

Abstract
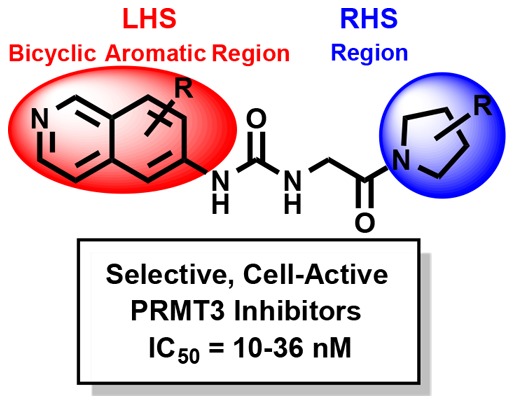
PRMT3 catalyzes the asymmetric dimethylation of arginine residues of various proteins. It is crucial for maturation of ribosomes and has been implicated in several diseases. We recently disclosed a highly potent, selective, and cell-active allosteric inhibitor of PRMT3, compound 4. Here, we report comprehensive structure–activity relationship studies that target the allosteric binding site of PRMT3. We conducted design, synthesis, and evaluation of novel compounds in biochemical, selectivity, and cellular assays that culminated in the discovery of 4 and other highly potent (IC50 values: ∼10–36 nM), selective, and cell-active allosteric inhibitors of PRMT3 (compounds 29, 30, 36, and 37). In addition, we generated compounds that are very close analogs of these potent inhibitors but displayed drastically reduced potency as negative controls (compounds 49–51). These inhibitors and negative controls are valuable chemical tools for the biomedical community to further investigate biological functions and disease associations of PRMT3.
Introduction
Protein arginine methyltransferase 3 (PRMT3) is a type I PRMT that catalyzes mono- and asymmetric dimethylation of arginine residues.1 Ribosomal protein S2 (rpS2) was identified as the major substrate of PRMT3 via its interaction with PRMT3 zinc finger domain in mammalian cells.2,3 PRMT3 plays a role in ribosome biosynthesis. However, the molecular mechanism by which PRMT3 influences ribosomal biosynthesis remains unclear.4 Very recently, an extraribosomal complex comprising PRMT3, rpS2, and human programmed cell-death 2-like (PDCD2L) protein was identified.5 While PRMT3 is localized exclusively in the cytoplasm,6 it has been shown that in cells treated with palmitic acid or T0901317 (a liver X receptor α (LXRα) agonist), PRMT3 colocalizes with LXRα in the cell nucleus, regulating hepatic lipogenesis.7 However, this effect appears to be independent of the PRMT3 methyltransferase activity. While rpS2 is the primary substrate of PRMT3, it is not the sole substrate. PRMT3 along with PRMT1 methylates the recombinant mammalian nuclear poly(A)-binding protein (PABPN1) and has been implicated in oculopharyngeal muscular dystrophy, which is caused by polyalanine expansion in PABPN1.8,9 A protein complex comprising the von Hippel–Lindau (VHL) tumor suppressor protein, PRMT3, and ARF (alternative reading frame) methylates p53.10 Importantly, the tumor suppressor DAL-1 (differentially expressed in adenocarcinoma of the lung, also known as 4.1B) interacts with PRMT3 and consequently inhibits its methyltransferase activity, suggesting a possible role of PRMT3 regulation in tumor growth.11 The interaction between DAL-1 and PRMT3 in the induction of apoptosis in MCF-7 cells suggests that this interaction is likely to be an important modulator of the apoptotic pathway and can be critical to controlling tumorigenesis in breast cancer cells.12 It has also been shown that PRMT3 methylates a histone peptide (H4 1–24) in vitro.13 Modified histone H4R3 is associated with increased transcription of a number of genes, including those under control of estrogen and androgen receptors.14−16 Furthermore, PRMT3 expression levels are elevated in myocardial tissues from patients with atherosclerosis, potentially implicating the involvement of PRMT3.17 Additionally, PRMT3 function has been reported to be essential for dendritic spine maturation in rats.18 A recent study suggests that PRMT3 mediates the preventive effects of irisin against lipogenesis and oxidative stress.19
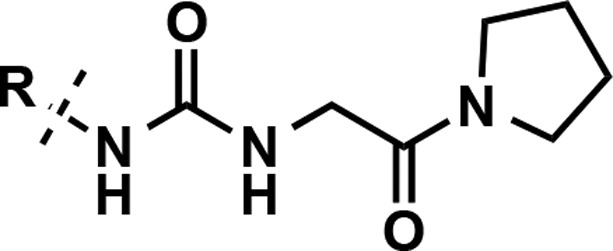
Our group embarked on research efforts to discover potent, selective, and cell-active inhibitors of PRMT3 as chemical tools for better understanding of biology and function of this understudied protein methyltransferase. We previously reported the discovery of a novel allosteric binding site of PRMT3 and the first selective, allosteric inhibitors of PRMT3 (compounds 1–3, Figure 1) and subsequently disclosed the discovery of SGC707 (4) (Figure 1), a highly potent, selective, and cell-active allosteric inhibitor of PRMT3.20−22 Here, we describe the design and synthesis of a large set of novel analogs and evaluation of these compounds in biochemical, selectivity, and cellular assays. This comprehensive structure–activity relationship (SAR) study resulted in the identification of multiple highly potent, selective, and cell-active allosteric inhibitors of PRMT3 including compound 4.
Figure 1.

Design and synthesis of highly potent inhibitors of PRMT3.
Results and Discussion
Our earlier efforts resulted in the identification of selective small-molecule inhibitors of PRMT3 (compounds 2 and 3) starting from a hit, compound 1 (Figure 1).20,21 X-ray crystal structures of 1 and 2 in complex with PRMT3 were obtained and showed that these inhibitors occupied a novel allosteric binding site (PDB ID: 3SMQ and 4HSG). These cocrystal structures revealed that the left-hand side (LHS) bicyclic benzothiadiazole moiety fits tightly in the allosteric pocket and the middle nitrogen atom forms a hydrogen bond with T466 (Figure 2). The urea linker is located at the entrance of the cavity and forms hydrogen bonds with the guanidine of R396 and the carboxylate of E422 (Figure 2). In addition, the right-hand side (RHS) moiety extends out of the allosteric binding pocket and makes hydrophobic interactions with a surface composed of the side chains from two different subunits of the PRMT3 homodimer. Both structural and SAR data clearly indicate that the middle urea region is essential for the affinity, and replacement of this group with its bioisosteres and more rigid analogs did not yield any inhibitors with improved potency.21 Therefore, in the current study the middle urea region of this scaffold was kept unmodified. However, further optimization of the RHS moiety led to the discovery of 3 (Figure 1).21 Herein, we further optimized both the LHS and RHS moieties of this scaffold to achieve improved potency for inhibiting PRMT3. First, we conducted a scaffold hopping exercise by using the benzothiadiazole ring of 3 as a query for the allosteric pocket. A hydrogen-bond constraint was imposed in this scaffold hopping study to preserve the important hydrogen-bond interaction between the inhibitors and the hydroxyl group of T466. As a result of this exercise,22 isoquinoline (compound 5), isobenzofuran-1-one (compound 6), and quinazoline (compound 7) groups were selected for experimental validation (Table 1).
Figure 2.
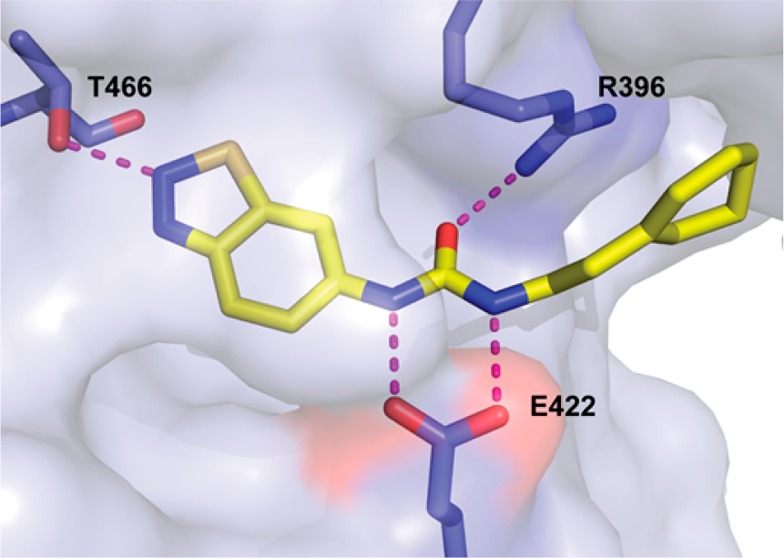
Reported cocrystal structure of compound 1 in complex with PRMT3 indicating key hydrogen bonding interactions (magenta dotted lines) (PDB ID: 3SMQ).20
Table 1. Inhibitors with Different LHS Bicyclic Ring Systems.
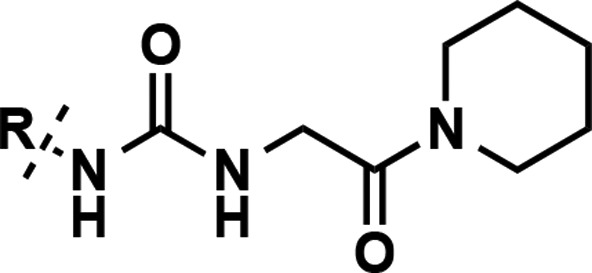
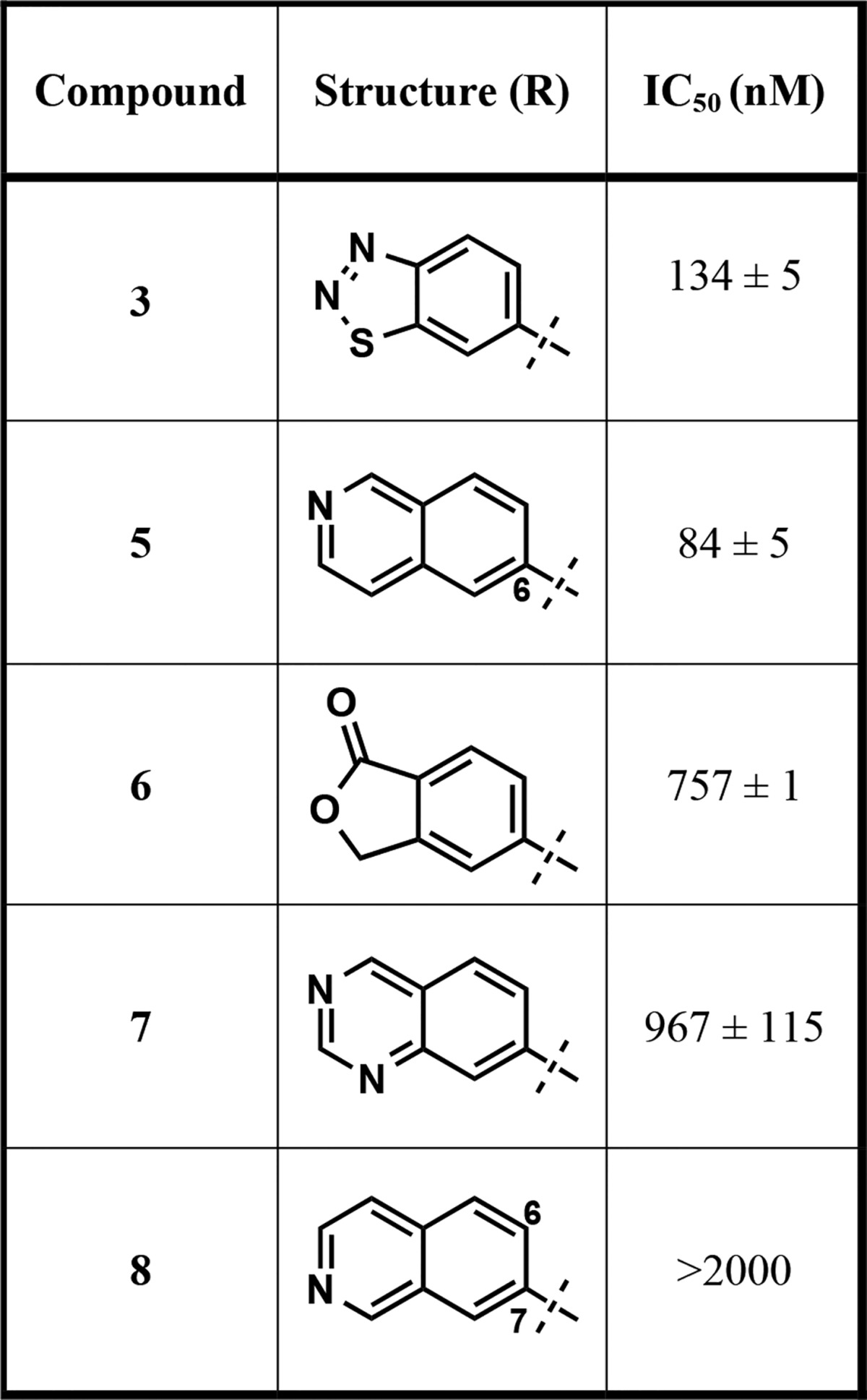
The RHS of compound 3, piperidineamide, was kept exactly the same in these newly synthesized analogs to accurately compare the effect of the different LHS bicyclic heteroaromatic rings on potency. The isoquinoline containing analog (compound 5) with IC50 of 84 ± 5 nM showed a small improvement over the parent compound, 3 (IC50 = 134 ± 5 nM). However, compounds 6 and 7 were around 6-fold less potent as compared to 3 (Table 1). Therefore, the isoquinoline bicyclic ring system was taken forward as the LHS moiety for further optimization. Compound 8 (Table 1) was also prepared to confirm the importance of the positioning and hydrogen bonding of the isoquinoline nitrogen with T466. This bicyclic ring is still an isoquinoline but substituted at the 7-position instead of the 6-position (isoquinoline numbering) in effect walking the nitrogen to the adjacent position of compound 5. As expected, this modification resulted in ablation of inhibitory activity.
