

Abstract
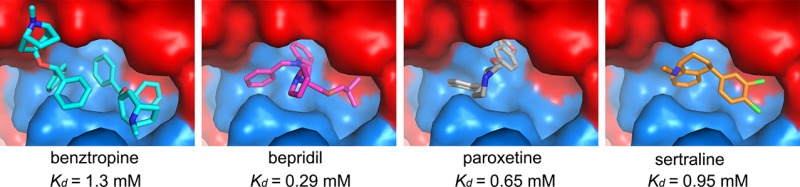
Here, we show that four chemically divergent approved drugs reported to inhibit Ebolavirus infection, benztropine, bepridil, paroxetine and sertraline, directly interact with the Ebolavirus glycoprotein. Binding of these drugs destabilizes the protein, suggesting that this may be the mechanism of inhibition, as reported for the anticancer drug toremifene and the painkiller ibuprofen, which bind in the same large cavity on the glycoprotein. Crystal structures show that the position of binding and the mode of interaction within the pocket vary significantly between these compounds. The binding constants (Kd) determined by thermal shift assay correlate with the protein–inhibitor interactions as well as with the antiviral activities determined by virus cell entry assays, supporting the hypothesis that these drugs inhibit viral entry by binding the glycoprotein and destabilizing the prefusion conformation. Details of the protein–inhibitor interactions of these complexes and their relation with binding affinity may facilitate the design of more potent inhibitors.
Introduction
Ebolavirus (EBOV) and Marburgvirus (MARV) belong to the family Filoviridae and cause severe, often fatal, diseases. There are currently no vaccines or drugs available to combat the diseases. The family Filoviridae comprises currently seven species in three genera (Cuevavirus, Ebolavirus, and Marburgvirus).1 Most of these viruses infect humans to cause severe hemorrhagic fever with a high fatality rate.2 The cost of the 2013–2016 outbreak of Ebolavirus in West Africa, to both human life and the economy, was unprecedented.3 Filoviruses are single-stranded negative-sense RNA viruses with a membrane envelope decorated by trimers of a glycoprotein (GP, cleaved by furin to form GP1 and GP2 subunits), which is solely responsible for host cell attachment, endosome entry, and membrane fusion.4−8 GP1 is responsible for binding to the receptor NPC1, and GP2 is a class I viral fusion protein catalyzing fusion of the viral and host membrane. The structures of Ebolavirus GP and its complexes with Fabs and receptor NPC1 have been previously described.9−11 In the prefusion state three copies of GP (each composed of GP1 and GP2 subunits) form the biological trimer with the receptor-binding site of GP1 being protected by a glycan cap (Figure 1). In the late endosome/lysosome the glycan cap is removed by cathepsin B/L to allow binding of the receptor, which subsequently triggers the uncoupling of GP2 from GP1 and leads to membrane fusion driven by large conformational changes, which ultimately lead to the radically different postfusion state.10,12 In the apo prefusion structure of EBOV GP, each subunit harbors a tunnel, and the three tunnels of the trimer join at the three-fold axis. The tunnel entrance is capped by a tight turn called the DFF lid.13
Figure 1.

Overall structure of EBOV GP and electron density maps. (A) Surface representation of the EBOV GP trimer; GP1 is in blue, GP2 in red, and the glycan cap in cyan. The bound bepridil at the entrance of a tunnel is shown as magenta sticks. (B) Close-up of the tunnel entrance with bound bepridil. (C) Slice of EBOV GP trimer perpendicular to the three-fold of the trimer as viewed toward the viral membrane showing the three tunnels, each from a monomer, connected at the three-fold; bepridils at the channel entrances are in magenta sticks. (D–G) Simulated annealing |Fo – Fc| omit electron density maps for benztropine (D), bepridil (E), paroxetine (F), and sertraline (G) contoured at 3.5σ; the orange density in (D) is contoured at 6σ showing that one benztropine molecule is less well ordered.
A surprisingly large number of Food and Drug Administration (FDA) approved drugs have been found to be active against Ebolavirus infection using either infectious Ebolavirus replication or pseudovirus entry assays.14−20 These drugs have various primary pharmacological targets and huge chemical diversity, and the mechanism of EBOV inhibition is largely unknown. Furthermore, given their weak inhibitory properties, these drugs may not be adequate to curtail EBOV infection. We have shown in previous studies that the anticancer drug toremifene and the painkiller ibuprofen both bind at the entrance of the tunnel of the EBOV GP pushing aside the DFF lid, and that inhibitor binding decreases the thermal stability of the GP.13 Here, we show, by crystallography, that another four FDA approved drugs that inhibit EBOV infection directly bind in the same cavity on the EBOV GP as toremifene. We also show, using thermal shift assay, that they decrease the thermal stability of the protein. Each of the drugs, due to their divergent chemical structures, exploits a different portion of the spatial volume of the cavity and makes distinct protein interactions. Structural analysis of these GP–drug complexes underlines key volumes of the binding cavity and protein–inhibitor interactions, which can help guide the design of more potent inhibitors.
