, Yao Y, Riffkin CD, Garnier JM, Khaw SL, Lessene G, Spencer A, Herold MJ, Roberts AW, Huang DC (2016) Hierarchy for targeting pro-survival BCL2 family proteins in multiple myeloma: pivotal role of MCL1. Blood 128(14): 1834–1844. [DOI] [PubMed] [Google Scholar]
- Green DR, Kroemer G (2004) The pathophysiology of mitochondrial cell death. Science 305(5684): 626–629. [DOI] [PubMed] [Google Scholar]
- Hebraud B, Magrangeas F, Cleynen A, Lauwers-Cances V, Chretien ML, Hulin C, Leleu X, Yon E, Marit G, Karlin L, Roussel M, Stoppa AM, Belhadj K, Voillat L, Garderet L, Macro M, Caillot D, Mohty M, Facon T, Moreau P, Attal M, Munshi N, Corre J, Minvielle S, Avet-Loiseau H (2015) Role of additional chromosomal changes in the prognostic value of t(4;14) and del(17p) in multiple myeloma: the IFM experience. Blood 125(13): 2095–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmeister CC, Poi M, Bowers MA, Zhao W, Phelps MA, Benson DM, Kraut EH, Farag S, Efebera YA, Sexton J, Lin TS, Grever M, Byrd JC (2014) A phase I trial of flavopiridol in relapsed multiple myeloma. Cancer Chemother Pharmacol 73(2): 249–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn DJ, Hunsucker SA, Chen Q, Voorhees PM, Orlowski M, Orlowski RZ (2009) Targeted inhibition of the immunoproteasome is a potent strategy against models of multiple myeloma that overcomes resistance to conventional drugs and nonspecific proteasome inhibitors. Blood 113(19): 4667–4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Kaufman JL, Gasparetto C, Mikhael J, Vij R, Pegourie B, Benboubker L, Facon T, Amiot M, Moreau P, Punnoose EA, Alzate S, Dunbar M, Xu T, Agarwal SK, Enschede SH, Leverson JD, Ross JA, Maciag PC, Verdugo M, Touzeau C (2017) Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood 130(22): 2401–2409. [DOI] [PubMed] [Google Scholar]
- Kumar S, Vij R, Kaufman JL, Mikhael J, Facon T, Pegourie B, Benboubker L, Gasparetto C, Amiot M, Moreau P, Alzate S (2016) Venetoclax monotherapy for relapsed/refractory multiple myeloma: safety and efficacy results from a phase I study. Blood (Abstract) 128: 488. [Google Scholar]
- Lauring J, Abukhdeir AM, Konishi H, Garay JP, Gustin JP, Wang Q, Arceci RJ, Matsui W, Park BH (2008) The multiple myeloma associated MMSET gene contributes to cellular adhesion, clonogenic growth, and tumorigenicity. Blood 111(2): 856–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loffler D, Brocke-Heidrich K, Pfeifer G, Stocsits C, Hackermuller J, Kretzschmar AK, Burger R, Gramatzki M, Blumert C, Bauer K, Cvijic H, Ullmann AK, Stadler PF, Horn F (2007) Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood 110(4): 1330–1333. [DOI] [PubMed] [Google Scholar]
- Marquez RT, Xu L (2012) Bcl-2:Beclin 1 complex: multiple, mechanisms regulating autophagy/apoptosis toggle switch. Am J Cancer Res 2(2): 214–221. [PMC free article] [PubMed] [Google Scholar]
- Morales AA, Kurtoglu M, Matulis SM, Liu J, Siefker D, Gutman DM, Kaufman JL, Lee KP, Lonial S, Boise LH (2011) Distribution of Bim determines Mcl-1 dependence or codependence with Bcl-xL/Bcl-2 in Mcl-1-expressing myeloma cells. Blood 118(5): 1329–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchtar E, Gatt ME, Rouvio O, Ganzel C, Chubar E, Suriu C, Tadmor T, Shevetz O, Lavi N, Shochat T, Cohen YC, Avivi I, Raanani P, Magen H (2016) Efficacy and safety of salvage therapy using Carfilzomib for relapsed or refractory multiple myeloma patients: a multicentre retrospective observational study. Br J Haematol 172(1): 89–96. [DOI] [PubMed] [Google Scholar]
- Nguyen M, Marcellus RC, Roulston A, Watson M, Serfass L, Murthy Madiraju SR, Goulet D, Viallet J, Belec L, Billot X, Acoca S, Purisima E, Wiegmans A, Cluse L, Johnstone RW, Beauparlant P, Shore GC (2007) Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci USA 104(49): 19512–19517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TK, Grant S (2014) Dinaciclib (SCH727965) inhibits the unfolded protein response through a CDK1- and 5-dependent mechanism. Mol Cancer Ther 13(3): 662–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oancea M, Mani A, Hussein MA, Almasan A (2004) Apoptosis of multiple myeloma. Int J Hematol 80(3): 224–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyajobi BO, Franchin G, Williams PJ, Pulkrabek D, Gupta A, Munoz S, Grubbs B, Zhao M, Chen D, Sherry B, Mundy GR (2003) Dual effects of macrophage inflammatory protein-1alpha on osteolysis and tumor burden in the murine 5TGM1 model of myeloma bone disease. Blood 102(1): 311–319. [DOI] [PubMed] [Google Scholar]