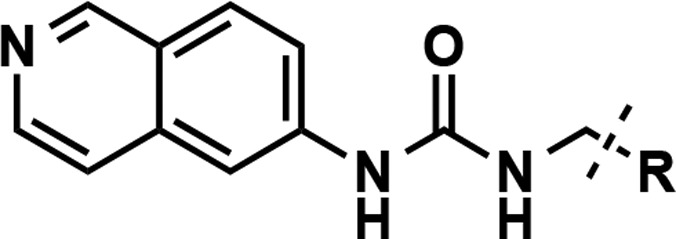
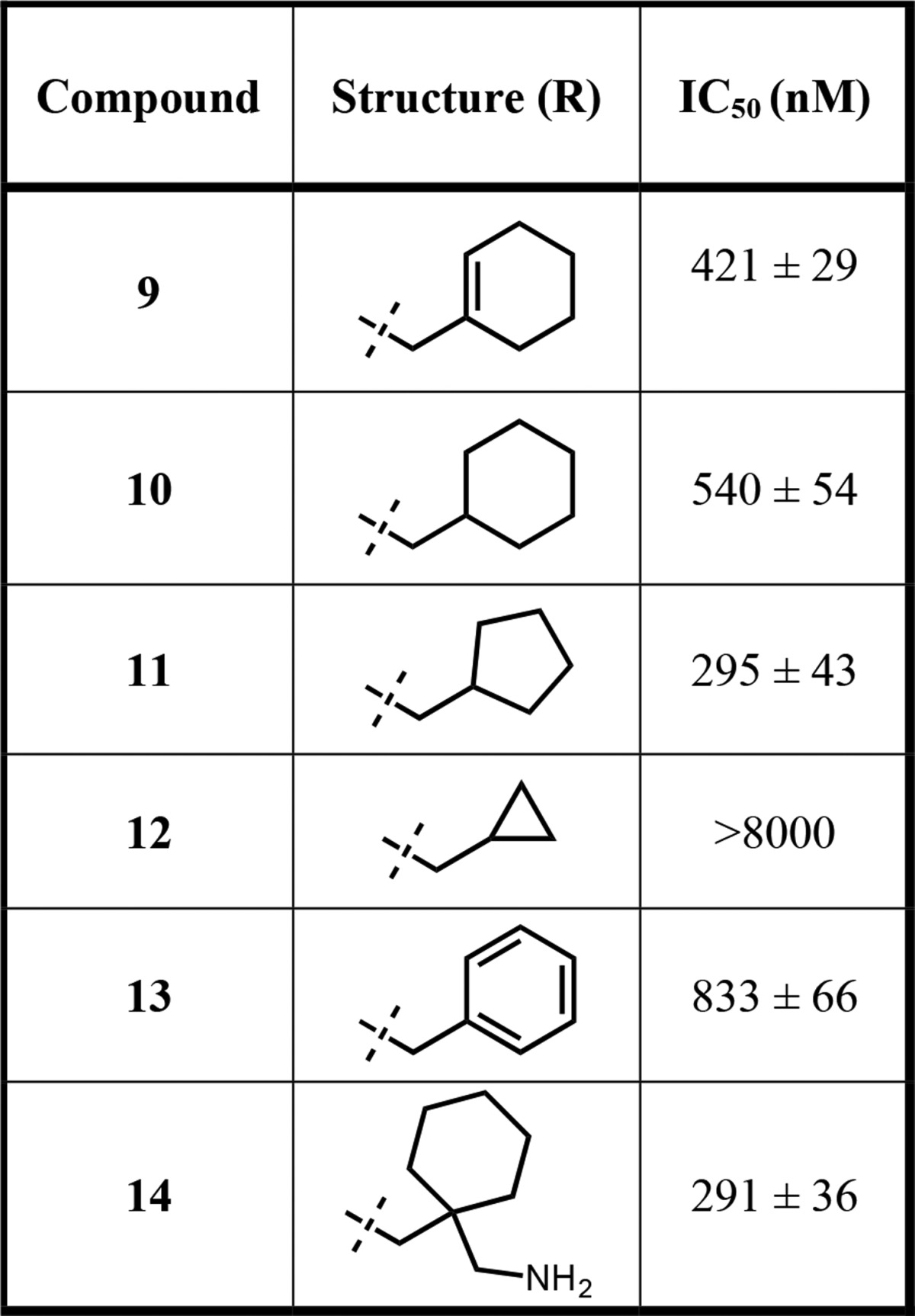
As the 6-substituted isoquinoline is an optimal LHS bicycle, we then turned our attention to optimizing the RHS moiety of the scaffold. We first revisited saturated aliphatic groups as the RHS functionality inspired from our initial studies (Table 2).20,21 Compound 9, a cyclohexenylethyl group containing analog, corresponding to the RHS region of the original hit (compound 1) was 5-fold less potent than 5, displaying IC50 of 421 ± 29 nM. As a logical extension of this compound, we also synthesized the fully saturated, cyclohexylethyl analog (10) as well as derivatives containing different ring sizes. Compound 10 (IC50 = 540 ± 54 nM) was very similar to 9 in potency. The cyclopentane-bearing analog (11) showed slight improvement in potency with IC50 of 295 ± 43 nM, while the cyclopropane-containing analog 12 was virtually inactive (IC50 > 8000 nM). These results indicated that hydrophobicity of the RHS moiety plays an important role in potency, and cyclohexyl and cylopentyl rings are preferred compared to the cyclopropyl group. Replacing the cyclohexyl group with a phenyl ring (compound 13) resulted in a decreased potency (IC50 = 833 ± 66 nM), underlying the importance of changes to the hydrophobicity of the RHS moiety. On the basis of these results, compound 10 was further investigated to improve the potency. A docking study with this compound hinted that a substituent at the C1 position of the cyclohexyl group would be a favorable position from which E422 might be possibly reached for further interactions. Therefore, compound 14, featuring a methyleneamine substituted cyclohexyl group, was synthesized. Interestingly, compound 14 displayed a modest improvement in potency with IC50 of 291 ± 36 nM, compared to compound 10. These compounds were synthesized following the previously published synthetic route.21
Table 2. Inhibitors with Saturated Aliphatic Groups as the RHS Moiety.
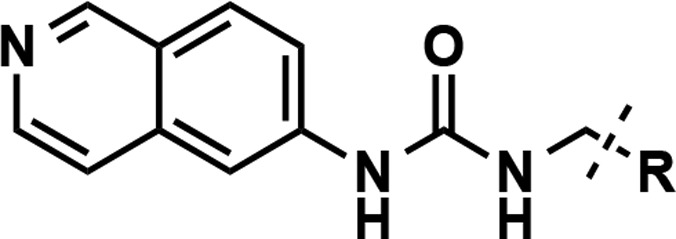
Since no significant improvement in potency was achieved with compounds 9–14 featuring aliphatic groups as the RHS moiety, we decided to keep the amide functionality as in compound 5 and further investigate the substituents on the nitrogen atom (Table 3) as the amide group could form direct or water-mediated contacts with the side chain of K392.21 We first synthesized new amide derivatives of compound 5, by replacing the six-membered piperidine ring with the four-membered azetidine ring (compound 15), five-membered pyrrolidine ring (compound 4), and seven-membered azepane ring (compound 16). While the azetidine amide analog 15 displayed only a slight improvement in potency (IC50 = 61 ± 8 nM), the pyrrolidine amide 4 (IC50 = 19 ± 1 nM) and azepane amide 16 (IC50 = 17 ± 2 nM) were ∼5-fold more potent than 5 in inhibiting PRMT3. The six-membered ring analogs of 5 such as 4,4-difluoro piperidine amide 17 (IC50 = 35 ± 1 nM) showed a more than 2-fold improvement over the unsubstituted piperidine amide 5. However, exchanging the piperidine ring with the 4-methylpiperizine ring (compound 18), which alters the electronic nature and polarity of the ring system, resulted in the loss of the inhibitory activity. We also designed and synthesized noncyclic di- and monosubstituted amide derivatives (compounds 19–26). The dimethyl and diethyl amide derivatives 19 and 20 displayed reduced potency with IC50 of 150 ± 11 and 114 ± 3 nM, respectively. The N-cyclopentyl, N-methyl amide 21 (IC50 = 87 ± 5 nM) was as potent as compound 5. The Weinreb amide derivative 22, however, was around 6-fold less potent than compound 5. The monosubstituted amides were also investigated (compounds 23–26). While N-methyl amide 23 displayed significantly weaker inhibitory effect, the cyclopropyl (24), cyclopentyl (25), and cyclohexyl (26) amide derivatives showed reduced potency as the ring size increased. Taken together, these results suggest that cyclic amides are preferred RHS moieties, which possess balanced steric and hydrophobic properties interacting with PRMT3. We, therefore, designated the pyrrolidine amide as the RHS moiety for the rest of the SAR study.
Table 3. Inhibitors Containing Different RHS Amide Moieties.

The compounds in Table 3 were synthesized via the general route shown in Scheme 1. The synthesis started with reacting commercially available 6-aminoisoquinoline and ethyl isocyanatoacetate to obtain the desired ethyl ester 27. The hydrolysis of the ethyl ester 27 resulted in the carboxylic acid 28, which was used as the key intermediate to perform amide coupling reactions with various amines to yield the desired amide analogs (4, 5, and 15–26). Detailed reaction conditions, yields, and characterization data for final compounds are reported in the Experimental Section.
Scheme 1. General Synthetic Route for the Preparation of Compounds Listed in Tables 3 and 4.

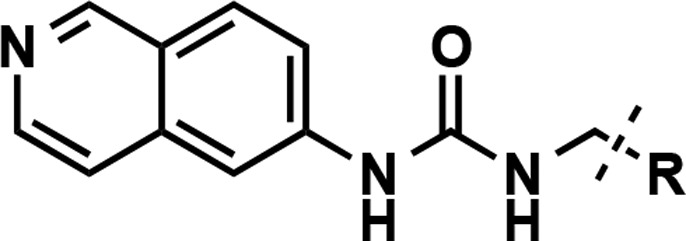
Next, we focused our attention on analogs of compound 4 (Table 4), containing substituted pyrrolidine (compounds 29–31) and fused bicyclic (compounds 32 and 33) and bridged bicyclic moieties (compounds 34 and 35). These compounds were again prepared according to the synthetic route outlined in Scheme 1. The 3,3,4,4-tetrafluoro pyrrolidine derivative (compound 29) as well as 2,5-dimethylpyrrolidine (a mixture of cis and trans isomers) (compound 30) analog showed very similar potency compared to compound 4. However, replacing pyrrolidine with l-proline (compound 31) resulted in around 9-fold drop in potency. The fused 5,5-bicyclic ring system (compound 32) was also tolerated, albeit with reduced potency (IC50 = 55 ± 5 nM). Interestingly, the benzene-fused pyrrolidine ring (isoindoline amide, compound 33) led to a complete loss of the inhibitory effect. Finally, 7-azabicyclo[2.2.1]heptane (compound 34) and 8-azabicyclo[3.2.1]octane (compound 35), two bridged bicyclic pyrrolidine containing moieties, did not result in any improvement of potency compared to compound 4, with IC50 of 46 ± 5 and 22 ± 4 nM, respectively. These results indicate that small substituents such as fluoro and methyl groups on the pyrrolidine ring as well as relatively flexible hydrophobic bicyclic ring systems are tolerated.
Table 4. Inhibitors with Modified Pyrrolidine Amide Moieties.