Results and Discussion
Identification of Drugs That Interact Directly with EBOV GP
Eight previously reported FDA approved drugs that inhibit EBOV entry were evaluated to determine if they directly interact with EBOV GP using thermal shift assays. The experiment was carried out at pH 5.2, which is close to the physiological pH of the late endosomal where the fusion takes place and also the pH at which the EBOV GP crystals were grown. The results show that four of these, the anticholinergic/antihistamine agent benztropine,15,16,21,22 antianginal bepridil,15,16 and antidepressants paroxetine15,23 and sertraline15,16 decrease the melting temperature (Tm) of EBOV GP by up to 6, 6, 4, and 3 °C, respectively, at 500 μM concentration (Figures 2 and 3; Figure S1 in Supporting Information). Other drugs15 tested, aripiprazole, astemizole, efavirenz and quinacrine, showed either marginal or no effect on the thermal stability of EBOV GP, suggesting weak or no direct binding. The binding constants, Kds, derived from the thermal-shift assay are 1.3 mM for benztropine, 0.29 mM for bepridil, 0.65 mM for paroxetine, and 0.95 mM for sertraline. Note that this assumes a 1:1 binding model; however, as described below, two molecules of benztropine bind to each binding pocket so the calculation may underestimate the strength of attachment (for instance, in the case of ordered binding). Compared with toremifene (ΔTm = 15 °C, Kd = 16 μM), the four inhibitors identified here in general are weaker binders and show correspondingly weaker thermal shifts and Kds (Table 1). The reported IC50 is 3 μM for sertraline compared to 5 μM for bepridil in Vero cells; however, bepridil provided better protection than sertraline in a mouse model.15 As negative controls we also performed the thermal shift analysis using two further inhibitors, strophanthin and U18666A. Strophanthin inhibits EBOV infection but is not an EBOV entry inhibitor,15 while U1866A inhibits viral entry by interacting with the receptor NPC1.24 As expected neither strophanthin nor U1866A alter the melting temperature of EBOV GP (Figure S1).
Figure 2.

Chemical structures. (A) Benztropine, (3-endo)-3-(diphenylmethoxy)-8-methyl-8-azabicyclo[3.2.1]octane. (B) Bepridil, 1-[2-(N-benzylanilino)-1-(isobutoxymethyl)ethyl]pyrrolidine. (C) Paroxetine, (3S,4R)-3-[(1,3-benzodioxol-5-yloxy)methyl]-4-(4-fluorophenyl)piperidine. (D) Sertraline, (1S,4S)-4-(3,4-dichlorophenyl)-N-methyl-1,2,3,4-tetrahydro-1-naphthalenamine. (E) Toremifene, 2-[4-[(Z)-4-chloro-1,2-diphenylbut-1-enyl]phenoxy]-N,N-dimethylethanamine.
Figure 3.

Summary of thermal shift assay. Shifts in melting temperature (ΔTm (°C) in absolute value) plotted against concentrations of benztropine (A), bepridil (B), paroxetine (C), and sertraline (D) at pH 5.2. Data are mean ± SD (n = 3). The dotted line in each panel indicates ΔTm at 0.5 mM of the drug. The affinity constant Kd is calculated by a ligand binding 1:1 saturation, fitted with the SigmaPlot version 13 (Systat Software Inc.). The raw data are shown in Figure S2.
Table 1. Binding Affinity (Kd), Protein Stability Change (ΔTm), and Half-Maximum Inhibitory Concentration (IC50).
| IC50 (μM)b |
||||
|---|---|---|---|---|
| inhibitor | Kd (μM) | ΔTm (°C)a | Vero E6 | HepG2 |
| toremifene | 16 | –15 | 0.162 (0.048) | 0.026 (0.0013) |
| benztropine | 1300 | –6 | 8.07 (0) | 2.82 (0.13) |
| bepridil | 290 | –6 | 5.08 (0.38) | 3.21 (0.15) |
| paroxetine | 650 | –4 | 7.45 (0.41) | 1.38 (0.076) |
| sertraline | 950 | –3 | 3.13 (0.24) | 1.44 (0.057) |
ΔTm is measured at inhibitor concentration of 500 μM.
IC50 values and standard deviations (in parentheses) are adapted from Johansen et al.15
Overall Structures of EBOV GP–Drug Complexes
The four drugs were separately prepared in crystallization liquor, and crystals of EBOV GP were soaked in these solutions before collecting X-ray diffraction data, which allowed structure determination, revealing that all four compounds bind EBOV GP (Experimental Section). X-ray data sets, all extending to 2.4 Å resolution or better and measured with high redundancy (Table S1), were collected at the Diamond synchrotron. The overall structures of the glycoprotein component of these four complexes are very similar to each other with rmsds less than 0.6 Å for all Cα atoms of the protein. Each bound drug has good electron density allowing its conformation to be defined (Figure 1). Significant conformational differences are observed at residues 46–52 of GP1, and 521–525 and 577–583 of GP2 (Figure S2). Residues 46–52 precede the disulfide bond (C53–C609) between GP1 and GP2 and have two conformations in the GP–paroxetine complex, one corresponding to that observed in apo GP, GP–toremifene, and GP–bepridil, and the other to that seen in GP–ibuprofen, GP–benztropine, and GP–sertraline. Residues 577–583 that link α3 and α4 move about 6 Å by rotation around the three-fold axis of the GP trimer in the structure of GP–benztropine. These two sets of structural changes do not appear to be related to inhibitor binding. The third place where large conformational differences are found is at the N-terminus of the fusion loop (residues 521–525), which is flexible and makes direct interactions with some of the drugs, such as toremifene.
Two Benztropine Molecules Bind within a Single Cavity in EBOV GP
The inhibitor-binding cavity, located between GP1 and GP2, is surrounded by residues from the β1−β2 hairpin, β3, β6, and β13 of GP1, and the stem of the fusion loop (β19-β20) and α3 of GP2. Two benztropine molecules (named A and B hereafter) bind in the cavity (Figures 1D and 4A). Benztropine has three rings each connected to a carbon atom acting as a hub (Figure 2). Molecule A has well-defined electron density and binds with one phenyl ring nestling in a subpocket adjacent to α3, delimited by side-chains of residues I38 and I43 of the β1−β2 hairpin, L184 and L186 of β13, and L554 and L558 of α3, while the second phenyl ring interacts with V66 of β3 and one edge of Y517 from β19 (Figures 1 and 4; Figure S3). The center of the molecule is sandwiched by L186 and M548, with all three rings making contact with M548. The second phenyl ring and the azabicyclo ring also make close contacts to both phenyl rings of the B molecule of benztropine, which has weaker electron density (Figure 1d; Figure S3A). One phenyl ring of benztropine B makes T-shaped stacking interactions with Y517 and extensive hydrophobic contacts with the side-chain of R64 and A101 on the floor of the binding site. The second phenyl ring points to the solvent and does not interact with any atom of the protein. The azabicyclo ring of benztropine B is not clearly defined in the electron density map (Figure 1D).