- Palumbo A, Avet-Loiseau H, Oliva S, Lokhorst HM, Goldschmidt H, Rosinol L, Richardson P, Caltagirone S, Lahuerta JJ, Facon T, Bringhen S, Gay F, Attal M, Passera R, Spencer A, Offidani M, Kumar S, Musto P, Lonial S, Petrucci MT, Orlowski RZ, Zamagni E, Morgan G, Dimopoulos MA, Durie BG, Anderson KC, Sonneveld P, San Miguel J, Cavo M, Rajkumar SV, Moreau P (2015) Revised international staging system for multiple myeloma: a report from international myeloma working group. J Clin Oncol 33(26): 2863–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips DC, Xiao Y, Lam LT, Litvinovich E, Roberts-Rapp L, Souers AJ, Leverson JD (2015) Loss in MCL-1 function sensitizes non-Hodgkin's lymphoma cell lines to the BCL-2-selective inhibitor venetoclax (ABT-199). Blood Cancer J 5: e368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podar K, Chauhan D, Anderson KC (2009) Bone marrow microenvironment and the identification of new targets for myeloma therapy. Leukemia 23(1): 10–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punnoose EA, Leverson JD, Peale F, Boghaert ER, Belmont LD, Tan N, Young A, Mitten M, Ingalla E, Darbonne WC, Oleksijew A, Tapang P, Yue P, Oeh J, Lee L, Maiga S, Fairbrother WJ, Amiot M, Souers AJ, Sampath D (2016) Expression profile of BCL-2, BCL-XL, and MCL-1 predicts pharmacological response to the BCL-2 selective antagonist venetoclax in multiple myeloma models. Mol Cancer Ther 15(5): 1132–1144. [DOI] [PubMed] [Google Scholar]
- Schoenwaelder SM, Jackson SP (2012) Bcl-xL-inhibitory BH3 mimetics (ABT-737 or ABT-263) and the modulation of cytosolic calcium flux and platelet function. Blood 119(5): 1320–1321, author reply 1321-2. [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A (2016) Cancer statistics, 2016. CA Cancer J Clin 66(1): 7–30. [DOI] [PubMed] [Google Scholar]
- Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DC, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, Lee J, Maecker HL, Marsh KC, Mason KD, Mitten MJ, Nimmer PM, Oleksijew A, Park CH, Park CM, Phillips DC, Roberts AW, Sampath D, Seymour JF, Smith ML, Sullivan GM, Tahir SK, Tse C, Wendt MD, Xiao Y, Xue JC, Zhang H, Humerickhouse RA, Rosenberg SH, Elmore SW (2013) ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med 19(2): 202–208. [DOI] [PubMed] [Google Scholar]
- Touzeau C, Dousset C, Bodet L, Gomez-Bougie P, Bonnaud S, Moreau A, Moreau P, Pellat-Deceunynk C, Amiot M, Le Gouill S (2011) ABT-737 induces apoptosis in mantle cell lymphoma cells with a Bcl-2high/Mcl-1low profile and synergizes with other antineoplastic agents. Clin Cancer Res 17(18): 5973–5981. [DOI] [PubMed] [Google Scholar]
- Touzeau C, Dousset C, Le Gouill S, Sampath D, Leverson JD, Souers AJ, Maiga S, Bene MC, Moreau P, Pellat-Deceunynck C, Amiot M (2014) The Bcl-2 specific BH3 mimetic ABT-199: a promising targeted therapy for t(11;14) multiple myeloma. Leukemia 28(1): 210–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, Roberts L, Tahir SK, Xiao Y, Yang X, Zhang H, Fesik S, Rosenberg SH, Elmore SW (2008) ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res 68(9): 3421–3428. [DOI] [PubMed] [Google Scholar]
- Usmani SZ, Rodriguez-Otero P, Bhutani M, Mateos MV, Miguel JS (2015) Defining and treating high-risk multiple myeloma. Leukemia 29(11): 2119–2125. [DOI] [PubMed] [Google Scholar]
- van de Donk NW, Moreau P, Plesner T, Palumbo A, Gay F, Laubach JP, Malavasi F, Avet-Loiseau H, Mateos MV, Sonneveld P, Lokhorst HM, Richardson PG (2016) Clinical efficacy and management of monoclonal antibodies targeting CD38 and SLAMF7 in multiple myeloma. Blood 127(6): 681–695. [DOI] [PubMed] [Google Scholar]
- Yecies D, Carlson NE, Deng J, Letai A (2010) Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood 115(16): 3304–3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Gojo I, Fenton RG (2002) Myeloid cell factor-1 is a critical survival factor for multiple myeloma. Blood 99(6): 1885–1893. [DOI] [PubMed] [Google Scholar]
- Zhu YX, Braggio E, Shi CX, Kortuem KM, Bruins LA, Schmidt JE, Chang XB, Langlais P, Luo M, Jedlowski P, LaPlant B, Laumann K, Fonseca R, Bergsagel PL, Mikhael J, Lacy M, Champion MD, Stewart AK (2014) Identification of cereblon-binding proteins and relationship with response and survival after IMiDs in multiple myeloma. Blood 124(4): 536–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.