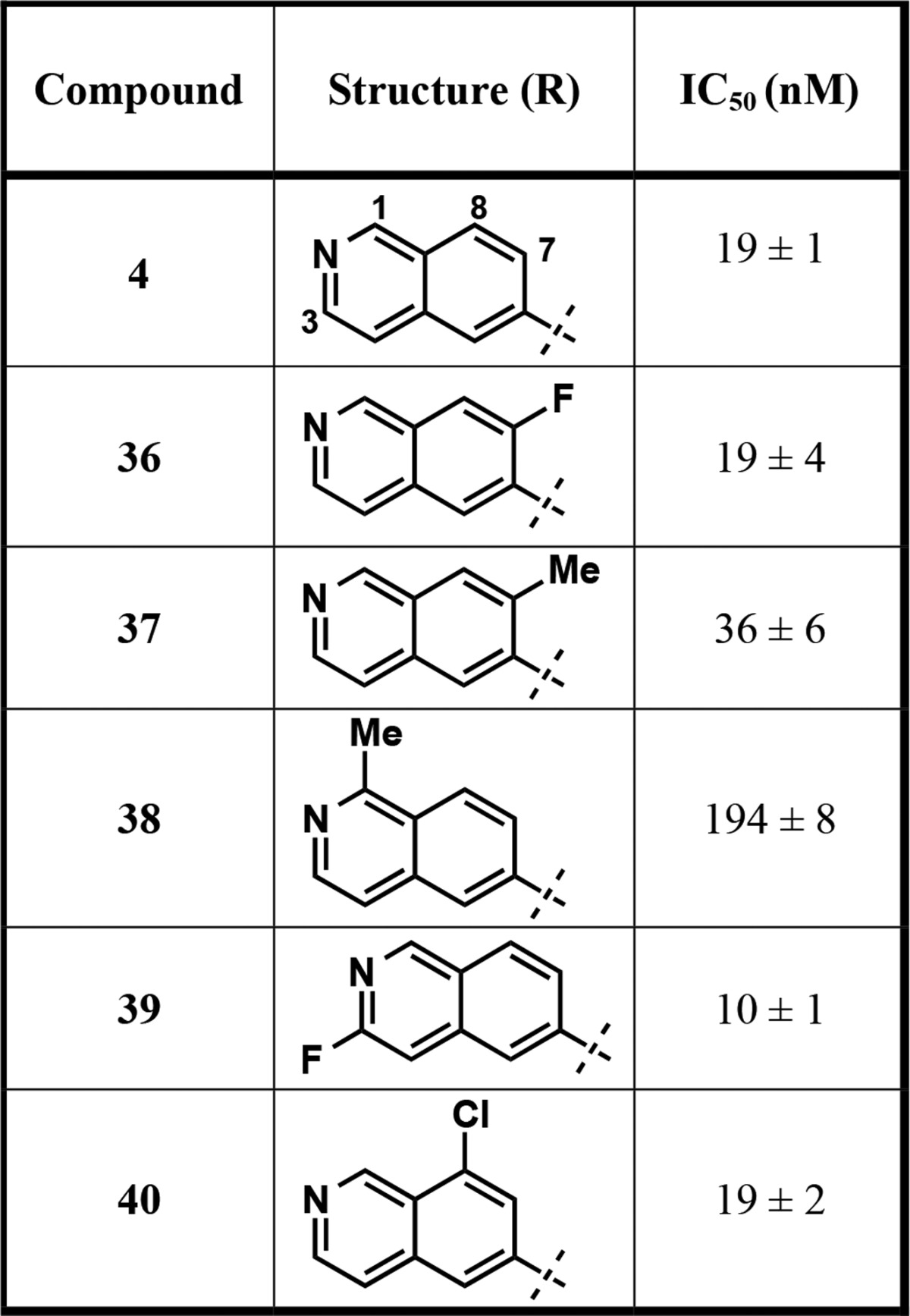
The results summarized in Table 4 and discussed above have shown that analogs of 4, namely, compounds 29, 30, 32, 34, and 35, potently inhibited PRMT3 with IC50 values of around 20–50 nM. Although these analogs did not display improved potency compared to compound 4, these substituted pyrrolidine groups are valuable alternatives to the unsubstituted pyrrolidine group (compound 4). After completing optimization of the RHS moiety, we further investigated the LHS isoquinoline ring. Analysis of the crystal structure of PRMT3 in complex with compound 4 (PDB ID: 4RYL)22 suggested that there is room in the binding pocket to tolerate a relatively small substituent at the 1-, 3-, 7-, and 8-positions of the isoquinoline ring system. Our structural analysis also suggested that a substituent at the 4- and 5-positions of the isoquinoline ring would not be tolerated. Therefore, we designed and synthesized the corresponding substituted isoquinoline analogs (compounds 36–40) to determine whether potency could further be enhanced (Table 5). For example, compounds 36 and 37 featuring small 7-fluoro and 7-methyl substituents were prepared. Compound 36 showed similar potency as 4, while 37, which has a slightly larger methyl substituent, was around 2-fold less potent. Compound 38, however, displayed significant potency loss (about 10-fold), indicating that the methyl group at the 1-position of the isoquinoline ring is not preferred. Interestingly, the 3-fluoro substituted analog 39 displayed almost 2-fold higher potency with IC50 of 10 ± 1 nM. This result suggests that the electronic modulation of the isoquinoline ring by a fluoro group does not have significant impact on the hydrogen bonding ability of the isoquinoline with T466 and a small substituent such as the fluoro group at the 3-position enhances potency, consistent with our structural analysis. In addition, we synthesized the 8-chloroisoquinoline derivative 40, which exhibited similar potency as compound 4. While the result obtained for compound 39 was consistent with our predication based on the analysis of the crystal structure of the PRMT3–compound 4 complex (PDB ID: 4RYL), it was surprising that a small substituent such as the fluoro group at the 7-position (compound 36) or the chloro group at the 8-position (compound 40) did not increase potency and a slightly larger substituent such as the methyl group at the 1- and 7-positions (compounds 37 and 38) reduced potency. More comprehensive structural analyses such as molecular dynamics simulation are needed to explain the SAR results. Nevertheless, the fluoro or chloro substituent could potentially improve metabolic stability of these compounds (36, 39, and 40), which are interesting alternative PRMT3 inhibitors to compound 4. Overall, these results have demonstrated that a small substituent at the isoquinoline ring can be tolerated but has limited impact on enhancing potency.
Table 5. Inhibitors with Substituted Isoquinolines.
The substituted 6-amino isoquinoline derivatives used for the synthesis of compounds 36–40 (Table 5) were not commercially available. Therefore, we devised synthetic routes and prepared these substituted 6-amino isoquinolines as shown in Scheme 2. The synthesis of 6-amino-7-fluoroisoquinoline (43), 6-amino-7-methylisoquinoline (44), and 6-amino-1-methylisoquinoline (45) started with reductive amination reactions of amino acetaldehyde dimethyl acetal with the corresponding 4-bromo aryl aldehyde or methyl aryl ketone to give amino dimethyl acetals 41 (Scheme 2A). The intermediates 41 were converted to the sulfonamides 42, via tosylation, which were then treated with aluminum chloride to yield the desired 6-bromoisoquinolines (43). These substituted 6-bromoisoquinolines were then converted to the desired 6-aminoisoquinolines (44–46) via aryl amination reactions (Scheme 2A). The 3-fluoro-6-aminoisoquinoline (47) was synthesized starting from the commercially available 3-amino-6-bromoisoquinoline in two steps via the Balz–Schiemann reaction23,24 followed by an aryl amination (Scheme 2B). As shown in Scheme 2C, 6-amino-8-chloroisoquinoline (48) was prepared in six steps. The commercially available 5-aminoisoquinoline was first acetylated and then chlorinated to install a chloro group at the 8-position. Bromination at the 6-position was achieved by using dibromoisocyanuric acid. Deacetylation followed by reductive diazotization resulted in 6-amino-8-chloroisoquinoline (48) (Scheme 2C). Intermediates 44–48 were then used to prepare compounds 36–40 according to the synthetic route outlined in Scheme 1.
Scheme 2. Synthetic Routes for Preparing Intermediates 44–48 for Synthesis of Compounds 36–40.

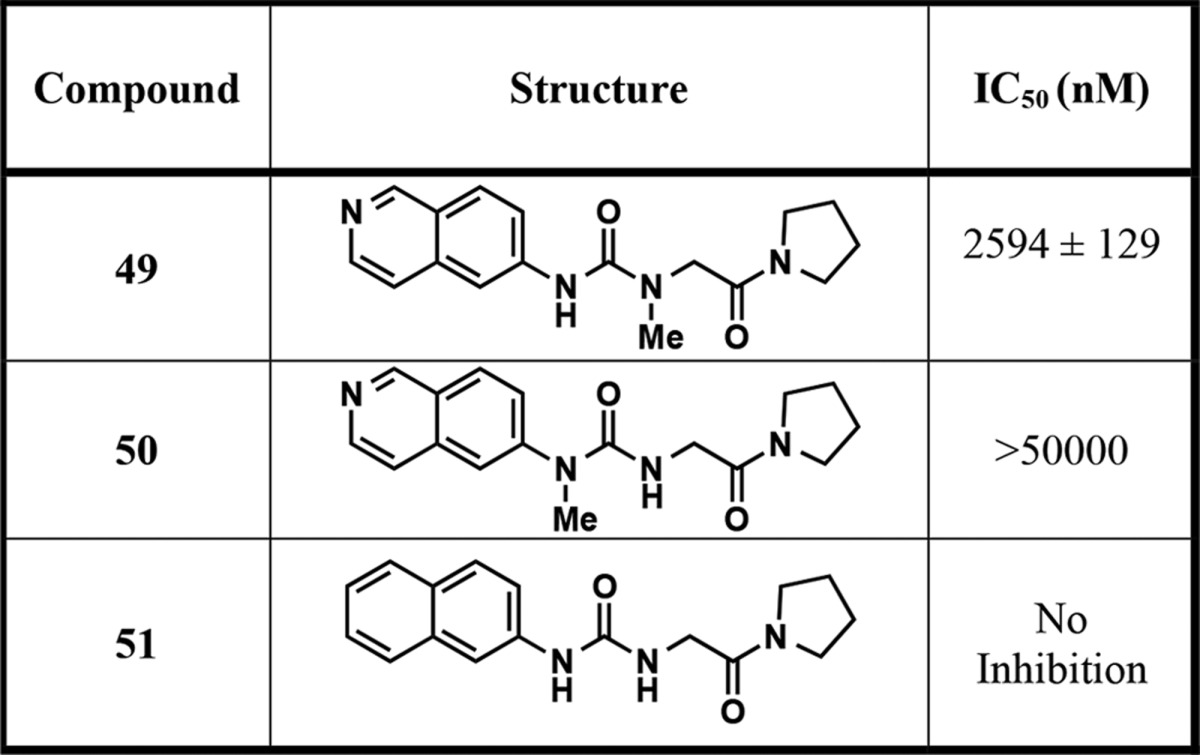
In addition, we designed and synthesized several close analogs of compound 4 to serve as negative controls for chemical biology studies. As described earlier, the middle urea region of these PRMT3 inhibitors forms the key hydrogen-bonding interactions with E422 of PRMT3. We therefore predicted that taking either of these hydrogen-bonding interactions away by methylating either nitrogen atom of the urea would drastically decrease PRMT3 inhibition. Indeed, as shown in Table 6, compound 49 displayed markedly diminished inhibitory activity (IC50 = 2594 ± 129 nM), while compound 50 was completely inactive (IC50 > 50 000 nM). Furthermore, the nitrogen atom in the isoquinoline ring of compound 4 forms a key hydrogen bond with T466 of PRMT3 in the crystal structure of the PRMT3–compound 4 complex (PDB ID: 4RYL). Thus, we replaced the isoquinoline ring of 4 with the naphthalene ring (compound 51 (XY1)),22 effectively removing the critical hydrogen bond with T466. As we reported previously,22 compound 51 displayed no inhibition of the PRMT3 catalytic activity in biochemical assays.
Table 6. Compounds Prepared as Negative Controls.
We previously reported that compound 4 was more than 200-fold selective for PRMT3 over 31 other methyltransferases and more than 250 kinases, GPCRs, ion channels, and transporters.22 Similarly, inhibitors 29, 30, and 36 were selective for PRMT3 over 31 other lysine methyltransferases, arginine methyltransferases, and DNA and RNA methyltransferases (Figure 3 and Supporting Information). In addition, 29, 30, and 36 were tested in a CEREP selectivity panel consisting of 55 protein targets (47 GPCRs, five ion channels, and three transporters) and did not show any significant off-target activities (% of inhibition <50% at 10 μM). It is of note that the cocrystal structure of compound 4 in complex with PRMT3 reported recently (PDB ID: 4RYL)22 clearly shows that this inhibitor binds the same allosteric pocket as earlier inhibitors (compounds 1 and 2 (PDB ID: 3SMQ and 4HSG)).
Figure 3.

Inhibitor 36 is highly selective for PRMT3 over 31 other methyltransferases. The selectivity data for compounds 29 and 30 are shown in the Supporting Information.
To establish the target engagement of PRMT3 inhibitors in cells (namely, inhibitors 4, 29, 36, and 37), we used an InCELL Hunter Assay, which measures intracellular binding of inhibitors to the methyltransferase domain of PRMT3 in cell lines expressing the methyltransferase domain of PRMT3 tagged with a short fragment of β-galactosidase (ePL). Binding of a compound to ePL–PRMT3 increases the fusion protein half-life. Inhibitors 4, 29, 36, and 37 stabilized PRMT3 in A549 cells, a human lung carcinoma cell line, with EC50 values of 2.0, 2.7, 1.6, and 4.9 μM, respectively (Figure 4, top). The same assay was performed in HEK293 cells and these compounds displayed EC50 values of 1.8, 3.1, 2.7, and 5.2 μM, respectively (Figure 4, bottom). Compound 51 was used as a negative control in these assays. As expected, no stabilization was observed with this compound.
Figure 4.