Figure 4.

Inhibitor-binding site. (A–D) Details of protein-inhibitor interactions of the GP–benztropine (A), GP–bepridil (B), GP–paroxetine (C), and GP–sertraline (D) complexes. Benztropene, bepridil, paroxetine, and sertraline are shown as cyan, magenta, gray, and orange sticks, respectively. Protein main-chains are shown as ribbons and side-chains as sticks (GP1, blue; GP2, red). Side-chains in the apo GP with significant conformation differences are shown as thinner gray sticks. Residues that interact with the drug (≤3.9 Å) in each panel are labeled. L68 and L515, which also line the binding-site and interact with bepridil and paroxetine (Figure S5), are not shown for clarity.
Interactions between EBOV GP and Bepridil
Bepridil is located in the binding cavity centered between L186 and Y517. Its phenyl ring binds deep and is fully buried in a hydrophobic subpocket interacting with the side-chains of V66, A101, L515, Y517, and L558. The side-chain of V66 rotates 110° upon inhibitor binding. The benzyl ring is flanked by R64 and T519 and also interacts with Y517 and A101. The isobutoxy group occupies the subpocket adjacent to α3, interacting with L186, M548, and L558. The pyrrolidine ring is exposed, and only makes contact with the hydroxyl group of Y517 (Figure 4B). Compared to GP–benztropine, bepridil occupies the key volumes in the binding cavity filled by the two benztropine molecules, the ethanamine moiety and the isobutoxy group overlapping with the two phenyl rings of benztropine A, while the benzyl and pyrrolidine rings partially overlap with the two phenyl rings of benztropine B (Figure S5).
Paroxetine Occupies Only Part of the Key Binding Volume
Paroxetine binds with its benzodioxol group overlapping the position of the phenyl ring of bepridil and makes interactions with not only the side-chains of V66, A101, L515, Y517, and L558 but also the main-chains of G67 and G102 and side-chain of L68. The side-chain of V66 adopts the conformation seen in the bepridil bound structure, and M548 rotates 140° around the Cβ–Cγ bond to interact with the yloxymethyl group of paroxetine. The fluorophenyl ring partially overlaps with the benzyl ring of bepridil, but is closer to R64, interacting with the side-chains of R64, T519, A101, and E100. There is an extra blob of electron density in front of M548, which presumably belongs to an unidentified small molecule of either a less-ordered paroxetine or a compound in the sample (purchased from Key Organics with purity >97%) (Figure 4C; Figure S3B). This volume in front of Y517 and M548 has a propensity to be occupied (including in the previously determined GP–toremifene and GP–ibuprofen structures13), presumably stabilizing the hydrophobic residues around it. We name this the FF volume since in apo GP it is filled by F193 and F194 of the DFF lid, and its importance for inhibitor binding was suggested from the analysis of the GP–toremifene complex where the chloroethyl group occupies part of the volume in front of M548 and substitution by an ethyl group in tamoxifen or a chlorine in clomiphene greatly reduces binding affinity.13 Here, we observe that benztropine A occupies only part of the volume in front of M548 and the second benztropine is required to fill the space in front of Y517, while vice versa, paroxetine binds in part of the volume in front of Y517, with an unidentified small molecule filling the space in front of M548 (Figures S3 and S5).
Interactions between EBOV GP and Sertraline
In contrast to benztropine and paroxetine, sertraline is bound largely in the FF volume. Its dichlorophenyl ring is located at the subpocket adjacent to the α3 helix, interacting with I38, L186, M548, and L558. The position of the dichlorophenyl ring overlaps one of the phenyl rings of benztropine A and the isobutoxy group of bepridil (Figure 4D; Figures S4 and S5). The tetrahydronaphthalene group binds in space between R64 and Y517, making contacts with V66, A101, and R64, and extensive T-shaped ring stacking interactions with Y517. Sertraline is the smallest molecule among the four drugs reported here, but it has better binding affinity and antiviral activity than benztropine (Table 1), suggesting that the volume it occupies in the binding cavity and the interactions with the protein are pivotal for inhibitor binding.
None of the Drugs Fully Exploits the Potential Protein–Inhibitor Interactions
The four drugs, despite their chemical diversity, all bind within the same rather extensive cavity of Zaire EBOV GP, which in the case of benztropine accommodates two molecules. Furthermore, the amino acids lining the cavity are highly conserved among the five known species of EBOVs, and so the compounds are likely to interact with GPs of all EBOV species. However, none of the drugs fully exploit the potential protein–inhibitor interactions within the binding pocket. The observed protein–drug interactions are predominantly hydrophobic with no hydrogen bond interactions. There is no significant structural change in the cavity upon drug binding except for some variations in side-chain conformation (Figure S5). Together with the two previously published structures, we have so far determined GP complex structures with six FDA approved drugs. The drug binding cavity has a volume of approximately 1000 Å3, while the molecular volumes of the six drugs ranges from the smallest of 188 Å3 of ibuprofen to the largest of 362 Å3 of toremifene (Figure 5). The total volume of the cavity sampled by the six drugs is 712 Å3 (excluding the less ordered benztropine B), of which only 25 Å3 is common to all. Bepridil and toremifene have the largest common volume of 234 Å3, while benztropine A and paroxetine, at 42 Å3, share the least. Of the six, toremifene remains the best inhibitor in terms of binding affinity and antiviral activity. Its Kd is 18-fold better than bepridil and at least 40-fold better than the other four (Table 1). We compared the four new complexes with GP–toremifene to explore the structure–activity relationship and potential for improvement of the drugs (Figure 6; Figure S6). Toremifene binds with its alkenyl group centered in the FF volume, making extensive interactions to Y517 from its three phenyl rings. This binding mode is most similar to that of bepridil. Ring A of toremifene overlaps with the phenyl ring and ring C with the benzyl ring of bepridil, while ring B partially overlaps with the pyrrolidine ring of bepridil, but makes more protein contacts. Benztropine and sertraline do not occupy the subpocket where the A and B phenyl rings of toremifene bind. The dimethylethanamine of toremifene, to which there is no corresponding group in the other drugs, extends into the main tunnel and is surrounded by polar/charged residues, including R64, E100, T519, T520, and D522. However, there is scope of improvement of toremifene to bind more tightly: (i) substitution of the chlorine by a six-membered ring to explore the subpocket adjacent to α3 occupied by a phenyl ring of benztropine A and the isobutoxy group of bepridil; (ii) modification of the dimethylethanamine group to make hydrogen bond interactions with nearby polar/charged residues.
Figure 5.

Molecular volumes. (A) Volume of EBOV GP drug-binding cavity exploited by the six FDA approved drugs, toremifene (yellow), bepridil (magenta), paroxetine (gray), sertraline (orange), ibuprofen (blue), and benztropine (green). (B) Matrix showing the molecular volumes (gray) of the drugs and common volumes (color coded by size from red to blue) shared by any pair of the drugs. All volumes are in Å3.
Figure 6.

Comparisons with GP–toremifene complex. (A–D) Comparing protein–inhibitor interactions of GP–toremifene with GP–benztropine (A), GP–bepridil (B), GP–paroxetine (C), and GP–sertraline (D). Benztropene, bepridil, paroxetine, and sertraline are shown as cyan, magenta, gray, and orange sticks, respectively, and their associated protein main-chains are shown as ribbons and side-chains as sticks (GP1, blue; GP2, red). Toremifene is drawn as yellow sticks, and side-chains in the GP–toremifene complex with large conformational differences are shown as thinner yellow sticks.
Do These Drugs Bind MARV GP?
Toremifene, bepridil, and sertraline have been found to inhibit MARV infection in vitro,15 and benztropine was able to block pseudo-MARV entry.21 However, MARV GP has only ∼27% sequence identity to EBOV GP (Ravn virus, which causes MARV disease, vs Zaire EBOV virus). The structure of MARV GP in complex with a monoclonal antibody has been published;25 however, there are no reports of complexes with putative drugs. Superposition of the structures of MARV and EBOV GP shows that there are significant structural differences at the inhibitor-binding site (Figure 7). The EBOV DFF lid and preceding loop are replaced by a helix in MARV GP, and 15 of the 19 residues that are involved in inhibitor interactions are substituted; in particular, the replacements Ala to Glu at 101 and Val to Met at 66 make the binding cavity much shallower, and the β1-β2 hairpin of MARV GP folds inward shortening the cavity, while residues Y517 and M548, which make key interactions with inhibitors, both become isoleucine.25 Therefore, the shape of the inhibitor-binding cavity in MARV GP is substantially different to that of EBOV GP. Superimpositions of the GP–inhibitor complexes onto the structure of MARV GP show the inhibitors make serious steric clashes with MARV GP (Figure 7). If these inhibitors are bound in the same site of MARV, then either their binding modes will differ dramatically or substantial structural changes are required. This observation, together with the fact that two molecules of benztropine bind quite differently in the same pocket, perhaps, illustrate the plasticity of binding when affinity derives simply from hydrophobic interactions without the orientational drivers of charge and hydrogen bond interactions.
Figure 7.

Differences between EBOV and MARV GPs at the inhibitor binding site. Superpositions of the inhibitor binding sites of EBOV GP–benztropine (A,B), GP–bepridil (C,D) and GP–sertraline (E,F) with that of MARV GP. In (A), (C, ) and (E), EBOV GPs are shown as surface representations, inhibitors as sticks, with MARV GP main-chain as gray ribbon and side-chains as gray sticks with labels. In (B), (D), and (F), MARV GP (excluding residues 172–180) is shown as a surface representation, with the EBOV GP complexes shown as ribbons and sticks and side-chains of EBOV GP labeled.