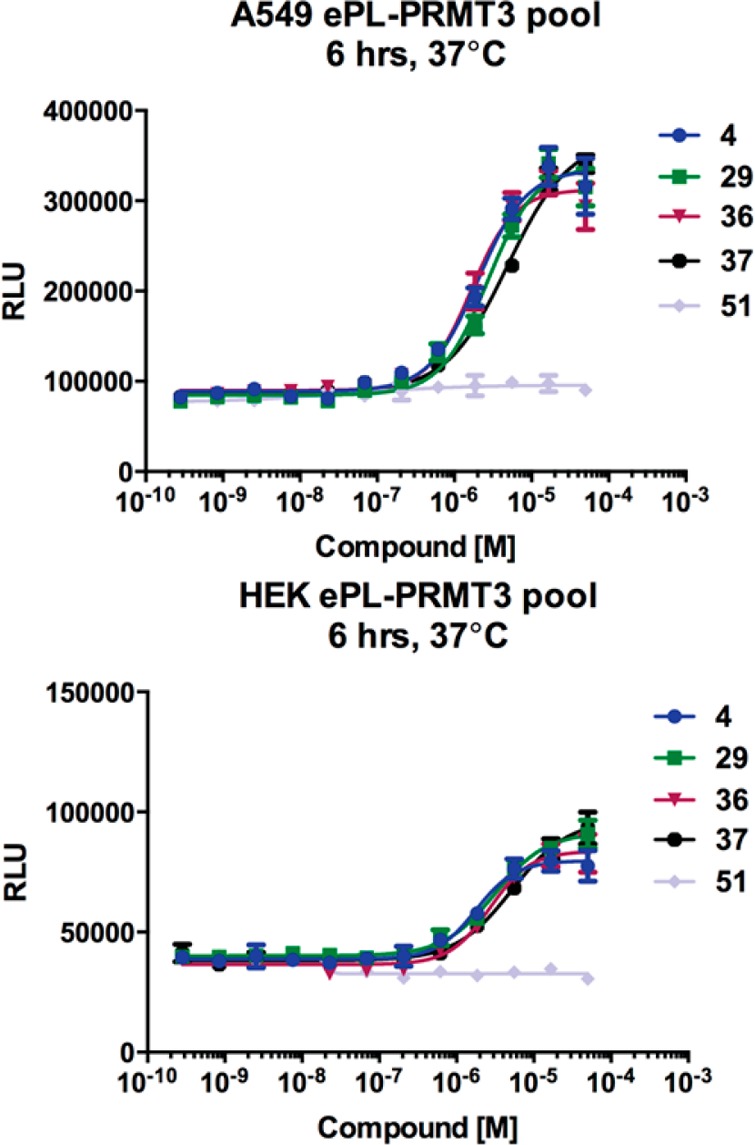
InCELL Hunter Assay results of compounds 4, 29, 36, 37, and 51 in A549 and HEK cells.
Furthermore, to establish whether these PRMT3 inhibitors can inhibit the PRMT3 catalytic activity in cells, we examined their effects on H4R3 asymmetric dimethylation. Since methylated arginine residues have relatively slow turnover, we overexpressed human Flag-tagged PRMT3 and followed the methylation of both endogenous H4 and exogenously introduced GFP-tagged H4. As we previously reported, overexpressed PRMT3 increased the endogenous H4R3me2a from the baseline levels, and compound 4 effectively inhibited this increase with an IC50 of 225 nM.22 The asymmetric dimethylation of exogenous H4R3 was also inhibited by compound 4 (IC50 = 91 nM), indicating that this inhibitor has robust cellular effect.22 Similarly, as shown in Figure 5, compounds 29, 30, and 36 inhibited the exogenous asymmetric dimethylation of H4R3 (IC50 = 240, 184, and 134 nM, respectively). The dependency on the transfected PRMT3 catalytic activity was determined by using the catalytically dead PRMT3 mutant (E335Q) that did not affect endogenous or exogenous H4R3me2a levels and therefore was used to establish the baseline levels of the mark. It is of note that effects of 1 μM compounds 29, 30, and 36 matched with that of the catalytically dead PRMT3 mutant E335Q.
Figure 5.

Cellular inhibitory activity of compounds 29, 30, and 36. HEK293 cells were cotransfected with FLAG tagged PRMT3 (wt) or its catalytic mutant (mt) and GFP-tagged histone H4 and treated with different concentrations of compounds, as indicated. Total cell lysates were collected 24 h post inhibitor treatment and analyzed for H4R3me2a, GFP, and FLAG levels by Western blotting. The graphs represent nonlinear fits of H4R3me2a fluorescence intensities normalized to GFP. The results are the averages of three replicates.
Conclusion
In summary, we conducted comprehensive SAR studies, starting from early inhibitors 1–3 and culminating in the discovery of highly potent (IC50 values = ∼10–36 nM), selective, and cell-active allosteric inhibitors of PRMT3 (inhibitors 4, 29, 30, 36, and 37). In addition, we generated compounds that are very close analogs of these potent inhibitors but that displayed drastically diminished potency as negative controls (compounds 49–51). The new inhibitors (compounds 29, 30, and 36) were highly selective for PRMT3 over 31 other methyltransferases and 55 other protein targets. In cell-based assays, compounds 29, 30, and 36 engaged PRMT3 and potently inhibited its methyltransferase activity (Figures 4 and 5). These inhibitors and negative controls are excellent chemical tools for the biomedical community to further investigate biological functions and disease associations of PRMT3.
Experimental Section
Chemistry General Procedures
Analytical thin-layer chromatography (TLC) was performed employing EMD Milipore 210–270 μm 60-F254 silica gel plates. The plates were visualized by exposure to UV light. Flash column chromatography was performed on a Teledyne ISCO CombiFlash Rf+ system equipped with a variable wavelength UV detector and a fraction collector using RediSep Rf normal phase silica columns. Nuclear magnetic resonance (NMR) spectra were acquired on a Bruker DRX-600 spectrometer or on a Varian Mercury spectrometer at 400 MHz. Chemical shifts are reported in parts per million (ppm, δ) scale relative to solvent residual peak (chloroform-d: 1H, 7.26 ppm; 13C, 77.16 ppm; methanol-d4: 1H, 3.31 ppm; 13C, 49.0 ppm). 1H NMR data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet, app = apparent), coupling constant, and integration. HPLC spectra for all compounds were acquired using an Agilent 6110 series system with a UV detector set to 254 nm. Samples were injected (5 μL) onto an Agilent Eclipse Plus, 4.6 Å, ∼50 mm, 1.8 μM, C18 column at room temperature, either with a linear gradient from 50% to 100% B (MeOH + 0.1% acetic acid) in 5.0 min followed by pumping 100% B for another 2 min with A being H2O + 0.1% acetic acid, or by a linear gradient from 10% to 100% B (MeOH + 0.1% acetic acid) in 5.0 min followed by pumping 100% B for another 2 min with A being H2O + 0.1% acetic acid. The flow rate was 1.0 mL/min. Mass spectrometry (MS) data were acquired in positive ion mode using an Agilent 6110 single-quadrupole mass spectrometer with an electrospray ionization (ESI) source. HRMS analysis was conducted on an Agilent Technologies G1969A high-resolution API-TOF mass spectrometer attached to an Agilent Technologies 1200 HPLC system. Samples were ionized by ESI in positive mode. All biologically evaluated compounds had >95% purity using the HPLC methods described above.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(2-oxo-2-(piperidin-1-yl)ethyl)urea (3)
Compound 3 was prepared according to previously published procedures.21
1-(Isoquinolin-6-yl)-3-(2-oxo-2-(pyrrolidin-1-yl)ethyl)urea (4)
Compound 4 was prepared according to previously published procedures.22
1-(Isoquinolin-6-yl)-3-(2-oxo-2-(piperidin-1-yl)ethyl)urea (5)
To a solution of isoquinolin-6-amine (50 mg, 0.347 mmol) in N,N-dimethylformamide (DMF) (1.6 mL) at room temperature was added N,N′-carbonyldiimidazole (CDI) (84 mg, 0.520 mmol). The resulting solution was stirred for 12 h prior to the addition of 2-amino-1-(piperidin-1-yl)ethan-1-one (99 mg, 0.694 mmol) and stirred for a further 6 h. Following dilution with water (20 mL), the aqueous layer was extracted with ethyl acetate (EtOAc) (3 × 20 mL), and the combined organic extracts were dried with anhydrous sodium sulfate. After filtration, all solvents were removed under reduced pressure, and the residue was purified by column chromatography on silica gel to afford title compound (5) (31 mg, 29% yield). 1H NMR (500 MHz, DMSO-d6) δ 9.08 (s, 1H), 8.35 (d, J = 5.7 Hz, 1H), 8.08 (br s, 1H), 7.98 (d, J = 8.9 Hz, 1H), 7.63 (d, J = 5.8 Hz, 1H), 7.02 (br s, 2H), 6.57 (t, J = 4.6 Hz, 1H), 4.02 (d, J = 4.7 Hz, 2H), 3.50–3.44 (m, 2H), 3.38–3.33 (m, 2H), 1.65–1.57 (m, 2H), 1.57–1.50 (m, 2H), 1.50–1.40 (m, 2H). m/z (HRMS) [M + H]+ for C17H21N4O2+: calculated 313.1659, found 313.1662.
1-(1-Oxo-1,3-dihydroisobenzofuran-5-yl)-3-(2-oxo-2-(piperidin-1-yl)ethyl)urea (6)
To a solution of 5-amino-3H-benzofuran-1-one (75 mg, 0.5 mmol, 1.0 equiv) in DMF (1.5 mL) was added CDI (90 mg, 0.55 mmol, 1.1 equiv), and the resulting mixture was stirred for 8 h at rt. 2-Amino-1-piperidin-1-ylethanone hydrochloride salt (134 mg, 0.75 mmol, 1.5 equiv) was then added followed by Hunig’s base (131 μL, 0.75 mmol, 1.5 equiv). After being stirred for 18 h at rt, the resulting mixture was diluted with water (25 mL) and extracted with EtOAc (3 × 25 mL). Combined organic layers were dried over sodium sulfate and concentrated under reduced pressure to give crude product, which was then purified by flash column chromatography to yield desired compound as white solid (39 mg, 25%). 1H NMR (600 MHz, methanol-d4) δ 7.85 (s, 1H), 7.73 (d, J = 8.5 Hz, 1H), 7.42 (dd, J = 8.5, 1.8 Hz, 1H), 5.31 (s, 2H), 4.10 (s, 2H), 3.57 (t, J = 5.6 Hz, 2H), 3.45 (t, J = 5.5 Hz, 2H), 1.74–1.50 (m, 6H). MS (ESI) m/z [M + H]+ for C16H20N3O4+: calculated 318.1, found 318.1.
1-(2-Oxo-2-(piperidin-1-yl)ethyl)-3-(quinazolin-7-yl)urea (7)
To a solution of quinazolin-7-amine (73 mg, 0.5 mmol, 1.0 equiv) in DMF (1.5 mL) was added CDI (90 mg, 0.55 mmol, 1.1 equiv), and the resulting mixture was stirred for 8 h at rt. 2-Amino-1-piperidin-1-ylethanone hydrochloride salt (134 mg, 0.75 mmol, 1.5 equiv) was then added followed by Hunig’s base (131 μL, 0.75 mmol, 1.5 equiv). After being stirred for 18 h at rt, the resulting mixture was diluted with water (25 mL) and extracted with EtOAc (3 × 25 mL). Combined organic layers were dried over sodium sulfate and concentrated under reduced pressure to give crude product, which was then purified by flash column chromatography to yield desired compound (10 mg, 6%). 1H NMR (600 MHz, methanol-d4) δ 9.29 (s, 1H), 9.06 (s, 1H), 8.22 (d, J = 2.1 Hz, 1H), 8.00 (d, J = 8.9 Hz, 1H), 7.71 (dd, J = 8.9, 2.1 Hz, 1H), 4.14 (s, 2H), 3.58 (t, J = 5.6 Hz, 2H), 3.47 (t, J = 5.5 Hz, 2H), 1.75–1.53 (m, 6H). MS (ESI) m/z [M + H]+ for C16H20N5O2+: calculated 314.2, found 314.2.
1-(Isoquinolin-7-yl)-3-(2-oxo-2-(piperidin-1-yl)ethyl)urea (8)
To a solution of isoquinolin-7-amine (50 mg, 0.347 mmol) in DMF (1.6 mL) at room temperature was added CDI (84 mg, 0.520 mmol). The resulting solution was stirred for 12 h prior to the addition of 2-amino-1-(piperidin-1-yl)ethan-1-one (99 mg, 0.694 mmol) and stirred for a further 6 h. Following dilution with water (20 mL), the aqueous layer was extracted with EtOAc (3 × 20 mL), and the combined organic extracts were dried with anhydrous sodium sulfate. After filtration, all solvents were removed under reduced pressure, and the residue was purified by column chromatography on silica gel to afford title compound (8) (45 mg, 42% yield). 1H NMR (500 MHz, DMSO-d6) δ 9.13 (s, 1H), 8.33 (d, J = 5.6 Hz, 1H), 8.22 (d, J = 1.9 Hz, 1H), 7.85 (d, J = 8.9 Hz, 1H), 7.69 (d, J = 5.6 Hz, 1H), 7.11 (br s, 1H), 6.92 (br s, 1H), 6.50 (t, J = 4.7 Hz, 1H), 4.03 (dd, J = 14.2, 5.9 Hz, 2H), 3.47–3.44 (m, 2H), 3.38–3.34 (m, 2H), 1.63–1.56 (m, 2H), 1.56–1.50 (m, 2H), 1.49–1.41 (m, 2H). m/z (HRMS) [M + H]+ for C17H21N4O2+: calculated 313.1659, found 313.1664.
General Procedures for the Preparation of Compounds 9–14 in Table 2
Compounds 9–14 shown in Table 2 were prepared according general procedures described below. To a solution of isoquinolin-6-amine (1.0 equiv) and triethylamine (TEA) (2 equiv) in DMF (1 mL/0.347 mmol) was added CDI (1.5 equiv), and the reaction mixture was allowed to stir at 25 °C for 4 h. To the reaction mixture was then added the corresponding amine (2 equiv), and the mixture was allowed to stir for additional 1 h. Then 50 mL of water and 50 mL of EtOAc were added to the reaction mixture. After extraction, the organic layer was washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography on silica gel eluting with 0–5% MeOH in DCM to give the product.
1-(2-(Cyclohex-1-en-1-yl)ethyl)-3-(isoquinolin-6-yl)urea (9)
Yellow oil (67 mg, 62% yield). 1H NMR (chloroform-d) δ: 9.07 (s, 1H), 8.40 (d, J = 5.8 Hz, 1H), 8.04(s, 1H), 7.81 (d, J = 8.8 Hz, 1H), 7.66 (s, 1H), 7.53 (d, J = 5.8 Hz, 1H), 7.44 (dd, J = 8.8, 2.0 Hz, 1H), 7.27 (s, 1H), 5.45 (s, 1H), 5.27–5.30 (m, 1H), 3.42–3.77 (m, 2H), 2.20–2.17 (m, 6H), 1.97–1.92 (m, 4H), 1.627–1.47 (m, 4H). MS (ESI) m/z [M + H]+ for C18H22N3O+: calculated 296.2, found 296.1.
1-(2-Cyclohexylethyl)-3-(isoquinolin-6-yl)urea (10)
Yellow oil (69 mg, 64% yield). 1H NMR (DMSO-d6) δ: 9.07 (s, 1H), 8.87 (s, 1H), 8.33 (d, J = 5.8 Hz, 1H), 8.06 (d, J = 1.8 Hz, 1H), 7.95 (d, J = 9.0 Hz, 1H), 7.60 (d, J = 6.0 Hz, 1H), 7.52 (dd, J = 8.8, 2.0 Hz, 1H), 6.28 (br t, J = 5.5 Hz, 1H), 3.11–3.24 (m, 2H), 1.58–1.76 (m, 5H), 1.11–1.39 (m, 6H), 0.83–0.98 (m, 2H). MS (ESI) m/z [M + H]+ for C18H24N3O+: calculated 298.2, found 298.1.
1-(2-Cyclopentylethyl)-3-(isoquinolin-6-yl)urea (11)
Light yellow oil (30 mg, 29% yield). 1H NMR (chloroform-d) δ: 9.04 (s, 1H), 8.36 (d, J = 5.8 Hz, 1H), 8.03 (s, 2H), 7.76 (d, J = 8.8 Hz, 1H), 7.34–7.56 (m, 2H), 7.27 (s, 1H), 3.19–3.40 (m, 2H), 1.64–1.82 (m, 3H), 1.40–1.64 (m, 6H), 0.95–1.16 (m, 2H), 0.01 (s, 1H). MS (ESI) m/z [M + H]+ for C17H22N3O+: calculated 284.2, found 284.1.
1-(2-Cyclopropylethyl)-3-(isoquinolin-6-yl)urea (12)
White solid (27 mg, 29% yield). 1H NMR (DMSO-d6) δ: 9.59 (s, 1H), 9.46 (s, 1H), 8.34–8.51 (m, 2H), 8.28 (d, J = 9.0 Hz, 1H), 8.12 (d, J = 6.5 Hz, 1H), 7.77 (dd, J = 9.0, 2.0 Hz, 1H), 6.71 (br t, J = 5.4 Hz, 1H), 3.11–3.27 (m, 3H), 1.38 (q, J = 7.0 Hz, 2H), 0.61–0.80 (m, 1H), 0.35–0.49 (m, 2H). MS (ESI) m/z [M + H]+ for C15H18N3O+: calculated 256.1, found 256.1.
1-(Isoquinolin-6-yl)-3-phenethylurea (13)
Light yellow oil (23 mg, 22% yield). 1H NMR (chloroform-d) δ: 8.95 (s, 1H), 7.89–8.47 (m, 3H), 7.74 (d, J = 8.8 Hz, 1H), 7.46 (d, J = 5.8 Hz, 1H), 7.37 (dd, J = 8.9, 1.9 Hz, 1H), 7.20–7.30 (m, 2H), 7.09–7.20 (m, 3H), 5.63 (br s, 1H), 3.55 (q, J = 6.8 Hz, 2H), 2.84 (t, J = 6.9 Hz, 2H). MS (ESI) m/z [M + H]+ for C18H18N3O+: calculated 292.1, found 292.1.
1-(2-(1-(Aminomethyl)cyclohexyl)ethyl)-3-(isoquinolin-6-yl)urea (14)
The general procedure was applied using tert-butyl ((1-(2-aminoethyl)cyclohexyl)methyl)carbamate as the amine (285 mg, 1.11 mmol) to give tert-butyl ((1-(2-(3-(isoquinolin-6-yl)ureido)ethyl)cyclohexyl)methyl)carbamate as a white solid. To the solution of tert-butyl ((1-(2-(3-(isoquinolin-6-yl)ureido)ethyl)cyclohexyl)methyl)carbamate (130 mg, 0.305 mmol) in DCM (1 mL) was added TFA (1.000 mL, 12.98 mmol). Then the reaction mixture was stirred at 25 °C for 0.5 h. To the mixture was added 5 mL of toluene, and it was then concentrated in vacuo. The residue was purified by preparative-HPLC to give the product as white solid (23 mg, 22% yield). 1H NMR (methanol-d4) δ 8.94–9.07 (m, 1H), 8.28 (br d, J = 5.8 Hz, 1H), 8.07 (br s, 1H), 7.88–7.99 (m, 1H), 7.49–7.69 (m, 2H), 3.26–3.44 (m, 2H), 3.02–3.26 (m, 2H), 2.65 (s, 2H), 1.14–1.78 (m, 13H). MS (ESI) m/z [M + H]+ for C19H27N4O+: calculated 327.2, found 327.2.
General Procedures for the Synthesis of Amides 15–35 in Tables 3 and 4
Synthesis of (Isoquinolin-6-ylcarbamoyl)glycine (28) (Scheme 1)
To a stirring solution of 6-aminoisoquinoline (1.2 g, 8.32 mmol, 1 equiv) in a mixture of dichloromethane and DMF (30 and 10 mL) was added ethyl isocyanatoacetate (2.80 mL, 25 mmol, 3.0 equiv), and the resulting mixture was stirred overnight at room temperature. After removal of volatiles, the crude mixture was purified by flash column chromatography (gradient from 100% dichloromethane to 10% methanol in dichloromethane) to yield the desired ethyl ester (27)22 as a pale yellow solid, which was resuspended in methanol (48 mL) and water (16 mL) followed by the addition of 1 N solution of NaOH (24 mL). The resulting clear mixture was then stirred at room temperature overnight. After concentration of the mixture under reduced pressure, the crude mixture was purified by reverse phase flash column chromatography (gradient from 100% water to 10% methanol in dichloromethane) to yield the desired acid 28 as a TFA salt (2.04 g, 57% over two steps).
To a stirring mixture of the above acid 28 (1.0 equiv) in DMF (0.8 mL/0.1 mmol) was added N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl) (1.5 equiv), 1-hydroxy-7-azabenzotriazole (HOAt) (1.5 equiv), and the corresponding amine (1.5 equiv) followed by N-methylmorpholine (NMM) (2 equiv), and the resulting mixture was stirred for 18 h at room temperature. The reaction was purified by either flash column chromatography or HPLC to give pure products.
1-(2-(Azetidin-1-yl)-2-oxoethyl)-3-(isoquinolin-6-yl)urea (15)
The reaction mixture was purified by HPLC to give pure product as a white solid (mono-TFA salt, 8 mg, 10%). 1H NMR (methanol-d4) δ 9.02 (d, J = 0.9 Hz, 1H), 8.29 (d, J = 5.9 Hz, 1H), 8.08 (d, J = 2.1 Hz, 1H), 7.97 (d, J = 9.0 Hz, 1H), 7.66–7.61 (m, 1H), 7.57 (dd, J = 8.9, 2.1 Hz, 1H), 4.35–4.27 (m, 2H), 4.07 (t, J = 7.8 Hz, 2H), 3.87 (s, 2H), 2.43–2.32 (m, 2H). MS (ESI) m/z [M + H]+ for C15H17N4O2+: calculated 285.1, found 285.2.
1-(2-(Azepan-1-yl)-2-oxoethyl)-3-(isoquinolin-6-yl)urea (16)
The title compound was obtained as a white solid (mono-TFA salt, 21 mg, 40%). 1H NMR (600 MHz, methanol-d4) δ 9.37 (s, 1H), 8.37 (d, J = 2.0 Hz, 1H), 8.31 (d, J = 6.6 Hz, 1H), 8.27 (d, J = 9.0 Hz, 1H), 8.07 (d, J = 6.6 Hz, 1H), 7.81 (dd, J = 9.0, 2.0 Hz, 1H), 4.17 (s, 2H), 3.61–3.56 (m, 2H), 3.56–3.51 (m, 2H), 1.86–1.82(m, 2H), 1.78–1.71 (m, 2H), 1.69–1.58 (m, 4H). 13C NMR (151 MHz, methanol-d4) δ 170.5, 156.7, 149.3, 146.9, 145.6, 142.1, 133.3, 132.1, 125.4, 124.2, 112.4, 48.1, 47.4, 42.5, 29.7, 28.5, 28.3, 27.8. MS (ESI) m/z [M + H]+ for C18H23N4O2+: calculated 327.2, found 327.2.
1-(2-(4,4-Difluoropiperidin-1-yl)-2-oxoethyl)-3-(isoquinolin-6-yl)urea (17)
The title compound was obtained as a white solid (mono-TFA salt, 51 mg, 66%). 1H NMR (400 MHz, methanol-d4) δ 9.39 (d, J = 0.9 Hz, 1H), 8.45 (d, J = 2.1 Hz, 1H), 8.37–8.27 (m, 2H), 8.14 (d, J = 6.7 Hz, 1H), 7.86 (dd, J = 9.1, 2.1 Hz, 1H), 4.21 (s, 2H), 3.75 (t, J = 6.1 Hz, 2H), 3.65 (t, J = 6.0 Hz, 2H), 2.14–1.96 (m, 4H). 13C NMR (151 MHz, methanol-d4) δ 169.5, 156.8, 149.3, 147.0, 145.7, 142.1, 132.2, 125.3, 124.3, 122.9, 112.5, 111.2, 42.6, 40.3, 35.1, 34.6. MS (ESI) m/z [M + H]+ for C17H19F2N4O2+: calculated 349.2, found 349.2.
1-(Isoquinolin-6-yl)-3-(2-(4-methylpiperazin-1-yl)-2-oxoethyl)urea (18)
The title compound was obtained as a light yellow solid (bis-TFA salt, 48 mg, 52%). 1H NMR (400 MHz, DMSO-d6) δ 9.96 (br s, 1H), 9.47 (s, 1H), 8.44 (d, J = 6.5 Hz, 1H), 8.32–8.28 (m, 2H), 8.10 (d, J = 6.5 Hz, 1H), 7.78 (dd, J = 9.0, 2.1 Hz, 1H), 6.90 (br s, 1H), 4.12 (br s, 2H), 3.40 (br s, 8H; these protons are obscured by residual water in DMSO), 2.83 (br s, 3H). MS (ESI) m/z [M + H]+ for C17H22N5O2+: calculated 328.2, found 328.2.
2-(3-(Isoquinolin-6-yl)ureido)-N,N-dimethylacetamide (19)
The title compound was obtained as a white solid (mono-TFA salt, 14.6 mg, 18%). 1H NMR (400 MHz, methanol-d4) δ 9.03 (s, 1H), 8.29 (d, J = 5.9 Hz, 1H), 8.10 (d, J = 2.1 Hz, 1H), 7.98 (d, J = 8.9 Hz, 1H), 7.65 (d, J = 5.9 Hz, 1H), 7.59 (dd, J = 8.9, 2.1 Hz, 1H), 4.13 (s, 2H), 3.08 (s, 3H), 2.99 (s, 3H). MS (ESI) m/z [M + H]+ for C14H17N4O2+: calculated 273.1, found 273.2.
N,N-Diethyl-2-(3-(isoquinolin-6-yl)ureido)acetamide (20)
The title compound was obtained as a white solid (34.1 mg, 81%). 1H NMR (400 MHz, methanol-d4) δ 9.05 (s, 1H), 8.30 (d, J = 5.9 Hz, 1H), 8.11 (s, 1H), 8.00 (d, J = 9.0 Hz, 1H), 7.67 (d, J = 6.0 Hz, 1H), 7.60 (dd, J = 8.9, 2.1 Hz, 1H), 4.14 (s, 2H), 3.42 (apparent p, J = 7.0 Hz, 4H), 1.27 (t, J = 7.2 Hz, 3H), 1.16 (t, J = 7.1 Hz, 3H). MS (ESI) m/z [M + H]+ for C16H21N4O2+: calculated 301.2, found 301.2.