Conclusions
Structures of two class 1 viral fusion proteins in complexes with small molecule inhibitors have been reported recently, the RSV fusion protein (RSV F) and influenza virus hemagglutinin (HA).26,27 Inhibitors of RSV F bind to a hydrophobic pocket adjacent to the fusion peptide around the three-fold symmetry axis with a stoichiometry of one inhibitor per RSV F trimer,26 while the inhibitor of flu-HA binds about 16 Å away from the fusion peptide with a stoichiometry of three inhibitors per HA trimer.27 For both of these inhibitors, binding stabilizes the fusion proteins, which contrasts with the EBOV GP inhibitors reported here that destabilize the protein by binding directly with the stem of the fusion loop with a stoichiometry of three inhibitors per trimer. Fusion proteins undergo huge structural rearrangement during fusion, supposedly triggered by low pH and receptor binding in the endo/lysosome, thus viral fusion can be inhibited by either stabilizing or destabilizing the fusion protein by direct binding of small molecule inhibitors. A large number of FDA approved drugs that inhibit EBOV entry have been reported, and various mechanisms of action have been proposed.28−32 We have now determined structures of EBOV GP in complexes with six such drugs. These drugs have five different primary pharmacological targets and contain both amphiphiles and cationic amphiphiles; nevertheless, they all bind within the same cavity of EBOV GP. All act to destabilize the GP as shown by thermal shift assay, and this correlates well with their antiviral activity, indicating that these drugs inhibit EBOV infection via the mechanism of action we proposed previously: inhibitor binding destabilizes GP and triggers premature release of GP2, thereby preventing fusion between the viral and endosome membranes.13
Experimental Section
Protein Expression and Purification
The previously described Zaire EBOV (strain Mayinga-76) recombinant glycoprotein extracellular domain construct13 with one extra mutation H613A was cloned in the mammalian expression vector pNeosec.33 The resulting plasmid pNeosec-GPΔ has the mucin domain deleted and is tagged with a foldon trimerization sequence from the bacteriophage T4 fibritin and six histidines at the C terminus. The endotoxin-free plasmid was transiently transfected into the human embryonic kidney HEK293T (ATCC CRL11268) cells with polyethylenmine (PEI, MW 25kd, Sigma, U.K.). For crystallization, the mannosidase inhibitor kifunensine (Cayman Chemical, Michigan, USA) was added to a final concentration of 5 μM. The conditioned media was collected 5 days after transfection and dialyzed against PBS. The His-tagged protein was captured with talon beads (Takara Bio Europe SAS, France) at 15 °C for 1 h with gentle shaking at 110 rpm. The beads were collected and washed in PBS with 5–10 mM imidazole. The protein was eluted with 200 mM imidazole in PBS and further purified by size exclusion chromatography with a Superdex 200 HiLoad 16/600 column (GE healthcare, Buckinghamshire, U.K.) and a buffer of 10 mM MES, pH 5.2, 150 mM NaCl.
Drugs Used in the Experiments
Toremifene (KS-5242), paroxetine hydrochloride hemihydrate (KS-1094), sertraline hydrochloride (KS-1111), aripiprazole (KS-1030), astemizole (KS-5171), efavirenz (KS-5380), and quinacrine dihydrochloride (KH-0002), all with specified purity of >97%, were purchased from Key Organics. Ibuprofen (Sigma-I4883), benztropine mesylate (Sigma-SML0847), bepridil hydrochloride (Sigma-B5016), strophanthin (Aldrich-S355445), and U18666A (Sigma-U3633), all with specified purity of ≥98%, were purchased from Sigma-Aldrich.
Thermal Shift Assay
Twenty-five microliters of solution containing 2 μM glycosylated EBOV GP protein, in a buffer of 25 mM sodium citrate at pH 5.2, 150 mM NaCl, and 6× SYPRO Orange dye (Thermo Fisher Scientific, UK), was mixed with 25 μL of compounds in 10% DMSO containing buffer. Each compound was initially dissolved in 100% DMSO and then diluted with buffer and DMSO to the desired concentration (except benztropine, which was mixed directly in buffer without DMSO). Then the samples were placed in a semiskirted 96-well PCR plate (4 Titude, Surrey, U.K.), sealed, and heated in an Mx3005p qPCR machine (Stratagene, Agilent Technologies, USA) from room temperature at a rate of 1 °C min–1 for 74 cycles. Fluorescence changes were monitored with excitation and emission wavelengths at 492 and 610 nm, respectively. Reference wells, i.e., solutions without drugs, but with same amount of DMSO, were used to compare the melting temperature (Tm). Experiments were carried out in triplicate.
Crystallization and Inhibitor Soaking
The protein was concentrated to 10–12 mg/mL. Crystallization was performed in nanoliter sitting drops (100 nL protein and 100 nL reservoir) dispensed from a Cartesian robot onto a 96-well Greiner plate.34 The crystals were grown in condition containing 9% (w/v) PEG 6000 and 0.1 M sodium citrate tribasic dihydrate at pH 5.2. We found microcrystal seeding is an reliable way to obtain reasonable sized crystals in most drops using a previously described method.35
GP and inhibitor complexes were obtained by crystal soaking experiments. The crystal soaking solutions were prepared by first dissolving the inhibitors in 100% DMSO and then diluting the dissolved inhibitors in 15% (w/v) PEG 6000 and 0.1 M sodium citrate tribasic dihydrate (pH 5.0) to a final DMSO concentration of 10% (except for benztropine, which did not require DMSO). The inhibitor concentration was typically from 1 to 10 mM depending on solubility. The crystals were soaked in the above solutions for different lengths of time, ranging from 5 to 20 min. Crystals soaked with inhibitors often resulted in weaker or no diffraction. To obtain the GP–inhibitor complexes, we usually had to soak a number of crystals in several concentrations of inhibitor for varying times.
X-ray Data Collection
The inhibitor-soaked crystals were transferred to solutions containing 75% inhibitor soaking solution and 25% (v/v) glycerol for a couple of seconds and then frozen in liquid nitrogen prior to data collection. All data were collected at 100 K with a frame size of 0.1° rotation using synchrotron X-rays and PILATUS 6 M detectors at Diamond Light Source, U.K. GP–benztropine and GP–sertraline data were acquired on beamline I02 with a beam size of 100 × 20 μm and a wavelength of 0.9795 Å. The shortest possible exposure time per frame was used.36,37 Three hundred and sixty degrees of data of GP–benztropine were collected from a single crystal with an exposure time of 0.04 s per frame and 40% beam transmission; 1410° of data were collected from five crystals with an exposure time of 0.04 s per frame and 50% beam transmission for GP–sertraline complex. GP–bepridil and GP–paroxetine data, both using 720° of data from two crystals, were collected on beamline I24 using an exposure time of 0.01 s per frame with a beam size of 50 × 50 μm2 and 30 to 50% beam transmission at wavelength of 0.9686 Å.