N-Cyclopentyl-2-(3-(isoquinolin-6-yl)ureido)-N-methylacetamide (21)
The title compound was obtained as a white solid (18.2 mg, 40%). 1H NMR (400 MHz, methanol-d4) δ 9.03 (s, 1H), 8.29 (d, J = 5.9 Hz, 1H), 8.10 (d, J = 2.1 Hz, 1H), 7.98 (d, J = 8.9 Hz, 1H), 7.65 (d, J = 5.9 Hz, 1H), 7.59 (dd, J = 8.9, 2.1 Hz, 1H), 4.92–4.90 (m, 1H), 4.27–4.25 (m, 1H), 4.21 (s, 1H), 4.11 (s, 1H), 2.93 (s, 1H), 2.86 (s, 1H), 1.94 (s, 1H), 1.86–1.55 (m, 7H). MS (ESI) m/z [M + H]+ for C18H23N4O2+: calculated 327.2, found 327.2.
2-(3-(Isoquinolin-6-yl)ureido)-N-methoxy-N-methylacetamide (22)
The title compound was obtained as a white solid (mono-TFA salt, 34.9 mg, 63%). 1H NMR (400 MHz, methanol-d4) δ 9.37 (d, J = 1.1 Hz, 1H), 8.33–8.28 (m, 2H), 8.25 (d, J = 9.1 Hz, 1H), 8.05 (d, J = 6.7 Hz, 1H), 7.79 (dd, J = 9.1, 2.1 Hz, 1H), 4.24 (s, 2H), 3.82 (s, 3H), 3.25 (s, 3H). MS (ESI) m/z [M + H]+ for C14H17N4O3+: calculated 289.1, found 289.1.
2-(3-(Isoquinolin-6-yl)ureido)-N-methylacetamide (23)
The reaction mixture was then purified by HPLC to give pure product as a white mono-TFA salt (4 mg, 8%). 1H NMR (400 MHz, methanol-d4) δ 9.41 (d, J = 0.9 Hz, 1H), 8.46 (d, J = 2.1 Hz, 1H), 8.36–8.27 (m, 2H), 8.16 (d, J = 6.7 Hz, 1H), 7.87 (dd, J = 9.1, 2.1 Hz, 1H), 3.92(s, 2H), 2.78 (s, 3H). MS (ESI) m/z [M + H]+ for C13H15N4O2+: calculated 259.1, found 259.1.
N-Cyclopropyl-2-(3-(isoquinolin-6-yl)ureido)acetamide (24)
The title compound was obtained as a white solid (58 mg, 78%). 1H NMR (400 MHz, DMSO-d6) δ 9.91 (s, 1H), 9.52 (s, 1H), 8.45 (d, J = 6.6 Hz, 1H), 8.44–8.28 (m, 2H), 8.18 (d, J = 6.6 Hz, 1H), 8.10 (d, J = 4.1 Hz, 1H), 7.79 (dd, J = 9.0, 2.0 Hz, 1H), 6.83 (t, J = 5.3 Hz, 1H), 3.74 (d, J = 5.2 Hz, 2H), 2.69–262 (m, 1H), 0.65–0.61 (m, 2H), 0.48–0.37 (m, 2H). MS (ESI) m/z [M + H]+ for C15H17N4O2+: calculated 285.1, found 285.1.
N-Cyclopentyl-2-(3-(isoquinolin-6-yl)ureido)acetamide (25)
The title compound was obtained as a white solid (15 mg, 38%). 1H NMR (400 MHz, methanol-d4) δ 9.04 (s, 1H), 8.30 (d, J = 5.9 Hz, 1H), 8.09 (d, J = 2.1 Hz, 1H), 7.99 (d, J = 8.9 Hz, 1H), 7.66 (d, J = 6.0 Hz, 1H), 7.59 (dd, J = 8.9, 2.1 Hz, 1H), 4.15 (p, J = 6.7 Hz, 1H), 3.88 (s, 2H), 2.00–1.89 (m, 2H), 1.73 (s, 2H), 1.68–1.56 (m, 2H), 1.49 (dq, J = 14.4, 8.3, 7.5 Hz, 2H). MS (ESI) m/z [M + H]+ for C17H21N4O2+: calculated 313.2, found 313.2.
N-Cyclohexyl-2-(3-(isoquinolin-6-yl)ureido)acetamide (26)
The title compound was obtained as a white solid (mono-TFA salt, 48.3 mg, 53%). 1H NMR (400 MHz, methanol-d4) δ 9.40 (s, 1H), 8.45 (d, J = 2.1 Hz, 1H), 8.34–8.31 (m, 2H), 8.14 (d, J = 6.7 Hz, 1H), 7.86 (dd, J = 9.0, 2.1 Hz, 1H), 3.91 (s, 2H), 3.73–3.69 (m, 1H), 1.90–1.87 (m, 2H), 1.78–1.75 (m, 2H), 1.66–1.63 (m, 1H), 1.42–1.19 (m, 5H). MS (ESI) m/z [M + H]+ for C15H23N4O2+: calculated 327.2, found 327.2.
1-(Isoquinolin-6-yl)-3-(2-oxo-2-(3,3,4,4-tetrafluoropyrrolidin-1-yl)ethyl)urea (29)
The title compound was obtained as a white solid (48.3 mg, 53%). 1H NMR (400 MHz, methanol-d4) δ 9.04 (s, 1H), 8.30 (d, J = 5.9 Hz, 1H), 8.11–8.07 (m, 1H), 7.98 (d, J = 8.9 Hz, 1H), 7.65 (d, J = 5.9 Hz, 1H), 7.59 (dd, J = 8.9, 2.1 Hz, 1H), 4.28 (t, J = 13.5 Hz, 2H), 4.08–4.01 (m, 4H). MS m/z (HRMS) [M + H]+ for C16H15F4N4O2+: calculated 371.1126, found 371.1153.
1-(2-(2,5-Dimethylpyrrolidin-1-yl)-2-oxoethyl)-3-(isoquinolin-6-yl)urea (30)
The title compound was obtained as a white solid (48.2 mg, 88%). The amine, 2,5-dimethylpyrrolidine, is used as mixture of cis and trans for the coupling reaction, and only the major product is reported below. 1H NMR (400 MHz, methanol-d4) δ 9.03 (br s, 1H), 8.29 (d, J = 5.9 Hz, 1H), 8.09 (d, J = 2.1 Hz, 1H), 7.98 (d, J = 8.9 Hz, 1H), 7.65 (d, J = 5.9 Hz, 1H), 7.59 (dd, J = 8.9, 2.2 Hz, 1H), 4.19–4.01 (m, 4H), 2.17–1.97 (m, 2H), 1.79–1.71 (m, 2H), 1.34 (d, J = 6.4 Hz, 3H), 1.33 (d, J = 6.4 Hz, 3H). MS m/z (HRMS) [M + H]+ for C18H23N4O2+: calculated 327.1816, found 371.1819.
Methyl(isoquinolin-6-ylcarbamoyl)glycyl-l-prolinate (31)
The title compound was obtained as a yellow solid (mono-TFA salt, 51 mg, 65%). 1H NMR (400 MHz, methanol-d4) δ 9.40 (s, 1H), 8. 39 (s, 1H), 8.32 (br s, 2H), 8.13 (s, 1H), 7.84 (s, 1H), 4.52 (dd, J = 8.7, 3.9 Hz, 1H), 4.23–4.08 (m, 2H), 3.81 (s, 1H), 3.75–3.57 (m, 4H), 2.32- 2.23 (m, 1H), 2.14–1.97 (m, 3H). MS (ESI) m/z [M + H]+ for C18H21N4O4+: calculated 357.2, found 357.2. [α]20D = −70 (c 1.6, CH3OH).
1-(2-(Hexahydrocyclopenta[c]pyrrol-2(1H)-yl)-2-oxoethyl)-3-(isoquinolin-6-yl)urea (32)
The title compound was obtained as a white solid (mono-TFA salt, 33 mg, 44%). 1H NMR (400 MHz, methanol-d4) δ 9.41 (s, 1H), 8.46 (s, 1H), 8.33 (d, J = 7.4 Hz, 2H), 8.15 (s, 1H), 7.87 (d, J = 9.1 Hz, 1H), 4.07 (s, 2H), 3.73–3.65 (m, 2H), 3.31–3.30 (m, 2H), 2.87–279 (m, 1H), 2.75–2.66 (m, 1H), 1.97–1.77 (m, 3H), 1.73–1.63 (m, 1H), 1.59–1.46 (m, 2H). MS (ESI) m/z [M + H]+ for C19H23N4O2+: calculated 339.2, found 339.2.
1-(2-(Isoindolin-2-yl)-2-oxoethyl)-3-(isoquinolin-6-yl)urea (33)
The title compound was obtained as a white solid (42 mg, 73%). 1H NMR (400 MHz, methanol-d4) δ 9.04 (s, 1H), 8.30 (d, J = 5.9 Hz, 1H), 8.12 (d, J = 2.1 Hz, 1H), 7.99 (d, J = 8.9 Hz, 1H), 7.63 (d, J = 5.8 Hz, 1H), 7.56 (dd, J = 8.9, 2.1 Hz, 1H), 7.36–7.31 (m, 3H), 6.65 (t, J = 4.8 Hz, 1H), 4.88 (s, 2H), 4.71 (s, 2H), 4.09 (d, J = 4.7 Hz, 2H). MS (ESI) m/z [M + H]+ for C20H19N4O2+: calculated 347.2, found 347.2.
1-(2-(7-Azabicyclo[2.2.1]heptan-7-yl)-2-oxoethyl)-3-(isoquinolin-6-yl)urea (34)
The title compound was obtained as a white solid (mono-TFA salt, 33 mg, 45%). 1H NMR (400 MHz, methanol-d4) δ 9.40 (br s, 1H), 8.43 (d, J = 2.1 Hz, 1H), 8.35–8.28 (m, 2H), 8.13 (d, J = 6.7 Hz, 1H), 7.85 (dd, J = 9.1, 2.1 Hz, 1H), 4.60 (t, J = 4.8 Hz, 1H), 4.41 (t, J = 4.8 Hz, 1H), 4.10 (s, 2H), 1.90 (q, J = 9.1, 7.0 Hz, 2H), 1.77 (s, 2H), 1.68–1.59 (m, 2H), 1.58–1.50 (m, 2H). MS (ESI) m/z [M + H]+ for C18H21N4O2+: calculated 325.2, found 325.2.
1-(2-(8-Azabicyclo[3.2.1]octan-8-yl)-2-oxoethyl)-3-(isoquinolin-6-yl)urea (35)
The title compound was obtained as a white solid (22 mg, 38%). 1H NMR (400 MHz, methanol-d4) δ 8.99 (s, 1H), 8.26 (d, J = 5.9 Hz, 1H), 8.05 (s, 1H), 7.93 (d, J = 9.0 Hz, 1H), 7.62–7.52 (m, 2H), 4.62–4.55 (m, 1H), 4.27 (d, J = 6.7 Hz, 1H), 4.18–4.02 (m, 2H), 2.15–2.03 (m, 1H), 1.97–1.70 (m, 6H), 1.62 (dd, J = 13.2, 5.4 Hz, 2H), 1.53 (d, J = 12.8 Hz, 1H). MS (ESI) m/z [M + H]+ for C19H23N4O2+: calculated 339.2, found 339.2.
1-(7-Fluoroisoquinolin-6-yl)-3-(2-oxo-2-(pyrrolidin-1-yl)ethyl)urea (36) (Scheme 2A; Table 5)
A mixture of aminoacetaldehyde dimethyl acetal (0.5 g, 2.46 mmol, 1.0 equiv) and 4-bromo-3-fluorobenzaldehyde (0.4 mL, 3.70 mmol, 1.5 equiv) in toluene (40 mL) in a round-bottom flask equipped with a reflux condenser and Dean–Stark trap was heated to reflux for 6 h. Then the reaction mixture was quenched with water (20 mL) and extracted with DCM (40 mL). After concentration of volatiles, crude mixture was dissolved in EtOH (20 mL), and NaBH4 (0.19 g, 4.92 mmol, 2 equiv) was added at room temperature and stirred overnight. The reaction was then quenched with water (50 mL) and extracted with EtOAc (50 mL × 3). Combined organic layers were washed with brine (50 mL × 2) and dried over Na2SO4 and concentrated under reduced pressure to give the desired amino acetal (41 (R1 = F, R2 = H) in Scheme 2A) (0.65 g, 90%). The amino acetal (0.35 g, 1.19 mmol) was resuspended in DCM (20 mL), and TEA (0.5 mL, 3.57 mmol, 3 equiv), TsCl (0.27 g, 1.43 mmol, 1.2 equiv), and DMAP (15 mg, 10 mol %) were added to give a clear solution, which was stirred at room temperature overnight. The reaction mixture was then suspended in water (20 mL) and extracted with DCM (50 mL × 3). Combined organic layers were washed with brine, dried over Na2SO4, and concentrated down. The crude mixture was purified by flash column chromatography to give the desired tosyl amine (0.51 g, 90%) (42 (R1 = F, R2 = H) in Scheme 2A).
To a flame-dried flask equipped with Teflon stir bar was added AlCl3 (0.27 g, 2.02 mmol, 4.5 equiv) under nitrogen atmosphere followed by the addition of the above solution of tosyl amine (0.2 g, 0.45 mmol, 1.0 equiv) in DCM (6 mL). The resulting solution was stirred under nitrogen at room temperature overnight. The reaction mixture was then cooled down to 0 °C, quenched with NaHCO3 (10 mL), and extracted with DCM (20 mL × 3). Combined organic layers were washed with brine (10 mL), dried over Na2SO4, and concentrated. The crude oil was then purified by flash column chromatography to yield desired 6-bromo-7-fluoroisouinoline (58 mg, 58%) (43 (R1 = F, R2 = H) in Scheme 2A).
To a flame-dried pressure vessel equipped with Teflon stirring bar was added CuI (10 mg, 0.05 mmol, 20 mol %), l-proline (12 mg, 0.10 mmol, 40 mol %), and K2CO3 (104 mg, 0.75 mmol, 3 equiv) as solid, and the vessel was flame-dried again under vacuum. Then DMSO (1 mL) solution of 6-bromo-7-fluoroisouinoline (58 mg, 0.25 mmol, 1 equiv) was added to the pressure vessel under nitrogen atmosphere followed by the addition of NH4OH (0.5 mL). The resulting suspension in a sealed vessel was then heated to 70 °C overnight. The reaction mixture was then cooled down to rt, suspended in water (5 mL) and EtOAc (10 mL), and further extracted with EtOAc (10 mL × 3). Combined organic layers were washed with brine (10 mL), dried over Na2SO4, and concentrated. The crude oil was then purified by flash column chromatography to yield desired 7-fluoroisouinoline-6-amine (44 (R1 = F, R2 = H) in Scheme 2A). This amine was then immediately dissolved in DCM/DMF (1 mL/0.3 mL), and ethyl isocyanato acetate (96 mg, 0.75 mmol, 3 equiv) was added. The resulting mixture was stirred at room temperature overnight, and after removal of volatiles, it was purified by flash column chromatography and immediately hydrolyzed to acid with 1 N NaOH (1 mL) in MeOH (1.5 mL) and water (0.5 mL) overnight. After purification by reverse phase chromatography, the desired acid was obtained as a white solid. To a stirring mixture of the acid (27 mg, 0.072 mmol) in THF (1.0 mL) was added pyrrolidine (10 μL, 0.122 mmol, 1.70 equiv) followed by EDC·HCl (23.4 mg, 0.122 mmol, 1.70 equiv), and the resulting mixture was stirred overnight at room temperature. Flash column chromatography yielded 1-(7-fluoroisoquinolin-6-yl)-3-(2-oxo-2-(pyrrolidin-1-yl)ethyl)urea (36) as a white solid (16.2 mg, 20% yield over four steps, starting from 6-bromo-7-fluoroisouinoline 43 (R1 = F, R2 = H)). 1H NMR (400 MHz, methanol-d4) δ 9.03 (s, 1H), 8.65 (d, J = 7.8 Hz, 1H), 8.31 (d, J = 5.8 Hz, 1H), 7.80 (d, J = 11.5 Hz, 1H), 7.67 (d, J = 5.9 Hz, 1H), 4.08 (br s, 2H), 3.53 (t, J = 6.8 Hz, 2H), 3.48 (t, J = 6.9 Hz, 2H), 2.07–2.00 (m, 2H), 1.95–1.88 (m, 2H). MS m/z (HRMS) [M + H]+ for C16H18FN4O2+: calculated 317.1408, found 317.1418.
1-(7-Methylisoquinolin-6-yl)-3-(2-oxo-2-(pyrrolidin-1-yl)ethyl)urea (37) (Scheme 2A; Table 5)
The same procedures as above were used for the synthesis of 1-(7-methylisoquinolin-6-yl)-3-(2-oxo-2-(pyrrolidin-1-yl)ethyl)urea starting from 4-bromo-3-methylbenzaldehyde. 6-Bromo-7-methylisouinoline (43 (R1 = Me, R2 = H); 50%, over the first four steps). Compound 37 was obtained in 20% (24 mg) over the last four steps as described above. 1H NMR (400 MHz, methanol-d4) δ 8.98 (s, 1H), 8.41 (s, 1H), 8.26 (d, J = 5.9 Hz, 1H), 7.86 (s, 1H), 7.63 (d, J = 5.9 Hz, 1H), 4.07 (s, 2H), 3.53 (t, J = 6.8 Hz, 2H), 3.48 (t, J = 6.9 Hz, 2H), 2.50 (s, 3H), 2.03 (p, J = 6.7 Hz, 2H), 1.91 (p, J = 6.6 Hz, 2H). MS m/z (HRMS) [M + H]+ for C17H21N4O2+: calculated 313.1659, found 313.1662.
1-(1-Methylisoquinolin-6-yl)-3-(2-oxo-2-(pyrrolidin-1-yl)ethyl)urea (38) (Scheme 2A; Table 5)
The same procedures as above were used for the synthesis of 1-(7-methylisoquinolin-6-yl)-3-(2-oxo-2-(pyrrolidin-1-yl)ethyl)urea (36) starting from 4′-bromoacetophenone. 6-Bromo-7-methylisouinoline (43 (R1 = Me, R2 = H)) was obtained in 27%, over the first four steps. Compound 38 was then obtained (2 mg, 2%) over the last four steps as described above. 1H NMR (400 MHz, methanol-d4) δ 8.17–8.09 (m, 2H), 8.05 (d, J = 2.2 Hz, 1H), 7.59 (dd, J = 9.1, 2.2 Hz, 1H), 7.50 (d, J = 6.0 Hz, 1H), 4.05 (s, 2H), 3.52–3.45 (m, 4H), 2.87 (s, 3H), 2.03 (p, J = 6.8 Hz, 2H), 1.91 (p, J = 7.1 Hz, 2H). MS (ESI) m/z [M + H]+ for C17H21N4O2+: calculated 313.2, found 313.2.
1-(3-Fluoroisoquinolin-6-yl)-3-(2-oxo-2-(pyrrolidin-1-yl)ethyl)urea (39) (Scheme 2B; Table 5)
To the mixture of 6-bromoisoquinolin-3-amine (158 mg, 0.71 mmol, 1.0 equiv) and HF·pyridine (10 mL) at 0 °C was added dropwise a solution of NaNO2 (243 mg, 3.54 mmol, 5 equiv) in water (5 mL). Upon completion of the addition, the cold bath was removed and the reaction was allowed to warm up to room temperature for 1.5 h at which point saturated NaHCO3 solution (45 mL) was added. The reaction mixture was diluted with DCM (20 mL) and saturated NH4Cl (10 mL) solution, and phases were separated and aqueous phase further extracted with DCM (20 mL × 3). Combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. Crude material was purified by flash column chromatography to yield 6-bromo-3-fluoroisoquinoline as a white solid (128 mg, 80%). The same procedure that was used for the synthesis of 36 (last four steps) was repeated to obtain the title compound 39 (6 mg, 5%). 1H NMR (400 MHz, methanol-d4) δ 8.79 (s, 1H), 8.10 (s, 1H), 7.99 (d, J = 8.9 Hz, 1H), 7.50 (dt, J = 8.9, 2.3 Hz, 1H), 7.23 (s, 1H), 4.05 (s, 2H), 3.53 (t, J = 6.8 Hz, 2H), 3.48 (t, J = 6.9 Hz, 2H), 2.02 (q, J = 6.8 Hz, 2H), 1.91 (p, J = 6.8 Hz, 2H). MS (ESI) m/z [M + H]+ for C16H18FN4O2+: calculated 317.1, found 317.2.
1-(8-Chloroisoquinolin-6-yl)-3-(2-oxo-2-(pyrrolidin-1-yl)ethyl)urea (40) (Scheme 2C; Table 5)
To a stirring mixture of 5-aminoisoquinoline (5.76 g, 40 mmol, 1 equiv) in pyridine (40 mL) was added acetic anhydride (5.7 mL, 60 mmol, 1.5 equiv), and the resulting reaction mixture was stirred at room temperature overnight. The precipitate formed was filtered and washed with cold hexanes (400 mL) in portions to give acetylated product (6.98 g, 94%). To a stirring mixture of 5-acetyl aminoisoquinoline (3.36 g, 18 mmol, 1 equiv) in DMF (30 mL) was added NCS (2.40 mL, 18 mmol, 1.0 equiv), and the resulting mixture was heated to 65 °C for 3 days. The reaction mixture was then diluted with EtOAc (50 mL), and water (50 mL) was added. After separation, the aqueous phase was extracted with EtOAc (50 mL × 3), and combined organic layers were then dried over Na2SO4 and concentrated under reduced pressure. Crude material was purified by flash column chromatography to yield desired compound (3.53 g, 88%).
To a solution of 5-acetylamino-8-chloroisoquinoline (1.0 g, 4.53 mmol, 1.0 equiv) in DMF (10 mL) was added dibromocyanuric acid (1.3 g, 4.53 mmol, 1 equiv), and the reaction mixture was heated to 65 °C for 3 h. After concentration, the obtained crude product resuspended in EtOH (45 mL), conc. HCl (9 mL) was added, and reaction mixture was heated to reflux for 3 h. The reaction mixture was then cooled down to rt and neutralized with 1 N NaOH to pH 7 and extracted with EtOAc (100 mL × 3). Combined organic layers were then dried over Na2SO4 and concentrated under reduced pressure. Crude material (300 mg, 26% over two steps) was used for the next step without further purification. Crude material (300 mg, 1.2 mmol, 1.0 equiv) was dissolved in EtOH (45 mL) followed by the addition of Ac2O (7.5 mL) and a solution of NaNO2 (in 15 mL water) and NaHSO3 (in 18 mL water). The resulting mixture was stirred at rt for 10 min, AcOH (7.5 mL) was added, and the reaction was allowed to stir at rt overnight. The reaction mixture was neutralized with 1 N NaOH (∼350 mL) to pH 8–9 and extracted with EtOAc (250 mL × 3). Combined organic layers were then dried over Na2SO4 and concentrated under reduced pressure. Crude material was purified by flash column chromatography to yield the desired compound (100 mg, 34%). The same procedures that were used for the synthesis of 36 (last four steps) were repeated to obtain the title compound 40 (51 mg, 30%). 1H NMR (400 MHz, methanol-d4) δ 9.60 (br s, 1H), 8.40 (d, J = 6.7 Hz, 1H), 8.29 (d, J = 2.0 Hz, 1H), 8.21 (d, J = 6.7 Hz, 1H), 8.12 (d, J = 1.9 Hz, 1H), 4.08 (s, 2H), 3.54–3.46 (m, 4H), 2.07–2.00 (m, 2H), 1.95–1.89 (m, 2H). MS (ESI) m/z [M + H]+ for C16H18ClN4O2+: calculated 333.1 and 335.1; found 333.1 and 335.1.
1-(Naphthalen-2-yl)-3-(2-oxo-2-(pyrrolidin-1-yl)ethyl)urea (51)
Compound 51 was prepared according to a previously published procedure.22
Synthesis and characterization data for compounds 49 and 50 are detailed in the Supporting Information.
PRMT3 Biochemical Assay
The radiometric scintillation proximity assays to evaluate the potency of the compounds were performed as described previously.22 The reactions were done under balanced conditions using the biotinylated histone H4 peptide (Tufts University Peptide Synthesis Core Facility, Boston, Ma) with the sequence of SGRGKGGKGLGKGGAKRHRKVLRDK-biotin) as substrate and [3H]S-adenosylmethionine (Waltham, MA, Cat# NET155 V001MC, specific activity range 12–18 Ci/mmol) as the methyl donor.
Selectivity Assays
The methyltransferase selectivity of 29, 30, and 36 was assessed at compound concentrations of 1, 5, and 20 μM as described previously.22,25−27
Cellular PRMT3 Assay
Compound effects in cells were determined as described previously (PMID: 27423858, 25728001). Briefly, HEK293 cells were grown in DMEM supplemented with 10% FBS, penicillin (100 units/mL), and streptomycin (100 μg/mL). The cells were cotransfected with FLAG-tagged PRMT3/mutantPRMT3 and GFP-tagged histone H4 (constructs described in PMID 25728001) using 293fectin Transfection Reagent (Invitrogen), following manufacturer instructions. Cells were lysed in lysis buffer (in mM: 20 Tris-HCl pH = 8, 150 NaCl, 1 EDTA, 10 MgCl2, 0.5% Triton-X100, 12.5 U/mL benzonase (Sigma), complete EDTA-free protease inhibitor cocktail (Roche)). After 3 min incubation at rt, SDS was added to a final 1% concentration. Lysates were separated on SDS PAGE, blotted, and probed with indicated antibodies: mouse anti-GFP (1:5000, Clontech #632381), mouse anti-H4 (1:1000, Abcam #174628), rabbit anti-H4R3me2a (1:1000 Active Motif #39705), and mouse anti-FLAG (1:5000, Sigma #F1804). The signal was read on an Odyssey scanner (LiCor) at 800 and 700 nm. Fluorescence intensity of H4R3me2a was quantified and normalized to GFP and H4 signals for exogenous and endogenous histones, respectively.