Data Processing, Structure Determination and Refinement
Diffraction images were indexed, integrated, and scaled with the automated data processing program Xia2–3dii or Xia2-Dials.38,39 The resolution of the diffraction data for these four complexes ranges from 2.07 to 2.40 Å with high redundancy. Each structure was initially phased with rigid-body refinement using the apo GP structure (PDB ID 5JQ3) by omitting residues 190–195 of GP1 and water molecules. The subsequent structure refinement used REFMAC540 or PHENIX,41 and models were rebuilt with COOT.42 All four models were refined to reasonable R-factors with good stereochemistry. Data collection and structure refinement statistics are given in Table S1. Structural comparisons used SHP,43 simulated annealing omit electron density maps were calculated with CNS,44 volumes of the drug-binding cavity and drug molecules were calculated with VOLUMES (Robert Esnouf, unpublished), and figures were prepared with PyMOL45 and LigPlot.46
Acknowledgments
The authors would like to thank Diamond Light Source for beamtime (proposal mx10627) and the staff of beamlines I02 and I24 for assistance with crystal testing and data collection. Y.Z. was supported by the Biostruct-X project (283570) funded by the EU seventh Framework Programme (FP7), J.R. by the Wellcome Trust, and D.I.S. by the UK Medical Research Council (MR/N00065X/1). This is a contribution from the UK Instruct Centre. The Wellcome Trust Centre for Human Genetics is supported by the Wellcome Trust (grant 090532/Z/09/Z).
Glossary
Abbreviations Used
- DMSO
dimethyl sulfoxide
- EBOV
Ebolavirus
- flu-HA
influenza virus hemagglutinin
- FDA
Food and Drug Administration
- GP
glycoprotein
- MARV
Marburgvirus
- PEI
polyethylenmine
- RSV
respiratory syncytial virus
- RSV F
respiratory syncytial virus fusion protein.
Supporting Information Available
. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.7b01249.
SMILES (CSV)
Raw data of thermal-shift assay, structural differences of the four GP–drug complexes, electron density maps for the bound benztropines and paroxetine, LigPlot diagrams of protein inhibitor interactions, comparisons of protein–drug interactions of the four complexes, comparisons of the binding mode of the current four drugs with toremifene, and X-ray data collection and structure refinement statistics (PDF)
Accession Codes
The coordinates and structure factors have been deposited with the RCSB Protein Data Bank under accession codes 6F6S, 6F5U, 6F6I, and 6F6N for the GP–benztropine, GP–bepridil, GP–paroxetine, and GP–sertraline, respectively.
Author Contributions
# These authors contributed equally to this work. J.R., Y.Z., and D.I.S. designed the project. J.R. and Y.Z. performed experiments. J.R., Y.Z., and D.I.S. analyzed the results together with E.E.F. and wrote the manuscript. All authors read and approved the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Kuhn J. H.; Becker S.; Ebihara H.; Geisbert T. W.; Johnson K. M.; Kawaoka Y.; Lipkin W. I.; Negredo A. I.; Netesov S. V.; Nichol S. T.; Palacios G.; Peters C. J.; Tenorio A.; Volchkov V. E.; Jahrling P. B. Proposal for a revised taxonomy of the family Filoviridae: classification, names of taxa and viruses, and virus abbreviations. Arch. Virol. 2010, 155, 2083–2103. 10.1007/s00705-010-0814-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony S. M.; Bradfute S. B. Filoviruses: One of these things is (not) like the other. Viruses 2015, 7, 5172–5190. 10.3390/v7102867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coltart C. E.; Lindsey B.; Ghinai I.; Johnson A. M.; Heymann D. L. The Ebola outbreak, 2013–2016: old lessons for new epidemics. Philos. Trans. R. Soc., B 2017, 372, 1–24. 10.1098/rstb.2016.0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carette J. E.; Raaben M.; Wong A. C.; Herbert A. S.; Obernosterer G.; Mulherkar N.; Kuehne A. I.; Kranzusch P. J.; Griffin A. M.; Ruthel G.; Dal Cin P.; Dye J. M.; Whelan S. P.; Chandran K.; Brummelkamp T. R. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 2011, 477, 340–343. 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacke M.; Bjorkholm P.; Hellwig A.; Himmels P.; de Almodovar C. R.; Brugger B.; Wieland F.; Ernst A. M. Inhibition of Ebola virus glycoprotein-mediated cytotoxicity by targeting its transmembrane domain and cholesterol. Nat. Commun. 2015, 6, 7688. 10.1038/ncomms8688. [DOI] [PubMed] [Google Scholar]
- Nanbo A.; Imai M.; Watanabe S.; Noda T.; Takahashi K.; Neumann G.; Halfmann P.; Kawaoka Y. Ebolavirus is internalized into host cells via macropinocytosis in a viral glycoprotein-dependent manner. PLoS Pathog. 2010, 6, e1001121. 10.1371/journal.ppat.1001121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed M. F.; Kolokoltsov A. A.; Albrecht T.; Davey R. A. Cellular entry of ebola virus involves uptake by a macropinocytosis-like mechanism and subsequent trafficking through early and late endosomes. PLoS Pathog. 2010, 6, e1001110. 10.1371/journal.ppat.1001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada A.; Robison C.; Goto H.; Sanchez A.; Murti K. G.; Whitt M. A.; Kawaoka Y. A system for functional analysis of Ebola virus glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 14764–14769. 10.1073/pnas.94.26.14764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. E.; Fusco M. L.; Hessell A. J.; Oswald W. B.; Burton D. R.; Saphire E. O. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature 2008, 454, 177–182. 