PRMT3 In-Cell Hunter Assay
This cellular assay was performed as described previously.22
Acknowledgments
This work was supported by grants R01GM122749, R01HD088626 and R01CA218600 (to J.J.) from the U.S. National Institutes of Health. F.L. was supported by the grant 21302134 (to F.L.) from the National Natural Science Foundation of China. The SGC is a registered charity (No. 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD Grant 115766], Janssen, Merck & Co., Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, Saõ Paulo Research Foundation-FAPESP, Takeda, and the Wellcome Trust. GPCR, kinase, and ion channel off-target selectivity screening was kindly supplied by Eurofins-Cerep.
Glossary
ABBREVIATIONS
- PRMT3
protein arginine methyltransferase 3
- rpS2
ribosomal protein S2
- PABPN1
recombinant mammalian nuclear poly(A)-binding protein
- LXRα
liver X receptor α
- DAL-1
differentially expressed in adenocarcinoma of the lung, also known as 4.1B
- SAR
structure–activity relationship
- LHS
left-hand side
- RHS
right-hand side
- GPCRs
G-protein coupled receptors
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.7b01674.
Author Present Address
○ Shanghai Institute for Advanced Immunochemical Studies, Shanghai Tech University, Pudong District, Shanghai 201210, China.
Author Present Address
◆ Deptartment of Chemical & Molecular Engineering/Applied Chemistry, Hanyang University, 55 Hanyangdaehak-ro, Sangnok-gu Ansan, Gyeonggi-do 15588, South Korea.
Author Present Address
□ School of Pharmacy, Yantai University, Yantai, 264005, P.R. China.
Author Contributions
¶ These authors contributed equally to this work.
The authors declare the following competing financial interest(s): K.Z., X.L., S.X., M.D., F.H., I.Z., Y.L., P.A., E.L., and Z.Y. are/were employees of Novartis. J.L. is an employee of DiscoveRx Corporation.
Supplementary Material
References
- Bedford M. T.; Clarke S. G. Protein Arginine Methylation in Mammals: Who, What, and Why. Mol. Cell 2009, 33, 1–13. 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiercz R.; Person M. D.; Bedford M. T. Ribosomal Protein S2 is A Substrate for Mammalian PRMT3 (Protein Arginine Methyltransferase 3). Biochem. J. 2005, 386, 85–91. 10.1042/BJ20041466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiercz R.; Cheng D.; Kim D.; Bedford M. T. Ribosomal Protein rpS2 is Hypomethylated in PRMT3-Deficient Mice. J. Biol. Chem. 2007, 282, 16917–16923. 10.1074/jbc.M609778200. [DOI] [PubMed] [Google Scholar]
- Bachand F.; Silver P. A. PRMT3 is A Ribosomal Protein Methyltransferase that Affects the Cellular Levels of Ribosomal Subunits. EMBO J. 2004, 23, 2641–2650. 10.1038/sj.emboj.7600265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry-Voyer A. M.; Bilodeau S.; Bergeron D.; Dionne K. L.; Port S. A.; Rouleau C.; Boisvert F. M.; Kehlenbach R. H.; Bachand F. Human PDCD2L Is an Export Substrate of CRM1 That Associates with 40S Ribosomal Subunit Precursors. Mol. Cell. Biol. 2016, 36, 3019–3032. 10.1128/MCB.00303-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J.; Gary J. D.; Clarke S.; Herschman H. R. Prmt 3, A Type I Protein Arginine N-Methyltransferase that Differs From PRMT1 in its Oligomerization, Subcellular Localization, Substrate Specificity, and Regulation. J. Biol. Chem. 1998, 273, 16935–16945. 10.1074/jbc.273.27.16935. [DOI] [PubMed] [Google Scholar]
- Kim D. I.; Park M. J.; Lim S. K.; Park J. I.; Yoon K. C.; Han H. J.; Gustafsson J. A.; Lim J. H.; Park S. H. PRMT3 Regulates Hepatic Lipogenesis Through Direct Interaction with LXRalpha. Diabetes 2015, 64, 60–71. 10.2337/db13-1394. [DOI] [PubMed] [Google Scholar]
- Fronz K.; Otto S.; Kolbel K.; Kuhn U.; Friedrich H.; Schierhorn A.; Beck-Sickinger A. G.; Ostareck-Lederer A.; Wahle E. Promiscuous Modification of the Nuclear Poly(A)-Binding Protein by Multiple Protein-Arginine Methyltransferases Does Not Affect The Aggregation Behavior. J. Biol. Chem. 2008, 283, 20408–20420. 10.1074/jbc.M802329200. [DOI] [PubMed] [Google Scholar]
- Tavanez J. P.; Bengoechea R.; Berciano M. T.; Lafarga M.; Carmo-Fonseca M.; Enguita F. J. Hsp70 Chaperones and Type I PRMTs are Sequestered at Intranuclear Inclusions Caused by Polyalanine Expansions in PABPN1. PLoS One 2009, 4, e6418. 10.1371/journal.pone.0006418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y.; Song M.; Hakala K.; Weintraub S. T.; Shiio Y. Proteomic Dissection of the von Hippel-Lindau (VHL) Interactome. J. Proteome Res. 2011, 10, 5175–5182. 10.1021/pr200642c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V.; Miranda T. B.; Jiang W.; Frankel A.; Roemer M. E.; Robb V. A.; Gutmann D. H.; Herschman H. R.; Clarke S.; Newsham I. F. DAL-1/4.1B Tumor Suppressor Interacts with Protein Arginine N-Methyltransferase 3 (PRMT3) and Inhibits its Ability to Methylate Substrates In Vitro and In Vivo. Oncogene 2004, 23, 7761–7771. 10.1038/sj.onc.1208057. [DOI] [PubMed] [Google Scholar]
- Jiang W.; Newsham I. F. The Tumor Suppressor DAL-1/4.1B and Protein Methylation Cooperate in Inducing Apoptosis in MCF-7 Breast Cancer Cells. Mol. Cancer 2006, 5, 4. 10.1186/1476-4598-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allali-Hassani A.; Wasney G. A.; Siarheyeva A.; Hajian T.; Arrowsmith C. H.; Vedadi M. Fluorescence-Based Methods For Screening Writers And Readers Of Histone Methyl Marks. J. Biomol. Screening 2012, 17, 71–84. 10.1177/1087057111422256. [DOI] [PubMed] [Google Scholar]
- Herrmann F.; Pably P.; Eckerich C.; Bedford M. T.; Fackelmayer F. O. Human Protein Arginine Methyltransferases in Vivo-Distinct Properties of Eight Canonical Members of the PRMT Family. J. Cell Sci. 2009, 122, 667–677. 10.1242/jcs.039933. [DOI] [PubMed] [Google Scholar]
- Wagner S.; Weber S.; Kleinschmidt M. A.; Nagata K.; Bauer U. M. SET-Mediated Promoter Hypoacetylation is A Prerequisite for Coactivation of the Estrogen-Responsive pS2 Gene by PRMT1. J. Biol. Chem. 2006, 281, 27242–27250. 10.1074/jbc.M605172200. [DOI] [PubMed] [Google Scholar]
- Obianyo O.; Causey C. P.; Jones J. E.; Thompson P. R. Activity-Based Protein Profiling of Protein Arginine Methyltransferase 1. ACS Chem. Biol. 2011, 6, 1127–1135. 10.1021/cb2001473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Niroomand F.; Liu Z.; Zankl A.; Katus H. A.; Jahn L.; Tiefenbacher C. P. Expression of Nitric Oxide Related Enzymes in Coronary Heart Disease. Basic Res. Cardiol. 2006, 101, 346–353. 10.1007/s00395-006-0592-5. [DOI] [PubMed] [Google Scholar]
- Miyata S.; Mori Y.; Tohyama M. PRMT3 is Essential for Dendritic Spine Maturation in Rat Hippocampal Neurons. Brain Res. 2010, 1352, 11–20. 10.1016/j.brainres.2010.07.033. [DOI] [PubMed] [Google Scholar]
- Park M. J.; Kim D. I.; Choi J. H.; Heo Y. R.; Park S. H. New Role of Irisin in Hepatocytes: The Protective Effect of Hepatic Steatosis In Vitro. Cell. Signalling 2015, 27, 1831–1839. 10.1016/j.cellsig.2015.04.010. [DOI] [PubMed] [Google Scholar]
- Siarheyeva A.; Senisterra G.; Allali-Hassani A.; Dong A.; Dobrovetsky E.; Wasney G. A.; Chau I.; Marcellus R.; Hajian T.; Liu F.; Korboukh I.; Smil D.; Bolshan Y.; Min J.; Wu H.; Zeng H.; Loppnau P.; Poda G.; Griffin C.; Aman A.; Brown P. J.; Jin J.; Al-awar R.; Arrowsmith C. H.; Schapira M.; Vedadi M. An Allosteric Inhibitor of Protein Arginine Methyltransferase 3. Structure 2012, 20, 1425–1435. 10.1016/j.str.2012.06.001. [DOI] [PubMed] [Google Scholar]
- Liu F.; Li F.; Ma A.; Dobrovetsky E.; Dong A.; Gao C.; Korboukh I.; Liu J.; Smil D.; Brown P. J.; Frye S. V.; Arrowsmith C. H.; Schapira M.; Vedadi M.; Jin J. Exploiting An Allosteric Binding Site of PRMT3 Yields Potent and Selective Inhibitors. J. Med. Chem. 2013, 56, 2110–2124. 10.1021/jm3018332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaniskan H. Ü.; Szewczyk M. M.; Yu Z.; Eram M. S.; Yang X.; Schmidt K.; Luo X.; Dai M.; He F.; Zang I.; Lin Y.; Kennedy S.; Li F.; Dobrovetsky E.; Dong A.; Smil D.; Min S. J.; Landon M.; Lin-Jones J.; Huang X. P.; Roth B. L.; Schapira M.; Atadja P.; Barsyte-Lovejoy D.; Arrowsmith C. H.; Brown P. J.; Zhao K.; Jin J.; Vedadi M. A Potent, Selective and Cell-Active Allosteric Inhibitor of Protein Arginine Methyltransferase 3 (PRMT3). Angew. Chem., Int. Ed. 2015, 54, 5166–5170. 10.1002/anie.201412154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balz G.; Schiemann G. On Aromatic Fluoric Compounds, I.: A New Method for its Representation. Ber. Dtsch. Chem. Ges. B 1927, 60, 1186–1190. 10.1002/cber.19270600539. [DOI] [Google Scholar]
- Zhu G. D.; Gong J.; Claiborne A.; Woods K. W.; Gandhi V. B.; Thomas S.; Luo Y.; Liu X.; Shi Y.; Guan R.; Magnone S. R.; Klinghofer V.; Johnson E. F.; Bouska J.; Shoemaker A.; Oleksijew A.; Stoll V. S.; De Jong R.; Oltersdorf T.; Li Q.; Rosenberg S. H.; Giranda V. L. Isoquinoline-Pyridine-Based Protein Kinase B/Akt Antagonists: SAR and In Vivo Antitumor Activity. Bioorg. Med. Chem. Lett. 2006, 16, 3150–3155. 10.1016/j.bmcl.2006.03.041. [DOI] [PubMed] [Google Scholar]
- Barsyte-Lovejoy D.; Li F.; Oudhoff M. J.; Tatlock J. H.; Dong A.; Zeng H.; Wu H.; Freeman S. A.; Schapira M.; Senisterra G. A.; Kuznetsova E.; Marcellus R.; Allali-Hassani A.; Kennedy S.; Lambert J. P.; Couzens A. L.; Aman A.; Gingras A. C.; Al-Awar R.; Fish P. V.; Gerstenberger B. S.; Roberts L.; Benn C. L.; Grimley R. L.; Braam M. J.; Rossi F. M.; Sudol M.; Brown P. J.; Bunnage M. E.; Owen D. R.; Zaph C.; Vedadi M.; Arrowsmith C. H. (R)-PFI-2 is a Potent and Selective Inhibitor of SETD7Methyltransferase Activity in Cells. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 12853–12858. 10.1073/pnas.1407358111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eram M. S.; Kuznetsova E.; Li F.; Lima-Fernandes E.; Kennedy S.; Chau I.; Arrowsmith C. H.; Schapira M.; Vedadi M. Kinetic Characterization of Human Histone H3 Lysine 36 Methyltransferases, ASH1L and SETD2. Biochim. Biophys. Acta, Gen. Subj. 2015, 1850, 1842–1848. 10.1016/j.bbagen.2015.05.013. [DOI] [PubMed] [Google Scholar]
- Eram M. S.; Bustos S. P.; Lima-Fernandes E.; Siarheyeva A.; Senisterra G.; Hajian T.; Chau I.; Duan S.; Wu H.; Dombrovski L.; Schapira M.; Arrowsmith C. H.; Vedadi M. Trimethylation of Histone H3 Lysine 36 by Human Methyltransferase PRDM9 Protein. J. Biol. Chem. 2014, 289, 12177–12188. 10.1074/jbc.M113.523183. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.