10.1038/nature07082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Shi Y.; Song J.; Qi J.; Lu G.; Yan J.; Gao G. F. Ebola viral glycoprotein bound to its endosomal receptor Niemann-Pick C1. Cell 2016, 164, 258–268. 10.1016/j.cell.2015.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Ren J.; Harlos K.; Stuart D. I. Structure of glycosylated NPC1 luminal domain C reveals insights into NPC2 and Ebola virus interactions. FEBS Lett. 2016, 590, 605–612. 10.1002/1873-3468.12089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissenhorn W.; Carfi A.; Lee K. H.; Skehel J. J.; Wiley D. C. Crystal structure of the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein ectodomain. Mol. Cell 1998, 2, 605–616. 10.1016/S1097-2765(00)80159-8. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Ren J.; Harlos K.; Jones D. M.; Zeltina A.; Bowden T. A.; Padilla-Parra S.; Fry E. E.; Stuart D. I. Toremifene interacts with and destabilizes the Ebola virus glycoprotein. Nature 2016, 535, 169–172. 10.1038/nature18615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen L. M.; Brannan J. M.; Delos S. E.; Shoemaker C. J.; Stossel A.; Lear C.; Hoffstrom B. G.; Dewald L. E.; Schornberg K. L.; Scully C.; Lehar J.; Hensley L. E.; White J. M.; Olinger G. G. FDA-approved selective estrogen receptor modulators inhibit Ebola virus infection. Sci. Transl. Med. 2013, 5, 190ra79. 10.1126/scitranslmed.3005471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen L. M.; DeWald L. E.; Shoemaker C. J.; Hoffstrom B. G.; Lear-Rooney C. M.; Stossel A.; Nelson E.; Delos S. E.; Simmons J. A.; Grenier J. M.; Pierce L. T.; Pajouhesh H.; Lehar J.; Hensley L. E.; Glass P. J.; White J. M.; Olinger G. G. A screen of approved drugs and molecular probes identifies therapeutics with anti-Ebola virus activity. Sci. Transl. Med. 2015, 7, 290ra89. 10.1126/scitranslmed.aaa5597. [DOI] [PubMed] [Google Scholar]
- Kouznetsova J.; Sun W.; Martinez-Romero C.; Tawa G.; Shinn P.; Chen C. Z.; Schimmer A.; Sanderson P.; McKew J. C.; Zheng W.; Garcia-Sastre A. Identification of 53 compounds that block Ebola virus-like particle entry via a repurposing screen of approved drugs. Emerging Microbes Infect. 2014, 3, e84. 10.1038/emi.2014.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yermolina M. V.; Wang J.; Caffrey M.; Rong L. L.; Wardrop D. J. Discovery, synthesis, and biological evaluation of a novel group of selective inhibitors of filoviral entry. J. Med. Chem. 2011, 54, 765–781. 10.1021/jm1008715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards M. R.; Pietzsch C.; Vausselin T.; Shaw M. L.; Bukreyev A.; Basler C. F. High-throughput minigenome system for identifying small-molecule inhibitors of Ebola virus replication. ACS Infect. Dis. 2015, 1, 380–387. 10.1021/acsinfecdis.5b00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantpadma M.; Kouznetsova J.; Wang H.; Huang R.; Kolokoltsov A.; Guha R.; Lindstrom A. R.; Shtanko O.; Simeonov A.; Maloney D. J.; Maury W.; LaCount D. J.; Jadhav A.; Davey R. A. Large-scale screening and identification of novel Ebola virus and Marburg virus entry inhibitors. Antimicrob. Agents Chemother. 2016, 60, 4471–4481. 10.1128/AAC.00543-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu A.; Li B.; Mills D. M.; Panchal R. G.; Cardinale S. C.; Butler M. M.; Peet N. P.; Majgier-Baranowska H.; Williams J. D.; Patel I.; Moir D. T.; Bavari S.; Ray R.; Farzan M. R.; Rong L.; Bowlin T. L. Identification of a small-molecule entry inhibitor for filoviruses. J. Virol. 2011, 85, 3106–3119. 10.1128/JVI.01456-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H.; Lear-Rooney C. M.; Johansen L.; Varhegyi E.; Chen Z. W.; Olinger G. G.; Rong L. Inhibition of Ebola and Marburg virus entry by G protein-coupled receptor antagonists. J. Virol. 2015, 89, 9932–9938. 10.1128/JVI.01337-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madrid P. B.; Panchal R. G.; Warren T. K.; Shurtleff A. C.; Endsley A. N.; Green C. E.; Kolokoltsov A.; Davey R.; Manger I. D.; Gilfillan L.; Bavari S.; Tanga M. J. Evaluation of Ebola virus inhibitors for drug repurposing. ACS Infect. Dis. 2015, 1, 317–326. 10.1021/acsinfecdis.5b00030. [DOI] [PubMed] [Google Scholar]
- Madrid P. B.; Chopra S.; Manger I. D.; Gilfillan L.; Keepers T. R.; Shurtleff A. C.; Green C. E.; Iyer L. V.; Dilks H. H.; Davey R. A.; Kolokoltsov A. A.; Carrion R. Jr.; Patterson J. L.; Bavari S.; Panchal R. G.; Warren T. K.; Wells J. B.; Moos W. H.; Burke R. L.; Tanga M. J. A systematic screen of FDA-approved drugs for inhibitors of biological threat agents. PLoS One 2013, 8, e60579. 10.1371/journal.pone.0060579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F.; Liang Q.; Abi-Mosleh L.; Das A.; De Brabander J. K.; Goldstein J. L.; Brown M. S. Identification of NPC1 as the target of U18666A, an inhibitor of lysosomal cholesterol export and Ebola infection. eLife 2015, 4, 1–16. 10.7554/eLife.12177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashiguchi T.; Fusco M. L.; Bornholdt Z. A.; Lee J. E.; Flyak A. I.; Matsuoka R.; Kohda D.; Yanagi Y.; Hammel M.; Crowe J. E. Jr.; Saphire E. O. Structural basis for Marburg virus neutralization by a cross-reactive human antibody. Cell 2015, 160, 904–912. 10.1016/j.cell.2015.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battles M. B.; Langedijk J. P.; Furmanova-Hollenstein P.; Chaiwatpongsakorn S.; Costello H. M.; Kwanten L.; Vranckx L.; Vink P.; Jaensch S.; Jonckers T. H.; Koul A.; Arnoult E.; Peeples M. E.; Roymans D.; McLellan J. S. Molecular mechanism of respiratory syncytial virus fusion inhibitors. Nat. Chem. Biol. 2016, 12, 87–93. 10.1038/nchembio.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadam R. U.; Wilson I. A. Structural basis of influenza virus fusion inhibition by the antiviral drug Arbidol. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 206–214. 10.1073/pnas.1617020114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. E.; Adhikary S.; Kolokoltsov A. A.; Davey R. A. Ebolavirus requires acid sphingomyelinase activity and plasma membrane sphingomyelin for infection. J. Virol. 2012, 86, 7473–7483. 10.1128/JVI.00136-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring G.; Rohrmann K.; Atenchong N.; Mittler E.; Becker S.; Dahlmann F.; Pohlmann S.; Vondran F. W.; David S.; Manns M. P.; Ciesek S.; von Hahn T. The clinically approved drugs amiodarone, dronedarone and verapamil inhibit filovirus cell entry. J. Antimicrob. Chemother. 2014, 69, 2123–2131. 10.1093/jac/dku091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H.; Du X.; Zhang J.; Zheng H.; Lu X.; Wu Q.; Li H.; Wang H.; Shi Y.; Gao G.; Zhou Z.; Tan D. X.; Li X. Selective inhibition of Ebola entry with selective estrogen receptor modulators by disrupting the endolysosomal calcium. Sci. Rep. 2017, 7, 41226. 10.1038/srep41226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker C. J.; Schornberg K. L.; Delos S. E.; Scully C.; Pajouhesh H.; Olinger G. G.; Johansen L. M.; White J. M. Multiple cationic amphiphiles induce a Niemann-Pick C phenotype and inhibit Ebola virus entry and infection. PLoS One 2013, 8, e56265. 10.1371/journal.pone.0056265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai Y.; Kolokoltsov A. A.; Chen C. C.; Tidwell M. W.; Bauta W. E.; Klugbauer N.; Grimm C.; Wahl-Schott C.; Biel M.; Davey R. A. Ebola virus. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science 2015, 347, 995–998. 10.1126/science.1258758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Ren J.; Padilla-Parra S.; Fry E. E.; Stuart D. I. Lysosome sorting of beta-glucocerebrosidase by LIMP-2 is targeted by the mannose 6-phosphate receptor. Nat. Commun. 2014, 5, 4321. 10.1038/ncomms5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter T. S.; Diprose J. M.; Mayo C. J.; Siebold C.; Pickford M. G.; Carter L.; Sutton G. C.; Berrow N. S.; Brown J.; Berry I. M.; Stewart-Jones G. B.; Grimes J. M.; Stammers D. K.; Esnouf R. M.; Jones E. Y.; Owens R. J.; Stuart D. I.; Harlos K. A procedure for setting up high-throughput nanolitre crystallization experiments. Crystallization workflow for initial screening, automated storage, imaging and optimization. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2005, 61, 651–657. 10.1107/S0907444905007808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter T. S.; Mancini E. J.; Kadlec J.; Graham S. C.; Assenberg R.; Ren J.; Sainsbury S.; Owens R. J.; Stuart D. I.; Grimes J. M.; Harlos K. Semi-automated microseeding of nanolitre crystallization experiments. Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 2008, 64, 14–18. 10.1107/S1744309107057260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen R. L.; Axford D.; Nettleship J. E.; Owens R. J.; Robinson J. I.; Morgan A. W.; Dore A. S.; Lebon G.; Tate C. G.; Fry E. E.; Ren J.; Stuart D. I.; Evans G. Outrunning free radicals in room-temperature macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2012, 68, 810–818. 10.1107/S0907444912012553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen R. L.; Paterson N.; Axford D.; Aishima J.; Schulze-Briese C.; Ren J.; Fry E. E.; Stuart D. I.; Evans G. Exploiting fast detectors to enter a new dimension in room-temperature crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2014, 70, 1248–1256. 10.1107/S1399004714005379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G.; Lobley C. M.; Prince S. M. Decision making in xia2. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, 69, 1260–1273. 10.1107/S0907444913015308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman D. G.; Winter G.; Gildea R. J.; Parkhurst J. M.; Brewster A. S.; Sauter N. K.; Evans G. Diffraction-geometry refinement in the DIALS framework. Acta Crystallogr. D Struct. Biol. 2016, 72, 558–575. 10.1107/S2059798316002187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubak P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 355–367. 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janowski P. A.; Moriarty N. W.; Kelley B. P.; Case D. A.; York D. M.; Adams P. D.; Warren G. L. Improved ligand geometries in crystallographic refinement using AFITT in PHENIX. Acta Crystallogr. D Struct. Biol. 2016, 72, 1062–1072. 10.1107/S2059798316012225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Stuart D. I.; Levine M.; Muirhead H.; Stammers D. K. Crystal structure of cat muscle pyruvate kinase at a resolution of 2.6 A. J. Mol. Biol. 1979, 134, 109–142. 10.1016/0022-2836(79)90416-9. [DOI] [PubMed] [Google Scholar]
- Brunger A. T. Version 1.2 of the Crystallography and NMR system. Nat. Protoc. 2007, 2, 2728–2733. 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- DeLano W. L.; Lam J. W. PyMOL: A communications tool for computational models. Abstracts of Papers of the American Chemical Society 2005, 230, U1371–U1372. [Google Scholar]
- Laskowski R. A.; Swindells M. B. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.